
Structural Studies Providing Insights into Production and Conformational Behavior of Amyloid-β Peptide Associated with Alzheimer’s Disease Development

 , ,
, , 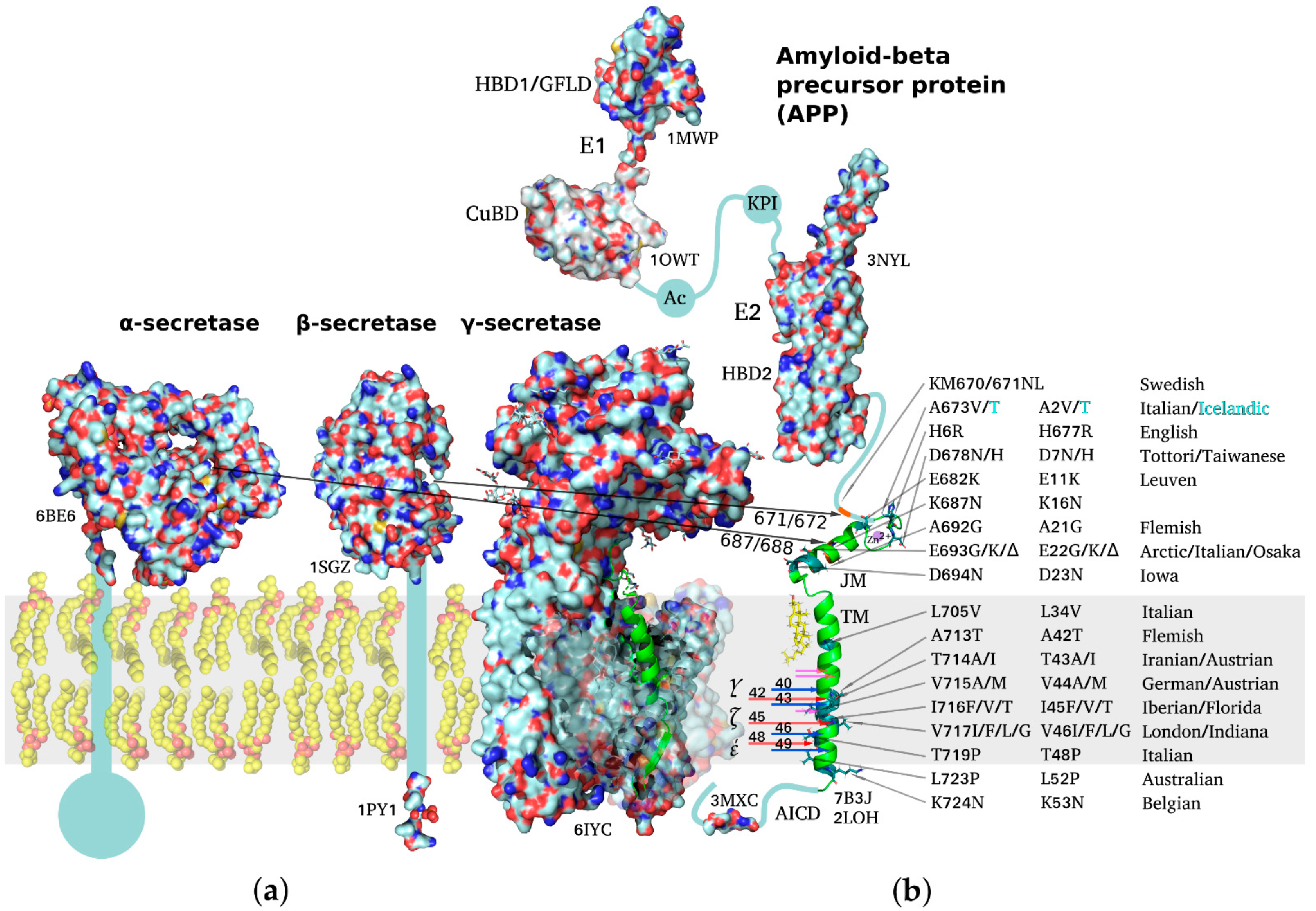{kind=link}
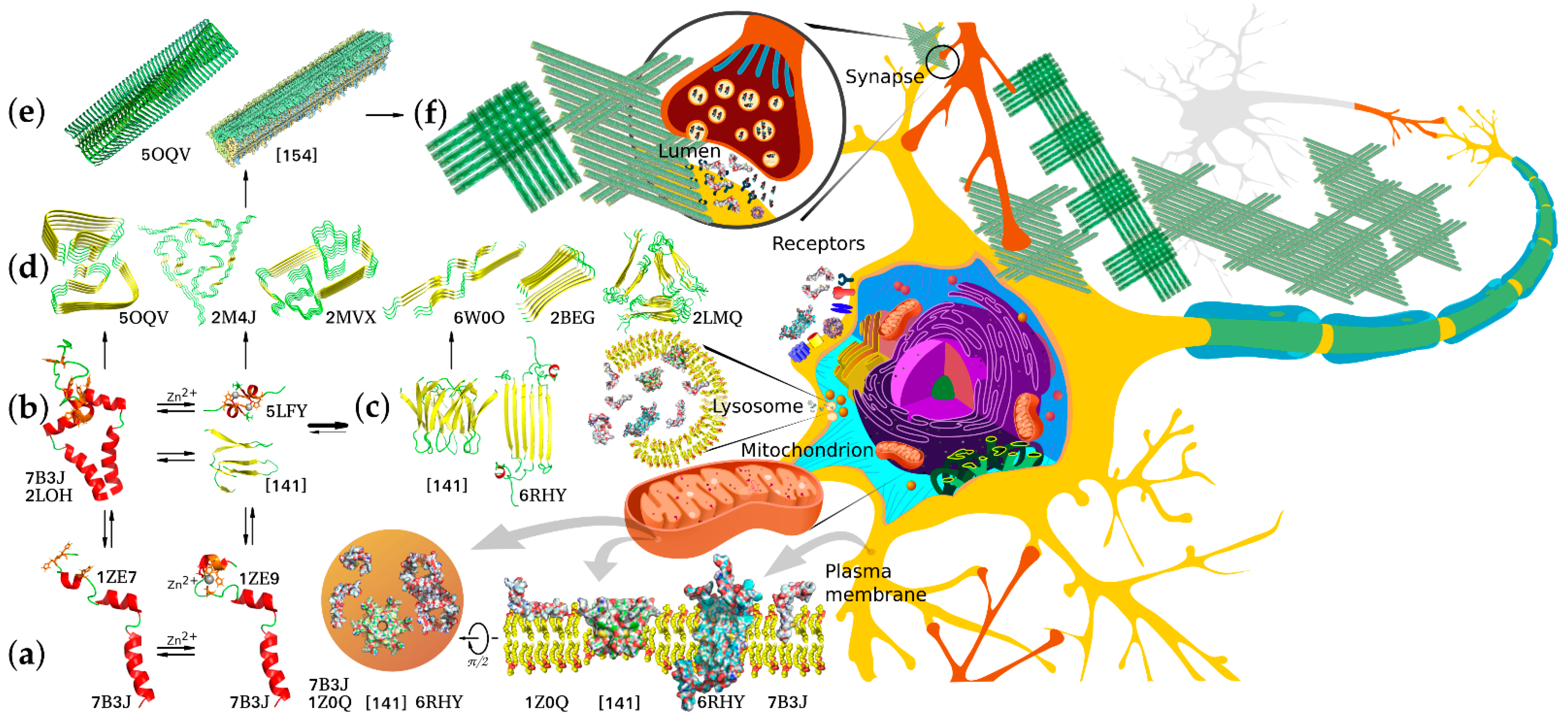{kind=link}
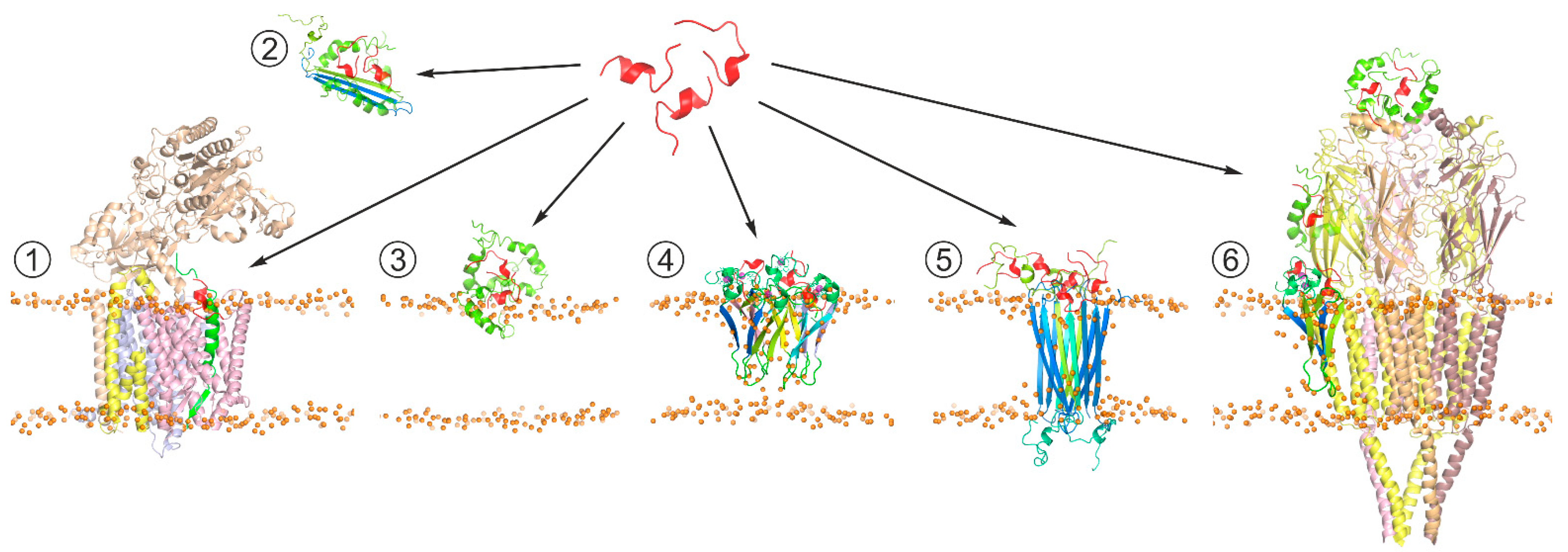{kind=link}
Abstract
:1. Introduction
2. Diverse Biological Function of the Amyloid Precursor Protein
3. Multidomain Structure of the Amyloid Precursor Protein
4. Proteolytical Processing of the Amyloid Precursor Protein
5. Aβ Peptide Functional Activities
6. Structural Properties of Aβ Monomers and Aggregates
7. Protein-Protein Interactions Targeting Pathological Traits of Aβ Forms and Aggregates
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s Association. 2016 Alzheimer’s Disease Facts and Figures. Alzheimers Dement. J. Alzheimers Assoc. 2016, 12, 459–509. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Alzheimer’s Disease: Striatal Amyloid Deposits and Neurofibrillary Changes. J. Neuropathol. Exp. Neurol. 1990, 49, 215–224. [Google Scholar] [CrossRef] [Green Version]
- Selkoe, D.J.; Hardy, J. The Amyloid Hypothesis of Alzheimer’s Disease at 25 Years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Grimm, M.O.W.; Mett, J.; Grimm, H.S.; Hartmann, T. APP Function and Lipids: A Bidirectional Link. Front. Mol. Neurosci. 2017, 10, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chew, H.; Solomon, V.A.; Fonteh, A.N. Involvement of Lipids in Alzheimer’s Disease Pathology and Potential Therapies. Front. Physiol. 2020, 11, 598. [Google Scholar] [CrossRef]
- Maltsev, A.V.; Bystryak, S.; Galzitskaya, O.V. The Role of β-Amyloid Peptide in Neurodegenerative Diseases. Ageing Res. Rev. 2011, 10, 440–452. [Google Scholar] [CrossRef]
- Cummings, J.L.; Kaufer, D. Neuropsychiatric Aspects of Alzheimer’s Disease: The Cholinergic Hypothesis Revisited. Neurology 1996, 47, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Chun, W.; Johnson, G.V.W. The Role of Tau Phosphorylation and Cleavage in Neuronal Cell Death. Front. Biosci. J. Virtual Libr. 2007, 12, 733–756. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Zhou, R.; Zhou, Q.; Guo, X.; Yan, C.; Ke, M.; Lei, J.; Shi, Y. Structural Basis of Notch Recognition by Human γ-Secretase. Nature 2019, 565, 192–197. [Google Scholar] [CrossRef]
- Hardy, J.; Allsop, D. Amyloid Deposition as the Central Event in the Aetiology of Alzheimer’s Disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Mudher, A.; Lovestone, S. Alzheimer’s Disease—Do Tauists and Baptists Finally Shake Hands? Trends Neurosci. 2002, 25, 22–26. [Google Scholar] [CrossRef]
- Chávez-Gutiérrez, L.; Szaruga, M. Mechanisms of Neurodegeneration—Insights from Familial Alzheimer’s Disease. Semin. Cell Dev. Biol. 2020, 105, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Szaruga, M.; Munteanu, B.; Lismont, S.; Veugelen, S.; Horré, K.; Mercken, M.; Saido, T.C.; Ryan, N.S.; De Vos, T.; Savvides, S.N.; et al. Alzheimer’s-Causing Mutations Shift Aβ Length by Destabilizing γ-Secretase-Aβn Interactions. Cell 2017, 170, 443–456.e14. [Google Scholar] [CrossRef] [Green Version]
- Lott, I.T.; Head, E. Alzheimer Disease and Down Syndrome: Factors in Pathogenesis. Neurobiol. Aging 2005, 26, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Selkoe, D.J. A Mechanistic Hypothesis for the Impairment of Synaptic Plasticity by Soluble Aβ Oligomers from Alzheimer’s Brain. J. Neurochem. 2020, 154, 583–597. [Google Scholar] [CrossRef]
- Giuffrida, M.L.; Caraci, F.; Pignataro, B.; Cataldo, S.; De Bona, P.; Bruno, V.; Molinaro, G.; Pappalardo, G.; Messina, A.; Palmigiano, A.; et al. Beta-Amyloid Monomers Are Neuroprotective. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 10582–10587. [Google Scholar] [CrossRef]
- Soscia, S.J.; Kirby, J.E.; Washicosky, K.J.; Tucker, S.M.; Ingelsson, M.; Hyman, B.; Burton, M.A.; Goldstein, L.E.; Duong, S.; Tanzi, R.E.; et al. The Alzheimer’s Disease-Associated Amyloid Beta-Protein Is an Antimicrobial Peptide. PLoS ONE 2010, 5, e9505. [Google Scholar] [CrossRef]
- Kumar, D.K.V.; Choi, S.H.; Washicosky, K.J.; Eimer, W.A.; Tucker, S.; Ghofrani, J.; Lefkowitz, A.; McColl, G.; Goldstein, L.E.; Tanzi, R.E.; et al. Amyloid-β Peptide Protects against Microbial Infection in Mouse and Worm Models of Alzheimer’s Disease. Sci. Transl. Med. 2016, 8, 340ra72. [Google Scholar] [CrossRef] [Green Version]
- Gosztyla, M.L.; Brothers, H.M.; Robinson, S.R. Alzheimer’s Amyloid-β Is an Antimicrobial Peptide: A Review of the Evidence. J. Alzheimers Dis. JAD 2018, 62, 1495–1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deyts, C.; Thinakaran, G.; Parent, A.T. APP Receptor? To Be or Not To Be. Trends Pharmacol. Sci. 2016, 37, 390–411. [Google Scholar] [CrossRef] [Green Version]
- Beel, A.J.; Mobley, C.K.; Kim, H.J.; Tian, F.; Hadziselimovic, A.; Jap, B.; Prestegard, J.H.; Sanders, C.R. Structural Studies of the Transmembrane C-Terminal Domain of the Amyloid Precursor Protein (APP): Does APP Function as a Cholesterol Sensor? Biochemistry 2008, 47, 9428–9446. [Google Scholar] [CrossRef] [Green Version]
- Duce, J.A.; Tsatsanis, A.; Cater, M.A.; James, S.A.; Robb, E.; Wikhe, K.; Leong, S.L.; Perez, K.; Johanssen, T.; Greenough, M.A.; et al. Iron-Export Ferroxidase Activity of β-Amyloid Precursor Protein Is Inhibited by Zinc in Alzheimer’s Disease. Cell 2010, 142, 857–867. [Google Scholar] [CrossRef] [Green Version]
- Miklós, I.; Zádori, Z. Positive Evolutionary Selection of an HD Motif on Alzheimer Precursor Protein Orthologues Suggests a Functional Role. PLoS Comput. Biol. 2012, 8, e1002356. [Google Scholar] [CrossRef] [PubMed]
- Wiese, M.; Antebi, A.; Zheng, H. Intracellular Trafficking and Synaptic Function of APL-1 in Caenorhabditis Elegans. PLoS ONE 2010, 5, e12790. [Google Scholar] [CrossRef] [Green Version]
- Poeck, B.; Strauss, R.; Kretzschmar, D. Analysis of Amyloid Precursor Protein Function in Drosophila Melanogaster. Exp. Brain Res. 2012, 217, 413–421. [Google Scholar] [CrossRef] [Green Version]
- Anliker, B.; Müller, U. The Functions of Mammalian Amyloid Precursor Protein and Related Amyloid Precursor-like Proteins. Neurodegener. Dis. 2006, 3, 239–246. [Google Scholar] [CrossRef]
- White, A.R.; Reyes, R.; Mercer, J.F.; Camakaris, J.; Zheng, H.; Bush, A.I.; Multhaup, G.; Beyreuther, K.; Masters, C.L.; Cappai, R. Copper Levels Are Increased in the Cerebral Cortex and Liver of APP and APLP2 Knockout Mice. Brain Res. 1999, 842, 439–444. [Google Scholar] [CrossRef]
- Grimm, M.O.W.; Grimm, H.S.; Pätzold, A.J.; Zinser, E.G.; Halonen, R.; Duering, M.; Tschäpe, J.A.; De Strooper, B.; Müller, U.; Shen, J.; et al. Regulation of Cholesterol and Sphingomyelin Metabolism by Amyloid-Beta and Presenilin. Nat. Cell Biol. 2005, 7, 1118–1123. [Google Scholar] [CrossRef]
- Muller, U.C.; Zheng, H. Physiological Functions of APP Family Proteins. Cold Spring Harb. Perspect. Med. 2012, 2, a006288. [Google Scholar] [CrossRef] [Green Version]
- Ring, S.; Weyer, S.W.; Kilian, S.B.; Waldron, E.; Pietrzik, C.U.; Filippov, M.A.; Herms, J.; Buchholz, C.; Eckman, C.B.; Korte, M.; et al. The Secreted -Amyloid Precursor Protein Ectodomain APPs Is Sufficient to Rescue the Anatomical, Behavioral, and Electrophysiological Abnormalities of APP-Deficient Mice. J. Neurosci. 2007, 27, 7817–7826. [Google Scholar] [CrossRef] [PubMed]
- Herms, J.; Anliker, B.; Heber, S.; Ring, S.; Fuhrmann, M.; Kretzschmar, H.; Sisodia, S.; Muller, U. Cortical Dysplasia Resembling Human Type 2 Lissencephaly in Mice Lacking All Three APP Family Members. EMBO J. 2004, 23, 4106–4115. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Cell Biology of the Amyloid Beta-Protein Precursor and the Mechanism of Alzheimer’s Disease. Annu. Rev. Cell Biol. 1994, 10, 373–403. [Google Scholar] [CrossRef]
- Gralle, M.; Ferreira, S.T. Structure and Functions of the Human Amyloid Precursor Protein: The Whole Is More than the Sum of Its Parts. Prog. Neurobiol. 2007, 82, 11–32. [Google Scholar] [CrossRef] [PubMed]
- Shih, I.-M.; Wang, T.-L. Notch Signaling, Gamma-Secretase Inhibitors, and Cancer Therapy. Cancer Res. 2007, 67, 1879–1882. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, D.M.; Acharya, M.; Leighton, P.L.A.; Wang, H.; Daude, N.; Wohlgemuth, S.; Shi, B.; Allison, W.T. Amyloid Beta Precursor Protein and Prion Protein Have a Conserved Interaction Affecting Cell Adhesion and CNS Development. PLoS ONE 2012, 7, e51305. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Brindisi, M.; Tang, J. Developing β-Secretase Inhibitors for Treatment of Alzheimer’s Disease. J. Neurochem. 2012, 120, 71–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamayev, R.; Matsuda, S.; Arancio, O.; D’Adamio, L. β- but Not γ-Secretase Proteolysis of APP Causes Synaptic and Memory Deficits in a Mouse Model of Dementia: SAPPβ/β-CTF and Not Aβ Cause Memory Deficits. EMBO Mol. Med. 2012, 4, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.M. Cholesterol and the Interaction of Proteins with Membrane Domains. Prog. Lipid Res. 2006, 45, 279–294. [Google Scholar] [CrossRef]
- Marenchino, M.; Williamson, P.T.F.; Murri, S.; Zandomeneghi, G.; Wunderli-Allenspach, H.; Meier, B.H.; Krämer, S.D. Dynamics and Cleavability at the Alpha-Cleavage Site of APP(684-726) in Different Lipid Environments. Biophys. J. 2008, 95, 1460–1473. [Google Scholar] [CrossRef] [Green Version]
- Richter, L.; Munter, L.-M.; Ness, J.; Hildebrand, P.W.; Dasari, M.; Unterreitmeier, S.; Bulic, B.; Beyermann, M.; Gust, R.; Reif, B. Amyloid Beta 42 Peptide (Aβ42)-Lowering Compounds Directly Bind to Aβ and Interfere with Amyloid Precursor Protein (APP) Transmembrane Dimerization. Proc. Natl. Acad. Sci. USA 2010, 107, 14597–14602. [Google Scholar] [CrossRef] [Green Version]
- Rossjohn, J.; Cappai, R.; Feil, S.C.; Henry, A.; McKinstry, W.J.; Galatis, D.; Hesse, L.; Multhaup, G.; Beyreuther, K.; Masters, C.L.; et al. Crystal Structure of the N-Terminal, Growth Factor-like Domain of Alzheimer Amyloid Precursor Protein. Nat. Struct. Biol. 1999, 6, 327–331. [Google Scholar] [CrossRef]
- Barnham, K.J.; McKinstry, W.J.; Multhaup, G.; Galatis, D.; Morton, C.J.; Curtain, C.C.; Williamson, N.A.; White, A.R.; Hinds, M.G.; Norton, R.S.; et al. Structure of the Alzheimer’s Disease Amyloid Precursor Protein Copper Binding Domain: A REGULATOR OF NEURONAL COPPER HOMEOSTASIS. J. Biol. Chem. 2003, 278, 17401–17407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadezhdin, K.D.; Bocharova, O.V.; Bocharov, E.V.; Arseniev, A.S. Dimeric Structure of Transmembrane Domain of Amyloid Precursor Protein in Micellar Environment. FEBS Lett. 2012, 586, 1687–1692. [Google Scholar] [CrossRef] [Green Version]
- Istrate, A.N.; Kozin, S.A.; Zhokhov, S.S.; Mantsyzov, A.B.; Kechko, O.I.; Pastore, A.; Makarov, A.A.; Polshakov, V.I. Interplay of Histidine Residues of the Alzheimer’s Disease Aβ Peptide Governs Its Zn-Induced Oligomerization. Sci. Rep. 2016, 6, 21734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, X.; Koelsch, G.; Wu, S.; Downs, D.; Dashti, A.; Tang, J. Human Aspartic Protease Memapsin 2 Cleaves the β-Secretase Site of β-Amyloid Precursor Protein. Proc. Natl. Acad. Sci. USA 2000, 97, 1456–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bittner, H.J.; Guixà-González, R.; Hildebrand, P.W. Structural Basis for the Interaction of the Beta-Secretase with Copper. Biochim. Biophys. Acta BBA Biomembr. 2018, 1860, 1105–1113. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Y.; Xu, H.; Zhang, Y. The γ-Secretase Complex: From Structure to Function. Front. Cell. Neurosci. 2014, 8. [Google Scholar] [CrossRef] [Green Version]
- Gralle, M.; Oliveira, C.L.P.; Guerreiro, L.H.; McKinstry, W.J.; Galatis, D.; Masters, C.L.; Cappai, R.; Parker, M.W.; Ramos, C.H.I.; Torriani, I.; et al. Solution Conformation and Heparin-Induced Dimerization of the Full-Length Extracellular Domain of the Human Amyloid Precursor Protein. J. Mol. Biol. 2006, 357, 493–508. [Google Scholar] [CrossRef]
- Svergun, D.I.; Petoukhov, M.V.; Koch, M.H. Determination of Domain Structure of Proteins from X-Ray Solution Scattering. Biophys. J. 2001, 80, 2946–2953. [Google Scholar] [CrossRef] [Green Version]
- Konarev, P.V.; Petoukhov, M.V.; Svergun, D.I. MASSHA—A Graphics System for Rigid-Body Modelling of Macromolecular Complexes against Solution Scattering Data. J. Appl. Crystallogr. 2001, 34, 527–532. [Google Scholar] [CrossRef]
- Scheidig, A.J.; Hynes, T.R.; Pelletier, L.A.; Wells, J.A.; Kossiakoff, A.A. Crystal Structures of Bovine Chymotrypsin and Trypsin Complexed to the Inhibitor Domain of Alzheimer’s Amyloid β-Protein Precursor (APPI) and Basic Pancreatic Trypsin Inhibitor (BPTI): Engineering of Inhibitors with Altered Specificities. Protein Sci. 1997, 6, 1806–1824. [Google Scholar] [CrossRef]
- Tsvetkov, P.O.; Kulikova, A.A.; Golovin, A.V.; Tkachev, Y.V.; Archakov, A.I.; Kozin, S.A.; Makarov, A.A. Minimal Zn(2+) Binding Site of Amyloid-β. Biophys. J. 2010, 99, L84–L86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozin, S.A.; Mezentsev, Y.V.; Kulikova, A.A.; Indeykina, M.I.; Golovin, A.V.; Ivanov, A.S.; Tsvetkov, P.O.; Makarov, A.A. Zinc-Induced Dimerization of the Amyloid-β Metal-Binding Domain 1-16 Is Mediated by Residues 11-14. Mol. Biosyst. 2011, 7, 1053–1055. [Google Scholar] [CrossRef]
- Nadezhdin, K.D.; Bocharova, O.V.; Bocharov, E.V.; Arseniev, A.S. Structural and Dynamic Study of the Transmembrane Domain of the Amyloid Precursor Protein. Acta Nat. 2011, 3, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, K.R. Folding and Stability of Alpha-Helical Integral Membrane Proteins. Chem. Rev. 2006, 106, 1931–1977. [Google Scholar] [CrossRef]
- Barrett, P.J.; Song, Y.; Van Horn, W.D.; Hustedt, E.J.; Schafer, J.M.; Hadziselimovic, A.; Beel, A.J.; Sanders, C.R. The Amyloid Precursor Protein Has a Flexible Transmembrane Domain and Binds Cholesterol. Science 2012, 336, 1168–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Götz, A.; Mylonas, N.; Högel, P.; Silber, M.; Heinel, H.; Menig, S.; Vogel, A.; Feyrer, H.; Huster, D.; Luy, B.; et al. Modulating Hinge Flexibility in the APP Transmembrane Domain Alters γ-Secretase Cleavage. Biophys. J. 2019, 116, 2103–2120. [Google Scholar] [CrossRef]
- Bocharov, E.V.; Nadezhdin, K.D.; Urban, A.S.; Volynsky, P.E.; Pavlov, K.V.; Efremov, R.G.; Arseniev, A.S.; Bocharova, O.V. Familial L723P Mutation Can Shift the Distribution between the Alternative APP Transmembrane Domain Cleavage Cascades by Local Unfolding of the Ε-Cleavage Site Suggesting a Straightforward Mechanism of Alzheimer’s Disease Pathogenesis. ACS Chem. Biol. 2019, 14, 1573–1582. [Google Scholar] [CrossRef]
- Itkin, A.; Salnikov, E.S.; Aisenbrey, C.; Raya, J.; Glattard, E.; Raussens, V.; Ruysschaert, J.-M.; Bechinger, B. Structural Characterization of the Amyloid Precursor Protein Transmembrane Domain and Its γ-Cleavage Site. ACS Omega 2017, 2, 6525–6534. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Gamache, E.; Rosenman, D.J.; Xie, J.; Lopez, M.M.; Li, Y.-M.; Wang, C. Familial Alzheimer’s Mutations within APPTM Increase Aβ42 Production by Enhancing Accessibility of ε-Cleavage Site. Nat. Commun. 2014, 5, 3037. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Tang, T.-C.; Reubins, G.; Fei, J.Z.; Fujimoto, T.; Kienlen-Campard, P.; Constantinescu, S.N.; Octave, J.-N.; Aimoto, S.; Smith, S.O. A Helix-to-Coil Transition at the Epsilon-Cut Site in the Transmembrane Dimer of the Amyloid Precursor Protein Is Required for Proteolysis. Proc. Natl. Acad. Sci. USA 2009, 106, 1421–1426. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, L.; Foster, L.; Straub, J.E.; Thirumalai, D. Impact of Membrane Lipid Composition on the Structure and Stability of the Transmembrane Domain of Amyloid Precursor Protein. Proc. Natl. Acad. Sci. USA 2016, 113, E5281–E5287. [Google Scholar] [CrossRef] [Green Version]
- Eggert, S.; Midthune, B.; Cottrell, B.; Koo, E.H. Induced Dimerization of the Amyloid Precursor Protein (APP) Leads to Decreased Amyloid-Beta Protein (Abeta) Production. J. Biol. Chem. 2009. [Google Scholar] [CrossRef] [Green Version]
- Hunter, S.; Brayne, C. Understanding the Roles of Mutations in the Amyloid Precursor Protein in Alzheimer Disease. Mol. Psychiatry 2018, 23, 81–93. [Google Scholar] [CrossRef]
- Langosch, D.; Scharnagl, C.; Steiner, H.; Lemberg, M.K. Understanding Intramembrane Proteolysis: From Protein Dynamics to Reaction Kinetics. Trends Biochem. Sci. 2015, 40, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.-C.; Hu, Y.; Kienlen-Campard, P.; El Haylani, L.; Decock, M.; Van Hees, J.; Fu, Z.; Octave, J.-N.; Constantinescu, S.N.; Smith, S.O. Conformational Changes Induced by the A21G Flemish Mutation in the Amyloid Precursor Protein Lead to Increased Aβ Production. Structure 2014, 22, 387–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polshakov, V.I.; Mantsyzov, A.B.; Kozin, S.A.; Adzhubei, A.A.; Zhokhov, S.S.; van Beek, W.; Kulikova, A.A.; Indeykina, M.I.; Mitkevich, V.A.; Makarov, A.A. A Binuclear Zinc Interaction Fold Discovered in the Homodimer of Alzheimer’s Amyloid-β Fragment with Taiwanese Mutation D7H. Angew. Chem. Int. Ed. Engl. 2017, 56, 11734–11739. [Google Scholar] [CrossRef] [PubMed]
- Götz, A.; Högel, P.; Silber, M.; Chaitoglou, I.; Luy, B.; Muhle-Goll, C.; Scharnagl, C.; Langosch, D. Increased H-Bond Stability Relates to Altered ε-Cleavage Efficiency and Aβ Levels in the I45T Familial Alzheimer’s Disease Mutant of APP. Sci. Rep. 2019, 9, 5321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramelot, T.A.; Gentile, L.N.; Nicholson, L.K. Transient Structure of the Amyloid Precursor Protein Cytoplasmic Tail Indicates Preordering of Structure for Binding to Cytosolic Factors. Biochemistry 2000, 39, 2714–2725. [Google Scholar] [CrossRef]
- Radzimanowski, J.; Simon, B.; Sattler, M.; Beyreuther, K.; Sinning, I.; Wild, K. Structure of the Intracellular Domain of the Amyloid Precursor Protein in Complex with Fe65-PTB2. EMBO Rep. 2008, 9, 1134–1140. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Koshiba, S.; Hayashi, F.; Tochio, N.; Tomizawa, T.; Kasai, T.; Yabuki, T.; Motoda, Y.; Harada, T.; Watanabe, S.; et al. Structure of the C-Terminal Phosphotyrosine Interaction Domain of Fe65L1 Complexed with the Cytoplasmic Tail of Amyloid Precursor Protein Reveals a Novel Peptide Binding Mode. J. Biol. Chem. 2008, 283, 27165–27178. [Google Scholar] [CrossRef] [Green Version]
- Shu, R.; Wong, W.; Ma, Q.H.; Yang, Z.Z.; Zhu, H.; Liu, F.J.; Wang, P.; Ma, J.; Yan, S.; Polo, J.M.; et al. APP Intracellular Domain Acts as a Transcriptional Regulator of MiR-663 Suppressing Neuronal Differentiation. Cell Death Dis. 2015, 6, e1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Copenhaver, P.F.; Kögel, D. Role of APP Interactions with Heterotrimeric G Proteins: Physiological Functions and Pathological Consequences. Front. Mol. Neurosci. 2017, 10, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, R.; Yang, G.; Shi, Y. Macromolecular Complex in Recognition and Proteolysis of Amyloid Precursor Protein in Alzheimer’s Disease. Curr. Opin. Struct. Biol. 2020, 61, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Seegar, T.C.M.; Killingsworth, L.B.; Saha, N.; Meyer, P.A.; Patra, D.; Zimmerman, B.; Janes, P.W.; Rubinstein, E.; Nikolov, D.B.; Skiniotis, G.; et al. Structural Basis for Regulated Proteolysis by the α-Secretase ADAM10. Cell 2017, 171, 1638–1648.e7. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, P.-H.; Koroniak, K.; Hogl, S.; Colombo, A.; Zeitschel, U.; Willem, M.; Volbracht, C.; Schepers, U.; Imhof, A.; Hoffmeister, A.; et al. Secretome Protein Enrichment Identifies Physiological BACE1 Protease Substrates in Neurons. EMBO J. 2012, 31, 3157–3168. [Google Scholar] [CrossRef]
- Weber, S.; Saftig, P. Ectodomain Shedding and ADAMs in Development. Development 2012, 139, 3693–3709. [Google Scholar] [CrossRef] [Green Version]
- Asai, M.; Hattori, C.; Szabó, B.; Sasagawa, N.; Maruyama, K.; Tanuma, S.; Ishiura, S. Putative Function of ADAM9, ADAM10, and ADAM17 as APP Alpha-Secretase. Biochem. Biophys. Res. Commun. 2003, 301, 231–235. [Google Scholar] [CrossRef]
- Kuhn, P.-H.; Wang, H.; Dislich, B.; Colombo, A.; Zeitschel, U.; Ellwart, J.W.; Kremmer, E.; Roßner, S.; Lichtenthaler, S.F. ADAM10 Is the Physiologically Relevant, Constitutive α-Secretase of the Amyloid Precursor Protein in Primary Neurons. EMBO J. 2010, 29, 3020–3032. [Google Scholar] [CrossRef] [Green Version]
- Vassar, R. ADAM10 Prodomain Mutations Cause Late-Onset Alzheimer’s Disease: Not Just the Latest FAD. Neuron 2013, 80, 250–253. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, R.R.; Holler, C.J.; Webb, R.L.; Li, F.; Beckett, T.L.; Murphy, M.P. BACE1 and BACE2 Enzymatic Activities in Alzheimer’s Disease. J. Neurochem. 2010, 112, 1045–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Xu, Q.; Cai, F.; Liu, X.; Wu, Y.; Song, W. BACE2, a Conditional β-Secretase, Contributes to Alzheimer’s Disease Pathogenesis. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willem, M.; Garratt, A.N.; Novak, B.; Citron, M.; Kaufmann, S.; Rittger, A.; DeStrooper, B.; Saftig, P.; Birchmeier, C.; Haass, C. Control of Peripheral Nerve Myelination by the Beta-Secretase BACE1. Science 2006, 314, 664–666. [Google Scholar] [CrossRef]
- Hu, X.; Hicks, C.W.; He, W.; Wong, P.; Macklin, W.B.; Trapp, B.D.; Yan, R. Bace1 Modulates Myelination in the Central and Peripheral Nervous System. Nat. Neurosci. 2006, 9, 1520–1525. [Google Scholar] [CrossRef]
- Hu, X.; He, W.; Luo, X.; Tsubota, K.E.; Yan, R. BACE1 Regulates Hippocampal Astrogenesis via the Jagged1-Notch Pathway. Cell Rep. 2013, 4, 40–49. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.Y.; Carey, B.W.; Wang, H.; Ingano, L.A.M.; Binshtok, A.M.; Wertz, M.H.; Pettingell, W.H.; He, P.; Lee, V.M.-Y.; Woolf, C.J.; et al. BACE1 Regulates Voltage-Gated Sodium Channels and Neuronal Activity. Nat. Cell Biol. 2007, 9, 755–764. [Google Scholar] [CrossRef]
- Sachse, C.C.; Kim, Y.H.; Agsten, M.; Huth, T.; Alzheimer, C.; Kovacs, D.M.; Kim, D.Y. BACE1 and Presenilin/γ-Secretase Regulate Proteolytic Processing of KCNE1 and 2, Auxiliary Subunits of Voltage-Gated Potassium Channels. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 2458–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venugopal, C.; Demos, C.M.; Rao, K.S.J.; Pappolla, M.A.; Sambamurti, K. Beta-Secretase: Structure, Function, and Evolution. CNS Neurol. Disord. Drug Targets 2008, 7, 278–294. [Google Scholar] [CrossRef] [Green Version]
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.; Maloney, J.; et al. A Mutation in APP Protects against Alzheimer’s Disease and Age-Related Cognitive Decline. Nature 2012, 488, 96–99. [Google Scholar] [CrossRef]
- Wolfe, M.S.; Xia, W.; Ostaszewski, B.L.; Diehl, T.S.; Kimberly, W.T.; Selkoe, D.J. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature 1999, 398, 513–517. [Google Scholar] [CrossRef]
- Xia, W. Intramembrane Proteolysis by Presenilin and Presenilin-like Proteases. J. Cell Sci. 2003, 116, 2839–2844. [Google Scholar] [CrossRef] [Green Version]
- Haapasalo, A.; Kovacs, D.M. The Many Substrates of Presenilin/γ-Secretase. J. Alzheimers Dis. JAD 2011, 25, 3–28. [Google Scholar] [CrossRef]
- Hemming, M.L.; Elias, J.E.; Gygi, S.P.; Selkoe, D.J. Identification of Beta-Secretase (BACE1) Substrates Using Quantitative Proteomics. PLoS ONE 2009, 4, e8477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.S.; Baker, G.B.; Todd, K.G.; Kar, S. Inhibition of β-Amyloid1-42 Internalization Attenuates Neuronal Death by Stabilizing the Endosomal-Lysosomal System in Rat Cortical Cultured Neurons. Neuroscience 2011, 178, 181–188. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B. Aph-1, Pen-2, and Nicastrin with Presenilin Generate an Active Gamma-Secretase Complex. Neuron 2003, 38, 9–12. [Google Scholar] [CrossRef] [Green Version]
- Shea, Y.-F.; Chu, L.-W.; Chan, A.O.-K.; Ha, J.; Li, Y.; Song, Y.-Q. A Systematic Review of Familial Alzheimer’s Disease: Differences in Presentation of Clinical Features among Three Mutated Genes and Potential Ethnic Differences. J. Formos. Med. Assoc. 2016, 115, 67–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfe, M.S. Substrate Recognition and Processing by γ-Secretase. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183016. [Google Scholar] [CrossRef]
- Wolfe, M.S. Structure, Mechanism and Inhibition of Gamma-Secretase and Presenilin-like Proteases. Biol. Chem. 2010, 391, 839–847. [Google Scholar] [CrossRef] [Green Version]
- Khan, I.; Krishnaswamy, S.; Sabale, M.; Groth, D.; Wijaya, L.; Morici, M.; Berger, I.; Schaffitzel, C.; Fraser, P.E.; Martins, R.N.; et al. Efficient Production of a Mature and Functional Gamma Secretase Protease. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Zhou, R.; Yang, G.; Guo, X.; Zhou, Q.; Lei, J.; Shi, Y. Recognition of the Amyloid Precursor Protein by Human γ-Secretase. Science 2019, 363. [Google Scholar] [CrossRef]
- Güner, G.; Lichtenthaler, S.F. The Substrate Repertoire of γ-Secretase/Presenilin. Semin. Cell Dev. Biol. 2020, 105, 27–42. [Google Scholar] [CrossRef]
- Copani, A. The Underexplored Question of β-Amyloid Monomers. Eur. J. Pharmacol. 2017, 817, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Parihar, M.S.; Brewer, G.J. Amyloid-β as a Modulator of Synaptic Plasticity. J. Alzheimers Dis. JAD 2010, 22, 741–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Megill, A.; He, K.; Kirkwood, A.; Lee, H.-K. Consequences of Inhibiting Amyloid Precursor Protein Processing Enzymes on Synaptic Function and Plasticity. Neural Plast. 2012, 2012, 272374. [Google Scholar] [CrossRef] [Green Version]
- Giuffrida, M.L.; Caraci, F.; De Bona, P.; Pappalardo, G.; Nicoletti, F.; Rizzarelli, E.; Copani, A. The Monomer State of Beta-Amyloid: Where the Alzheimer’s Disease Protein Meets Physiology. Rev. Neurosci. 2010, 21. [Google Scholar] [CrossRef] [PubMed]
- Plant, L.D.; Boyle, J.P.; Smith, I.F.; Peers, C.; Pearson, H.A. The Production of Amyloid β Peptide Is a Critical Requirement for the Viability of Central Neurons. J. Neurosci. 2003, 23, 5531–5535. [Google Scholar] [CrossRef]
- Barry, A.E.; Klyubin, I.; Mc Donald, J.M.; Mably, A.J.; Farrell, M.A.; Scott, M.; Walsh, D.M.; Rowan, M.J. Alzheimer’s Disease Brain-Derived Amyloid-β-Mediated Inhibition of LTP in Vivo Is Prevented by Immunotargeting Cellular Prion Protein. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 7259–7263. [Google Scholar] [CrossRef]
- Han, S.-H.; Kim, Y.H.; Mook-Jung, I. RAGE: The Beneficial and Deleterious Effects by Diverse Mechanisms of Actions. Mol. Cells 2011, 31, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Rhein, V.; Song, X.; Wiesner, A.; Ittner, L.M.; Baysang, G.; Meier, F.; Ozmen, L.; Bluethmann, H.; Dröse, S.; Brandt, U.; et al. Amyloid-Beta and Tau Synergistically Impair the Oxidative Phosphorylation System in Triple Transgenic Alzheimer’s Disease Mice. Proc. Natl. Acad. Sci. USA 2009, 106, 20057–20062. [Google Scholar] [CrossRef] [Green Version]
- Hane, F.; Leonenko, Z. Effect of Metals on Kinetic Pathways of Amyloid-β Aggregation. Biomolecules 2014, 4, 101–116. [Google Scholar] [CrossRef] [Green Version]
- Caughey, B.; Lansbury, P.T. PROTOFIBRILS, PORES, FIBRILS, AND NEURODEGENERATION: Separating the Responsible Protein Aggregates from The Innocent Bystanders. Annu. Rev. Neurosci. 2003, 26, 267–298. [Google Scholar] [CrossRef] [PubMed]
- Crescenzi, O.; Tomaselli, S.; Guerrini, R.; Salvadori, S.; D’Ursi, A.M.; Temussi, P.A.; Picone, D. Solution Structure of the Alzheimer Amyloid Beta-Peptide (1-42) in an Apolar Microenvironment. Similarity with a Virus Fusion Domain. Eur. J. Biochem. 2002, 269, 5642–5648. [Google Scholar] [CrossRef]
- Tomaselli, S.; Esposito, V.; Vangone, P.; van Nuland, N.A.J.; Bonvin, A.M.J.J.; Guerrini, R.; Tancredi, T.; Temussi, P.A.; Picone, D. The α-to-β Conformational Transition of Alzheimer’s Aβ-(1-42) Peptide in Aqueous Media Is Reversible: A Step by Step Conformational Analysis Suggests the Location of β Conformation Seeding. ChemBioChem 2006, 7, 257–267. [Google Scholar] [CrossRef] [Green Version]
- Economou, N.J.; Giammona, M.J.; Do, T.D.; Zheng, X.; Teplow, D.B.; Buratto, S.K.; Bowers, M.T. Amyloid β-Protein Assembly and Alzheimer’s Disease: Dodecamers of Aβ42, but Not of Aβ40, Seed Fibril Formation. J. Am. Chem. Soc. 2016, 138, 1772–1775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigemitsu, Y.; Iwaya, N.; Goda, N.; Matsuzaki, M.; Tenno, T.; Narita, A.; Hoshi, M.; Hiroaki, H. Nuclear Magnetic Resonance Evidence for the Dimer Formation of Beta Amyloid Peptide 1–42 in 1,1,1,3,3,3-Hexafluoro-2-Propanol. Anal. Biochem. 2016, 498, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Mehrazma, B.; Rauk, A. Exploring Amyloid-β Dimer Structure Using Molecular Dynamics Simulations. J. Phys. Chem. A 2019, 123, 4658–4670. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.N.; Long, H.W.; Mu, Y.; Chew, L.Y. The Toxicity of Amyloid ß Oligomers. Int. J. Mol. Sci. 2012, 13, 7303–7327. [Google Scholar] [CrossRef] [Green Version]
- Sengupta, U.; Nilson, A.N.; Kayed, R. The Role of Amyloid-β Oligomers in Toxicity, Propagation, and Immunotherapy. EBioMedicine 2016, 6, 42–49. [Google Scholar] [CrossRef] [Green Version]
- Rauk, A. Why Is the Amyloid Beta Peptide of Alzheimer’s Disease Neurotoxic? Dalton Trans. 2008, 1273–1282. [Google Scholar] [CrossRef]
- Kandel, N.; Zheng, T.; Huo, Q.; Tatulian, S.A. Membrane Binding and Pore Formation by a Cytotoxic Fragment of Amyloid β Peptide. J. Phys. Chem. B 2017, 121, 10293–10305. [Google Scholar] [CrossRef]
- Shirwany, N.A.; Payette, D.; Xie, J.; Guo, Q. The Amyloid Beta Ion Channel Hypothesis of Alzheimer’s Disease. Neuropsychiatr. Dis. Treat. 2007, 3, 597–612. [Google Scholar]
- Arispe, N.; Pollard, H.B.; Rojas, E. Giant Multilevel Cation Channels Formed by Alzheimer Disease Amyloid Beta-Protein [A Beta P-(1-40)] in Bilayer Membranes. Proc. Natl. Acad. Sci. USA 1993, 90, 10573–10577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Scala, C.; Yahi, N.; Boutemeur, S.; Flores, A.; Rodriguez, L.; Chahinian, H.; Fantini, J. Common Molecular Mechanism of Amyloid Pore Formation by Alzheimer’s β-Amyloid Peptide and α-Synuclein. Sci. Rep. 2016, 6, 28781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venko, K.; Novič, M.; Stoka, V.; Žerovnik, E. Prediction of Transmembrane Regions, Cholesterol, and Ganglioside Binding Sites in Amyloid-Forming Proteins Indicate Potential for Amyloid Pore Formation. Front. Mol. Neurosci. 2021, 14, 619496. [Google Scholar] [CrossRef]
- Wärmländer, S.K.T.S.; Österlund, N.; Wallin, C.; Wu, J.; Luo, J.; Tiiman, A.; Jarvet, J.; Gräslund, A. Metal-binding to the Amyloid-β Peptides in the Presence of Biomembranes: Potential Mechanisms of Cell Toxicity. J. Biol. Inorg. Chem. JBIC Publ. Soc. Biol. Inorg. Chem. 2019, 24, 1189–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, Y.; Takiguchi, M.; Tamano, H.; Takeda, A. Extracellular Zn2+-Dependent Amyloid-Β1-42 Neurotoxicity in Alzheimer’s Disease Pathogenesis. Biol. Trace Elem. Res. 2021, 199, 53–61. [Google Scholar] [CrossRef]
- Bode, D.C.; Baker, M.D.; Viles, J.H. Ion Channel Formation by Amyloid-Β42 Oligomers but Not Amyloid-Β40 in Cellular Membranes. J. Biol. Chem. 2017, 292, 1404–1413. [Google Scholar] [CrossRef] [Green Version]
- Serra-Batiste, M.; Ninot-Pedrosa, M.; Puig, E.; Ciudad, S.; Gairí, M.; Carulla, N. Preparation of a Well-Defined and Stable β-Barrel Pore-Forming Aβ42 Oligomer. In Amyloid Proteins; Sigurdsson, E.M., Calero, M., Gasset, M., Eds.; Springer: New York, NY, USA, 2018; Volume 1779, pp. 13–22. ISBN 978-1-4939-7815-1. [Google Scholar]
- Di Scala, C.; Troadec, J.-D.; Lelièvre, C.; Garmy, N.; Fantini, J.; Chahinian, H. Mechanism of Cholesterol-Assisted Oligomeric Channel Formation by a Short Alzheimer β-Amyloid Peptide. J. Neurochem. 2014, 128, 186–195. [Google Scholar] [CrossRef]
- Bocharov, E.V.; Mineev, K.S.; Pavlov, K.V.; Akimov, S.A.; Kuznetsov, A.S.; Efremov, R.G.; Arseniev, A.S. Helix-Helix Interactions in Membrane Domains of Bitopic Proteins: Specificity and Role of Lipid Environment. Biochim. Biophys. Acta BBA Biomembr. 2017, 1859, 561–576. [Google Scholar] [CrossRef]
- Bocharov, E.V.; Sharonov, G.V.; Bocharova, O.V.; Pavlov, K.V. Conformational Transitions and Interactions Underlying the Function of Membrane Embedded Receptor Protein Kinases. Biochim. Biophys. Acta BBA Biomembr. 2017, 1859, 1417–1429. [Google Scholar] [CrossRef]
- Pavlov, K.V.; Akimov, S.A.; Batishchev, O.V.; Chekashkina, K.V.; Bashkirov, P.V.; Bocharov, E.V. Protein-Lipid Interplay in Vital Biological Functions. In Protein-Lipid Interactions: Perspectives, Techniques and Challenges; Catalá, A., Ed.; Nova Science Publishers: Harpark, NY, USA, 2018; pp. 133–199. ISBN 978-1-5361-3125-3. [Google Scholar]
- Limbocker, R.; Chia, S.; Ruggeri, F.S.; Perni, M.; Cascella, R.; Heller, G.T.; Meisl, G.; Mannini, B.; Habchi, J.; Michaels, T.C.T.; et al. Trodusquemine Enhances Aβ 42 Aggregation but Suppresses Its Toxicity by Displacing Oligomers from Cell Membranes. Nat. Commun. 2019, 10, 225. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.G.; Cappai, R.; Barnham, K.J. The Redox Chemistry of the Alzheimer’s Disease Amyloid Beta Peptide. Biochim. Biophys. Acta 2007, 1768, 1976–1990. [Google Scholar] [CrossRef] [Green Version]
- Džinić, T.; Dencher, N.A. Oxygen Concentration and Oxidative Stress Modulate the Influence of Alzheimer’s Disease Aβ1-42 Peptide on Human Cells. Oxid. Med. Cell. Longev. 2018, 2018, 7567959. [Google Scholar] [CrossRef]
- Shafrir, Y.; Durell, S.; Arispe, N.; Guy, H.R. Models of Membrane-Bound Alzheimer’s Abeta Peptide Assemblies. Proteins 2010, 78, 3473–3487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Yin, T.; Peng, Q.; Kong, L.; Li, C.; Tang, D.; Yin, X. Simultaneous Monitoring of Amyloid-β (Aβ) Oligomers and Fibrils for Effectively Evaluating the Dynamic Process of Aβ Aggregation. ACS Sens. 2019, 4, 471–478. [Google Scholar] [CrossRef]
- Shea, D.; Hsu, C.-C.; Bi, T.M.; Paranjapye, N.; Childers, M.C.; Cochran, J.; Tomberlin, C.P.; Wang, L.; Paris, D.; Zonderman, J.; et al. α-Sheet Secondary Structure in Amyloid β-Peptide Drives Aggregation and Toxicity in Alzheimer’s Disease. Proc. Natl. Acad. Sci. USA 2019, 116, 8895–8900. [Google Scholar] [CrossRef] [Green Version]
- Ono, K.; Yamada, M. Low-n Oligomers as Therapeutic Targets of Alzheimer’s Disease. J. Neurochem. 2011, 117, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Kreutzer, A.G.; Hamza, I.L.; Spencer, R.K.; Nowick, J.S. X-ray Crystallographic Structures of a Trimer, Dodecamer, and Annular Pore Formed by an Aβ17-36 β-Hairpin. J. Am. Chem. Soc. 2016, 138, 4634–4642. [Google Scholar] [CrossRef] [PubMed]
- Lendel, C.; Bjerring, M.; Dubnovitsky, A.; Kelly, R.T.; Filippov, A.; Antzutkin, O.N.; Nielsen, N.C.; Härd, T. A Hexameric Peptide Barrel as Building Block of Amyloid-β Protofibrils. Angew. Chem. Int. Ed. Engl. 2014, 53, 12756–12760. [Google Scholar] [CrossRef] [PubMed]
- Norlin, N.; Hellberg, M.; Filippov, A.; Sousa, A.A.; Gröbner, G.; Leapman, R.D.; Almqvist, N.; Antzutkin, O.N. Aggregation and Fibril Morphology of the Arctic Mutation of Alzheimer’s Aβ Peptide by CD, TEM, STEM and in Situ AFM. J. Struct. Biol. 2012, 180, 174–189. [Google Scholar] [CrossRef] [Green Version]
- Ciudad, S.; Puig, E.; Botzanowski, T.; Meigooni, M.; Arango, A.S.; Do, J.; Mayzel, M.; Bayoumi, M.; Chaignepain, S.; Maglia, G.; et al. Aβ(1-42) Tetramer and Octamer Structures Reveal Edge Conductivity Pores as a Mechanism for Membrane Damage. Nat. Commun. 2020, 11, 3014. [Google Scholar] [CrossRef]
- Nguyen, H.L.; Krupa, P.; Hai, N.M.; Linh, H.Q.; Li, M.S. Structure and Physicochemical Properties of the Aβ42 Tetramer: Multiscale Molecular Dynamics Simulations. J. Phys. Chem. B 2019, 123, 7253–7269. [Google Scholar] [CrossRef]
- Snyder, S.W.; Ladror, U.S.; Wade, W.S.; Wang, G.T.; Barrett, L.W.; Matayoshi, E.D.; Huffaker, H.J.; Krafft, G.A.; Holzman, T.F. Amyloid-beta aggregation: Selective inhibition of aggregation in mixtures of amyloid with different chain lengths. Biophys. J. 1994, 67, 1216–1228. [Google Scholar] [CrossRef] [Green Version]
- Jarrett, J.T.; Berger, E.P.; Lansbury, P.T., Jr. The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: Implications for the pathogenesis of Alzheimer’s disease. Biochemistry 1993, 32, 4693–4697. [Google Scholar] [CrossRef] [PubMed]
- Poma, A.B.; Chwastyk, M.; Cieplak, M. Elastic moduli of biological fibers in a coarse-grained model: Crystalline cellulose and β-amyloids. Phys. Chem. Chem. Phys. 2017, 19, 28195–28206. [Google Scholar] [CrossRef] [PubMed]
- Poma, A.B.; Guzman, H.V.; Li, M.S.; Theodorakis, P.E. Mechanical and thermodynamic properties of Aβ42, Aβ40, and α-synuclein fibrils: A coarse-grained method to complement experimental studies. Beilstein J. Nanotechnol. 2019, 10, 500–513. [Google Scholar] [CrossRef] [Green Version]
- Ruggeri, F.S.; Adamcik, J.; Jeong, J.S.; Lashuel, H.A.; Mezzenga, R.; Dietler, G. Influence of the β-sheet content on the mechanical properties of aggregates during amyloid fibrillization. Angew. Chem. Int. Ed. Engl. 2015, 54, 2462–2466. [Google Scholar] [CrossRef] [PubMed]
- Lührs, T.; Ritter, C.; Adrian, M.; Riek-Loher, D.; Bohrmann, B.; Döbeli, H.; Schubert, D.; Riek, R. 3D Structure of Alzheimer’s Amyloid-β (1–42) Fibrils. Proc. Natl. Acad. Sci. USA 2005, 102, 17342–17347. [Google Scholar] [CrossRef] [Green Version]
- Paravastu, A.K.; Leapman, R.D.; Yau, W.-M.; Tycko, R. Molecular Structural Basis for Polymorphism in Alzheimer’s β-Amyloid Fibrils. Proc. Natl. Acad. Sci. USA 2008, 105, 18349–18354. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.-X.; Qiang, W.; Yau, W.-M.; Schwieters, C.D.; Meredith, S.C.; Tycko, R. Molecular Structure of β-Amyloid Fibrils in Alzheimer’s Disease Brain Tissue. Cell 2013, 154, 1257–1268. [Google Scholar] [CrossRef] [Green Version]
- Kodali, R.; Williams, A.D.; Chemuru, S.; Wetzel, R. Aβ(1–40) Forms Five Distinct Amyloid Structures Whose β-Sheet Contents and Fibril Stabilities Are Correlated. J. Mol. Biol. 2010, 401, 503–517. [Google Scholar] [CrossRef] [Green Version]
- Goodsell, D.S.; Dutta, S.; Zardecki, C.; Voigt, M.; Berman, H.M.; Burley, S.K. The RCSB PDB “Molecule of the Month”: Inspiring a Molecular View of Biology. PLoS Biol. 2015, 13, e1002140. [Google Scholar] [CrossRef]
- Gremer, L.; Schölzel, D.; Schenk, C.; Reinartz, E.; Labahn, J.; Ravelli, R.B.G.; Tusche, M.; Lopez-Iglesias, C.; Hoyer, W.; Heise, H.; et al. Fibril Structure of Amyloid-β(1–42) by Cryo–Electron Microscopy. Science 2017, 358, 116–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schenk, D.; Basi, G.S.; Pangalos, M.N. Treatment Strategies Targeting Amyloid β-Protein. Cold Springer Harb. Perspect. Med. 2012, 2, a006387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Fu, A.K.Y.; Ip, N.Y. Synaptic Dysfunction in Alzheimer’s Disease: Mechanisms and Therapeutic Strategies. Pharmacol. Ther. 2019, 195, 186–198. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s Disease Drug Development Pipeline: 2020. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2020, 6, e12050. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Chahinian, H.; Yahi, N. Progress toward Alzheimer’s Disease Treatment: Leveraging the Achilles’ Heel of Aβ Oligomers? Protein Sci. Publ. Protein Soc. 2020, 29, 1748–1759. [Google Scholar] [CrossRef]
- Heller, G.T.; Aprile, F.A.; Michaels, T.C.T.; Limbocker, R.; Perni, M.; Ruggeri, F.S.; Mannini, B.; Löhr, T.; Bonomi, M.; Camilloni, C.; et al. Small-Molecule Sequestration of Amyloid-β as a Drug Discovery Strategy for Alzheimer’s Disease. Sci. Adv. 2020, 6. [Google Scholar] [CrossRef]
- Di, L. Strategic Approaches to Optimizing Peptide ADME Properties. AAPS J. 2015, 17, 134–143. [Google Scholar] [CrossRef]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The Future of Peptide-Based Drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef]
- Funke, S.A.; Willbold, D. Peptides for Therapy and Diagnosis of Alzheimer’s Disease. Curr. Pharm. Des. 2012, 18, 755–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhang, X.; Ladiwala, A.R.A.; Du, D.; Yadav, J.K.; Tessier, P.M.; Wright, P.E.; Kelly, J.W.; Buxbaum, J.N. Mechanisms of Transthyretin Inhibition of β-Amyloid Aggregation in Vitro. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 19423–19433. [Google Scholar] [CrossRef]
- Stein, T.D.; Johnson, J.A. Lack of Neurodegeneration in Transgenic Mice Overexpressing Mutant Amyloid Precursor Protein Is Associated with Increased Levels of Transthyretin and the Activation of Cell Survival Pathways. J. Neurosci. Off. J. Soc. Neurosci. 2002, 22, 7380–7388. [Google Scholar] [CrossRef] [Green Version]
- Volpina, O.M.; Samokhin, A.N.; Koroev, D.O.; Nesterova, I.V.; Volkova, T.D.; Medvinskaya, N.I.; Nekrasov, P.V.; Tatarnikova, O.G.; Kamynina, A.V.; Balasanyants, S.M.; et al. Synthetic Fragment of Receptor for Advanced Glycation End Products Prevents Memory Loss and Protects Brain Neurons in Olfactory Bulbectomized Mice. J. Alzheimers Dis. JAD 2018, 61, 1061–1076. [Google Scholar] [CrossRef]
- Kamynina, A.; Esteras, N.; Koroev, D.O.; Angelova, P.R.; Volpina, O.M.; Abramov, A.Y. Activation of RAGE Leads to the Release of Glutamate from Astrocytes and Stimulates Calcium Signal in Neurons. J. Cell. Physiol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Barykin, E.P.; Garifulina, A.I.; Tolstova, A.P.; Anashkina, A.A.; Adzhubei, A.A.; Mezentsev, Y.V.; Shelukhina, I.V.; Kozin, S.A.; Tsetlin, V.I.; Makarov, A.A. Tetrapeptide Ac-HAEE-NH2 Protects A4β2 NAChR from Inhibition by Aβ. Int. J. Mol. Sci. 2020, 21, 6272. [Google Scholar] [CrossRef]
- Funke, S.A.; Willbold, D. Mirror Image Phage Display—A Method to Generate D-Peptide Ligands for Use in Diagnostic or Therapeutical Applications. Mol. Biosyst. 2009, 5, 783–786. [Google Scholar] [CrossRef]
- Wiesehan, K.; Buder, K.; Linke, R.P.; Patt, S.; Stoldt, M.; Unger, E.; Schmitt, B.; Bucci, E.; Willbold, D. Selection of D-Amino-Acid Peptides That Bind to Alzheimer’s Disease Amyloid Peptide Abeta1-42 by Mirror Image Phage Display. Chembiochem. Eur. J. Chem. Biol. 2003, 4, 748–753. [Google Scholar] [CrossRef]
- Ziehm, T.; Brener, O.; van Groen, T.; Kadish, I.; Frenzel, D.; Tusche, M.; Kutzsche, J.; Reiß, K.; Gremer, L.; Nagel-Steger, L.; et al. Increase of Positive Net Charge and Conformational Rigidity Enhances the Efficacy of D-Enantiomeric Peptides Designed to Eliminate Cytotoxic Aβ Species. ACS Chem. Neurosci. 2016, 7, 1088–1096. [Google Scholar] [CrossRef]
- Willbold, D.; Kutzsche, J. Do We Need Anti-Prion Compounds to Treat Alzheimer’s Disease? Molecules 2019, 24, 2237. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N. The multifaceted roles of intrinsic disorder in protein complexes. FEBS Lett. 2015, 589, 2498–2506. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urban, A.S.; Pavlov, K.V.; Kamynina, A.V.; Okhrimenko, I.S.; Arseniev, A.S.; Bocharov, E.V. Structural Studies Providing Insights into Production and Conformational Behavior of Amyloid-β Peptide Associated with Alzheimer’s Disease Development. Molecules 2021, 26, 2897. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26102897
Urban AS, Pavlov KV, Kamynina AV, Okhrimenko IS, Arseniev AS, Bocharov EV. Structural Studies Providing Insights into Production and Conformational Behavior of Amyloid-β Peptide Associated with Alzheimer’s Disease Development. Molecules. 2021; 26(10):2897. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26102897
Chicago/Turabian StyleUrban, Anatoly S., Konstantin V. Pavlov, Anna V. Kamynina, Ivan S. Okhrimenko, Alexander S. Arseniev, and Eduard V. Bocharov. 2021. "Structural Studies Providing Insights into Production and Conformational Behavior of Amyloid-β Peptide Associated with Alzheimer’s Disease Development" Molecules 26, no. 10: 2897. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26102897