
New Cardenolides from Biotransformation of Gitoxigenin by the Endophytic Fungus Alternaria eureka 1E1BL1: Characterization and Cytotoxic Activities

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results and Discussion
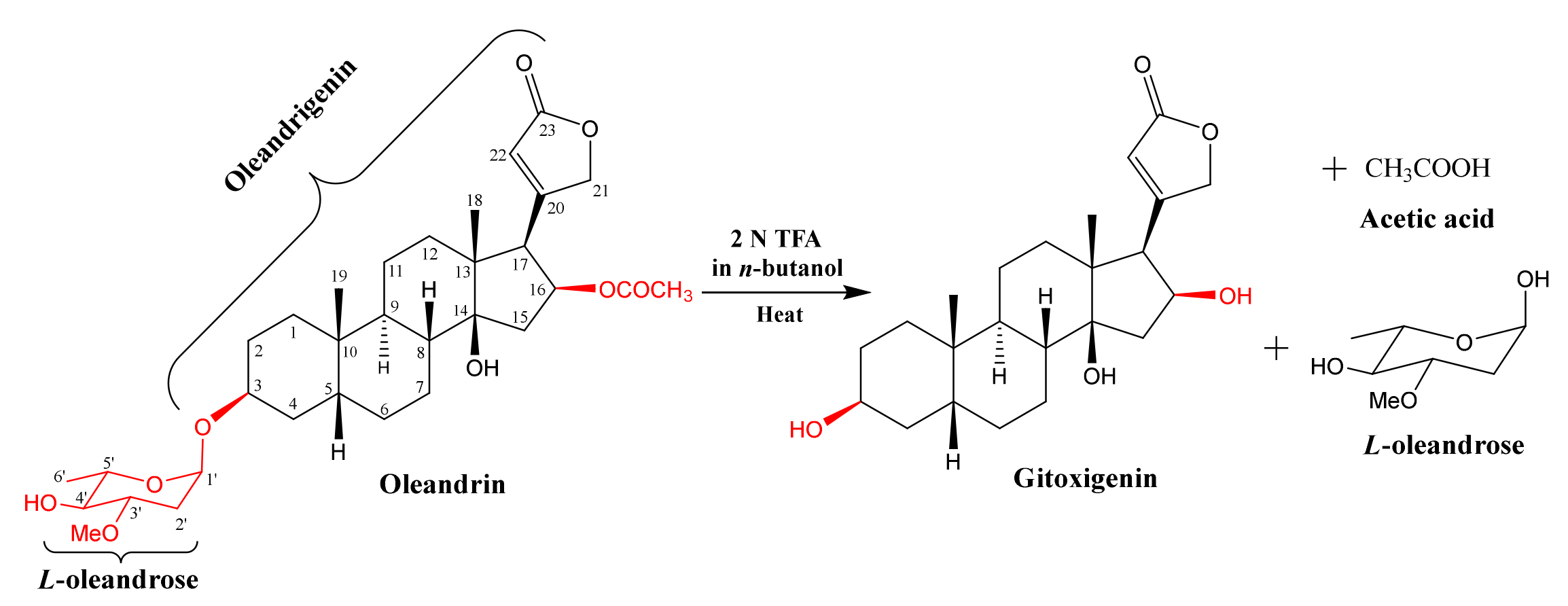
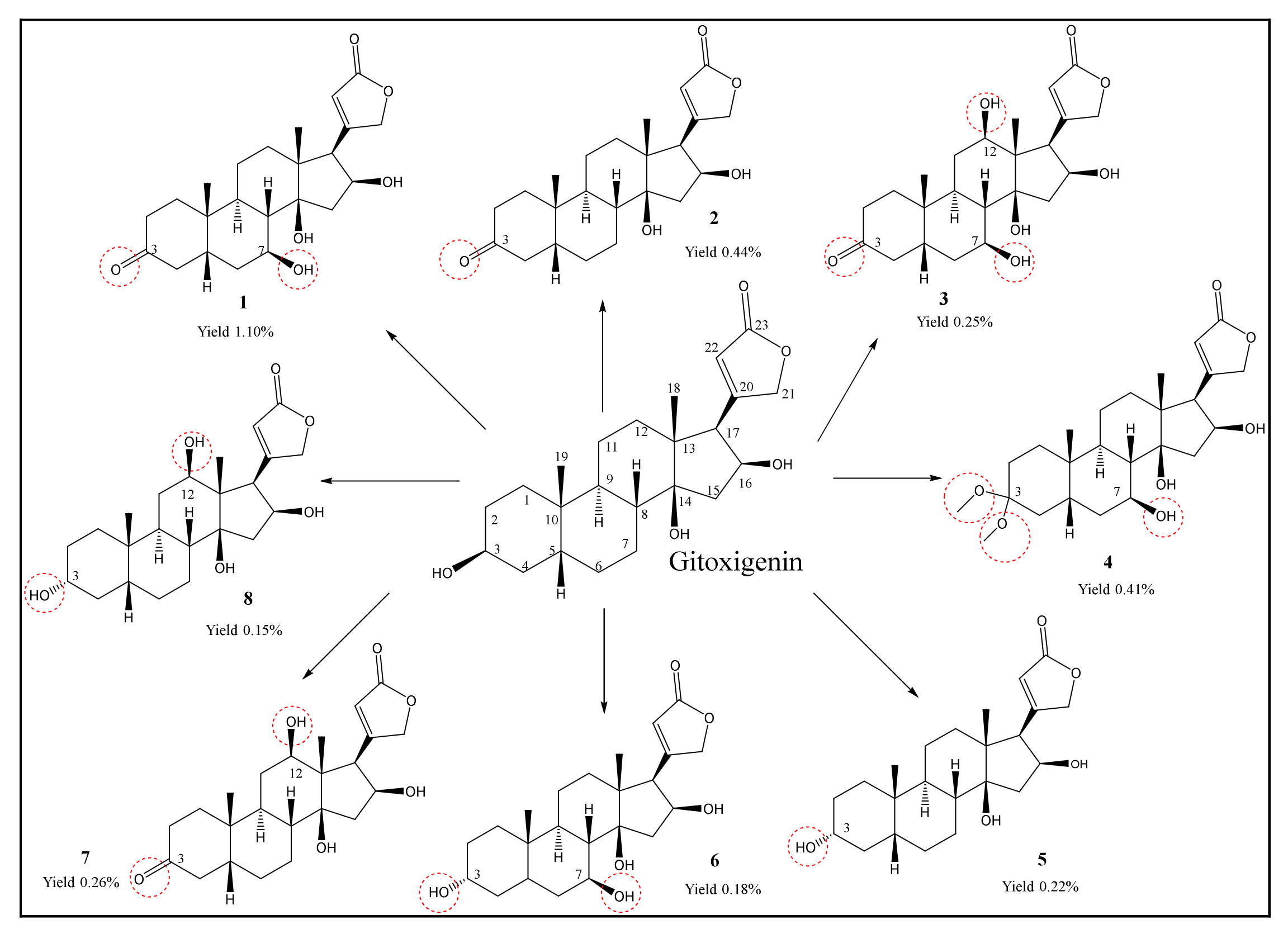
2.1. Isolation and Purification of Oleandrin from N. oleander L. and Preparation of Substrate Gitoxigenin and Obtaining Biotransformation Products
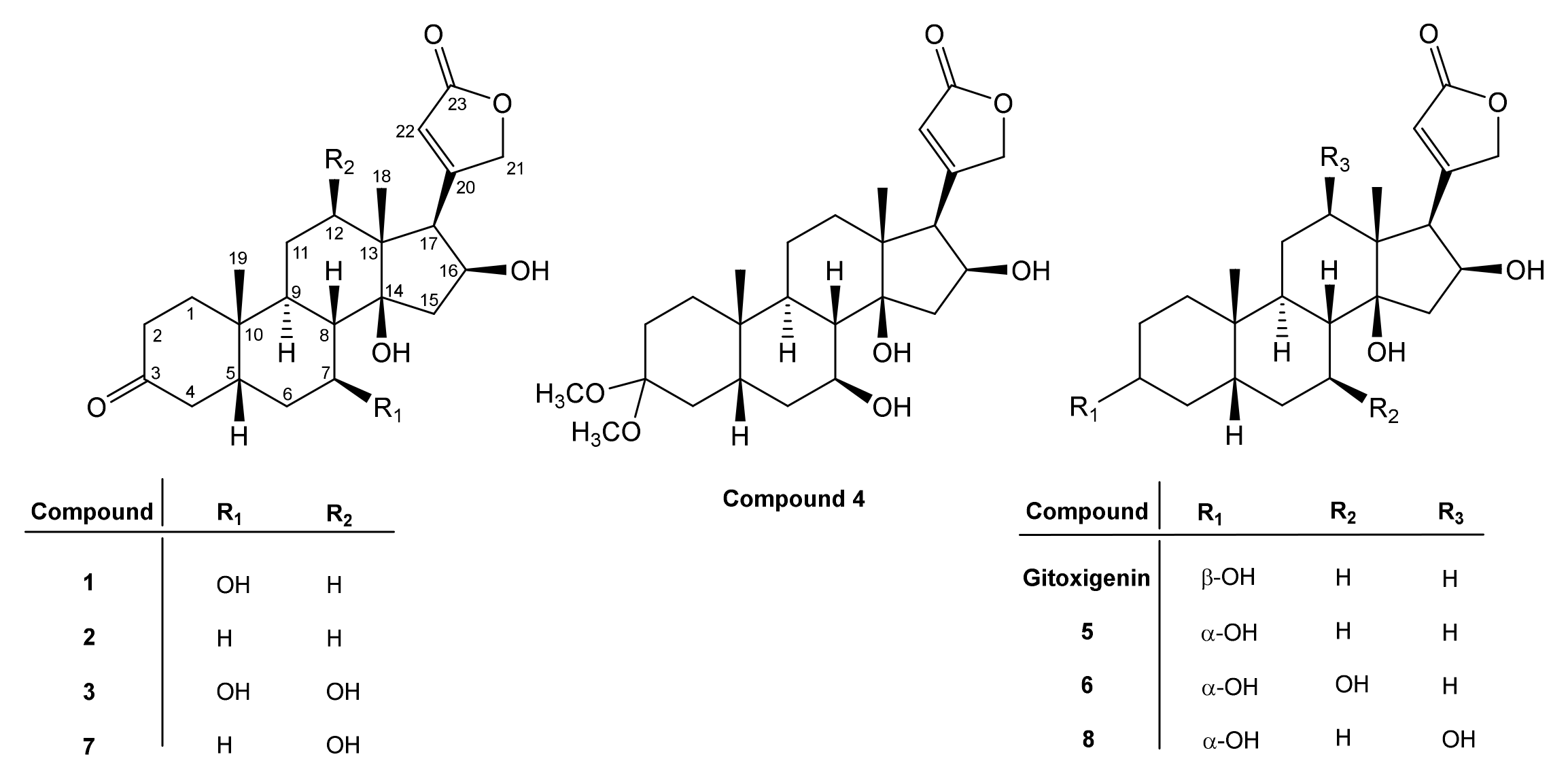
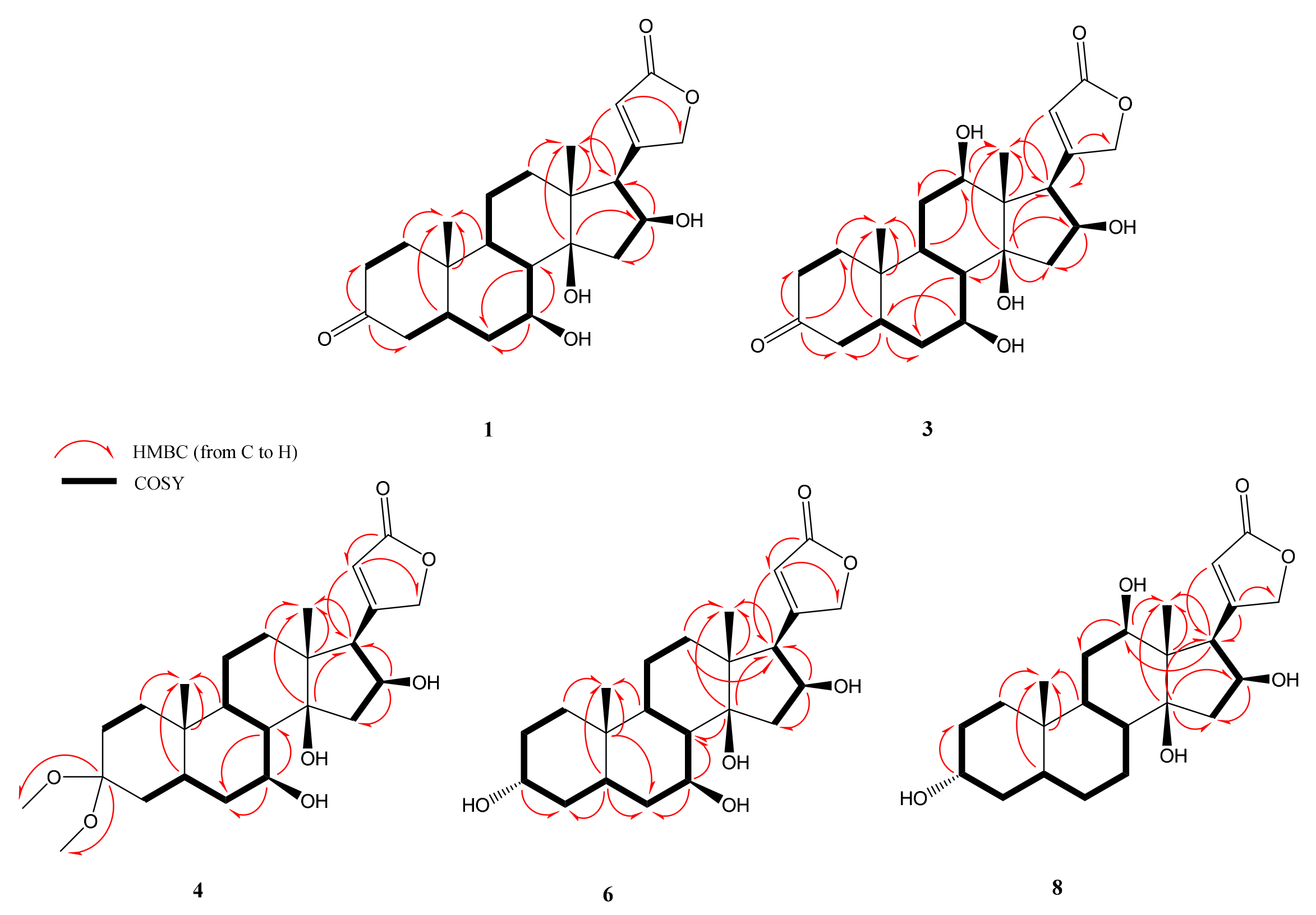
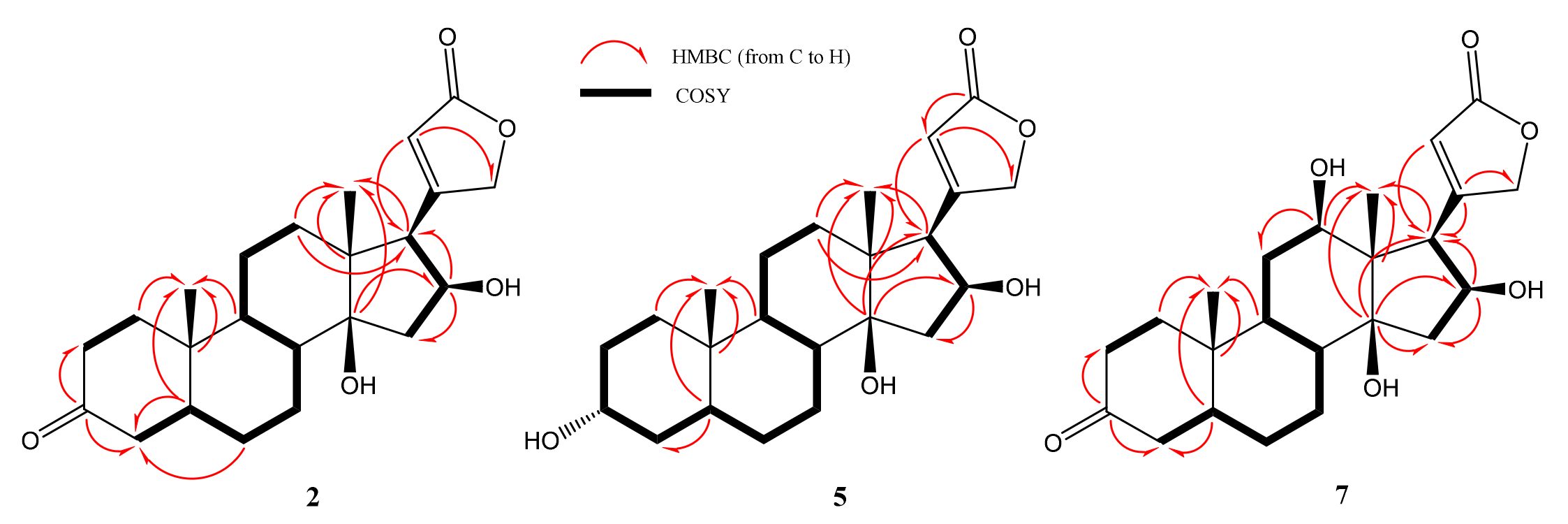
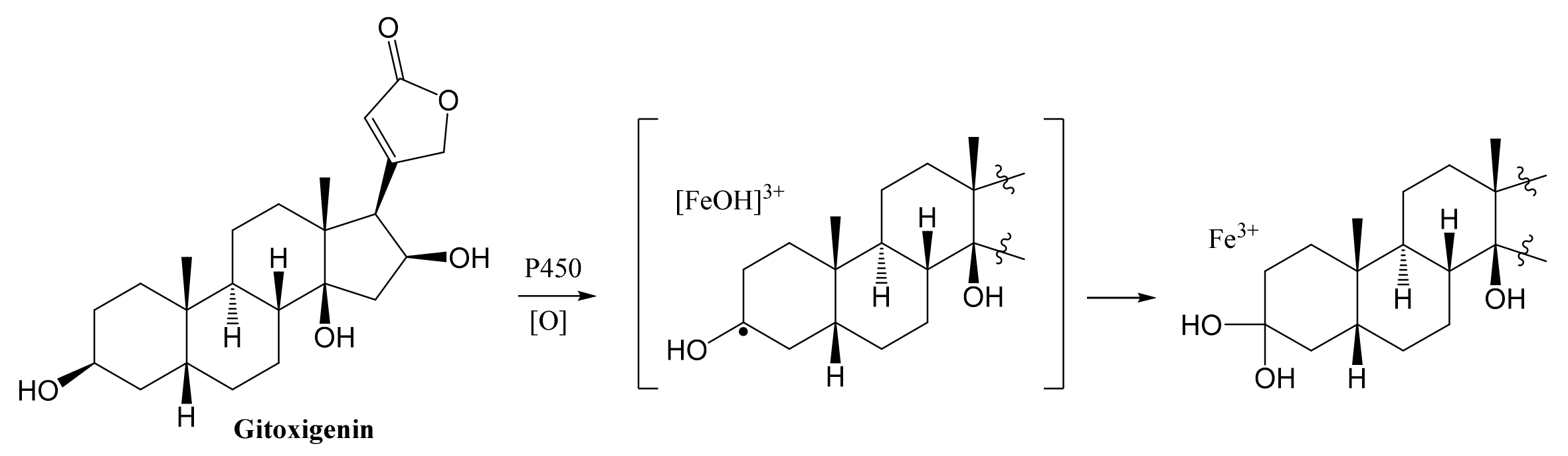
2.2. Structural Elucidations of Biotransformation Products
2.3. MTT Cell Viability Assay
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Preparation of Gitoxigenin, the Substrate, by Acid-Catalysed Hydrolysis of Oleandrin
3.4. Microbial Biotransformation Procedure
3.5. Isolation and Purification
3.6. Compound Characterization
3.7. MTT Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Venisetty, R.; Ciddi, V. Application of microbial biotransformation for the new drug discovery using natural drugs as substrates. Curr. Pharm. Biotechnol. 2003, 4, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Gilchrist, C.L.M.; Phan, C.-S.; Lacey, H.J.; Vuong, D.; Moggach, S.A.; Lacey, E.; Piggott, A.M.; Chooi, Y.-H. Biosynthesis of a new benzazepine alkaloid nanangelenin A from Aspergillus nanangensis Involves an unusual l-kynurenine-incorporating NRPS catalyzing regioselective lactamization. J. Am. Chem. Soc. 2020, 142, 7145–7152. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.-W.; Jamieson, C.S.; Wang, G.; Yan, Y.; Zhou, J.; Houk, K.N.; Tang, Y. A Polyketide cyclase that forms medium-ring lactones. J. Am. Chem. Soc. 2021, 143, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Adam, S.; Franz, L.; Milhim, M.; Bernhardt, R.; Kalinina, O.V.; Koehnke, J. Characterization of the stereoselective P450 enzyme BotCYP enables the in vitro biosynthesis of the Bottromycin core scaffold. J. Am. Chem. Soc. 2020, 142, 20560–20565. [Google Scholar] [CrossRef] [PubMed]
- Monagas, M.; Urpi-Sarda, M.; Sánchez-Patán, F.; Llorach, R.; Garrido, I.; Gómez-Cordovés, C.; Andres-Lacueva, C.; Bartolomé, B. Insights into the metabolism and microbial biotransformation of dietary flavan-3-ols and the bioactivity of their metabolites. Food Funct. 2010, 1, 233–253. [Google Scholar] [CrossRef] [Green Version]
- Bianchini, L.F.; Arruda, M.F.C.; Vieira, S.R.; Campelo, P.M.S.; Gregio, A.M.T.; Rosa, E.A.R. Microbial biotransformation to obtain new antifungals. Front. Microbiol. 2015, 6, 1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahir, K.; Nazir, S.; Li, B.; Khan, A.U.; Khan, Z.U.H.; Gong, P.Y.; Khan, S.U.; Ahmad, A. Nerium oleander leaves extract mediated synthesis of gold nanoparticles and its antioxidant activity. Mater. Lett. 2015, 156, 198–201. [Google Scholar] [CrossRef]
- Abdou, R.H.; Basha, W.A.; Khalil, W.F. Subacute toxicity of Nerium oleander ethanolic extract in mice. Toxicol. Res. 2019, 35, 233–239. [Google Scholar] [CrossRef]
- Dunn, D.E.; He, D.N.; Yang, P.; Johansen, M.; Newman, R.A.; Lo, D.C. In vitro and in vivo neuroprotective activity of the cardiac glycoside oleandrin from Nerium oleander in brain slice-based stroke models. J. Neurochem. 2011, 119, 805–814. [Google Scholar] [CrossRef]
- Ozel, H.Z. Extracts of Nerium Species, Methods of Preparation, and Use Therefore. U.S. Patent 5135745A, 4 August 1992. [Google Scholar]
- Rashan, L.J.; Franke, K.; Khine, M.M.; Kelter, G.; Fiebig, H.H.; Neumann, J.; Wessjohann, L.A. Characterization of the anticancer properties of monoglycosidic cardenolides isolated from Nerium oleander and Streptocaulon tomentosum. J. Ethnopharmacol. 2011, 134, 781–788. [Google Scholar] [CrossRef]
- Aslanipour, B.; Alan, M. Therapeutic aspects of some extracts and purified cardiac glycosides obtained from Nerium oleander L. In Drug Development for Cancer and Diabetes. A Path to 2030; Apple Academic Press: New York, NY, USA, 2020; p. 79. [Google Scholar]
- Newman, R.; Sastry, K.J.; Arav-Boger, R.; Cai, H.; Matos, R.; Harrod, R. Antiviral effects of oleandrin. J. Exp. Pharmacol. 2020, 12, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Wasfi, I.A.; Zorob, O.; Al Katheeri, N.A.; Al Awadhi, A.M. A fatal case of oleandrin poisoning. Forensic Sci. Int. 2008, 179, e31–e36. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Yu, F.; Zeng, P.; Zhang, T.; Huang, H.; Chen, W.; Wu, B. Circadian sensitivity to the cardiac glycoside oleandrin is associated with diurnal intestinal P-glycoprotein expression. Biochem. Pharmacol. 2019, 169, 113622. [Google Scholar] [CrossRef]
- Plante, K.S.; Plante, J.A.; Fernandez, D.; Mirchandani, D.; Bopp, N.; Aguilar, P.V.; Sastry, K.J.; Newman, R.A.; Weaver, S.C. Prophylactic and therapeutic inhibition of in vitro SARS-CoV-2 replication by oleandrin. bioRxiv 2020. [Google Scholar] [CrossRef]
- Arao, T.; Fuke, C.; Takaesu, H.; Nakamoto, M.; Morinaga, Y.; Miyazaki, T. Simultaneous determination of cardenolides by sonic spray ionization liquid chromatography-ion trap mass spectrometry—a fatal case of oleander poisoning. J. Anal. Toxicol. 2002, 26, 222–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grote-Levi, L.M. Identification and Functional Characterization of Anti-Fibrotic Natural Compounds In Vitro. Ph.D. Thesis, Medizinische Hochschule Hannover, Hannover, Germany, 2018. [Google Scholar]
- Milutinovic, S.; Heynen-Genel, S.; Chao, E.; Dewing, A.; Solano, R.; Milan, L.; Barron, N.; He, M.; Diaz, P.W.; Matsuzawa, S.-I.; et al. Cardiac glycosides activate the tumor suppressor and viral restriction factor promyelocytic leukemia protein (PML). PLoS ONE 2016, 11, e0152692. [Google Scholar] [CrossRef]
- Petschenka, G.; Fei, C.S.; Araya, J.J.; Schröder, S.; Timmermann, B.N.; Agrawal, A.A. Relative selectivity of plant cardenolides for Na+/K+-ATPases from the monarch butterfly and non-resistant insects. Front. Plant Sci. 2018, 9, 1424. [Google Scholar] [CrossRef] [Green Version]
- López-Lázaro, M.; De La Peña, N.P.; Pastor, N.; Martín-Cordero, C.; Navarro, E.; Cortés, F.; Ayuso, M.J.; Toro, M.V. Anti-tumour activity of Digitalis purpurea L. subsp. heywoodii. Planta Medica 2003, 69, 701–704. [Google Scholar] [CrossRef]
- Karuppannan, A.K.; Wu, K.X.; Qiang, J.; Chu, J.J.-H.; Kwang, J. Natural compounds inhibiting the replication of Porcine reproductive and respiratory syndrome virus. Antivir. Res. 2012, 94, 188–194. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Xiao, L.Y.; Wu, P.C.; Chen, Y.K.; Lo, S.; Hu, S.C.S.; Chen, Y.H.; Chiu, C.C.C.; Yuan, S.S.F. Orabase-formulated gentian violet effectively improved oral potentially malignant disorder in vitro and in vivo. Biochem. Pharmacol. 2020, 171, 113713. [Google Scholar] [CrossRef]
- Laird, G.M.; Eisele, E.E.; Rabi, S.A.; Nikolaeva, D.; Siliciano, R.F. A novel cell-based high-throughput screen for inhibitors of HIV-1 gene expression and budding identifies the cardiac glycosides. J. Antimicrob. Chemother. 2013, 69, 988–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Low, J.S.Y.; Wu, K.X.; Chen, K.C.; Ng, M.M.-L.; Chu, J.J.H. Narasin, a novel antiviral compound that blocks dengue virus protein expression. Antivir. Ther. 2011, 16, 1203–1218. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Gore, J.C.; Xie, J. High throughput screening for colorectal cancer specific compounds. Comb. Chem. High Throughput Screen. 2016, 19, 180–188. [Google Scholar] [CrossRef]
- Veliky, I.A.; Jones, A.; Ozubko, R.S.; Przybylska, M.; Ahmed, F.P. 5β-hydroxygitoxigenin, a product of gitoxigenin produced by Daucus carota culture. Phytochemistry 1980, 19, 2111–2112. [Google Scholar] [CrossRef]
- Titus, E. The metabolism of cardiac lactones by microorganisms. In Advances in applied Microbiology; Elsevier: Amsterdam, The Netherlands, 1961; pp. 279–292. [Google Scholar]
- Ekiz, G.; Duman, S.; Bedir, E. Biotransformation of cyclocanthogenol by the endophytic fungus Alternaria eureka 1E1BL1. Phytochemistry 2018, 151, 91–98. [Google Scholar] [CrossRef]
- Özçınar, Ö.; Tag, O.; Yusufoglu, H.; Kivçak, B.; Bedir, E.; Kıvcak, B. Biotransformation of neoruscogenin by the endophytic fungus Alternaria eureka. J. Nat. Prod. 2018, 81, 1357–1367. [Google Scholar] [CrossRef]
- Ekiz, G.; Yılmaz, S.; Yusufoglu, H.; Kırmızıbayrak, P.B.; Bedir, E. Microbial transformation of cycloastragenol and astragenol by endophytic fungi isolated from astragalus species. J. Nat. Prod. 2019, 82, 2979–2985. [Google Scholar] [CrossRef]
- Duman, S.; Ekiz, G.; Yılmaz, S.; Yusufoglu, H.; Kırmızıbayrak, P.B.; Bedir, E. Telomerase activators from 20(27)-octanor-cycloastragenol via biotransformation by the fungal endophytes. Bioorg. Chem. 2021, 109, 104708. [Google Scholar] [CrossRef]
- Karakoyun, Ç.; Küçüksolak, M.; Bilgi, E.; Doğan, G.; Çömlekçi, Y.E.; Bedir, E. Five new cardenolides transformed from oleandrin and nerigoside by Alternaria eureka 1E1BL1 and Phaeosphaeriasp. 1E4CS-1 and their cytotoxic activities. Phytochem. Lett. 2021, 41, 152–157. [Google Scholar] [CrossRef]
- Kamiya, T.; Yamano, T. Microbiological transformation of a cardiac aglycone. Oxidation of hydroxyl group of gitoxigenin. Chem. Pharm. Bull. 1961, 9, 579–580. [Google Scholar] [CrossRef] [Green Version]
- Nozaki, Y. Transformations of cardio-active steroids by microorganisms: Part I. microbial dehydrogenation of 3-hydroxyl group of digitalis cardiac aglycone. Agric. Biol. Chem. 1961, 25, 461–465. [Google Scholar] [CrossRef]
- Kawaguchi, K.; Hirotani, M.; Furuya, T. Biotransformation of digitoxigenin by cell suspension cultures of Strophanthus divaricatus. Phytochemistry 1991, 30, 1503–1506. [Google Scholar] [CrossRef]
- Repke, K.; Klesczewski, S. On the absorption spectra of cardiac poisons in sulfuric acid. Naunyn Schmiedebergs Arch. Exp. Pathologie Pharmakol. 1960, 239, 131–143. [Google Scholar]
- Nawa, H.; Uchibaysahi, M.; Kamiya, T.; Yamano, T.; Arai, H.; Abe, M.; Uchibayashi, M.; Nawa, M.U.H. Microbiological transformation of a cardiac aglycone. Nat. Cell Biol. 1959, 184, 469–470. [Google Scholar] [CrossRef] [PubMed]
- Yamada, A. Studies on the derivatives of cardiac aglycones of digitalis. Chem. Pharm. Bull. 1960, 8, 18–24. [Google Scholar] [CrossRef] [Green Version]
- Repke, K.; Samuels, L.T. Enzymatic basis for epimerization of cardiotonic steroids at carbon 3 in rat liver. Biochemistry 1964, 3, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Hennebert, O.; Montes, M.; Favre-Réguillon, A.; Chermette, H.; Ferroud, C.; Morfin, R. Epimerase activity of the human 11β-hydroxysteroid dehydrogenase type 1 on 7-hydroxylated C19-steroids. J. Steroid Biochem. Mol. Biol. 2009, 114, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Templeton, J.F.; Cheung, A.H.T.; Sham, R.C.; Watson, T.R.; Jie, K. Ring-A oxygenated derivatives of 5α-and 5β-cardenolides. J. Chem. Soc. 1983, 251–256. [Google Scholar] [CrossRef]
- Bell-Parikh, L.C.; Guengerich, F.P. Kinetics of cytochrome P450 2E1-catalyzed oxidation of ethanol to acetic acid via acetaldehyde. J. Biol. Chem. 1999, 274, 23833–23840. [Google Scholar] [CrossRef] [Green Version]
- Nagel, R.; Peters, R.J. Diverging mechanisms: Cytochrome-P450-catalyzed demethylation and γ-Lactone formation in bacterial gibberellin biosynthesis. Angew. Chem. 2018, 130, 6190–6193. [Google Scholar] [CrossRef]
- Fuska, J.; Proksa, B.; Khandlová, A.; Šturdíkova, M. Microbial transformations of cardioglycosides. Appl. Microbiol. Biotechnol. 1987, 26, 313–317. [Google Scholar] [CrossRef]
- Murphy, J.E. Diginatin—A new cardioactive glycoside from Digitalis lanata. J. Am. Pharm. Assoc. 1955, 44, 719–722. [Google Scholar] [CrossRef] [PubMed]
- Linde, H.; Murphy, J.E.; Meyer, K. Chemischer Beweis der 12β-HO-Gruppe in Diginatigenin. Helv. Chim. Acta 1959, 42, 2040–2043. [Google Scholar] [CrossRef]
- Ashley, J.; Brown, B.; Okita, G.; Wright, S. The metabolites of cardiac glycosides in human urine. J. Biol. Chem. 1958, 232, 315–322. [Google Scholar] [CrossRef]
- Shiratori, O. Growth inhibitory effect of cardiac glycosides and aglycones on neoplastic cells in vitro and in vivo studies. Gann Jpn. J. Cancer Res. 1967, 58, 521–528. [Google Scholar]
- Doskotch, R.W.; Malik, M.Y.; Hufford, C.D.; Malik, S.N.; Trent, J.E.; Kubelka, W. Antitumor agents V: Cytotoxic cardenolides from Cryptostegia grandiflora (Roxb.). R. Br. J. Pharm. Sci. 1972, 61, 570–573. [Google Scholar] [CrossRef]
- Ryer, A.I.; Marie, F. Process for the Isolation of Oleandrin. U.S. Patent 2438418A, 23 March 1948. [Google Scholar]
- Ekiz, G. Research on Bioactive Secondary Metabolite Profile of Septofusidium Berolinense and Biotranformation of Cycloartane Type Saponins by Endophytic Fungi. Ph.D. Thesis, Department of Bioengineering, Ege University, Izmir, Turkey, 2016. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | Gitoxigenin | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1.22 m; 1.27 m | 1.41 m; 1.93 m | 1.45 m; 2.00 m | 1.48 m; 2.05 dd (14.2,4.5) | 1.25 m; 1.65 m | 1.04 td (14.2,3.5); 1.82 m | 1.06 d (14.5); 1.80 m | 1.45 m; 2.11 m | 1.11 td (14.2, 3.4); 1.85 m |
| 2 | 1.24 m; 1.36 m | 2.12 m; 2.21 td (14.4, 5.3) | 2.20 d (16.1); 2.32 dd (14.5, 6.4) | 2.12 m; 2.47 td (14.5, 5.1) | 1.26 m; 1.77 m | 1.37 m; 1.65 m | 1.65 q (12.5); 1.96 m | 2.12 m; 2.57 dd (14.4, 5.5) | 1.39 m; 1.69 m |
| 3 | 3.82 t (2.8) | - | - | - | - | 3.56 m | 3.85 m | - | 3.59 m |
| 4 | 1.09 m; 1.67 td (13.7, 3.2) | 2.10 m; 2.43 m | 2.06 m; 2.62 m | 2.12 m; 2.66 t (14.3) | 1.55 m; 1.77 m | 1.49 m; 1.74 m | 1.92 m; 1.99 m | 1.97 m; 2.81 t (14.3) | 1.53 m; 1.70 m |
| 5 | 1.54 m | 1.86 m | 1.84 m | 1.88 m | 1.67 m | 1.40 m | 1.51 m | 1.82 m | 1.45 m |
| 6 | 0.96 m; 1.63 m | 1.50 m; 1.83 m | 1.37 m; 1.90 m | 1.58 m; 1.90 m | 1.57 m; 1.92 m | 1.35 m; 1.88 m | 1.80 m; 2.14 td (12.2, 4.9) | 1.37 m; 1.91 m | 1.38 m; 1.91 m |
| 7 | 0.97 m; 1.57 m | 3.84 td (10.6, 4.9) | 1.24 m; 1.91 m | 3.91 td (11.6, 11.2, 4.7) | 3.96 m | 1.29 m; 1.84 m * | 4.26 m | 1.31 m; 1.91 m | 1.31 m; 1.84 m |
| 8 | 1.34 m | 1.69 dd (11.8, 10.4) | 1.62 m | 1.71 m | 1.72 m | 1.59 m | 1.95 m | 1.65 m | 1.60 m |
| 9 | 1.37 m | 1.58 m | 1.69 m | 1.88 m | 1.54 m | 1.70 m | 1.77 m | 1.89 m | 1.72 m |
| 10 | - | - | - | - | - | - | - | - | - |
| 11 | 0.94 m; 1.11 m | 1.31 m; 1.37 m | 1.32 m; 1.46 m | 1.45 m; 1.63 m | 1.25 m; 1.41 m | 1.21 m; 1.42 m * | 1.25 m; 1.35 m | 1.30 m; 1.66 m | 1.30 m; 1.61 m |
| 12 | 1.11 m; 1.26 m | 1.25 m; 1.61 m | 1.35 m; 1.69 m | 3.36 dd (11.8, 4.1) | 1.25 m; 1.65 m | 1.39 m; 1.55 m | 1.31 m; 1.49 m | 3.40 dd (11.8, 4.1) | 3.34 m |
| 13 | - | - | - | - | - | - | - | - | - |
| 14 | - | - | - | - | - | - | - | - | - |
| 15 | 1.49 dd (15.0, 2.4); 2.35 dd (14.9, 8.5) | 1.95 m; 2.36 dd (14.2, 6.4) | 1.90 m; 2.42 dd (14.5, 6.4) | 1.97 m; 2.59 dd (14.5, 7.5) | 2.05 m; 2.40 dt (14.4, 4.8) | 1.72 m; 2.63 dd (14.9, 8.5) | 2.27 d (13.8); 2.67 m | 1.79 m; 2.49 dd (14.9, 7.6) | 1.82 m; 2.43 dd (14.7, 7.6) |
| 16 | 4.43 td (8.3, 2.4) | 4.41 t (6.5) | 4.54 t (6.5) | 4.58 m | 4.44 brs | 4.66 td (8.2, 2.3) | 4.94 m | 4.59 s | 4.60 td (7.6, 1.7) |
| 17 | 2.88 d (7.9) | 2.91 d (6.9) | 2.96 d (7.1) | 3.58 d (7.6) | 2.91 dd (7.0, 3.0) | 3.14 d (7.9) | 3.29 d (8.0) | 3.59 d (7.6) | 3.59 m |
| 18 | 0.66 s | 0.90 s | 0.98 s | 0.89 s | 0.96 s | 0.92 s | 1.11 s | 0.89 s | 0.87 s |
| 19 | 0.66 s | 0.98 s | 1.02 s | 1.06 s | 0.97 s | 0.93 s | 0.93 s | 1.06 s | 0.97 s |
| 20 | - | - | - | - | - | - | - | - | - |
| 21 | 4.96 d (18.4); 4.91 m | 4.87 d (18.3); 5.02 d (18.3) | 4.90 d (18.1); 5.05 d (18.1) | 5.03 dd (18.5, 1.8); 5.14 dd (18.5, 1.9) | 4.89 m; 5.06 m | 5.11 dd (18.4, 1.8); 5.17 dd (18.5, 1.8) | 5.57 m; 5.69 m | 5.04 dd (18.3, 1.8); 5.18 dd (18.4, 1.9) | 5.04 dd (18.5, 1.8); 5.19 dd (18.5, 1.9) |
| 22 | 5.71 s | 5.88 s | 5.97 s | 6.01 s | 5.97 s | 5.94 s | 6.25 s | 6.03 s | 6.03 s |
| 23 | - | - | - | - | - | - | - | - | - |
| 3-O-Me | - | - | - | - | 3.12 s | - | - | - | - |
| 3-O-Me | - | - | - | - | 3.19 s | - | - | - | - |
| Solvent | CD3OD/a drop of pyridine-d5 | CDCl3/a drop of CD3OD | CDCl3 | CD3OD | CDCl3 | CD3OD | Pyridine-d5 | CD3OD | CD3OD |
| Position | Gitoxigenin | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 30.7 t | 36.0 t | 36.7 t | 37.6 t | 32.4 t | 36.2 t | 35.7 t | 37.8 t | 36.2 t |
| 2 | 28.5 t | 36.8 t | 37.2 t | 38.9 t | 27.5 t | 31.3 t | 31.8 t | 37.9 t | 31.2 t |
| 3 | 67.6 d | 212.7 s | 212.7 s | 214.5 s | 100.4 s | 72.3 d | 71.2 d | 215.9 s | 72.2 d |
| 4 | 34.1 t | 42.8 t | 42.2 t | 43.7 t | 34.5 t | 37.0 t | 38.7 t | 43.0 t | 37.0 t |
| 5 | 37.3 d | 43.7 d | 43.6 d | 45.1 d | 40.1 d | 43.1 d | 43.1 d | 45.4 d | 43.1 d |
| 6 | 27.7 t | 35.9 t | 26.6 t | 37.2 t | 37.1 t | 28.2 t | 38.5 t | 27.7 t | 28.2 t |
| 7 | 22.4 t | 69.1 d | 21.5 t | 70.3 d | 70.9 d | 22.6 t * | 70.5 d | 22.4 t | 23.0 t |
| 8 | 42.8 d | 45.6 d | 41.7 d | 46.8 d | 46.4 d | 43.0 d | 47.3 d | 42.1 d | 42.4 d |
| 9 | 36.5 d | 35.8 d | 36.7 d | 33.8 d | 35.0 d | 37.4 d | 36.2 d | 34.4 d | 34.3 d |
| 10 | 36.3 s | 35.0 s | 35.4 s | 36.0 s | 35.1 s | 35.9 s | 35.4 s | 36.2 s | 35.8 s |
| 11 | 22.0 t | 21.3 t | 21.2 t | 30.7 t | 21.4 t | 21.9 t * | 21.5 t | 30.7 t | 30.5 t |
| 12 | 40.8 t | 41.2 t | 41.5 t | 75.9 d | 42.0 t | 40.9 t | 40.2 t | 76.3 d | 76.6 d |
| 13 | 51.3 s | 49.5 s | 49.8 s | 57.2 s | 49.4 s | 51.3 s | 50.5 s | 57.6 s | 57.6 s |
| 14 | 85.5 s | 86.0 s | 85.8 s | 87.0 s | 86.6 s | 85.6 s | 85.4 s | 86.3 s | 86.4 s |
| 15 | 43.8 t | 42.1 t | 42.2 t | 44.4 t | 42.4 t | 43.8 t | 45.5 t | 43.4 t | 43.5 t |
| 16 | 73.0 d | 73.0 d | 73.2 d | 73.6 d | 73.7 d | 73.1 d | 72.7 d | 73.4 d | 73.4 d |
| 17 | 59.6 d | 58.2 d | 58.1 d | 55.0 d | 58.5 d | 59.7 d | 59.9 d | 54.9 d | 54.9 d |
| 18 | 17.1 q | 16.8 q | 16.9 q | 10.5 q | 17.0 q | 17.0 q | 17.4 q | 10.5 q | 10.5 q |
| 19 | 24.3 q | 22.4 q | 22.7 q | 22.7 q | 23.2 q | 23.8 q | 23.8 q | 22.8 q | 23.7 q |
| 20 | 173.8 s | 169.9 s | 168.8 s | 173.1 s | 169.0 s | 173.7 s | 173.0 s | 173.2 s | 173.3 s |
| 21 | 77.9 t | 75.9 t | 75.7 t | 77.7 t | 75.6 t | 77.9 t | 77.2 t | 77.7 t | 77.7 t |
| 22 | 120.6 d | 119.5 d | 120.0 d | 120.7 d | 119.7 d | 120.6 d | 120.5 d | 120.6 d | 120.6 d |
| 23 | 177.3 s | 175.3 s | 174.5 s | 177.5 s | 174.7 s | 177.4 s | 175.1 s | 177.6 s | 177.4 s |
| 3-O-Me | - | - | - | - | 47.7 q (×2) | - | - | - | - |
| Compound | A549 | DU 145 | PANC-1 | MIA PaCa-2 | HEK-293 | MRC-5 |
|---|---|---|---|---|---|---|
| 1 | >10 | >10 | >10 | >10 | >10 | >10 |
| 2 | >10 | >10 | >10 | >10 | >10 | >10 |
| 3 | >10 | >10 | >10 | >10 | >10 | >10 |
| 4 | >10 | >10 | >10 | >10 | >10 | >10 |
| 5 | >10 | >10 | >10 | >10 | >10 | >10 |
| 6 | >10 | >10 | >10 | >10 | >10 | >10 |
| 7 | >10 | >10 | 6.24 | >10 | >10 | >10 |
| 8 | 8.25 | >10 | 1.95 | 3.4 | >10 | 9.35 |
| Gitoxigenin | 2.19 | 4.08 | 1.53 | 3.1 | 7.6 | 0.815 |
| Oleandrin | 0.0426 | 0.0210 | 0.0387 | 0.0350 | 0.0730 | 0.0173 |
| Doxorubicin | 0.6200 | 0.1900 | >10 | 0.5900 | 0.5870 | 0.8650 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bedir, E.; Karakoyun, Ç.; Doğan, G.; Kuru, G.; Küçüksolak, M.; Yusufoğlu, H. New Cardenolides from Biotransformation of Gitoxigenin by the Endophytic Fungus Alternaria eureka 1E1BL1: Characterization and Cytotoxic Activities. Molecules 2021, 26, 3030. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26103030
Bedir E, Karakoyun Ç, Doğan G, Kuru G, Küçüksolak M, Yusufoğlu H. New Cardenolides from Biotransformation of Gitoxigenin by the Endophytic Fungus Alternaria eureka 1E1BL1: Characterization and Cytotoxic Activities. Molecules. 2021; 26(10):3030. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26103030
Chicago/Turabian StyleBedir, Erdal, Çiğdem Karakoyun, Gamze Doğan, Gülten Kuru, Melis Küçüksolak, and Hasan Yusufoğlu. 2021. "New Cardenolides from Biotransformation of Gitoxigenin by the Endophytic Fungus Alternaria eureka 1E1BL1: Characterization and Cytotoxic Activities" Molecules 26, no. 10: 3030. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26103030