4.1. Chemistry
4.1.1. General Information
All reagents were purchased from commercial sources (Sigma-Aldrich, TCI, Acros or Buchler) and used without further purification. The solvents used were of analytical grade. The 1H and 13C NMR spectra were recorded at room temperature on a Bruker Avance 300, Bruker Avance 400 or Bruker Avance 600, operating at 300 MHz, 400 MHz or 600 MHz for 1H and 75 MHz, 100 MHz or 150 MHz for 13C. Chemical shifts (δ) were reported as relative to the tetramethylsilane peak set at 0.00 ppm. In the case of multiplets, the signals were reported as intervals. The signals were abbreviated as s, singlet; d, doublet; t, triplet; q, quadruplet; and m, multiplet. Mass spectra were recorded on a Finnigan MAT 8400-MSS and Finnigan MAT 4515. High resolution mass spectra were recorded on a Finnigan MAT 95 XP. The reactions were monitored by TLC and performed on silica gel plates 40 × 80 mm Polygram Sil G/UV254 (Macherey-Nagel). Visualization on TLC was achieved by UV light. Column chromatography was performed with Merck silica gel 60 (70–200 mesh).
The numbering of the Cinchona core followed the rules of Paul Rabe, with the side chain numbered based on the IUPAC rules.
![Molecules 26 03357 i001]()
4.1.2. Synthesis of the Azido-Derivatives 8–13
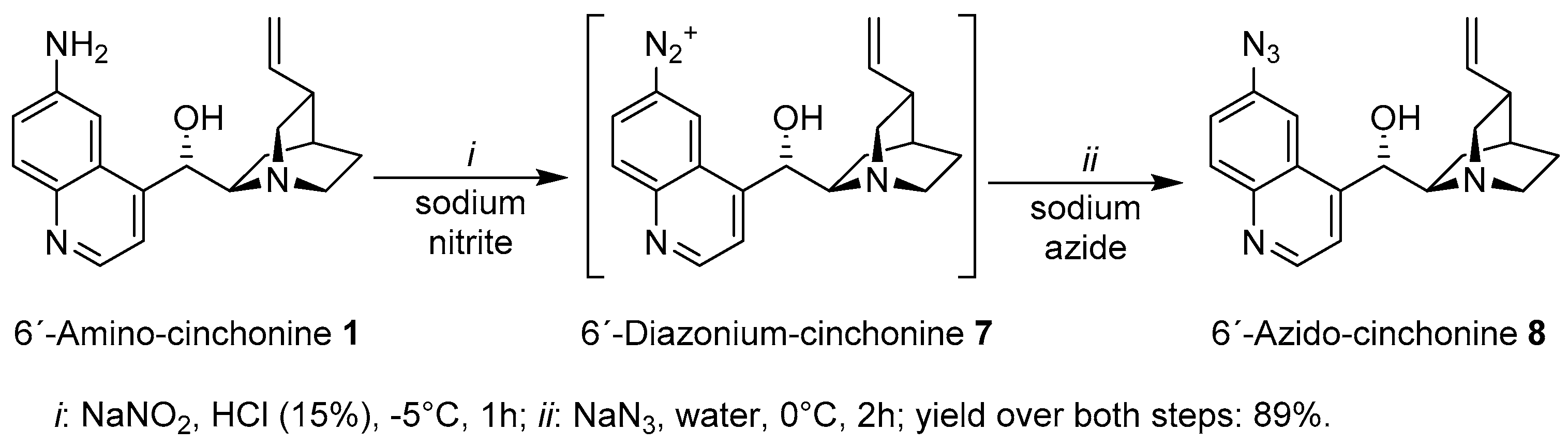
General procedure: a dropwise a solution of NaNO2 (1.2 eq) in H2O (0.33 g/mL) was added to a mixture of Cinchona amine (1.0 eq) and 15% HCl (0.1 g amine/mL) cooled at −5 °C. After the addition was complete, the reaction mixture was stirred at this temperature for 60 min. A solution of NaN3 (1.7 eq) in H2O (0.2 g/mL) was added dropwise to the reaction mixture at 0 °C. After the addition was finished, the reaction mixture was maintained at 0 °C for 2 h and then stirred at room temperature for 12 h. The product was precipitated by addition of NaOH (20%) until pH 10. Upon filtration, the solid was washed with distilled water and dried. Crystallization from DCM presented the Cinchona azides as yellow solids in good yields (70–90%)
(S)-(6-Azidoquinolin-4-yl)((1S,2R,4S,5R)-5-vinylquinuclidin-2-yl)methanol (8): the title compound was synthesized from 1 (25.0 g, 81 mmol) to afford 8 (25.8 g, yield: 95%) as a yellow-orange solid. 1H-NMR (MeOH-d4, 400 MHz) δ 8.79 (d, J = 4.6 Hz, H-2′), 8.08 (d, J = 9.05 Hz, H-8′), 7.82 (d, J = 2.4 Hz, H-5′), 7.74 (d, J = 4.6 Hz, H-3′), 7.52 (dd, J = 9.05 Hz, J = 2.45 Hz, H-7′), 6.17 (ddd, J = 17.4 Hz, J = 10.3 Hz, J = 7.7 Hz, H-10), 5.66–5.52 (m, H-9), 5.17–5.08 (m, H-11, H-11), 3.50–3.43 (m, H-2), 3.35–3.32 (m, H-8), 2.96–2.74 (m, H-2, H-6, H-6), 2.37–2.29 (m, H-3), 2.23–2.16 (m, H-7), 1.93–1.68 (m, H-4), 1.66–1.51 (m, H-5, H-5), and 1.31–1.18 (m, H-7); 13C-NMR (MeOH-d4, 100 MHz) δ 151.27 (Cq, C-4′), 150.32 (CH, C-2′), 146.64 (Cq, C-8a′); 141.73 (CH, C-10); 140.31 (Cq, C-6′), 132.14 (CH; C-8′), 128.03 (Cq, C-4a′), 123.87 (CH, C-7′), 120.87 (CH; C-3′), 115.15 (CH2, C-11), 112.90 (CH, C-5′), 72.49 (CH; C-9), 61.27 (CH, C-8), 50.82 (CH2, C-6), 50.19 (CH2, C-2), 41.29 (CH, C-3), 29.65 (CH, C-4), 27.20 (CH2, C-5), and 22.25 (CH2, C-7); H,H-Cosy, HSQC and HMBC were made. Melting point: the substance decomposed at T = 173 °C; TLC (TBME: MeOH: ammonia 25%/100:10:1): rf-value = 0.60; IR: see spectrum in the supporting information; HR-MS (ESI, MeOH) was calculated for C19H21N5O1 + H+: 336.18189 Da, found 336.18185 Da.
(R)-(6-Azidoquinolin-4-yl)((1S,2S,4S,5R)-5-vinylquinuclidin-2-yl)methanol (9): the title compound was synthesized from 2 (25.0 g, 81 mmol) to afford 9 (24.1 g, yield: 89%) as a yellow-orange solid. 1H-NMR (MeOH-d4, 400 MHz) δ 8.76 (d, J = 4.57 Hz, H-2′), 8.05 (d, J = 9.05 Hz, H-8′), 7.82 (d, J = 2.4 Hz, H-5′), 7.71 (d, J = 4.57 Hz, H-3′), 7.52 (dd, J = 9.05 Hz, J = 2.4 Hz, H-7′), 5.78 (ddd, J = 17.7 Hz, J = 10.7 Hz, J = 7.7 Hz, H-10), 5.53–5.49 (m, H-9), 5.02–4.94 (m, H-11, H-11), 3.61–3.51 (m, H-6), 3.12–2.94 (m, H-2, H-8), 2.72–2.49 (m, H-2, H-6), 2.48–2.18 (m; H-3), 1.93–1.81 (m,H-5, H-7), 1.80–1.78 (m, H-4), and 1.72–1.50 (m, H-5, H-7); 13C-NMR (MeOH-d4, 100 MHz) δ 151.22 (Cq, C-4′), 150.27 (CH, C-2′), 146.62 (Cq, C-8a′); 142.78 (CH, C-10); 140.24 (Cq, C-6′), 132.15 (CH, C-8′), 127.95 (Cq, C-4a′), 123.25 (CH, C-7′), 120.93 (CH, C-3′), 114.90 (CH2, C-11), 112.99 (CH, C-5′), 72.40 (CH, C-9), 61.53 (CH, C-8), 57.59 (CH2, C-2), 43.82 (CH2, C-6), 40.95 (CH, C-3), 29.18 (CH, C-4), 28.25 (CH2, C-5), and 22.61 (CH2, C-7); H,H-Cosy, HSQC and HMBC were made. Melting point: the substance decomposed at T = 144 °C; TLC (TBME: MeOH: ammonia 25%/100:10:1): rf-value = 0.62; IR: see spectrum in the supporting information; HR-MS (ESI, MeOH) was calculated for C19H21N5O1 + H+: 336.18189 Da, found 336.18183 Da.
(S)-(6-Azidoquinolin-4-yl)((1S,2R,4S,5R)-5-ethylquinuclidin-2-yl)methanol (10): the title compound was synthesized from 3 (5.0 g, 16.0 mmol) to afford 10 (4.3 g, yield: 79%) as a yellowish solid. 1H-NMR (DMSO-d6, 400 MHz) δ 8.78 (d, J = 4.5 Hz, H-2′), 8.04 (d, J = 9.0 Hz, H-8′), 7.86 (d, J = 2.5 Hz, H-5′), 7.54 (d, J = 4.5 Hz, H-3′), 7.48 (dd, J = 9.0 Hz, J = 2.5 Hz, H-7′), 5.20–5.16 (m, H-9), 2.96–2.88 (m, H-8), 2.68–2.62 (m, H-6, H-6), 2.52–2.44 (m, H-2, H-2, H-3), 2.00–2.76 (m, H-7), 1.65–1.62 (m, H-4); 1.53–1.30 (m, H-5, H-5, H-7, H-10, H-10), and 0.86 (t, J = 7.2 Hz, H-11, H-11, H-11); 13C-NMR (DMSO-d6, 100 MHz) δ 150.30 (Cq, C-4′), 149.50 (CH, C-2′), 145.62 (Cq, C-8a′), 136.85 (Cq, C-6′), 131.70 (CH, C-8′), 126.94 (Cq, C-4a′), 121.56 (CH, C-7′), 119.76 (CH, C-3′), 112.15 (CH, C-5′), 70.73 (CH, C-9), 61.14 (CH, C-8), 49.89 (CH2, C-6), 49.11 (CH2, C-2), 37.08 (CH, C-3), 27.11 (CH2, C-5), 26.01 (CH, C-4), 25.03 (CH2, C-10), 24.01 (CH2, C-7), and 11.90 (CH3, C-11); H,H-Cosy, HSQC and HMBC were made. Melting point: the substance decomposed at T = 191 °C; TLC (TBME: MeOH: ammonia 25%/100:10:1): rf-value = 0.50; IR: see spectrum in the supporting information; HR-MS (ESI, MeOH) calculated for C19H23N5O1 + H+: 338.19754 Da, found 338.19756 Da.
(R)-(6-Azidoquinolin-4-yl)((1S,2S,4S,5R)-5-ethylquinuclidin-2-yl)methanol (11): the title compound was synthesized from 4 (5.0 g, 16 mmol) to afford 8 (4.2 g, yield: 78%) as a yellow solid. 1H-NMR (DMSO-d6, 400 MHz) δ 8.78 (d, J = 4.45 Hz, H-2′), 8.05 (d, J = 9.0 Hz, H-8′), 7.91 (d, J = 2.4 Hz, H-5′), 7.55 (d, J = 4.45 Hz, H-3′), 7.51 (dd, J = 9.0 Hz, J = 2.4 Hz, H-7′), 5.18–5.13 (m, H-9), 3.21–2.98 (m, H-6, H-8), 2.87–2.77 (m, H-2), 2.45–2.31 (m, H-6), 2.18–2.06 (m, H-2), 1.71–1.50 (m; H-4, H-5, H-7, H-7), 1.40–1.15 (m, H-3, H-5, H-10, H-10), and 0.86 (t, J = 7.1 Hz, H-11, H-11, H-11); 13C-NMR (DMSO-d6, 100 MHz) δ 149.87 (CH, C-2′), 149.50 (Cq, C-4′), 145.75 (Cq, C-8a′); 136.92 (Cq, C-6′), 131.79 (CH, C-8′), 126.87 (Cq, C-4a′), 121.45 (CH, C-7′), 119.97 (CH, C-3′), 112.73 (CH, C-5′), 71.44 (CH, C-9),60.59 (CH, C-8), 57.53 (CH2, C-2), 41.64 (CH2, C-6), 37.14 (CH, C-3), 28.18 (CH2, C-5), 27.20 (CH2, C-10), 25.14 (CH, C-4), 24.35 (CH2, C-7), and 12.04 (CH3, C-11); H,H-Cosy, HSQC and HMBC were made. Melting point: the substance decomposed at T = 163 °C; TLC (TBME: MeOH: ammonia 25%/100:10:1): rf-value = 0.54; IR: see spectrum in the supporting information; HR-MS (ESI, MeOH) calculated for C19H23N5O1 + H+: 338.19754 Da, found 338.19757 Da.
(S)-(6-Azidoquinolin-4-yl)((1S,2R,4S,5S)-5-ethynylquinuclidin-2-yl)methanol (12): the title compound was synthesized from 5 (1.7 g, 5.5 mmol) to afford 12 (1.8 g, yield: 97%) as a yellow solid. 1H-NMR (CDCl3, 300MHz) δ 8.73 (d, J = 4.5 Hz, H-2′), 8.07 (d, J = 9.0 Hz, H-8′), 7.68 (d, J = 2.3 Hz, H-5′), 7.53 (d, J = 4.5 Hz, H-3′), 7.36 (dd, J = 9.0 Hz, J = 2.3 Hz, H-7′), 5.58–5.48 (m, H-9), 3.28–3.20 (m, H-2), 3.18–3.09 (m, H-8), 3.03–2.94 (m, H-2), 2.84–2.74 (m, H-6), 2.68–2.55 (m, H-6), 2.51–2.44 (m, H-3), 2.28–2.16 (m, H-7), 2.15 (d, J = 2.45 Hz, H-11), 2.07–1.93 (m, H-4), and 1.60–1.41 (m, H-5, H-5, H-7); 13C-NMR (CDCl3, 75MHz) δ 149.50 (CH, C-2′), 147.93 (Cq, C-4′), 146.11 (Cq, C-8a′), 138.16 (Cq, C-6′), 132.22 (CH, C-8′), 126.81 (Cq, C-4a′), 121.60 (CH, C-7′), 119.47 (CH, C-3′), 111.73 (CH, C-5′), 87.31 (CH, C-11), 72.01 (CH, C-9), 69.31 (Cq, C-10), 60.25 (CH, C-8), 50.28 (CH2, C-6), 49.41 (CH2, C-2), 28.03 (CH, C-3), 27.86 (CH, C-4), 25.11 (CH2, C-5), and 23.51 (CH2, C-7); H,H-Cosy, HSQC and HMBC were made. Melting point: the substance decomposed at T = 112 °C; TLC (TBME: MeOH: ammonia 25%/100:10:1): rf-value = 0.55; IR: see spectrum in the supporting information; HR-MS (ESI, MeOH) calculated for C19H19N5O1 + H+: 334.16624 Da, found 334.16647 Da.
(R)-(6-Azidoquinolin-4-yl)((1S,2S,4S,5S)-5-ethynylquinuclidin-2-yl)methanol (13): the title compound was synthesized from 6 (1.7 g, 5.5 mmol) to afford 13 (1.4 g, yield: 76%) as a yellow solid. 1H-NMR (CDCl3, 300 MHz) δ 8.74 (d, J = 4.5 Hz, H-2′), 8.06 (d, J = 9.0 Hz, H-8′), 7.95 (d, J = 2.5 Hz, H-5′), 7.51 (d, J = 4.5 Hz, H-3′), 7.21 (dd, J = 9.0 Hz, J = 2.5 Hz, H-7′), 5.61–5.55 (m, H-9), 3.42–3–3.34 (m, H-6), 3.24–3.16 (m, H-8), 2.98–2.88 (m, H-2), 2.75–2.61 (m, H-6), 2.61–2.48 (m, H-2), 2.44–2.37 (m, H-3), 2.16 (d, J = 2.4 Hz, H-11), 2.06–1.94 (m,H-5, H-7), 1.90–1.89 (m, H-4), and 1.81–1.59 (m, H-5, H-7); 13C-NMR (CDCl3, 75MHz) δ 149.65 (CH, C-2′), 148.02 (Cq, C-4′), 146.31 (Cq, C-8a′), 138.27 (Cq, C-6′), 131.97 (CH, C-8′), 126.86 (Cq, C-4a′), 121.53 (CH, C-7′), 119.67 (CH, C-3′), 111.68 (CH, C-5′), 87.26 (CH, C-11), 71.87 (CH, C-9), 69.21 (Cq, C-10), 60.62 (CH, C-8), 56.31 (CH2, C-2), 49.42 (CH2, C-6), 28.17 (CH, C-3), 27.96 (CH, C-4), 24.94 (CH2, C-5), and 23.47 (CH2, C-7); H,H-Cosy, HSQC and HMBC were made. Melting point: the substance decomposed at T = 96 °C; TLC (TBME: MeOH: ammonia 25%/100:10:1): rf-value = 0.57; IR: see spectrum in the supporting information; HR-MS (ESI, MeOH) calculated for C19H19N5O1 + H+: 334.16624 Da, found 334.16633 Da.
4.1.3. Synthesis of the Silylated Triazole-Derivatives 14, 16, 18 and 20
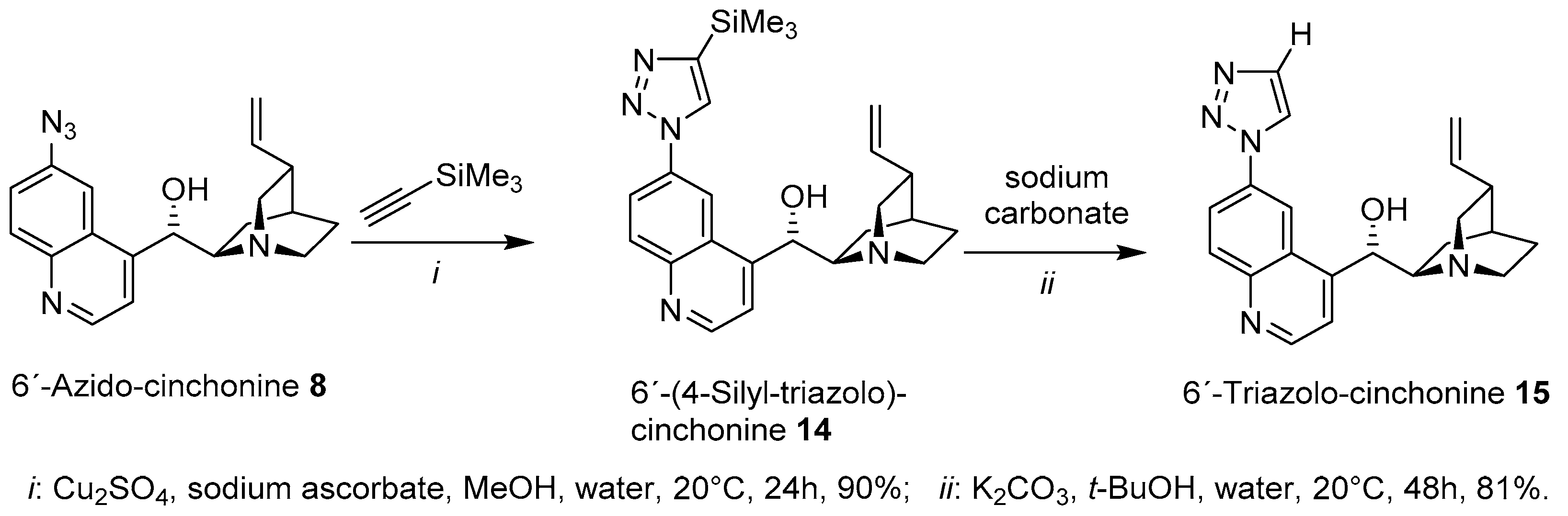
General procedure: in a round-bottom flask the corresponding Cinchona azide (1 eq.) and trimethylsilyl acetylene (1.5 eq.) were suspended in an H2O: MeOH (1:1) mixture to deliver a substrate concentration of 0.1 M Cu2SO4 (0.2 eq., 20 mol%) and sodium ascorbate (0.4 eq., 40 mol%). The reaction mixture was stirred at room temperature until complete conversion of the azide (12–24 h). After that, the reaction was quenched by adding an aqueous solution of ammonium chloride and ammonium hydroxide (pH 10/12). The mixture was extracted with DCM (three times), and the combined organic layers were dried over anhydrous sodium sulphate, filtered and concentrated in vacuo. The residues obtained were purified by flash column chromatography using MTBE: MeOH as a gradient elution to afford the corresponding TMS-triazole derivatives.
(S)-(6-(4-(Trimethylsilyl)-1H-1,2,3-triazol-1-yl)quinolin-4-yl)((1S,2R,4S,5R)-5-vinylquinuclidin-2-yl)methanol (14): the title compound was synthesized from 8 (1.0 g, 3.0 mmol) to afford 14 (1.15 g, yield: 90%) as a colourless wax. 1H-NMR (CDCl3, 300 MHz) δ 8.77 (bs, H-Triazole-5), 8.49 (d, J = 4.4 Hz, H-2′), 8.10–8.06 (m, H-5′, H-7′), 7.87 (d, J = 9.7 Hz, H-8′), 7.35 (d, J = 4.4 Hz, H-3′), 6.32 (bs, H-9), 6.06 (ddd, J = 17.5 Hz, J = 10.1 Hz, J = 7.4 Hz, H-10), 5.24–5.18 (m, H-11, H-11), 4.22–3.96 (m, H-2), 3.34–3.08 (m, H-2, H-8), 3.05–2.88 (m, H-6), 2.58–2.42 (m, H-3), 2.37–2.21 (m, H-6), 1.96–1.87 (m, H-4), 1.85–1.71 (m, H-7), 1.69–1.54 (m, H-5), 1.15–1.00 (m, H-5), and 0.93–0.79 (m, H-7) 0.46 (s, 9H, -Si(CH3)3); 13C-NMR (CDCl3, 75MHz) δ 150.13 (CH, C-2′), 147.41 (Cq, C-4′), 146.93 (Cq, C-Triazole-4), 146.62 (Cq, C-8a′); 137.55 (CH, C-10); 134.68 (Cq, C-6′), 131.88 (CH; C-8′), 128.35 (CH, C-Triazole-5), 124.52 (Cq, C-4a′), 121.47 (CH, C-7′), 119.27 (CH; C-3′), 116.78 (CH2, C-11), 110.91 (CH, C-5′), 67.67 (CH, C-9), 60.37 (CH, C-8), 49.61 (CH2, C-6), 48.71 (CH2, C-2), 38.28 (CH, C-3), 27.67 (CH, C-4), 24.31 (CH2, C-5), 19.00 (CH2, C-7), and -1.06 (CH3, -SiMe3); H,H-Cosy, HSQC and HMBC were made. Melting point: the substance decomposed at T = 147 °C; TLC (TBME: MeOH: ammonia 25%/100:10:1): rf-value = 0.19; IR: see spectrum in the supporting information; HR-MS (ESI, MeOH) calculated for C24H31N5O1Si1 + H+: 434.23706 Da, found 434.23733 Da.
(R)-(6-(4-(Trimethylsilyl)-1H-1,2,3-triazol-1-yl)quinolin-4-yl)((1S,2S,4S,5R)-5-vinylquinuclidin-2-yl)methanol (16): the title compound was synthesized from 9 (1.0 g, 3.0 mmol) to afford 16 (1.2 g, yield: 92%) as a colourless wax. 1H-NMR (CDCl3, 300 MHz) δ 8.61 (d, J = 4.4 Hz, H-2′), 8.34 (bs, H-Triazole-5), 8.28 (d, J = 1.6 Hz, H-5′), 7.97 (d, J = 9.1 Hz, H-8′), 7.90 (dd, J = 9.1 Hz, J = 1.6 Hz, H-7′), 7.52 (d, J = 4.4 Hz, H-3′), 5.87–5.75 (m, H-9), 5.69 (ddd, J = 17.5 Hz, J = 10.3 Hz, J = 7.4 Hz, H-10), 5.00–4.85 (m, H-11, H-11), 3.77–3.57 (m, H-6), 3.17–3.03 (m, H-2, H-8), 2.78–2.62 (m, H-2, H-6), 2.41–2.28 (m; H-3), 1.92–1.79 (m, H-4, H-7), 1.62–1.48 (m, H-5, H-5), 0.92–0.81 (m, H-7) 0.41 (s, 9H, -Si(CH3)3); 13C-NMR (CDCl3, 75 MHz) δ 150.44 (CH, C-2′), 149.23 (Cq, C-4′), 147.56 (Cq, C-Triazole-4), 146.96 (Cq, C-8a′); 140.20 (CH, C-10); 134.34 (Cq, C-6′), 131.78 (CH, C-8′), 127.79 (CH, C-Triazole-5), 125.50 (Cq, C-4a′), 121.54 (CH, C-7′), 119.57 (CH, C-3′), 115.03 (CH2, C-11), 112.93 (CH, C-5′), 69.83 (CH, C-9), 60.52 (CH, C-8), 56.12 (CH2, C-2), 43.07 (CH2, C-6), 39.15 (CH, C-3), 29.63 (CH, C-4), 27.52 (CH2, C-5), 21.12 (CH2, C-7), and -1.14 (CH3, -SiMe3); H,H-Cosy, HSQC and HMBC were made. Melting point: the substance decomposed at T = 113 °C; TLC (TBME: MeOH: ammonia 25%/100:10:1): rf-value = 0.15; IR: see spectrum in the supporting information; HR-MS (ESI, MeOH) calculated for C24H31N5O1Si1 + H+: 434.23706 Da, found 434.23742 Da.
(S)-(6-(4-(Trimethylsilyl)-1H-1,2,3-triazol-1-yl)quinolin-4-yl)((1S,2R,4S,5R)-5-ethylquinuclidin-2-yl)methanol (18): the title compound was synthesized from 10 (0.5 g, 1.5 mmol) to afford 18 (0.6 g, yield: 94%) as a colourless honey-type liquid. 1H-NMR (CDCl3, 300 MHz) δ 8.77 (bs, H-Triazole-5), 8.47 (d, J = 4.4 Hz, H-2′), 8.13–8.09 (m, H-5′, H-7′), 7.81 (d, J = 9.7 Hz, H-8′), 7.34 (d, J = 4.4 Hz, H-3′), 6.38 (bs, H-9), 4.19–3.92 (m, H-2), 3.28–3.04 (m, H-2, H-8), 3.08–2.91 (m, H-6), 2.52–2.37 (m, H-3), 2.31–2.17 (m, H-6), 1.92–1.85 (m, H-4), 1.81–1.70 (m, H-7), 1.61–1.49 (m, H-5), 1.29–1.15 (m, H-5, H-10, H-10), 0.95–0.83 (m, H-7) 0.79 (t, J = 7.2 Hz, H-11, H-11, H-11), and 0.42 (s, 9H, -Si(CH3)3); 13C-NMR (CDCl3, 75MHz) δ 150.20 (CH, C-2′), 148.27 (Cq, C-4′), 147.43 (Cq, C-Triazole-4), 146.70 (Cq, C-8a′); 134.39 (Cq, C-6′), 131.74 (CH; C-8′), 128.03 (CH, C-Triazole-5), 124.98 (Cq, C-4a′), 121.33 (CH, C-7′), 119.33 (CH; C-3′), 111.88 (CH, C-5′), 68.77 (CH, C-9), 60.33 (CH, C-8), 50.42 (CH2, C-6), 49.67 (CH2, C-2), 36.38 (CH, C-3), 26.88 (CH2, C-10), 25.58 (CH, C-4), 24.69 (CH2, C-5), 19.48 (CH2, C-7), 11.75 (CH3, C-11), and -1.15 (CH3, -SiMe3); H,H-Cosy, HSQC and HMBC were made. Melting point: the substance decomposed at T = 166 °C; TLC (TBME: MeOH: ammonia 25%/100:10:1): rf-value = 0.11; IR: see spectrum in the supporting information; HR-MS (ESI, MeOH) calculated for C24H33N5O1Si1 + H+: 436.25271 Da, found 436.25296 Da.
(R)-(6-(4-(Trimethylsilyl)-1H-1,2,3-triazol-1-yl)quinolin-4-yl)((1S,2S,4S,5R)-5-ethylquinuclidin-2-yl)methanol (20): the title compound was synthesized from 11 (0.5 g, 1.5 mmol) to afford 20 (0.57 g, yield: 89%) as a honey-type liquid. 1H-NMR (CDCl3, 300MHz) δ 8.63 (d, J = 4.5 Hz, H-2′), 8.35 (bs, H-Triazole-5), 8.24 (d, J = 1.9 Hz, H-5′), 8.00 (d, J = 9.1 Hz, H-8′), 7.95 (dd, J = 9.1 Hz, J = 1.9 Hz, H-7′), 7.51 (d, J = 4.5 Hz, H-3′), 5.84–5.73 (m, H-9), 5.69 3.73–3.57 (m, H-6), 3.16–3.02 (m, H-2, H-8), 2.77–2.63 H-6), 2.47–2.33 (m, H-2), 1.93–1.77 (m; H-4, H-7), 1.57–1.40 (m, H-3, H-5, H-7), 1.29–1.15 (m, H-5, H-10, H-10), 0.78 (t, J = 7.3 Hz, H-11, H-11, H-11), and 0.43 (s, 9H, -Si(CH3)3); 13C-NMR (CDCl3, 75 MHz) δ 150.50 (CH, C-2′), 149.18 (Cq, C-4′), 147.58 (Cq, C-Triazole-4), 147.04 (Cq, C-8a′); 134.45 (Cq, C-6′), 131.89 (CH, C-8′), 127.83 (CH, C-Triazole-5), 125.49 (Cq, C-4a′), 121.64 (CH, C-7′), 119.53 (CH, C-3′), 112.84 (CH, C-5′), 69.96 (CH, C-9), 60.36 (CH, C-8), 57.97 (CH2, C-2), 43.23 (CH2, C-6), 36.95 (CH, C-3), 27.39 (CH2, C-5), 27.37 (CH2, C-10), 25.18 (CH, C-4), 20,76 (CH2, C-7), 11.88 (CH3, C-11), and -1.11 (CH3, -SiMe3); H,H-Cosy, HSQC and HMBC were made. Melting point: the substance decomposed at T = 136 °C; TLC (TBME: MeOH: ammonia 25%/100:10:1): rf-value = 0.16; IR: see spectrum in the supporting information; HR-MS (ESI, MeOH) calculated for C24H33N5O1Si1 + H+: 436.25271 Da, found 436.25305 Da.
4.1.4. Synthesis of the Triazole-Derivatives 15, 17, 19 and 21 via Desilylation
General procedure: a purified Cinchona TMS-triazole (1.00 eq.) was suspended in a mixture tBu-OH/H2O (1:1 ratio) and K2CO3 (2.0 equiv) was added. The reaction mixture was rigorously stirred for 48 h. Upon completion of the reaction, EtOAc was added and the organic layer was separated, washed with water, brine, dried over Na2SO4, filtered and concentrated in vacuo. The residues obtained were purified by flash column chromatography using MTBE: MeOH as a gradient elution to afford the corresponding triazole products.
(S)-(6-(1H-1,2,3-Triazol-1-yl)quinolin-4-yl)((1S,2R,4S,5R)-5-vinylquinuclidin-2-yl)methanol (15): the title compound was synthesized from 14 (1.0 g, 2.3 mmol) to afford 15 (672 mg, yield: 81%) as a white solid. 1H-NMR (CDCl3, 600 MHz) δ 8.68 (d, J = 4.5 Hz, H-2′), 8.34 (d, J = 2.2 Hz, H-5′), 8.23 (d, J = 0.8 Hz, H-Triazole-5), 8.04 (d, J = 9.0 Hz, H-8′), 7.93 (dd, J = 9.0 Hz, J = 2.2 Hz, H-7′), 7.77 (d, J = 0.8 Hz, H-Triazole-4), 7.58 (d, J = 4.5 Hz, H-3′), 6.04 (ddd, J = 16.7 Hz, J = 10.3 Hz, J = 7.5 Hz, H-10), 5.69–5.66 (m, H-9), 5.08–5.01 (m, H-11, H-11), 3.33–3.25 (m, H-2), 3.06–3.00 (m, H-8), 2.88–2.78 (m, H-2, H-6), 2.72–2.64 (m, H-6), 2.26–2.19 (m, H-3), 2.12–2.03 (m, H-7), 1.80–1.75 (m, H-4), 1.56–1.45 (m, H-5, H-5), and 1.28–1.20 (m, H-7); 13C-NMR (CDCl3, 150 MHz) δ 150.71 (CH, C-2′), 150.24 (Cq, C-4′), 147.13 (Cq, C-8a′); 140.30 (CH, C-10); 134.54 (CH, C-Triazole-4), 134.22 (Cq, C-6′), 131.86 (CH; C-8′), 125.78 (Cq, C-4a′), 122.07 (CH, C-Triazole-5), 121.41 (CH, C-7′), 119.60 (CH; C-3′), 114.73 (CH2, C-11), 113.70 (CH, C-5′), 71.11 (CH, C-9), 60.25 (CH, C-8), 49.73 (CH2, C-6), 49.11 (CH2, C-2), 39.78 (CH, C-3), 28.01 (CH, C-4), 26.16 (CH2, C-5), and 21.42 (CH2, C-7); H,H-Cosy, HSQC and HMBC were made. Melting point: the substance decomposed at T = 117 °C; TLC (TBME: MeOH: ammonia 25%/100:10:1): rf-value = 0.56; IR: see spectrum in the supporting information; HR-MS (ESI, MeOH) calculated for C21H23N5O1 + H+: 362.19754 Da, found 362.19785 Da.
(R)-(6-(1H-1,2,3-Triazol-1-yl)quinolin-4-yl)((1S,2S,4S,5R)-5-vinylquinuclidin-2-yl)methanol (17): the title compound was synthesized from 16 (1.0 g, 2.3 mmol) to afford 17 (725 mg, yield: 87%) as a white solid. 1H-NMR (CDCl3, 600 MHz) δ 8.59 (d, J = 4.5 Hz, H-2′), 8.49 (bs, H-Triazole-5), 8.37 (d, J = 1.8 Hz, H-5), 8.00 (dd, J = 9.1 Hz, J = 1.8 Hz, H-7′), 7.95 (d, J = 9.1 Hz, H-8′), 7.74 (bs, H-Triazole-4), 7.59 (d, J = 4.5 Hz, H-3′), 5.77–5.68 (m, H-9), 5.70 (ddd, J = 17.4 Hz, J = 10.3 Hz, J = 7.5 Hz, H-10), 4.96–4.85 (m, H-11, H-11), 3.67–3.53 (m, H-6), 3.12–2.98 (m, H-2, H-8), 2.73–2.58 (m, H-2, H-6), 2.35–2.25 (m; H-3), 1.92–1.76 (m, H-4, H-5, H-7), and 1.60–1.48 (m, H-5, H-7); 13C-NMR (CDCl3, 150 MHz) δ 150.19 (CH, C-2′), 150.09 (Cq, C-4′), 146.62 (Cq, C-8a′); 140.77 (CH, C-10); 134.14 (CH, C-Triazole-4), 134.12 (Cq, C-6′), 131.31 (CH; C-8′), 125.31 (Cq, C-4a′), 122.07 (CH, C-Triazole-5), 121.14 (CH, C-7′), 119.27 (CH; C-3′), 114.41 (CH2, C-11), 112.73 (CH, C-5′), 70.03 (CH, C-9), 60.22 (CH, C-8), 56.06 (CH2, C-2), 42.70 (CH2, C-6), 39.06 (CH, C-3), 27.29 (CH, C-4), 26.73 (CH2, C-5), and 20.92 (CH2, C-7); H,H-Cosy, HSQC and HMBC were made. Melting point: the substance decomposed at T = 92 °C; TLC (TBME: MeOH: ammonia 25%/100:10:1): rf-value = 0.63; IR: see spectrum in the supporting information; HR-MS (ESI, MeOH) calculated for C21H23N5O1 + H+: 362.19754 Da, found 362.19796 Da.
(S)-(6-(1H-1,2,3-Triazol-1-yl)quinolin-4-yl)((1S,2R,4S,5R)-5-ethylquinuclidin-2-yl)methanol (19): the title compound was synthesized from 18 (0.5 g, 1.15 mmol) to afford 19 (359 mg, yield: 86%) as an off-white solid. 1H-NMR (CDCl3, 300 MHz) δ 8.54 (d, J = 4.5 Hz, H-2′), 8.26 (d, J = 2.2 Hz, H-5′), 8.35 (d, J = 0.8 Hz, H-Triazole-5), 8.98 (d, J = 9.0 Hz, H-8′), 7.65 (dd, J = 9.0 Hz, J = 2.2 Hz, H-7′), 7.98 (d, J = 0.8 Hz, H-Triazole-4), 7.94 (d, J = 4.5 Hz, H-3′), 5.87–5.77 (m, H-9), 3.31–3.20 (m, H-2), 3.11–3.08 (m, H-8), 2.85–2.73 (m, H-2, H-6), 2.69–2.58 (m, H-6), 2.21–2.15 (m, H-3), 2.32–2.23 (m, H-7), 1.85–1.80 (m, H-4), 1.51–1.40 (m, H-5, H-5, H-10, H-10), 1.22–1.14 (m, H-7), and 0.81 (t, J = 7.2 Hz, H-11, H-11, H-11); 13C-NMR (CDCl3, 75 MHz) δ 13C-NMR (CDCl3, 75MHz) δ 150.81 (CH, C-2′), 149.80 (Cq, C-4′), 147.27 (Cq, C-8a′), 134.62 (CH, H-Triazole-4), 134.30 (Cq, C-6′), 132.07 (CH; C-8′), 125.75 (Cq, C-4a′), 122.12 (CH, H-Triazole-5), 121.40 (CH, C-7′), 119.60 (CH; C-3′), 113.56 (CH, C-5′), 70.93 (CH, C-9), 60.38 (CH, C-8), 50.74 (CH2, C-6), 49.87 (CH2, C-2), 37.12 (CH, C-3), 26.76 (CH, C-4), 26.02 (CH2, C-10), 25.08 (CH2, C-5), 21.09 (CH2, C-7), and 11.92 (CH3, C-11); H,H-Cosy, HSQC and HMBC were made. Melting point: Substance decompose at T = 143 °C; TLC (TBME: MeOH: ammonia 25%/100:10:1): rf-value = 0.45; IR: see spectrum in the supporting information; HR-MS (ESI, MeOH) calculated for C21H25N5O1 + H+: 364.21319 Da, found 364.21347 Da.
(R)-(6-(1H-1,2,3-Triazol-1-yl)quinolin-4-yl)((1S,2S,4S,5R)-5-ethylquinuclidin-2-yl)methanol (21): the title compound was synthesized from 20 (0.5 g, 1.15 mmol) to afford 21 (346 mg, yield: 84%) as an off-white solid. 1H-NMR (CDCl3, 300 MHz) δ 8.68 (d, J = 4.5 Hz, H-2′), 8.37(d, J = 2.3 Hz, H-5), 8.29 (d, J = 1.0 Hz, H-Triazole-5), 8.05 (d, J = 9.1 Hz, H-8′), 7.96 (dd, J = 9.1 Hz, J = 2.3 Hz, H-7′), 7.80 (d, J = 1.0 Hz, H-Triazole-4), 7.57 (d, J = 4.57 Hz, H-3′), 5.69–5.59 (m, H-9), 3.47–3.41 (m, H-6), 3.11–2.96 (m, H-2, H-8), 2.65–2.55 (m, H-6), 2.35–2.29 (m, H-2), 1.79–1.71 (m; H-4, H-5, H-7), 1.60–1.41 (m, H-3, H-10, H-10), 1.34–1.18 (m, H-5, H-7), and 0.79 (t, J = 7.3 Hz, H-11, H-11, H-11); 13C-NMR (CDCl3, 75MHz) δ 150.79 (CH, C-2′), 150.07 (Cq, C-4′), 147.25 (Cq, C-8a′); 134.60 (CH, H-Triazole-4), 134.36 (Cq, C-6′), 132.01 (CH, C-8′), 125.80 (Cq, C-4a′), 122.27 (CH, H-Triazole-5), 121.49 (CH, C-7′), 119.65 (CH, C-3′), 113.51 (CH, C-5′), 70.85 (CH, C-9), 60.39 (CH, C-8), 58.18 (CH2, C-2), 43.03 (CH2, C-6), 37.20 (CH, C-3), 27.88 (CH2, C-5), 27.52 (CH2, C-10), 25.27 (CH, C-4), 21.70 (CH2, C-7), and 11.96 (CH3, C-11); H,H-Cosy, HSQC and HMBC were made. Melting point: the substance decomposed at T = 106 °C; TLC (TBME: MeOH: ammonia 25%/100:10:1): rf-value = 0.58; IR: see spectrum in the supporting information; HR-MS (ESI, MeOH) calculated for C21H25N5O1 + H+: 364.21319 Da, found 364.21358 Da.
4.1.5. Synthesis of the Quinuclidine Containing Triazole-Derivatives 24–31
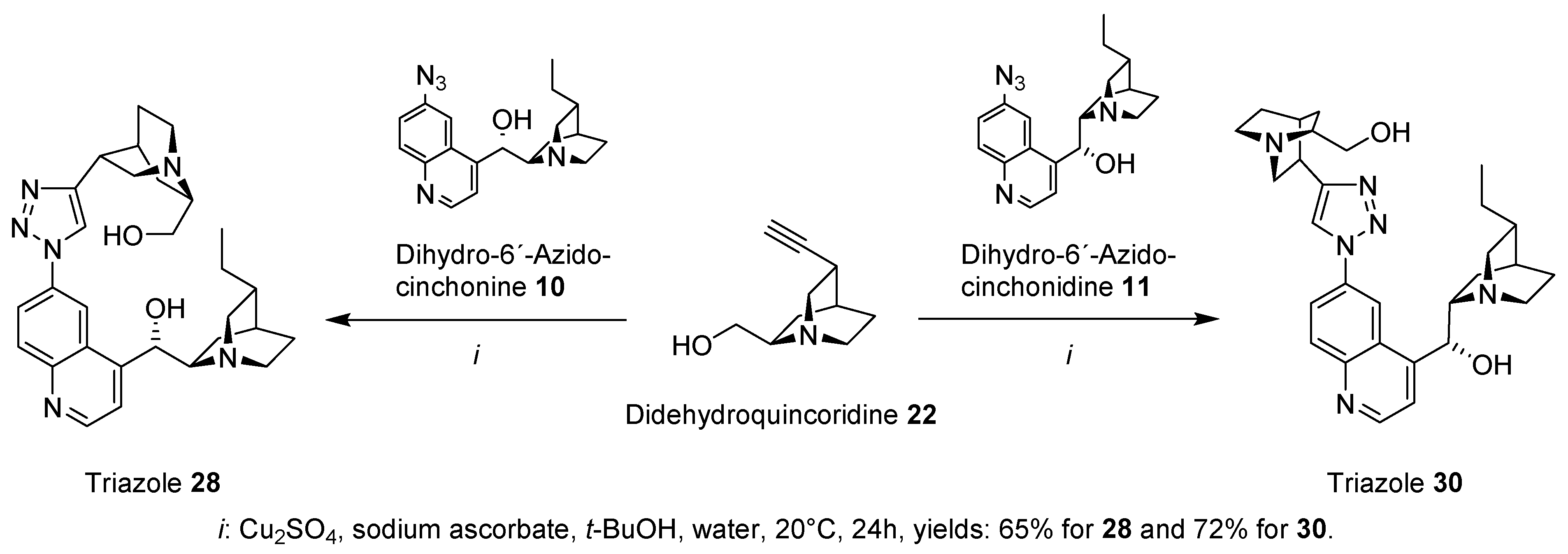
General procedure: a dropwise a solution of NaNO2(1.2 eq) in H2O (0.33 g/mL) was added to a mixture of Cinchona amine (1.0 eq) and 15% HCl (0.1g amine/mL) cooled at −5 °C. After the completion of the addition, the reaction mixture was stirred at this temperature for 60 min. A solution of NaN3 (1.7 eq) in H2O (0.2 g/mL) was added dropwise to the reaction mixture at 0 °C. After the addition was finished, the reaction mixture was maintained at 0 °C for 2 h and then stirred at room temperature for 12 h. The product was precipitated by the addition of NaOH (20%) until pH 10. Upon filtration, the solid was washed with distilled water and dried. Crystallization from DCM presented the Cinchona azides as yellow solids in good yields (70–90%).
(S)-(6-(4-((1S,3R,4S,6R)-6-(Hydroxymethyl)quinuclidin-3-yl)-1H-1,2,3-triazol-1-yl)quinolin-4-yl)((1S,2R,4S,5R)-5-vinylquinuclidin-2-yl)methanol (24): the title compound was synthesized from 8 (1.0 g, 3.0 mmol) to afford 24 (925 mg, yield: 62%) as a reddish solid. 1H-NMR (CDCl3, 400 MHz) δ 8.87–8.72 (1 H), 8.41–8.28 (2 H), 8.39–7.54 (2 H), 7.69–7.58 (1 H), 6.27–6.56 (1 H), 5.73–5.65 (1 H), 5.21–5.11 (2 H), 3.75–3.29 (4 H), 3.11–2.75 (6 H), 2.89–2.75 (2 H), 2.46–2.33 (1 H), 2.11–2.01 (2 H), 1.78–1.35 (7 H), 1.29–1.15 (1 H), and 0.89–0.76 (1 H); 13C-NMR (CDCl3, 100 MHz) δ 151.20 (Cq), 150.54 (CH), 150.34 (Cq), 146.96 (Cq); 140.45 (CH), 134.55 (CH), 131.79 (CH), 125.49 (Cq), 121.19 (CH), 119.36 (CH), 119.22 (CH), 114.73 (CH2), 112.50 (CH), 70.67 (CH), 62.64 (CH2), 59.94 (CH), 57.30 (CH), 49.83 (CH2), 49.30 (CH2), 48.89 (CH2), 49.77 (CH2), 39.98 (CH), 33.36 (CH), 28.15 (CH), 27.55 (CH2), 26.35 (CH), 26.16 (CH2), 23.71 (CH2), and 20.08 (CH2); Melting point: the substance decomposed at T = 126 °C; TLC (TBME: MeOH: ammonia 25%/100:10:1): rf-value = 0.21; IR: see spectrum in the supporting information; HR-MS (ESI, MeOH) calculated for C29H36N6O2 + H+: 501.29725 Da, found 501.29739 Da.
(S)-(6-(4-((1S,3R,4S,6S)-6-(Hydroxymethyl)quinuclidin-3-yl)-1H-1,2,3-triazol-1-yl)quinolin-4-yl)((1S,2R,4S,5R)-5-vinylquinuclidin-2-yl)methanol (25): the title compound was synthesized from 8 (1.0 g, 3.0 mmol) to afford 25 (1.0 g, yield: 68%) as a pink solid. 1H-NMR (CDCl3, 400 MHz) δ 8.71–8.64 (1 H), 8.37–8.25 (2 H), 8.00–7.88 (2 H), 7.62–7.56 (1 H), 6.16–6.04 (1 H), 5.82–5.70 (1 H), 5.13–5.01 (2 H), 3.64–3.19 (4 H), 3.16–2.82 (6 H), 2.80–2.69 (2 H), 2.32–2.21 (1 H), 2.19–2.08 (2 H), 1.82–1.43 (7 H), 1.25–1.13 (1 H), and 0.94–0.83 (1 H); 13C-NMR (CDCl3, 100 MHz) δ 151.73 (Cq), 150.36 (CH), 150.24 (Cq), 146.82 (Cq); 140.17 (CH), 134.35 (CH), 131.42 (CH), 125.60 (Cq), 121.08 (CH), 119.35 (CH), 119.12 (CH), 114.71 (CH2), 112.86 (CH), 70.3 (CH), 63.33 (CH2), 59.59 (CH), 57.28 (CH), 54.75 (CH2), 49.61 (CH2), 49.20 (CH2), 40.44 (CH2), 39.75 (CH), 33.02 (CH), 27.96 (CH), 27.20 (CH2), 27.06 (CH), 25.97 (CH2), 24.89 (CH2), and 20.92 (CH2). Melting point: the substance decomposed at T = 119 °C; TLC (TBME: MeOH: ammonia 25%/100:10:1): rf-value = 0.20; IR: see spectrum in the supporting information; HR-MS (ESI, MeOH) calculated for C29H36N6O2 + H+: 501.29725 Da, found 501.29742 Da.
(R)-(6-(4-((1S,3R,4S,6R)-6-(Hydroxymethyl)quinuclidin-3-yl)-1H-1,2,3-triazol-1-yl)quinolin-4-yl)((1S,2S,4S,5R)-5-vinylquinuclidin-2-yl)methanol (26): the title compound was synthesized from 9 (1.0 g, 3.0 mmol) to afford 26 (1.1 g, yield: 74%) as a light red solid.1 H-NMR (CDCl3, 300 MHz) δ 8.61–8.54 (1 H), 8.67–8.54 (2 H), 8.13–7.87 (2 H), 7.68–7.46 (1 H), 6.96–6.64 (1 H), 5.34–5.23 (1 H), 5.17–5.03 (2 H), 3.78–3.35 (4 H), 3.26–2.92 (6 H), 2.63–2.46 (2 H), 2.39–2.26 (1 H), 2.12–2.01 (2 H), 1.76–1.32 (7 H), 1.19–1.07 (1 H), and 0.97–0.85 (1 H); 13C-NMR (CDCl3, 75 MHz) δ 152.08 (Cq), 150.67 (CH), 148.43 (Cq), 147.67 (Cq); 141.21 (CH), 133.96 (CH), 132.54 (CH), 126.31 (Cq), 122.45 (CH), 120.08 (CH), 119.23 (CH), 115.76 (CH2), 111.47 (CH), 69.93 (CH), 62.48 (CH2), 61.13 (CH2), 60.67 (CH), 57.79 (CH2), 56.36 (CH2), 54.76 (CH2), 42.98 (CH2), 40.54 (CH), 34.65 (CH), 27.87 (CH), 26.03 (CH), 25.53 (CH2), 24.52 (CH2), 21.73 (CH), and 19.26 (CH2). Melting point: the substance decomposed at T = 110 °C; TLC (TBME: MeOH: ammonia 25%/100:10:1): rf-value = 0.16; IR: see spectrum in the supporting information; HR-MS (ESI, MeOH) calculated for C29H36N6O2 + H+: 501.29725 Da, found 501.29747 Da.
(R)-(6-(4-((1S,3R,4S,6S)-6-(Hydroxymethyl)quinuclidin-3-yl)-1H-1,2,3-triazol-1-yl)quinolin-4-yl)((1S,2S,4S,5R)-5-vinylquinuclidin-2-yl)methanol (27): the title compound was synthesized from 9 (1.0 g, 3.0 mmol) to afford 27 (1.15 mg, yield: 76%) as a pink solid. 1H-NMR (CDCl3, 300 MHz) δ 8.71–8.64 (1 H), 8.37–8.25 (2 H), 8.00–7.88 (2 H), 7.62–7.56 (1 H), 6.16–6.04 (1 H), 5.82–5.70 (1 H), 5.13–5.01 (2 H), 3.64–3.19 (4 H), 3.16–2.82 (6 H), 2.80–2.69 (2 H), 2.32–2.21 (1 H), 2.19–2.08 (2 H), 1.82–1.43 (7 H), 1.25–1.13 (1 H), and 0.94–0.83 (1 H); 13C-NMR (CDCl3, 75 MHz) δ 151.99 (Cq), 150.65 (CH), 147.30 (Cq), 147.26 (Cq); 140.48 (CH), 133.16 (CH), 132.17 (CH), 125.47 (Cq), 121.58 (CH), 119.79 (CH), 119.33 (CH), 115.07 (CH2), 112.09 (CH), 69.33 (CH), 63.43 (CH2), 60.54 (CH2), 60.34 (CH), 57.43 (CH2), 55.89 (CH2), 54.16 (CH2), 43.25 (CH2), 40.19 (CH), 33.11 (CH), 27.49 (CH), 26.96 (CH), 26.79 (CH2), 24.71 (CH2), 21.00 (CH), and 19.88 (CH2). Melting point: the substance decomposed at T = 103 °C; TLC (TBME: MeOH: ammonia 25%/100:10:1): rf-value = 0.12; IR: see spectrum in the supporting information; HR-MS (ESI, MeOH) calculated for C29H36N6O2 + H+: 501.29725 Da, found 501.29753 Da.
(S)-(6-(4-((1S,3R,4S,6R)-6-(Hydroxymethyl)quinuclidin-3-yl)-1H-1,2,3-triazol-1-yl)quinolin-4-yl)((1S,2R,4S,5R)-5-ethylquinuclidin-2-yl)methanol (28): the title compound was synthesized from 10 (0.5 g, 1.5 mmol) to afford 28 (490 mg, yield: 65%) as an off-white solid. 1H-NMR (CDCl3, 300 MHz) δ 8.75–8.54 (1 H), 8.28–8.14 (2 H), 8.01–7.82 (2 H), 7.59–7.54 (1 H), 5.73–5.61 (1 H), 3.79–3.27 (4 H), 3.21–2.88 (6 H), 2.81–2.61 (2 H), 2.41–2.32 (1 H), 2.26–2.18 (2 H), 1.89–1.31 (9 H), 1.21–1.12 (1 H), and 0.99–0.81 (4 H); 13C-NMR (CDCl3, 75 MHz) δ 152.71 (Cq), 150.98 (CH), 150.37 (Cq), 147.63 (Cq), 135.01 (CH), 132.23 (CH), 126.29 (Cq), 121.78 (CH), 119.73 (CH), 118.61 (CH), 113.93 (CH), 71.35 (CH), 62.67 (CH2), 60.59 (CH), 58.79 (CH), 55.23 (CH2), 50.69 (CH2), 49.13 (CH2), 41.65 (CH2), 40.72 (CH), 33.19 (CH), 28.17 (CH), 27.64 (CH2), 27.13 (CH), 26.27 (CH2), 25.93 (CH2), 24.63 (CH2), 21.47 (CH2), and 12.65 (CH3). Melting point: the substance decomposed at T = 145 °C; TLC (TBME: MeOH: ammonia 25%/100:10:1): rf-value = 0.19; IR: see spectrum in the supporting information; HR-MS (ESI, MeOH) calculated for C29H38N6O2 + H+: 503.31290 Da, found 503.31304 Da.
(S)-(6-(4-((1S,3R,4S,6S)-6-(Hydroxymethyl)quinuclidin-3-yl)-1H-1,2,3-triazol-1-yl)quinolin-4-yl)((1S,2R,4S,5R)-5-ethylquinuclidin-2-yl)methanol (29): the title compound was synthesized from 10 (0.5 g, 1.5 mmol) to afford 29 (580 mg, yield: 78%) as an off-white solid; 1H-NMR (CDCl3, 400 MHz) δ 9.03–8.73 (1 H), 8.41–8.29 (2 H), 8.13–7.91 (2 H), 7.68–7.59 (1 H), 6.23–5.81 (1 H), 3.69–3.23 (4 H), 3.17–2.81 (6 H), 2.75–2.34 (2 H), 2.32–2.23 (1 H), 2.16–2.01 (2 H), 1.87–1.34 (9 H), 1.27–1.14 (1 H), and 0.91–0.87 (4 H); 13C-NMR (CDCl3, 100 MHz) δ 151.54 (Cq), 151.12 (CH), 150.27 (Cq), 146.12(Cq), 135.69 (CH), 130.19 (CH), 125.94 (Cq), 121.98 (CH), 120.15 (CH), 119.72 (CH), 113.26 (CH), 71.3 (CH), 63.99 (CH2), 60.57 (CH), 57.52 (CH), 55.75 (CH2), 49.69 (CH2), 49.13 (CH2), 41.64 (CH2), 40.73 (CH), 33.92 (CH), 27.56 (CH), 27.21 (CH2), 27.16 (CH), 25.47 (CH2), 25.22 (CH2), 24.83 (CH2), 21.62 (CH2), and 12.35 (CH3). Melting point: the substance decomposed at T = 138 °C; TLC (TBME: MeOH: ammonia 25%/100:10:1): rf-value = 0.21; IR: see spectrum in the supporting information; HR-MS (ESI, MeOH) calculated for C29H38N6O2 + H+: 503.31290 Da, found 503.31298 Da.
(R)-(6-(4-((1S,3R,4S,6R)-6-(Hydroxymethyl)quinuclidin-3-yl)-1H-1,2,3-triazol-1-yl)quinolin-4-yl)((1S,2S,4S,5R)-5-ethylquinuclidin-2-yl)methanol (30): the title compound was synthesized from 11 (0.5 g, 1.5 mmol) to afford 30 (535 mg, yield: 72%) as an off-white solid. 1H-NMR (CDCl3, 300 MHz) δ 8.62–8.49 (1 H), 8.38–8.27 (2 H), 7.99–7.81 (2 H), 7.71–7.60 (1 H), 5.89–5.65 (1 H), 5.17–4.89 (2 H), 3.73–3.29 (4 H), 3.21–2.94 (6 H), 2.81–2.73 (2 H), 2.54–2.36 (1 H), 2.28–2.17 (2 H), 1.93–1.49 (9 H), 1.29–1.14 (1 H), and 0.97–0.73 (4 H); 13C-NMR (CDCl3, 75 MHz) δ 152.21 (Cq), 151.25 (CH), 147.91 (Cq), 146.66 (Cq); 133.73 (CH), 131.17 (CH), 125.97 (Cq), 122.68 (CH), 120.19 (CH), 119.13 (CH), 112.34 (CH), 69.78 (CH), 64.41 (CH2), 61.64 (CH2), 60.52 (CH), 58.67 (CH2), 56.37 (CH2), 54.73 (CH2), 44.87 (CH2), 40.36 (CH), 33.57 (CH), 28.19 (CH), 27.91 (CH), 26.71 (CH2), 25.52 (CH2), 24.11 (CH2), 21.68 (CH), 20.18 (CH2), and 12.37 (CH3). Melting point: the substance decomposed at T = 132 °C; TLC (TBME: MeOH: ammonia 25%/100:10:1): rf-value = 0.14; IR: see spectrum in the supporting information; HR-MS (ESI, MeOH) calculated for C29H38N6O2 + H+: 503.31290 Da, found 503.31312 Da.
(R)-(6-(4-((1S,3R,4S,6S)-6-(Hydroxymethyl)quinuclidin-3-yl)-1H-1,2,3-triazol-1-yl)quinolin-4-yl)((1S,2S,4S,5R)-5-ethylquinuclidin-2-yl)methanol (31): the title compound was synthesized from 11 (0.5 g, 1.5 mmol) to afford 31 (610 mg, yield: 82%) as an off-white solid. 1H-NMR (CDCl3, 300 MHz) δ 8.89–8.71 (1 H), 8.39–8.31 (2 H), 8.23–8.12 (2 H), 7.73–7.66 (1 H), 5.79–5.65 (1 H), 5.56–5.23 (2 H), 3.81–3.39 (4 H), 3.25–2.93 (6 H), 2.85–2.71 (2 H), 2.54–2.35 (1 H), 2.23–2.13 (2 H), 1.97–1.51 (9 H), 1.33–1.21 (1 H), and 0.89–0.68 (4 H); 13C-NMR (CDCl3, 75 MHz) δ 152.00 (Cq), 150.69 (CH), 150.35 (Cq), 147.34 (Cq); 135.10 (CH), 132.16 (CH), 125.65 (Cq), 121.59 (CH), 119.93 (CH), 119.42 (CH), 112.46 (CH), 69.27 (CH), 63.47 (CH2), 60.59 (CH2), 60.33 (CH), 57.67 (CH2), 57.43 (CH2), 54.13 (CH2), 40.22 (CH2), 36.83 (CH), 33.10 (CH), 27.40 (CH), 27.00 (CH), 26.89 (CH2), 25.16 (CH2), 24.79 (CH2), 20.99 (CH), 20.10 (CH2), and 11.83 (CH3). Melting point: the substance decomposed at T = 121 °C; TLC (TBME: MeOH: ammonia 25%/100:10:1): rf-value = 0.14; IR: see spectrum in the supporting information; HR-MS (ESI, MeOH) calculated for C29H38N6O2 + H+: 503.31290 Da, found 503.31306 Da.
4.1.6. X-ray Structure Determination
The single crystals were mounted on a Hampton loop using perfluoroether oil and placed in the cold nitrogen gas stream on the diffractometer [
29]. The data were collected on a Rigaku Oxford Diffraction Synergy-S using mirror-focused CuK
α radiation. The reflections were indexed, integrated, and appropriate absorption corrections were applied as implemented in the CrysAlisPro software package [
30]. The structures were solved employing the program SHELXT and refined an isotropically for all non-hydrogen atoms by full-matrix least squares on all F2 using SHELXL software [
31,
32]. Carbon bound hydrogen atoms were refined employing a riding model; methyl groups were treated as rigid bodies and were allowed to rotate about the E–CH3 bond. Nitrogen and oxygen bound hydrogen atoms were located in the difference Fourier map and were refined freely for
10 and
11, whilst for
28 geometrical restraints and constraints for the displacement parameters were employed. For
28, the structure contains a partly occupied HCl and two partly occupied units of water. The absolute structure parameter suggests numerically a very small contribution of an inversion twined component. As the only heavy atom (Cl1) is only partly occupied, a twin refinement was considered not reliable and was not conducted. During refinement and analysis of the crystallographic data, the programs OLEX2 and Diamond were used [
33,
34]. Unless noted otherwise, the shown ellipsoids represent the 50% probability level and hydrogen atoms are displayed with an arbitrary radius.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}