
Chalcogen Bonding in Co-Crystals: Activation through 1,4-Perfluorophenylene vs. 4,4′-Perfluorobiphenylene Cores


Abstract
:1. Introduction
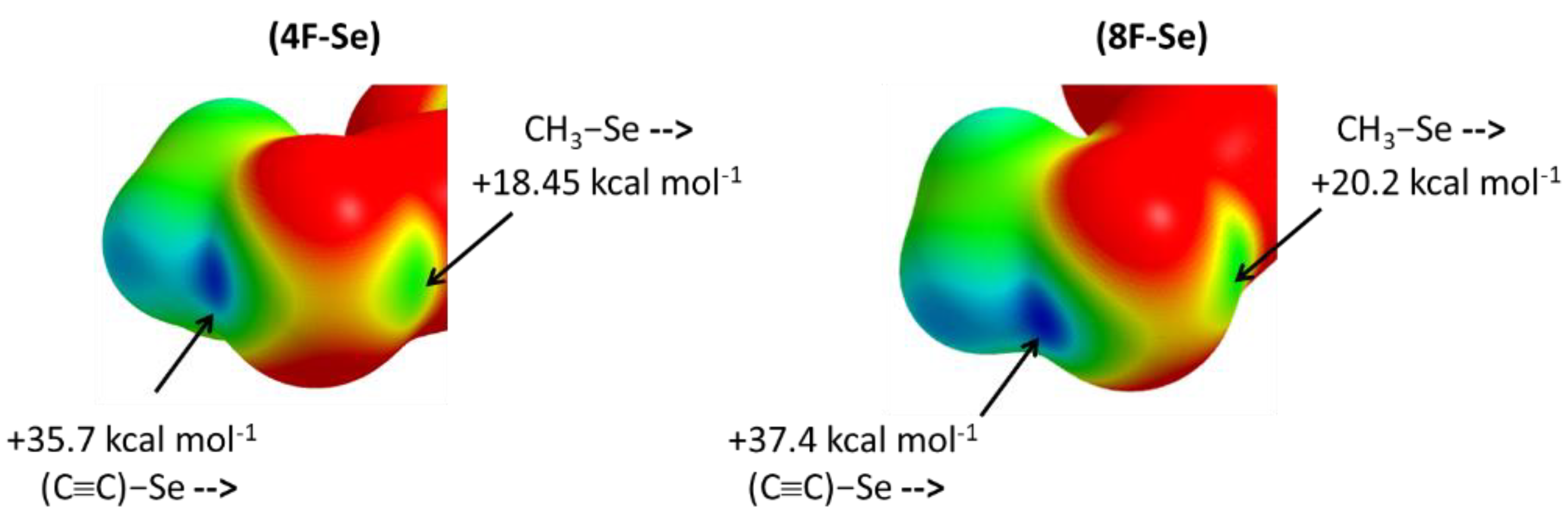
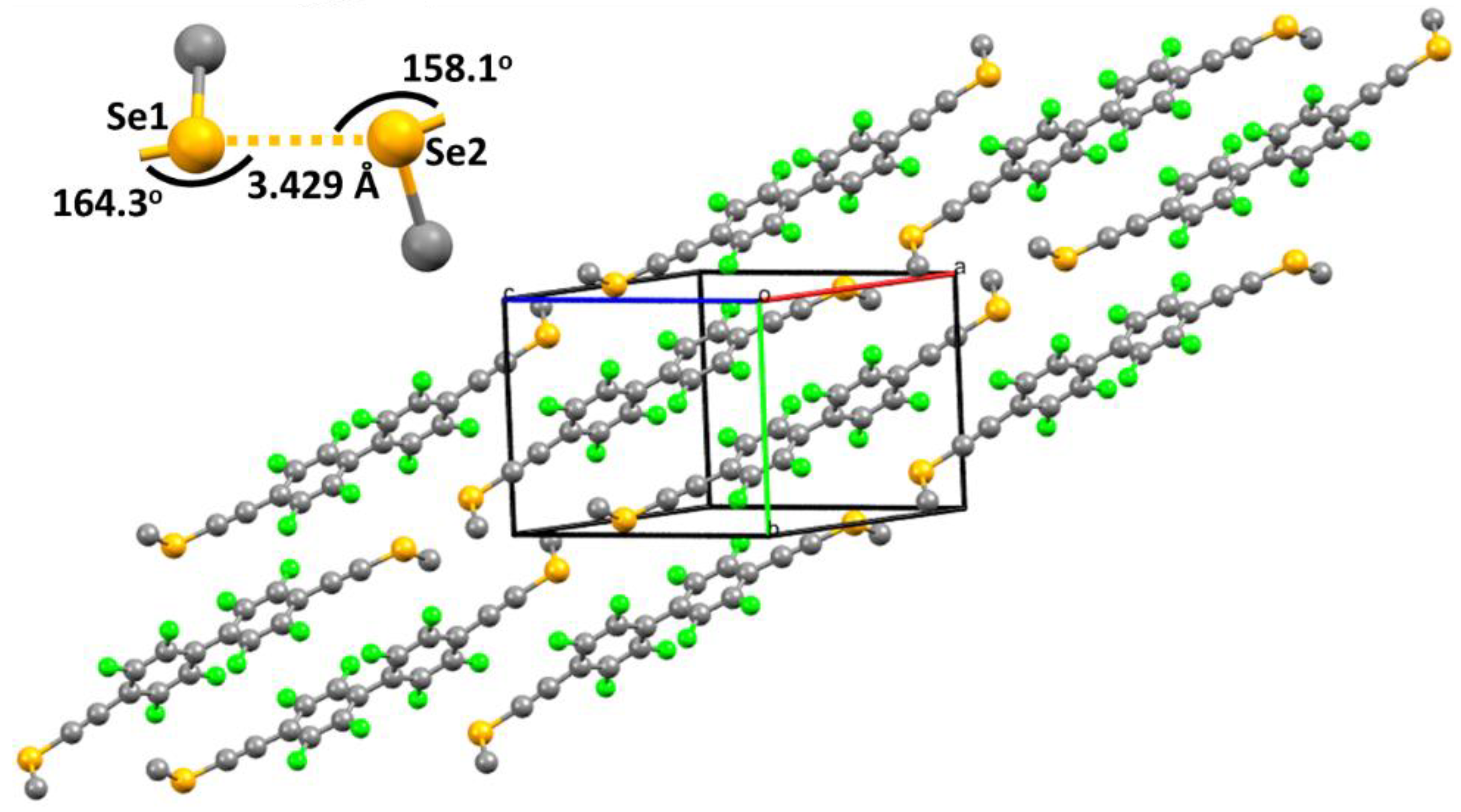
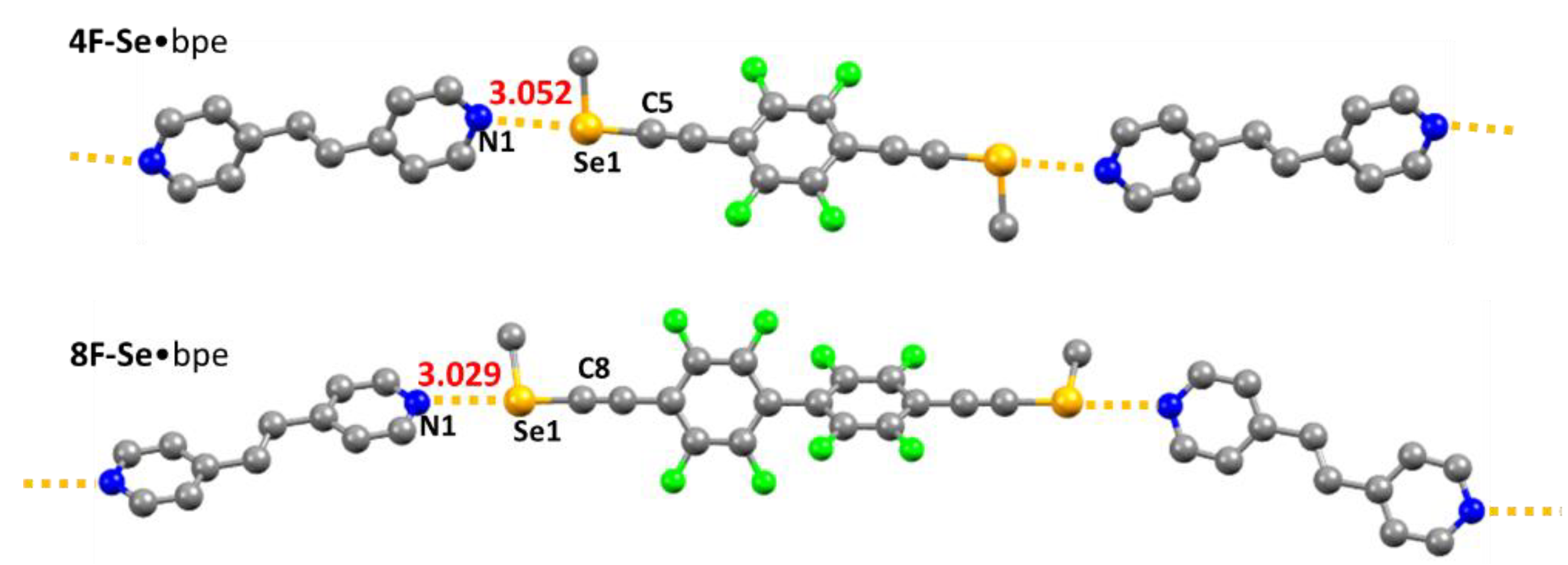
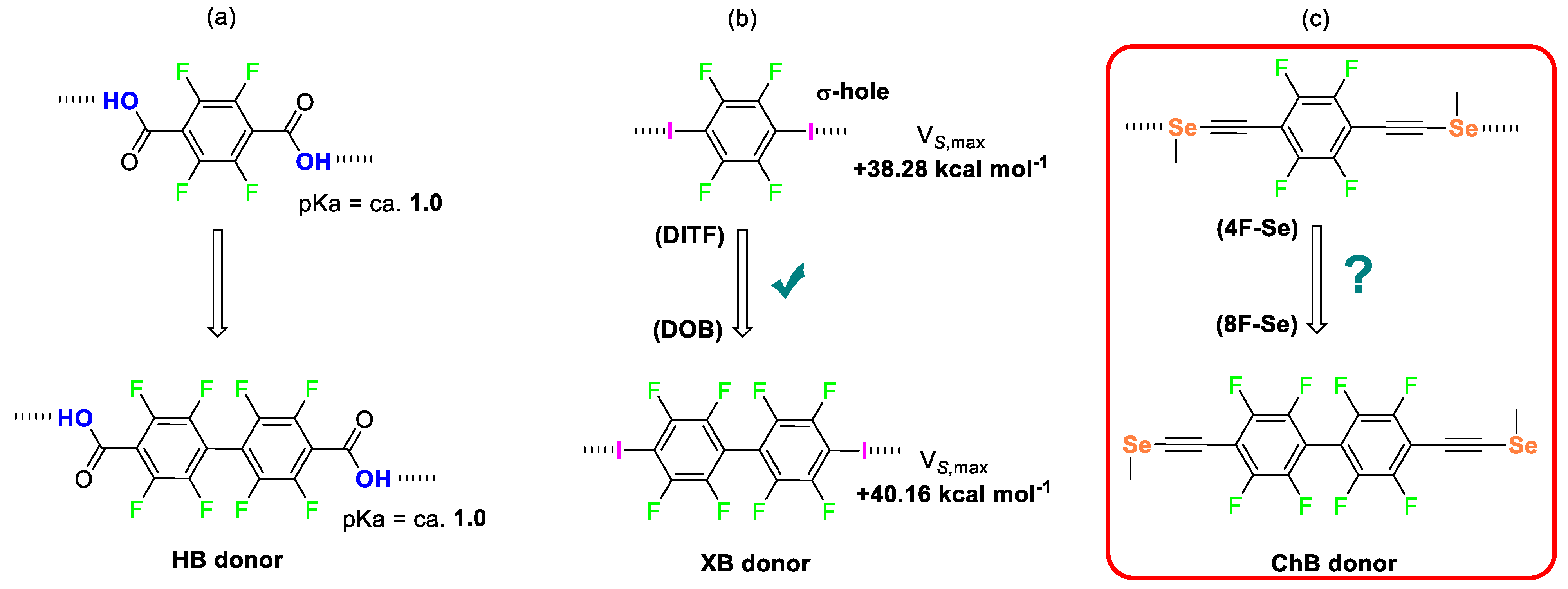
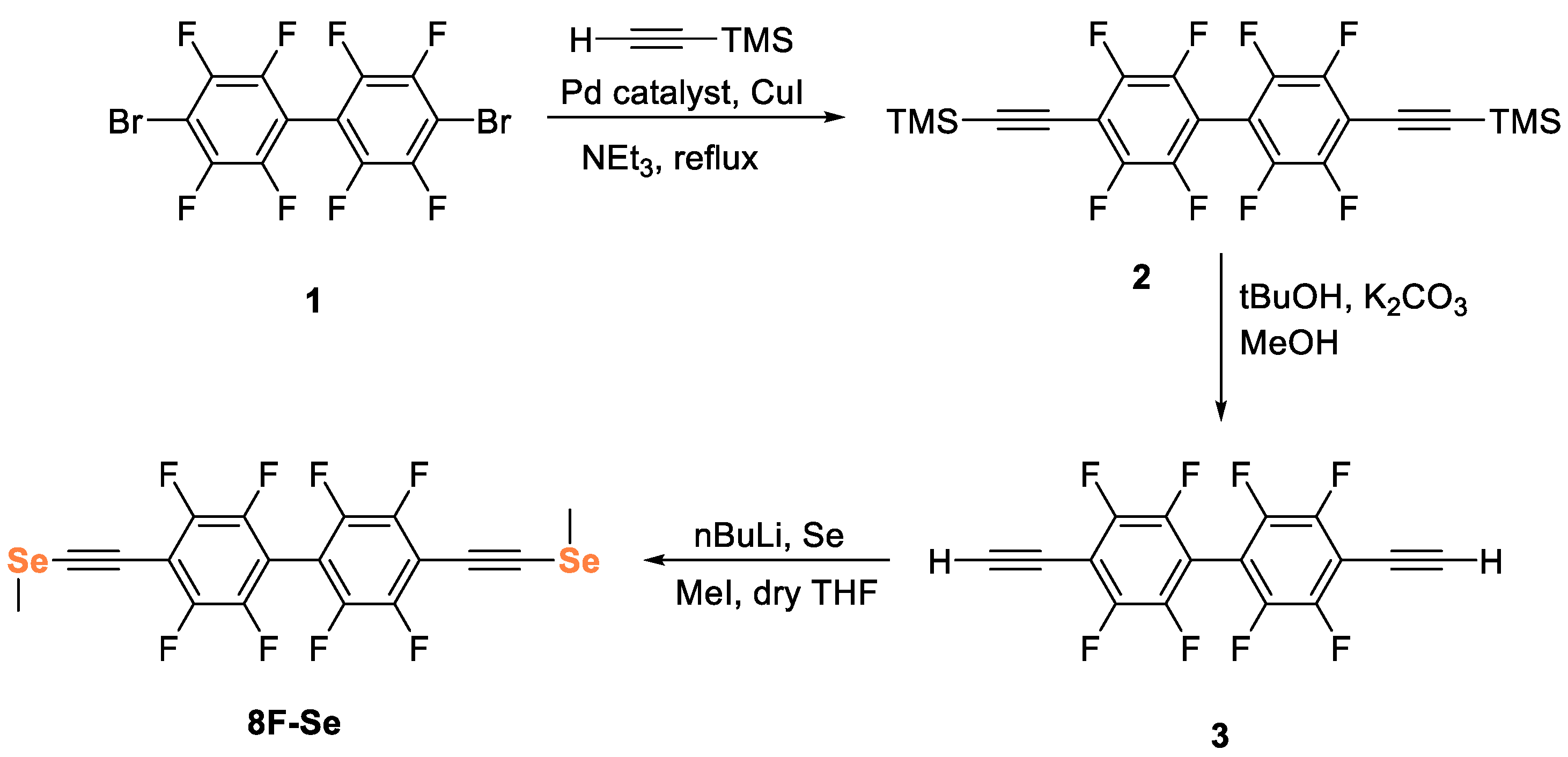
2. Results and Discussions
3. Materials and Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Desiraju, G.R. Crystal Engineering: From Molecule to Crystal. J. Am. Chem. Soc. 2013, 135, 9952–9967. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the Halogen Bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [Green Version]
- Gilday, L.C.; Robinson, S.W.; Barendt, T.A.; Langton, M.J.; Mullaney, B.R.; Beer, P.D. Halogen Bonding in Supramolecular Chemistry. Chem. Rev. 2015, 115, 7118–7195. [Google Scholar] [CrossRef]
- Priimagi, A.; Cavallo, G.; Metrangolo, P.; Resnati, G. The Halogen Bond in the Design of Functional Supramolecular Materials: Recent Advances. Acc. Chem. Res. 2013, 46, 2686–2695. [Google Scholar] [CrossRef] [Green Version]
- Fourmigué, M. Halogen Bonding: Recent Advances. Curr. Op. Solid State Mater. Sc. 2009, 13, 36–45. [Google Scholar] [CrossRef]
- Aakeroy, C.B.; Bryce, D.L.; Desiraju, G.R.; Frontera, A.; Legon, A.C.; Nicotra, F.; Rissanen, K.; Scheiner, S.; Terraneo, G.; Metrangolo, P.; et al. Definition of the Chalcogen Bond (IUPAC Recommendations 2019). Pure Appl. Chem. 2019, 91, 1889–1892. [Google Scholar] [CrossRef]
- Vogel, L.; Wonner, P.; Huber, S.M. Chalcogen Bonding: An Overview. Angew. Chem. Int. Ed. 2019, 58, 1880–1891. [Google Scholar] [CrossRef]
- Fourmigué, M.; Dhaka, A. Chalcogen Bonding in Crystalline Diselenides and Selenocyanates: From Molecules of Pharmaceutical Interest to Conducting Materials. Coord. Chem. Rev. 2020, 403, 213084. [Google Scholar] [CrossRef]
- Wang, W.; Ji, B.; Zhang, Y. Chalcogen Bond: A Sister Noncovalent Bond to Halogen Bond. J. Phys. Chem. A 2009, 113, 8132–8135. [Google Scholar] [CrossRef]
- Scilabra, P.; Terraneo, G.; Resnati, G. The Chalcogen Bond in Crystalline Solids: A World Parallel to Halogen Bond. Acc. Chem. Res. 2019, 52, 1313–1324. [Google Scholar] [CrossRef] [PubMed]
- Alikhani, E.; Fuster, F.; Madebene, B.; Grabowski, S.J. Topological Reaction Sites–Very Strong Chalcogen Bonds. Phys. Chem. Chem. Phys. 2014, 16, 2430–2442. [Google Scholar] [CrossRef] [PubMed]
- Burchell, C.J.; Kilian, P.; Slawin, A.M.Z.; Woollins, J.D.; Tersago, K.; Van Alsenoy, C.; Blockhuys, F. E2(CN)2 (E = S, Se) and Related Compounds. Inorg. Chem. 2006, 45, 710–716. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, A.F.; Vargas-Baca, I.; Mansour, S.; Mahmoudkhani, A.H. The Nature of the Supramolecular Association of 1,2,5-Chalcogenadiazoles. J. Am. Chem. Soc. 2005, 127, 3184–3190. [Google Scholar] [CrossRef] [PubMed]
- Garrett, G.E.; Gibson, G.L.; Straus, R.N.; Seferos, D.S.; Taylor, M.S. Chalcogen Bonding in Solution: Interactions of Benzotelluradiazoles with Anionic and Uncharged Lewis Bases. J. Am. Chem. Soc. 2015, 137, 4126–4133. [Google Scholar] [CrossRef]
- Ho, P.C.; Szydlowski, P.; Sinclair, J.; Elder, P.J.W.; Kübel, J.; Gendy, C.; Lee, L.M.; Jenkins, H.; Britten, J.F.; Morim, D.R.; et al. Supramolecular Macrocycles Reversibly Assembled by Te … O Chalcogen Bonding. Nature Comm. 2016, 7, 1–10. [Google Scholar] [CrossRef]
- Ho, P.C.; Rafique, J.; Lee, J.; Lee, L.M.; Jenkins, H.A.; Britten, J.F.; Braga, A.L.; Vargas-Baca, I. Synthesis and Structural Characterisation of the Aggregates of Benzo-1,2-Chalcogenazole 2-Oxides. Dalton Trans. 2017, 46, 6570–6579. [Google Scholar] [CrossRef]
- Werz, D.B.; Gleiter, R.; Rominger, F. Nanotube Formation Favored by Chalcogen−Chalcogen Interactions. J. Am. Chem. Soc. 2002, 124, 10638–10639. [Google Scholar] [CrossRef]
- Gleiter, R.; Haberhauer, G.; Werz, D.B.; Rominger, F.; Bleiholder, C. From Noncovalent Chalcogen–Chalcogen Interactions to Supramolecular Aggregates: Experiments and Calculations. Chem. Rev. 2018, 118, 2010–2041. [Google Scholar] [CrossRef] [PubMed]
- Huynh, H.-T.; Jeannin, O.; Fourmigué, M. Organic Selenocyanates as Strong and Directional Chalcogen Bond Donors for Crystal Engineering. Chem. Commun. 2017, 53, 8467–8469. [Google Scholar] [CrossRef] [PubMed]
- Jeannin, O.; Huynh, H.-T.; Riel, A.M.S.; Fourmigué, M. Chalcogen Bonding Interactions in Organic Selenocyanates: From Cooperativity to Chelation. New J. Chem. 2018, 42, 10502–10509. [Google Scholar] [CrossRef]
- Riel, A.M.S.; Huynh, H.-T.; Jeannin, O.; Berryman, O.; Fourmigué, M. Organic Selenocyanates as Halide Receptors: From Chelation to One-Dimensional Systems. Cryst. Growth Des. 2019, 19, 1418–1425. [Google Scholar] [CrossRef]
- Huynh, H.-T.; Jeannin, O.; Aubert, E.; Espinosa, E.; Fourmigué, M. Chalcogen Bonding Interactions in Chelating, Chiral Bis(Selenocyanates). New J. Chem. 2021, 45, 76–84. [Google Scholar] [CrossRef]
- Xu, Y.; Kumar, V.; Bradshaw, M.J.Z.; Bryce, D.L. Chalcogen-Bonded Cocrystals of Substituted Pyridine N-Oxides and Chalcogenodiazoles: An X-Ray Diffraction and Solid-State NMR Investigation. Cryst. Growth Des. 2020, 20, 7910–7920. [Google Scholar] [CrossRef]
- Kumar, V.; Xu, Y.; Bryce, D.L. Double Chalcogen Bonds: Crystal Engineering Stratagems via Diffraction and Multinuclear Solid-State Magnetic Resonance Spectroscopy. Chem. Eur. J. 2020, 26, 3275–3286. [Google Scholar] [CrossRef]
- Dhaka, A.; Jeannin, O.; Jeon, I.-R.; Aubert, E.; Espinosa, E.; Fourmigué, M. Activating Chalcogen Bonding (ChB) in Alkylseleno/Alkyltelluroacetylenes toward Chalcogen Bonding Directionality Control. Angew. Chem. Int. Ed. 2020, 132, 23789–23793. [Google Scholar] [CrossRef]
- Dhaka, A.; Jeannin, O.; Aubert, E.; Espinosa, E.; Fourmigué, M. Supramolecular Rectangles through Directional Chalcogen Bonding. Chem. Commun. 2021, 57, 4560–4563. [Google Scholar] [CrossRef] [PubMed]
- Capucci, D.; Balestri, D.; Mazzeo, P.P.; Pelagatti, P.; Rubini, K.; Bacchi, A. Liquid Nicotine Tamed in Solid Forms by Cocrystallization. Cryst. Growth Des. 2017, 17, 4958–4964. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Wijethunga, T.K.; Haj, M.A.; Desper, J.; Moore, C. The Structural Landscape of Heteroaryl-2-Imidazoles: Competing Halogen- and Hydrogen-Bond Interactions. CrystEngComm 2014, 16, 7218. [Google Scholar] [CrossRef]
- Zhang, Z.; Kim, D.S.; Lin, C.-Y.; Zhang, H.; Lammer, A.D.; Lynch, V.M.; Popov, I.; Miljanić, O.Š.; Anslyn, E.V.; Sessler, J.L. Expanded Porphyrin-Anion Supramolecular Assemblies: Environmentally Responsive Sensors for Organic Solvents and Anions. J. Am. Chem. Soc. 2015, 137, 7769–7774. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Programs for the Refinement of Crystal Structures; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Rev. A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keith, T.A. AIMAll, Version 19.10.12; TK Gristmill Software; Todd A. Keith: Overland Park, KS, USA, 2019; Available online: aim.tkgristmill.com (accessed on 13 December 2019).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Co-Crystal | T (K) | Se•••N dist. (Å) | RR | C−(Se)•••N Angle (°) |
|---|---|---|---|---|
| 4F-Se•bpe | 296 | 3.052 (2) | 0.88 | 172.1 (1) |
| 8F-Se•bpe | 296 | 3.029 (4) | 0.87 | 176.7 (1) |
| 4F-Se•bpe | 150 | 3.005 (2) | 0.87 | 172.7 (1) |
| 8F-Se•bpe | 150 | 2.958 (2) | 0.85 | 177.3 (1) |
| Title 1 | 8F-Se | 4F-Se•bpe (RT) | 4F-Se.bpe (150 K) | 8F-Se•bpe (RT) | 8F-Se•bpe (150 K) |
|---|---|---|---|---|---|
| CCDC number | 2090416 | 2090417 | 2090418 | 2090419 | 2090420 |
| Formula | C18H6F8Se2 | C24H18F4N2Se2 | C24H18F4N2Se2 | C30H18F8N2Se2 | C30H18F8N2Se2 |
| FW | 532.15 | 568.32 | 568.32 | 716.38 | 716.38 |
| Crystal system | triclinic | monoclinic | monoclinic | monoclinic | monoclinic |
| Space group | P21/n | P21/n | C2/c | C2/c | |
| a/Å | 8.4551 (6) | 12.6631 (8) | 12.6624 (16) | 22.529 (2) | 21.575 (2) |
| b/Å | 8.6842 (5) | 5.7143 (4) | 5.6541 (8) | 6.2237 (6) | 6.1938 (5) |
| c/Å | 12.4001 (9) | 16.6845 (13) | 16.341 (2) | 21.539 (3) | 21.412 (2) |
| α/° | 91.956 (2) | 90.00 | 90.00 | 90.00 | 90.00 |
| β /° | 108.704 (2) | 105.503 (4) | 105.367 (4) | 105.862 (7) | 105.762 (3) |
| γ /° | 94.837 (2) | 90.00 | 90.00 | 90.00 | 90.00 |
| V/Å3 | 857.52 (10) | 1163.38 (14) | 1128.1 (3) | 2905.1 (6) | 2753.7 (4) |
| Z | 2 | 2 | 2 | 4 | 4 |
| Dc/g cm−3 | 2.061 | 1.622 | 1.673 | 1.638 | 1.728 |
| T/K | 150 (2) | 296 (2) | 150 (2) | 296 (2) | 150 (2) |
| μ / mm−1 | 4.395 | 3.224 | 3.325 | 2.620 | 2.764 |
| F(000) | 508 | 560 | 560 | 1408 | 1408 |
| Refl. collected | 40049 | 7490 | 7242 | 13879 | 18466 |
| Refl. indep. (Rint) | 3944 (0.0583) | 2635 (0.0489) | 2570 (0.0409) | 3295 (0.0439) | 3152 (0.0426) |
| Refl. Obs. [I > 2σ(I)] | 3449 | 1779 | 1985 | 2005 | 2568 |
| GOF on F2 | 1.076 | 1.018 | 1.046 | 1.014 | 1.126 |
| R1 [I > 2σ(I)] (all) | 0.0264 (0.0332) | 0.0453 (0.080) | 0.0373 (0.0559) | 0.0419 (0.0842) | 0.0333 (0.0452) |
| wR2 [I > 2σ(I)] (all) | 0.0608 (0.0651) | 0.0966 (0.107) | 0.0795 (0.0853) | 0.1031 (0.1224) | 0.0680 (0.0725) |
| ∆ρmax,min/e Å−3 | +0.508, −0.933 | +0.899, −0.606 | +0.561, −0.514 | 0.546, −0.297 | +0.573, −0.461 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dhaka, A.; Jeannin, O.; Aubert, E.; Espinosa, E.; Fourmigué, M. Chalcogen Bonding in Co-Crystals: Activation through 1,4-Perfluorophenylene vs. 4,4′-Perfluorobiphenylene Cores. Molecules 2021, 26, 4050. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26134050
Dhaka A, Jeannin O, Aubert E, Espinosa E, Fourmigué M. Chalcogen Bonding in Co-Crystals: Activation through 1,4-Perfluorophenylene vs. 4,4′-Perfluorobiphenylene Cores. Molecules. 2021; 26(13):4050. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26134050
Chicago/Turabian StyleDhaka, Arun, Olivier Jeannin, Emmanuel Aubert, Enrique Espinosa, and Marc Fourmigué. 2021. "Chalcogen Bonding in Co-Crystals: Activation through 1,4-Perfluorophenylene vs. 4,4′-Perfluorobiphenylene Cores" Molecules 26, no. 13: 4050. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26134050