
Optimization of Sample Preparation for Metabolomics Exploration of Urine, Feces, Blood and Saliva in Humans Using Combined NMR and UHPLC-HRMS Platforms

 , , ,
, , ,
Abstract
:
1. Introduction
2. Results and Discussion
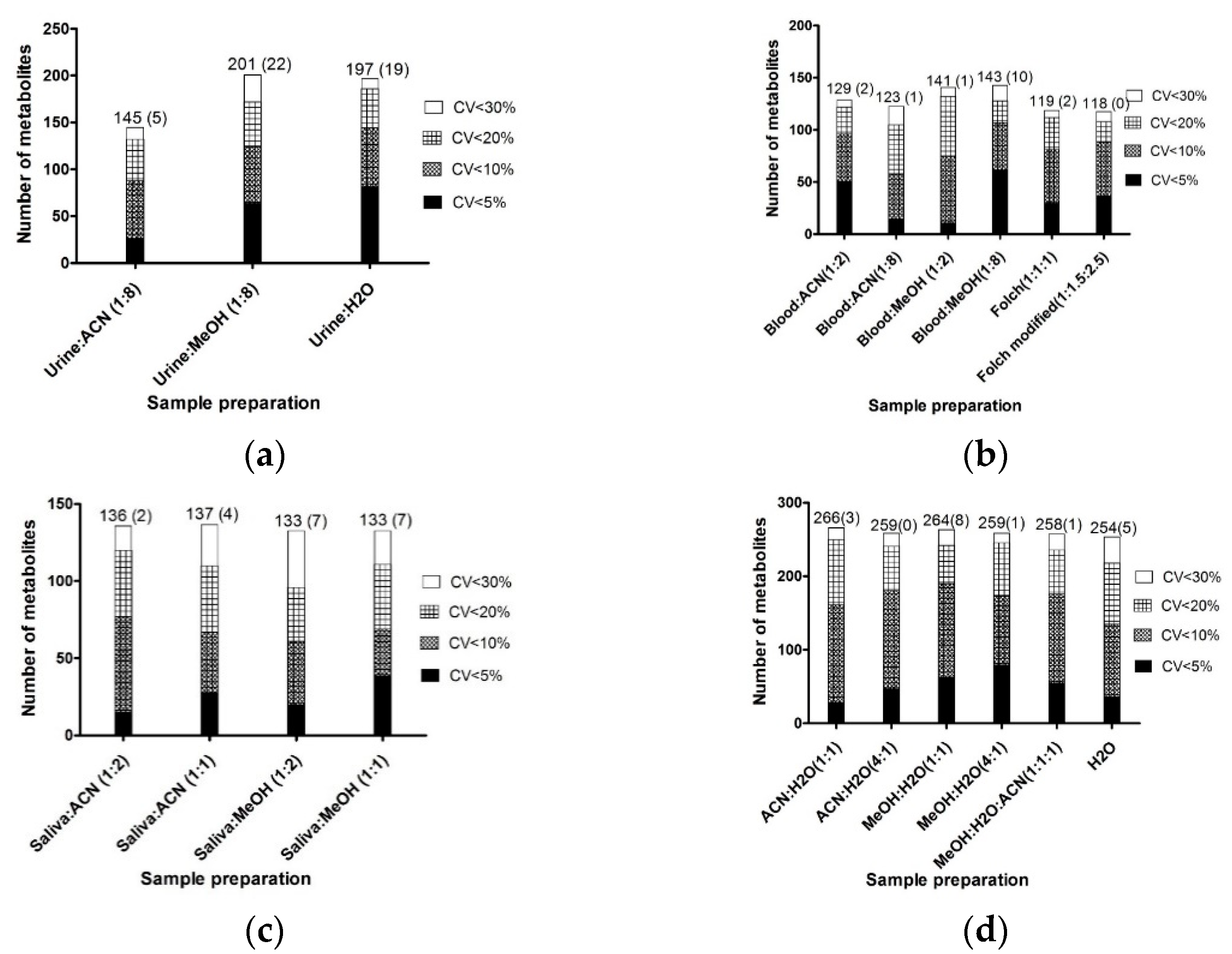
2.1. Optimization of Sample Preparations
2.1.1. Urine
2.1.2. Blood
2.1.3. Saliva
2.1.4. Feces
2.2. Overall Synthesis on Optimization of Sample Preparation
2.3. Intraday Precision
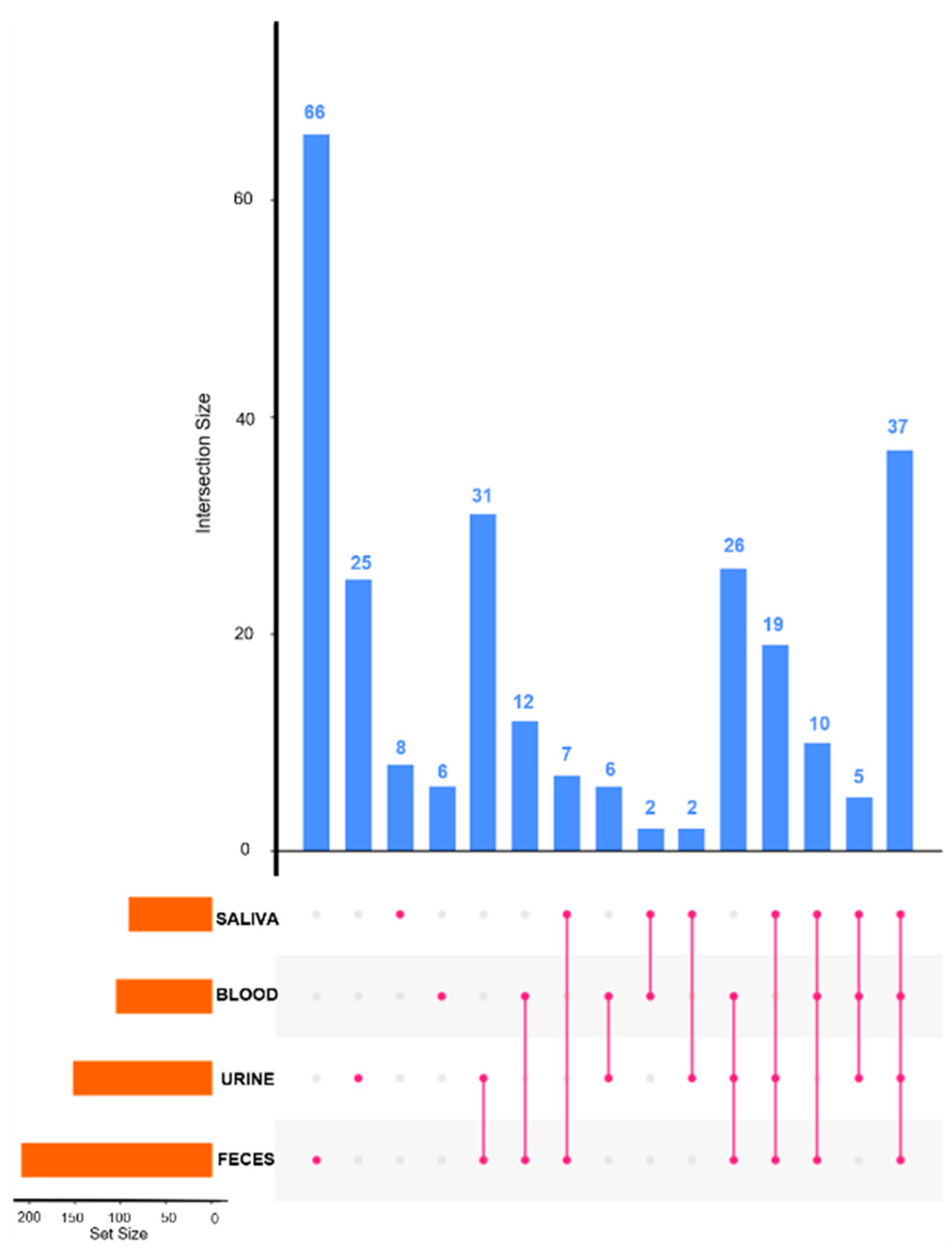
2.4. Platform Complementarity
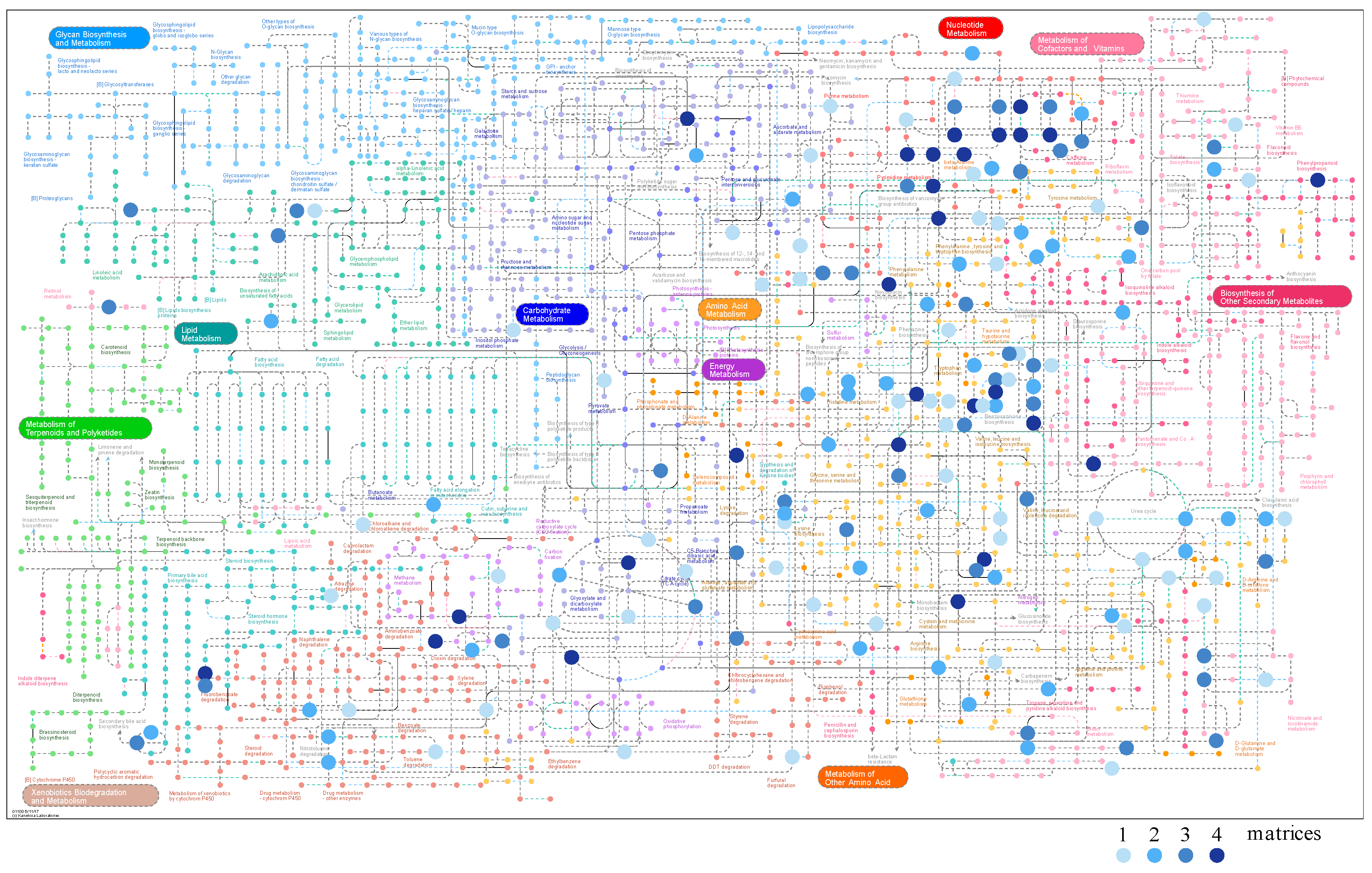
2.5. Matrix Complementarity, Metabolic Coverage, and Mapping
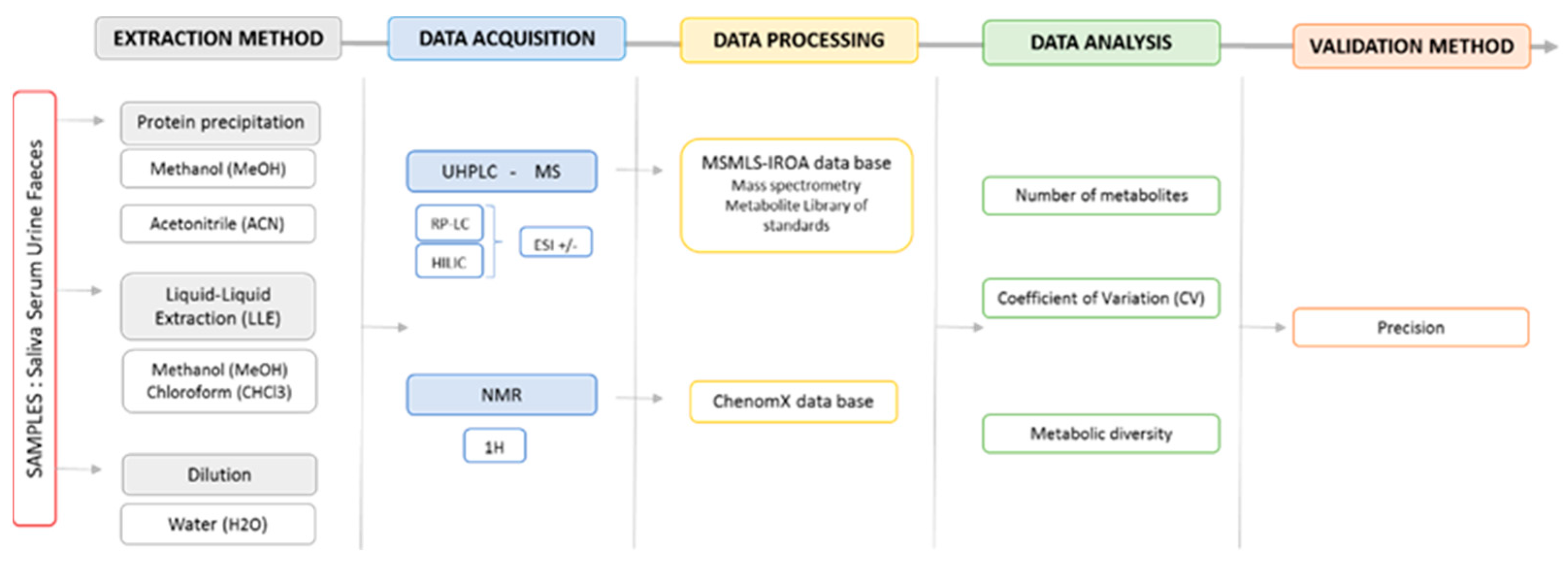
3. Materials and Methods
3.1. Sample Preparations
3.1.1. Urine
3.1.2. Blood
3.1.3. Saliva
3.1.4. Feces
3.2. Data Acquisition
3.2.1. UHPLC-MS
3.2.2. NMR
3.3. Data Processing
3.3.1. UHPLC-MS
3.3.2. 1H-NMR
3.4. Data Fusion
3.5. Data Analysis
3.6. Intraday Precision
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Holmes, E.; Wilson, I.D.; Nicholson, J.K. Metabolic Phenotyping in Health and Disease. Cell 2008, 134, 714–717. [Google Scholar] [CrossRef] [Green Version]
- Collino, S.; Martin, F.-P.J.; Rezzi, S. Clinical metabolomics paves the way towards future healthcare strategies. Br. J. Clin. Pharmacol. 2013, 75, 619–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wishart, D.S. Emerging applications of metabolomics in drug discovery and precision medicine. Nat. Rev. Drug Discov. 2016, 15, 473–484. [Google Scholar] [CrossRef]
- Newgard, C.B. Metabolomics and Metabolic Diseases: Where Do We Stand? Cell Metab. 2017, 25, 43–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallares-Méndez, R.; Aguilar-Salinas, C.A.; Cruz-Bautista, I.; Del Bosque-Plata, L. Metabolomics in diabetes, a review. Ann. Med. 2016, 48, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Miani, M.; Le Naour, J.; Waeckel-Enee, E.; Verma, S.C.; Straube, M.; Emond, P.; Ryffel, B.; van Endert, P.; Sokol, H.; Diana, J. Gut Microbiota-Stimulated Innate Lymphoid Cells Support beta-Defensin 14 Expression in Pancreatic Endocrine Cells, Preventing Autoimmune Diabetes. Cell Metab. 2018, 28, 557–572e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urpi-Sarda, M.; Almanza-Aguilera, E.; Llorach, R.; Vázquez-Fresno, R.; Estruch, R.; Corella, D.; Sorli, J.; Carmona, F.; Sanchez-Pla, A.; Salas-Salvadó, J.; et al. Non-targeted metabolomic biomarkers and metabotypes of type 2 diabetes: A cross-sectional study of PREDIMED trial participants. Diabetes Metab. 2019, 45, 167–174. [Google Scholar] [CrossRef]
- Jansson, J.; Willing, B.; Lucio, M.; Fekete, A.; Dicksved, J.; Halfvarson, J.; Tysk, C.; Schmitt-Kopplin, P. Metabolomics reveals metabolic biomarkers of Crohn’s disease. PLoS ONE 2009, 4, e6386. [Google Scholar] [CrossRef] [Green Version]
- Scoville, E.A.; Allaman, M.M.; Brown, C.T.; Motley, A.K.; Horst, S.N.; Williams, C.S.; Koyama, T.; Zhao, Z.; Adams, D.W.; Beaulieu, D.B.; et al. Alterations in lipid, amino acid, and energy metabolism distinguish Crohn’s disease from ulcerative colitis and control subjects by serum metabolomic profiling. Metabolomics 2018, 14, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, I.; Roy, B.C.; Khan, S.A.; Septer, S.; Umar, S. Microbiome, Metabolome and Inflammatory Bowel Disease. Microorganisms 2016, 4, 20. [Google Scholar] [CrossRef] [Green Version]
- Ni, Y.; Xie, G.; Jia, W. Metabonomics of Human Colorectal Cancer: New Approaches for Early Diagnosis and Biomarker Discovery. J. Proteome Res. 2014, 13, 3857–3870. [Google Scholar] [CrossRef] [PubMed]
- Euceda, L.R.; Andersen, M.K.; Tessem, M.-B.; Moestue, S.A.; Grinde, M.T.; Bathen, T.F. NMR-Based Prostate Cancer Metabolomics. Breast Cancer 2018, 1786, 237–257. [Google Scholar] [CrossRef]
- Hoang, G.; Udupa, S.; Le, A. Application of metabolomics technologies toward cancer prognosis and therapy. Int. Rev. Cell Mol. Biol. 2019, 347, 191–223. [Google Scholar] [CrossRef] [PubMed]
- Goonewardena, S.N.; Prevette, L.E.; Desai, A. Metabolomics and Atherosclerosis. Curr. Atheroscler. Rep. 2010, 12, 267–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Wu, X.; Ke, C.; Yin, M.; Li, Z.; Fan, L.; Zhang, W.; Zhang, H.; Zhao, F.; Zhou, X.; et al. Identification of Potential Biomarkers for Ovarian Cancer by Urinary Metabolomic Profiling. J. Proteome Res. 2013, 12, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Rauschert, S.; Kirchberg, F.F.; Marchioro, L.; Hellmuth, C.; Uhl, O.; Koletzko, B. Early Programming of Obesity Throughout the Life Course: A Metabolomics Perspective. Ann. Nutr. Metab. 2017, 70, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Bagheri, M.; Farzadfar, F.; Qi, L.; Yekaninejad, M.S.; Chamari, M.; Zeleznik, O.A.; Kalantar, Z.; Ebrahimi, Z.; Sheidaie, A.; Koletzko, B.; et al. Obesity-Related Metabolomic Profiles and Discrimination of Metabolically Unhealthy Obesity. J. Proteome Res. 2018, 17, 1452–1462. [Google Scholar] [CrossRef]
- Orozco, J.S.; Hertz-Picciotto, I.; Abbeduto, L.; Slupsky, C.M. Metabolomics analysis of children with autism, idiopathic-developmental delays, and Down syndrome. Transl. Psychiatry 2019, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Li, L. Sample normalization methods in quantitative metabolomics. J. Chromatogr. A 2016, 1430, 80–95. [Google Scholar] [CrossRef]
- Pezzatti, J.; Boccard, J.; Codesido, S.; Gagnebin, Y.; Joshi, A.; Picard, D.; González-Ruiz, V.; Rudaz, S. Implementation of liquid chromatography–high resolution mass spectrometry methods for untargeted metabolomic analyses of biological samples: A tutorial. Anal. Chim. Acta 2020, 1105, 28–44. [Google Scholar] [CrossRef]
- Zhang, A.; Sun, H.; Wang, X. Serum metabolomics as a novel diagnostic approach for disease: A systematic review. Anal. Bioanal. Chem. 2012, 404, 1239–1245. [Google Scholar] [CrossRef] [PubMed]
- Bouatra, S.; Aziat, F.; Mandal, R.; Guo, A.C.; Wilson, M.R.; Knox, C.; Bjorndahl, T.C.; Krishnamurthy, R.; Saleem, F.; Liu, P.; et al. The Human Urine Metabolome. PLoS ONE 2013, 8, e73076. [Google Scholar] [CrossRef] [Green Version]
- Duarte, I.F.; Diaz, S.O.; Gil, A.M. NMR metabolomics of human blood and urine in disease research. J. Pharm. Biomed. Anal. 2014, 93, 17–26. [Google Scholar] [CrossRef]
- Zhao, L.; Ni, Y.; Su, M.; Li, H.; Dong, F.; Chen, W.; Wei, R.; Zhang, L.; Guiraud, S.P.; Martin, F.-P.; et al. High Throughput and Quantitative Measurement of Microbial Metabolome by Gas Chromatography/Mass Spectrometry Using Automated Alkyl Chloroformate Derivatization. Anal. Chem. 2017, 89, 5565–5577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breban, M.; Tap, J.; Leboime, A.; Said-Nahal, R.; Langella, P.; Chiocchia, G.; Furet, J.-P.; Sokol, H. Faecal microbiota study reveals specific dysbiosis in spondyloarthritis. Ann. Rheum. Dis. 2017, 76, 1614–1622. [Google Scholar] [CrossRef]
- Dunn, W.B.; Bailey, N.J.C.; Johnson, H.E. Measuring the metabolome: Current analytical technologies. Analyst 2005, 130, 606–625. [Google Scholar] [CrossRef]
- Beckonert, O.; Keun, H.C.; Ebbels, T.; Bundy, J.; Holmes, E.; Lindon, J.; Nicholson, J. Metabolic profiling, metabolomic and metabonomic procedures for NMR spectroscopy of urine, plasma, serum and tissue extracts. Nat. Protoc. 2007, 2, 2692–2703. [Google Scholar] [CrossRef]
- Gowda, G.A.N.; Raftery, D. Quantitating Metabolites in Protein Precipitated Serum Using NMR Spectroscopy. Anal. Chem. 2014, 86, 5433–5440. [Google Scholar] [CrossRef]
- Gowda, G.A.N.; Gowda, Y.N.; Raftery, D. Expanding the Limits of Human Blood Metabolite Quantitation Using NMR Spectroscopy. Anal. Chem. 2015, 87, 706–715. [Google Scholar] [CrossRef] [Green Version]
- Rico, E.; González, O.; Blanco, M.E.; Alonso, R.M. Evaluation of human plasma sample preparation protocols for untargeted metabolic profiles analyzed by UHPLC-ESI-TOF-MS. Anal. Bioanal. Chem. 2014, 406, 7641–7652. [Google Scholar] [CrossRef]
- Dunn, W.B. Mass spectrometry in systems biology an introduction. Methods Enzymol. 2011, 500, 15–35. [Google Scholar] [PubMed]
- Stevens, V.L.; Hoover, E.; Wang, Y.; Zanetti, K.A. Pre-Analytical Factors that Affect Metabolite Stability in Human Urine, Plasma, and Serum: A Review. Metabolites 2019, 9, 156. [Google Scholar] [CrossRef] [Green Version]
- Karu, N.; Deng, L.; Slae, M.; Guo, A.C.; Sajed, T.; Huynh, H.; Wine, E.; Wishart, D.S. A review on human fecal metabolomics: Methods, applications and the human fecal metabolome database. Anal. Chim. Acta 2018, 1030, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.H.; Karlsson, E.; Sarda, S.; Iddon, L.; Iqbal, M.; Meng, X.; Harding, J.R.; Stachulski, A.V.; Nicholson, J.; Wilson, I.D.; et al. Integrated HPLC-MS and 1H-NMR spectroscopic studies on acyl migration reaction kinetics of model drug ester glucuronides. Xenobiotica 2009, 40, 9–23. [Google Scholar] [CrossRef] [PubMed]
- De Paepe, E.; Van Meulebroek, L.; Rombouts, C.; Huysman, S.; Verplanken, K.; Lapauw, B.; Wauters, J.; Hemeryck, L.Y.; Vanhaecke, L. A validated multi-matrix platform for metabolomic fingerprinting of human urine, feces and plasma using ultra-high performance liquid-chromatography coupled to hybrid orbitrap high-resolution mass spectrometry. Anal. Chim. Acta 2018, 1033, 108–118. [Google Scholar] [CrossRef]
- Guan, Z.; Wu, J.; Wang, C.; Zhang, F.; Wang, Y.; Wang, M.; Zhao, M.; Zhao, C. Investigation of the preventive effect of Sijunzi decoction on mitomycin C-induced immunotoxicity in rats by 1H NMR and MS-based untargeted metabolomic analysis. J. Ethnopharmacol. 2018, 210, 179–191. [Google Scholar] [CrossRef] [PubMed]
- McHugh, C.E.; Flott, T.L.; Schooff, C.R.; Smiley, Z.; Puskarich, M.; Myers, D.D.; Younger, J.G.; Jones, A.E.; Stringer, K.A. Rapid, Reproducible, Quantifiable NMR Metabolomics: Methanol and Methanol: Chloroform Precipitation for Removal of Macromolecules in Serum and Whole Blood. Metabolites 2018, 8, 93. [Google Scholar] [CrossRef] [Green Version]
- Wallner-Liebmann, S.; Tenori, L.; Mazzoleni, A.; Dieber-Rotheneder, M.; Konrad, M.; Hofmann, P.; Luchinat, C.; Turano, P.; Zatloukal, K. Individual Human Metabolic Phenotype Analyzed by 1H NMR of Saliva Samples. J. Proteome Res. 2016, 15, 1787–1793. [Google Scholar] [CrossRef]
- Schulz, A.; Lang, R.; Behr, J.; Hertel, S.; Reich, M.; Kümmerer, K.; Hannig, M.; Hannig, C.; Hofmann, T. Targeted metabolomics of pellicle and saliva in children with different caries activity. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Deda, O.; Gika, H.G.; Wilson, I.; Theodoridis, G.A. An overview of fecal sample preparation for global metabolic profiling. J. Pharm. Biomed. Anal. 2015, 113, 137–150. [Google Scholar] [CrossRef]
- Romick-Rosendale, L.; Goodpaster, A.M.; Hanwright, P.J.; Patel, N.B.; Wheeler, E.T.; Chona, D.L.; Kennedy, M.A. NMR-based metabonomics analysis of mouse urine and fecal extracts following oral treatment with the broad-spectrum antibiotic enrofloxacin (Baytril). Magn. Reson. Chem. 2009, 47, S36–S46. [Google Scholar] [CrossRef]
- Saric, J.; Wang, Y.; Li, J.; Coen, M.; Utzinger, J.; Marchesi, J.; Keiser, J.; Veselkov, K.; Lindon, J.C.; Nicholson, J.; et al. Species Variation in the Fecal Metabolome Gives Insight into Differential Gastrointestinal Function. J. Proteome Res. 2008, 7, 352–360. [Google Scholar] [CrossRef]
- Xu, W.; Chen, D.; Wang, N.; Zhang, T.; Zhou, R.; Huan, T.; Lu, Y.; Su, X.; Xie, Q.; Li, L.; et al. Development of High-Performance Chemical Isotope Labeling LC–MS for Profiling the Human Fecal Metabolome. Anal. Chem. 2014, 87, 829–836. [Google Scholar] [CrossRef] [PubMed]
- Moosmang, S.; Pitscheider, M.; Sturm, S.; Seger, C.; Tilg, H.; Halabalaki, M.; Stuppner, H. Metabolomic analysis—Addressing NMR and LC-MS related problems in human feces sample preparation. Clin. Chim. Acta 2019, 489, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Lex, A.; Gehlenborg, N.; Strobelt, H.; Vuillemot, R.; Pfister, H. UpSet: Visualization of Intersecting Sets. IEEE Trans. Vis. Comput. Graph. 2014, 20, 1983–1992. [Google Scholar] [CrossRef]
- Letertre, M.P.M.; Dervilly, G.; Giraudeau, P. Combined Nuclear Magnetic Resonance Spectroscopy and Mass Spectrometry Approaches for Metabolomics. Anal. Chem. 2021, 93, 500–518. [Google Scholar] [CrossRef] [PubMed]
- Rathahao-Paris, E.; Alves, S.; Junot, C.; Tabet, J.-C. High resolution mass spectrometry for structural identification of metabolites in metabolomics. Metabolomics 2015, 12, 1–15. [Google Scholar] [CrossRef]
- Hounoum, B.M.; Blasco, H.; Nadal-Desbarats, L.; Diémé, B.; Montigny, F.; Andres, C.; Emond, P.; Mavel, S. Analytical methodology for metabolomics study of adherent mammalian cells using NMR, GC-MS and LC-HRMS. Anal. Bioanal. Chem. 2015, 407, 8861–8872. [Google Scholar] [CrossRef]
- Guitton, Y.; Tremblay-Franco, M.; Le Corguillé, G.; Martin, J.-F.; Pétéra, M.; Roger-Mele, P.; Delabrière, A.; Goulitquer, S.; Monsoor, M.; Duperier, C.; et al. Create, run, share, publish, and reference your LC–MS, FIA–MS, GC–MS, and NMR data analysis workflows with the Workflow4Metabolomics 3.0 Galaxy online infrastructure for metabolomics. Int. J. Biochem. Cell Biol. 2017, 93, 89–101. [Google Scholar] [CrossRef] [Green Version]
- Giacomoni, F.; Le Corguillé, G.; Monsoor, M.; Landi, M.; Pericard, P.; Pétéra, M.; Duperier, C.; Tremblay-Franco, M.; Martin, J.-F.; Jacob, D.; et al. Workflow4Metabolomics: A collaborative research infrastructure for computational metabolomics. Bioinformatics 2015, 31, 1493–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing Mass Spectrometry Data for Metabolite Profiling Using Nonlinear Peak Alignment, Matching, and Identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Wishart, D.S.; Xia, J. Using MetaboAnalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis. Curr. Protoc. Bioinform. 2019, 68, e86. [Google Scholar] [CrossRef] [PubMed]
- Darzi, Y.; Letunic, I.; Bork, P.; Yamada, T. iPath3.0: Interactive pathways explorer v3. Nucleic Acids Res. 2018, 46, W510–W513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudzik, D.; Barbas-Bernardos, C.; García, A.; Barbas, C. Quality assurance procedures for mass spectrometry untargeted metabolomics. A review. J. Pharm. Biomed. Anal. 2018, 147, 149–173. [Google Scholar] [CrossRef]
- Naz, S.; Vallejo, M.; García, A.; Barbas, C. Method validation strategies involved in non-targeted metabolomics. J. Chromatogr. A 2014, 1353, 99–105. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Urine | Blood | Saliva | Feces | |
|---|---|---|---|---|
| Sample preparation chosen | Urine/MeOH (1:8) | Blood/MeOH (1:8) | Saliva/ACN (1:2) | MeOH/H2O (1:1) |
| Number of metabolites | 151 | 104 | 90 | 208 |
| CV mean % (min–max) | 7% (2–53%) | 8% (3–53%) | 7% (2–26%) | 11% (3–40%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martias, C.; Baroukh, N.; Mavel, S.; Blasco, H.; Lefèvre, A.; Roch, L.; Montigny, F.; Gatien, J.; Schibler, L.; Dufour-Rainfray, D.; et al. Optimization of Sample Preparation for Metabolomics Exploration of Urine, Feces, Blood and Saliva in Humans Using Combined NMR and UHPLC-HRMS Platforms. Molecules 2021, 26, 4111. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26144111
Martias C, Baroukh N, Mavel S, Blasco H, Lefèvre A, Roch L, Montigny F, Gatien J, Schibler L, Dufour-Rainfray D, et al. Optimization of Sample Preparation for Metabolomics Exploration of Urine, Feces, Blood and Saliva in Humans Using Combined NMR and UHPLC-HRMS Platforms. Molecules. 2021; 26(14):4111. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26144111
Chicago/Turabian StyleMartias, Cécile, Nadine Baroukh, Sylvie Mavel, Hélène Blasco, Antoine Lefèvre, Léa Roch, Frédéric Montigny, Julie Gatien, Laurent Schibler, Diane Dufour-Rainfray, and et al. 2021. "Optimization of Sample Preparation for Metabolomics Exploration of Urine, Feces, Blood and Saliva in Humans Using Combined NMR and UHPLC-HRMS Platforms" Molecules 26, no. 14: 4111. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26144111