
Quadruple Hydrogen Bond-Containing A-AB-A Triblock Copolymers: Probing the Influence of Hydrogen Bonding in the Central Block


Abstract
:
1. Introduction
2. Results and Discussion
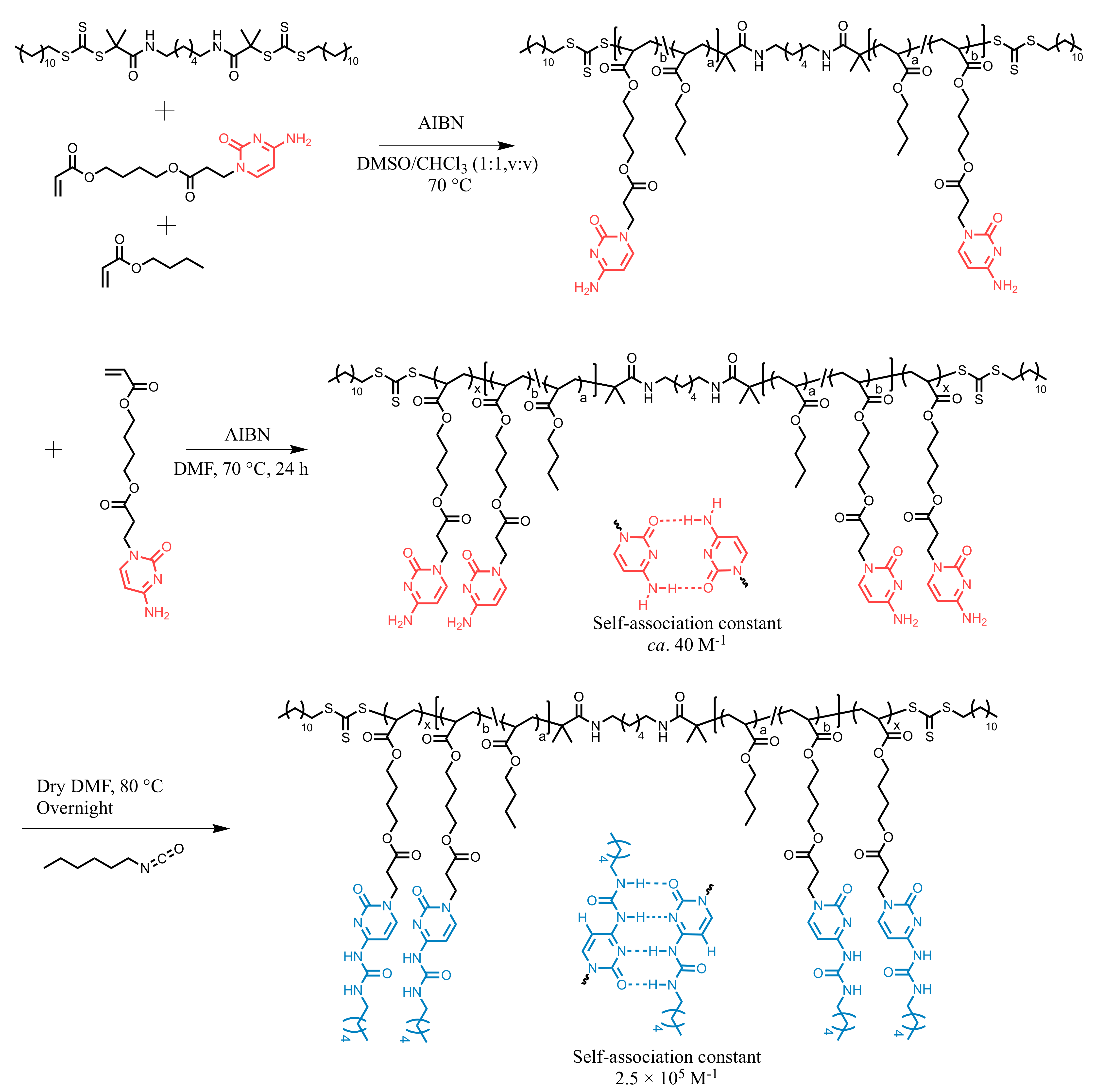
2.1. Synthesis of CyA and UCyA Triblock Copolymer
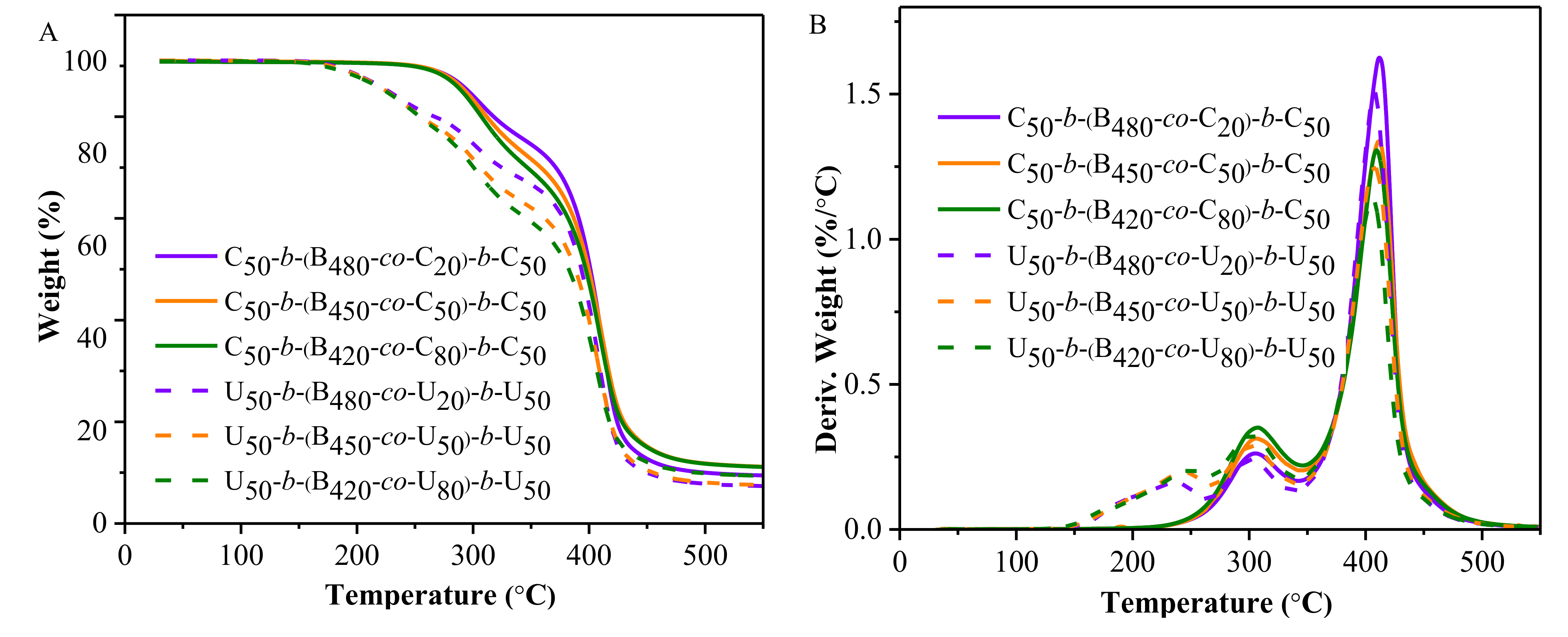
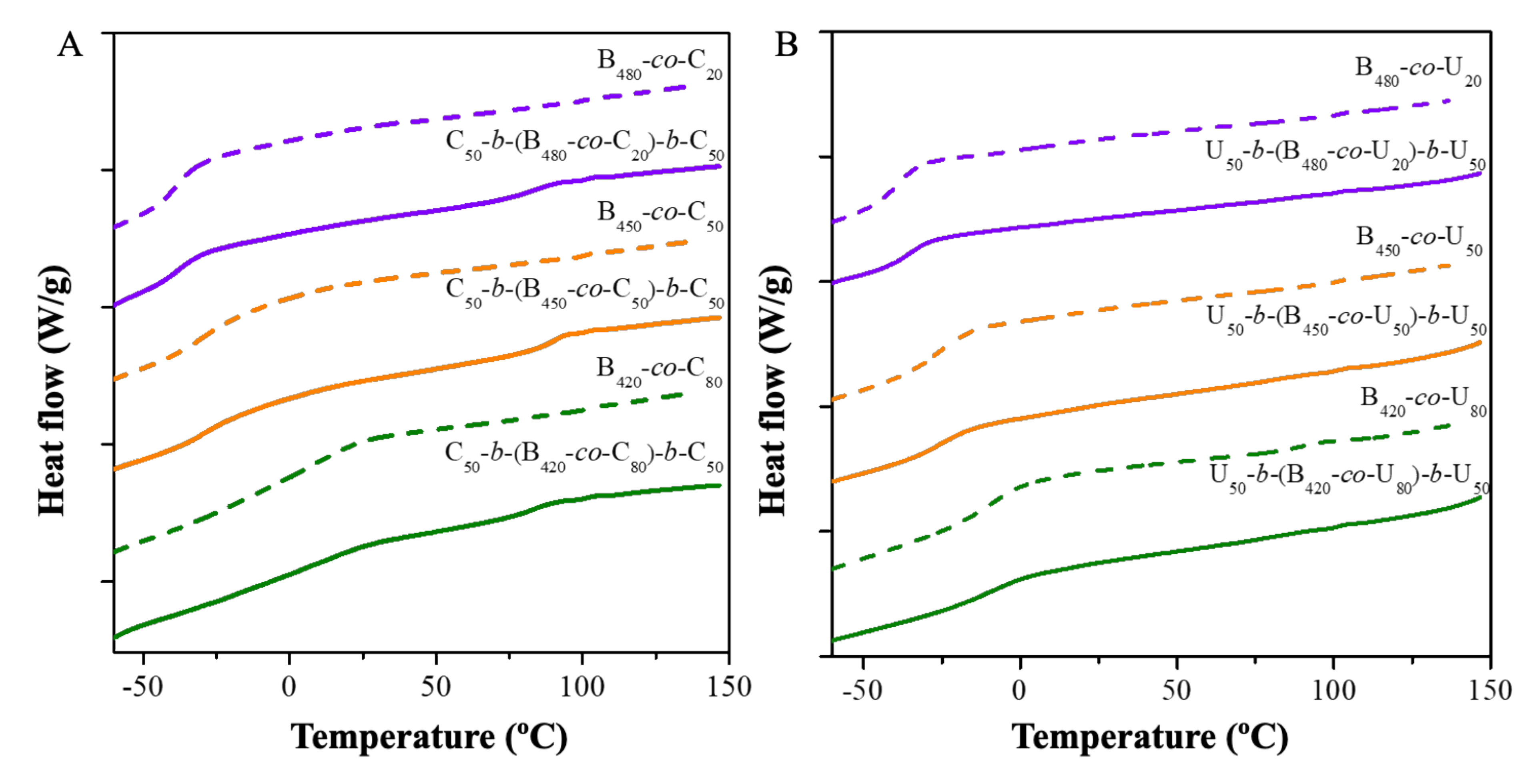
2.2. Thermal Analysis
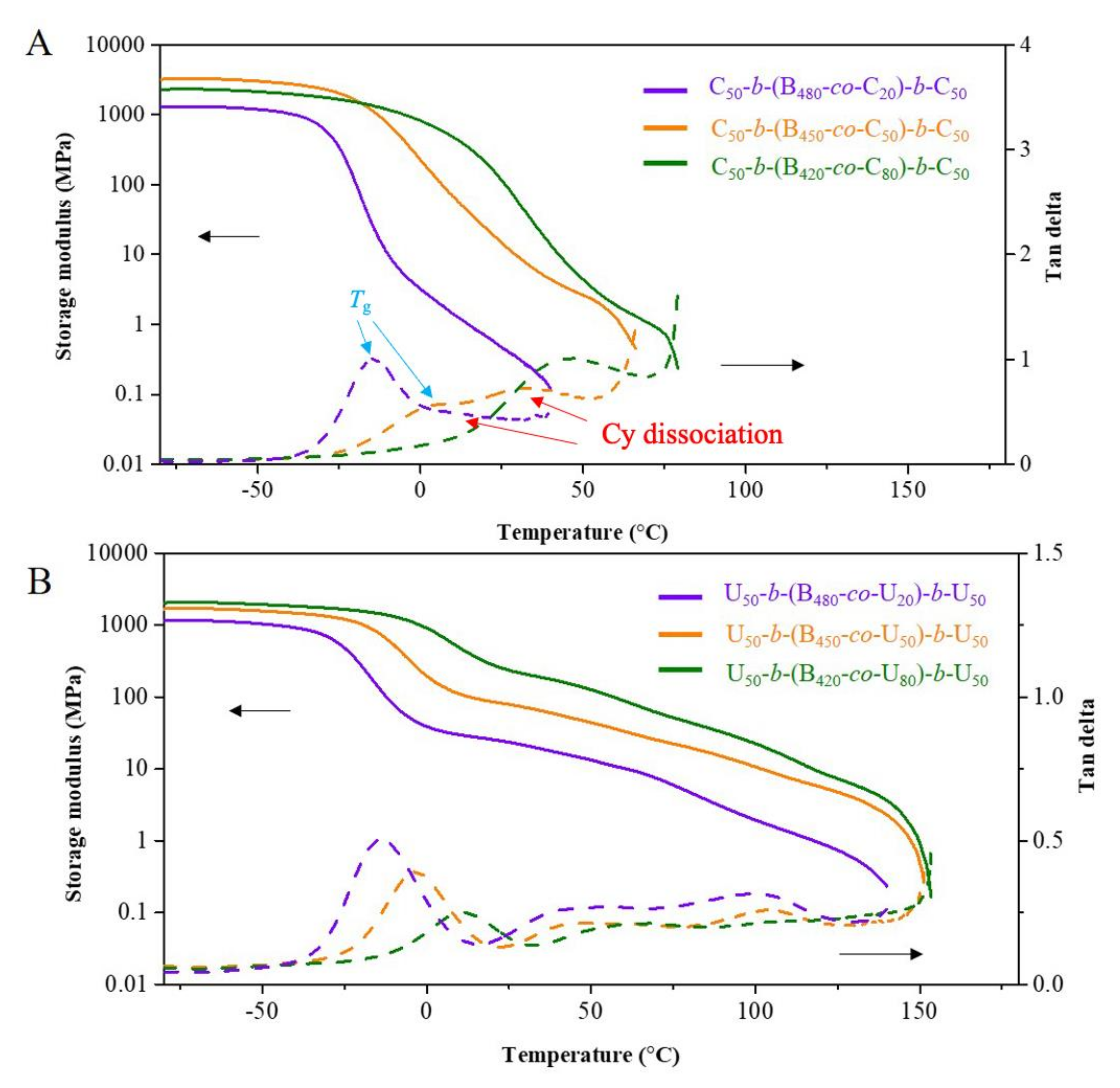
2.3. Thermomechanical Analysis
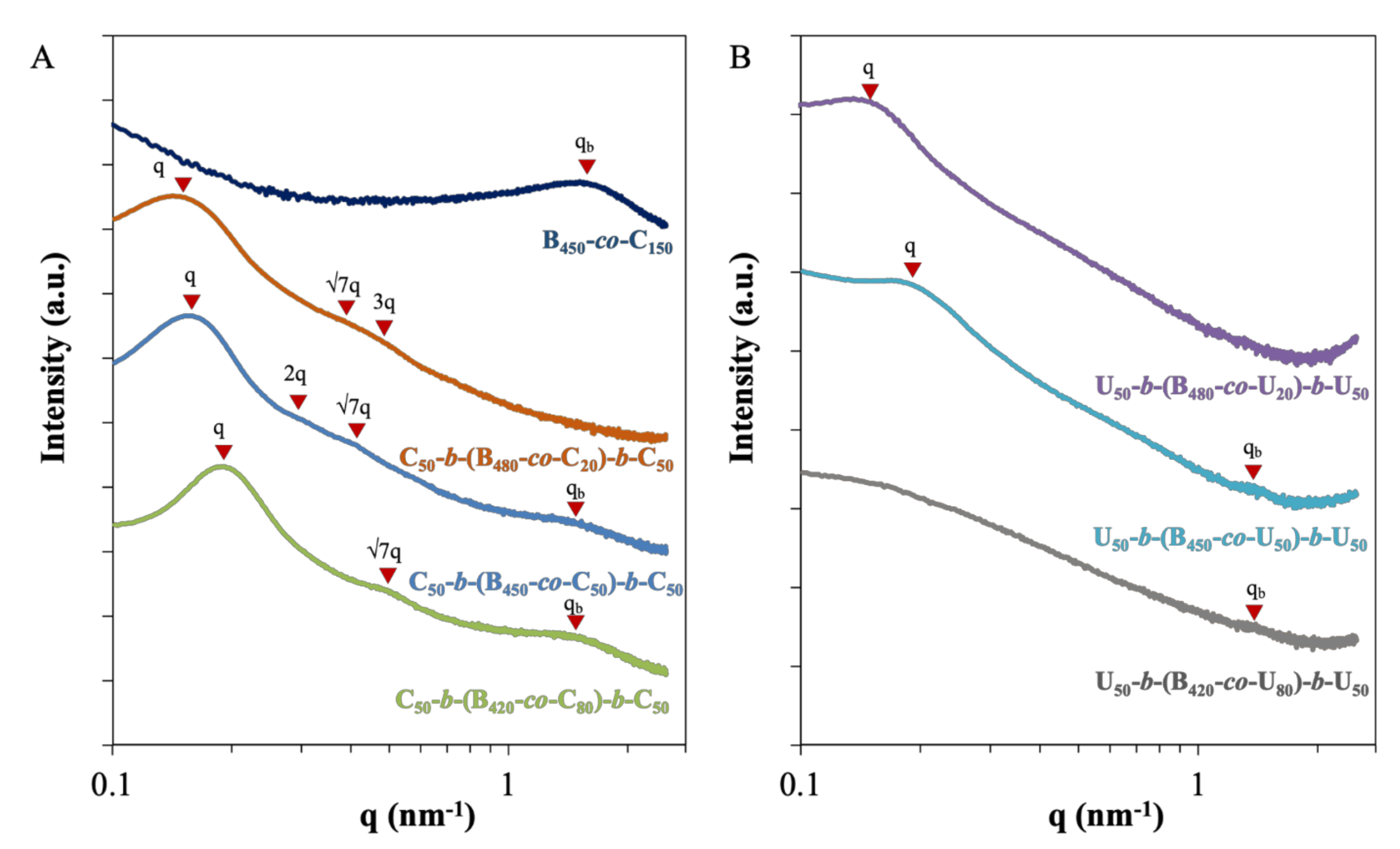
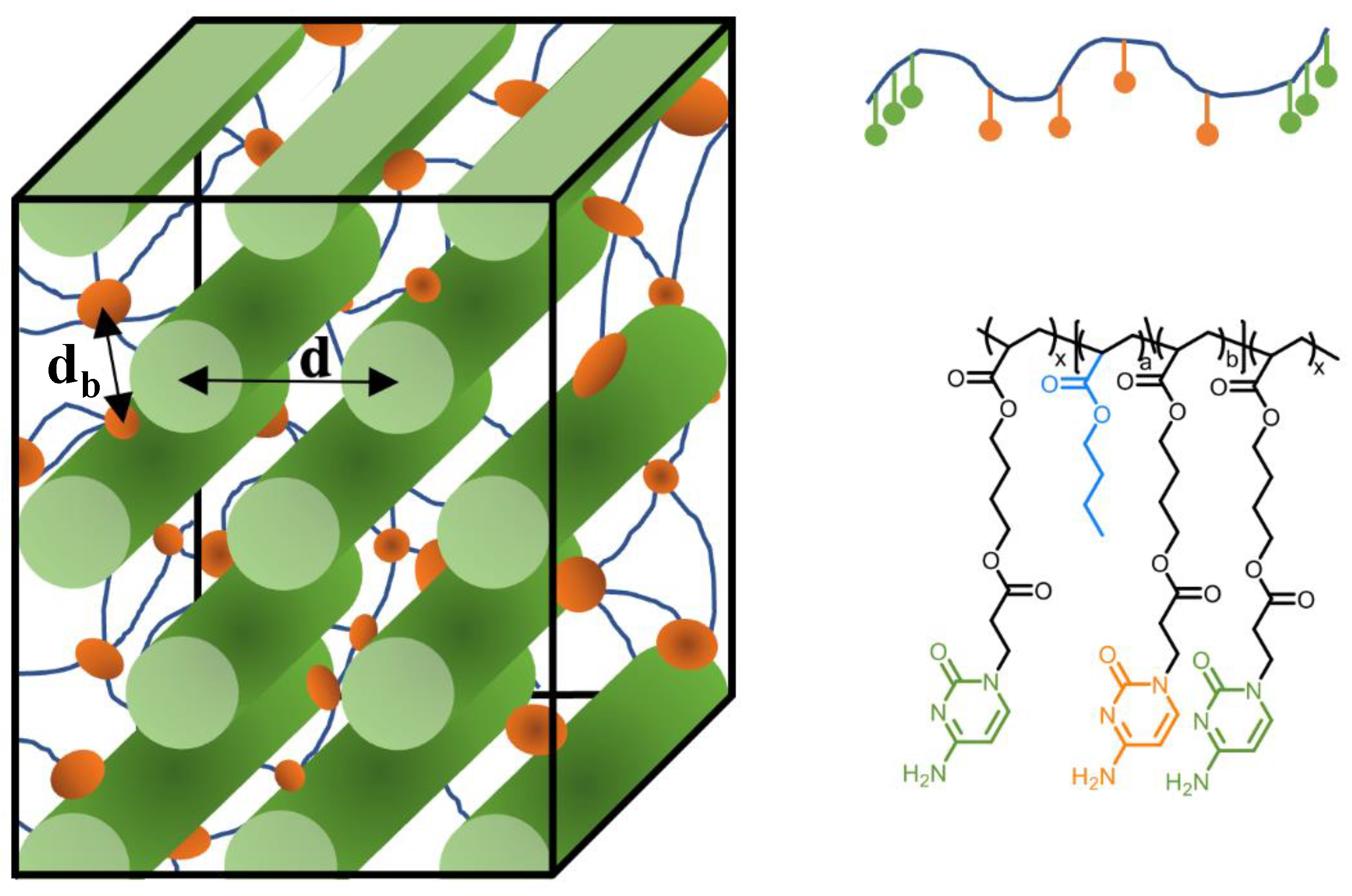
2.4. Morphological Characterization
3. Materials and Methods
3.1. Materials
3.2. Synthesis of Poly(nBA-co-CyA) Difunctional Macro-CTA (Scheme 1)
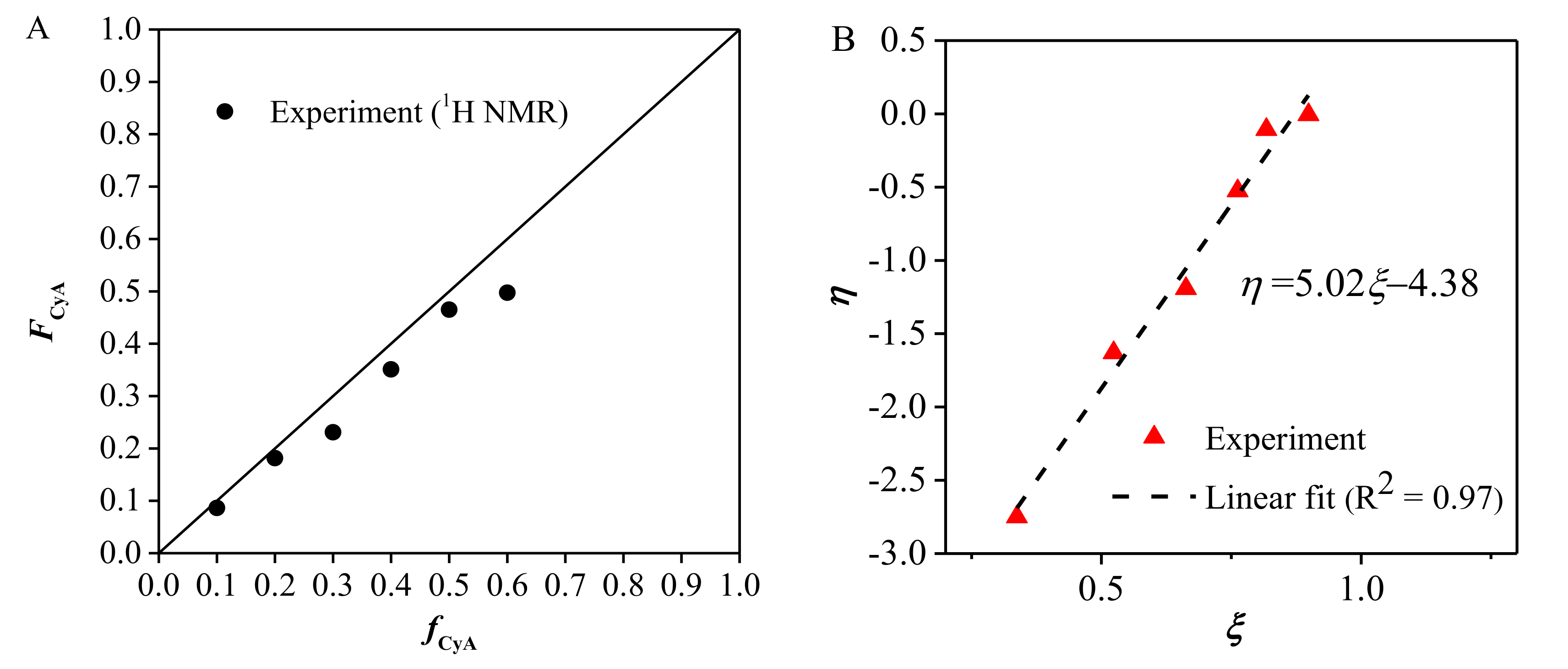
3.3. Reactivity Ratio Determination
3.4. Chain Extension of Poly(nBA-co-CyA) with CyA (Scheme 1)
3.5. Synthesis of Poly(UCyA-b-(nBA-co-UCyA)-b-UCyA) Triblock Copolymers (Scheme 1)
3.6. Polymer Film Preparation and Annealing Conditions
3.7. Instrumentation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Pekkanen, A.M.; Zawaski, C.; Stevenson, A.T., Jr.; Dickerman, R.; Whittington, A.R.; Williams, C.B.; Long, T.E. Poly (ether ester) ionomers as water-soluble polymers for material extrusion additive manufacturing processes. ACS Appl. Mater. Interfaces 2017, 9, 12324–12331. [Google Scholar] [CrossRef]
- Chen, X.; Zawaski, C.E.; Spiering, G.A.; Liu, B.; Orsino, C.M.; Moore, R.B.; Williams, C.B.; Long, T.E. Quadruple Hydrogen Bonding Supramolecular Elastomers for Melt Extrusion Additive Manufacturing. ACS Appl. Mater. Interfaces 2020, 12, 32006–32016. [Google Scholar] [CrossRef]
- Thermoplastic Elastomer (TPE) Market Size & Share Report. 2016. Available online: https://www.grandviewresearch.com/industry-analysis/thermoplastic-elastomers-market (accessed on 9 March 2021).
- Lu, W.; Wang, Y.; Wang, W.; Cheng, S.; Zhu, J.; Xu, Y.; Hong, K.; Kang, N.G.; Mays, J. All acrylic-based thermoplastic elastomers with high upper service temperature and superior mechanical properties. Polym. Chem. 2017, 8, 5741–5748. [Google Scholar] [CrossRef]
- Kraus, G.; Rollmann, K.W. Dynamic viscoelastic behavior of ABA block polymers and the nature of the domain boundary. J. Polym. Sci. Polym. Phys. Ed. 1976, 14, 1133–1148. [Google Scholar] [CrossRef]
- Chen, X.; Talley, S.J.; Haag, J.V.; Spiering, G.A.; Liu, B.; Drummey, K.J.; Murayama, M.; Moore, R.B.; Long, T.E. Doubly Charged ABA Triblock Copolymers: Thermomechanically Robust Physical Network and Hierarchical Microstructures. Macromolecules 2019, 52, 9168–9176. [Google Scholar] [CrossRef]
- Wang, W.; Schlegel, R.; White, B.T.; Williams, K.; Voyloy, D.; Steren, C.A.; Goodwin, A.; Coughlin, E.B.; Gido, S.; Beiner, M.; et al. High Temperature Thermoplastic Elastomers Synthesized by Living Anionic Polymerization in Hydrocarbon Solvent at Room Temperature. Macromolecules 2016, 49, 2646–2655. [Google Scholar] [CrossRef]
- Huang, X.; Nakagawa, S.; Houjou, H.; Yoshie, N. Insights into the Role of Hydrogen Bonds on the Mechanical Properties of Polymer Networks. Macromolecules 2021, 54, 4070–4080. [Google Scholar] [CrossRef]
- Tee, H.T.; Koynov, K.; Reichel, T.; Wurm, F.R. Noncovalent Hydrogen Bonds Tune the Mechanical Properties of Phosphoester Polyethylene Mimics. ACS Omega 2019, 4, 9324–9332. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.; Zhang, M.; Dixit, N.; Moore, R.B.; Long, T.E. Nucleobase Self-Assembly in Supramolecular Adhesives. Macromolecules 2012, 45, 805–812. [Google Scholar] [CrossRef]
- Varlas, S.; Hua, Z.; Jones, J.R.; Thomas, M.; Foster, J.C.; O’Reilly, R.K. Complementary nucleobase interactions drive the hierarchical self-assembly of core-shell bottlebrush block copolymers toward cylindrical supramolecules. Macromolecules 2020, 53, 9747–9757. [Google Scholar] [CrossRef]
- Hua, Z.; Keogh, R.; Li, Z.; Wilks, T.R.; Chen, G.; O’Reilly, R.K. Reversibly Manipulating the Surface Chemistry of Polymeric Nanostructures via a “Grafting To” Approach Mediated by Nucleobase Interactions. Macromolecules 2017, 50, 3662–3670. [Google Scholar] [CrossRef]
- Mather, B.D.; Baker, M.B.; Beyer, F.L.; Berg, M.A.G.; Green, M.D.; Long, T.E. Supramolecular Triblock Copolymers Containing Complementary Nucleobase Molecular Recognition. Macromolecules 2007, 40, 6834–6845. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, D.; Liu, H.; Yue, L.; Bai, Y.; He, J. A nucleobase-inspired super adhesive hydrogel with desirable mechanical, tough and fatigue resistant properties based on cytosine and ε-caprolactone. Eur. Polym. J. 2020, 133, 109741. [Google Scholar] [CrossRef]
- Chen, Y.N.; Peng, L.; Liu, T.; Wang, Y.; Shi, S.; Wang, H. Poly(vinyl alcohol)-Tannic Acid Hydrogels with Excellent Mechanical Properties and Shape Memory Behaviors. ACS Appl. Mater. Interfaces 2016, 8, 27199–27206. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Pitet, L.M.; Wyss, H.M.; Vos, M.; Dankers, P.Y.W.; Meijer, E.W. Tough stimuli-responsive supramolecular hydrogels with hydrogen-bonding network junctions. J. Am. Chem. Soc. 2014, 136, 6969–6977. [Google Scholar] [CrossRef]
- Yang, Q.; Zheng, W.; Zhao, W.; Peng, C.; Ren, J.; Yu, Q.; Hu, Y.; Zhang, X. One-way and two-way shape memory effects of a high-strain cis-1,4-polybutadiene–polyethylene copolymer based dynamic network via self-complementary quadruple hydrogen bonding. Polym. Chem. 2019, 10, 718–726. [Google Scholar] [CrossRef]
- Zhang, K.; Aiba, M.; Fahs, G.B.; Hudson, A.G.; Chiang, W.D.; Moore, R.B.; Ueda, M.; Long, T.E. Nucleobase-functionalized acrylic ABA triblock copolymers and supramolecular blends. Polym. Chem. 2015, 6, 2434–2444. [Google Scholar] [CrossRef] [Green Version]
- Lafitte, V.G.H.H.; Aliev, A.E.; Horton, P.N.; Hursthouse, M.B.; Bala, K.; Golding, P.; Hailes, H.C. Quadruply Hydrogen Bonded Cytosine Modules for Supramolecular Applications. J. Am. Chem. Soc. 2006, 128, 6544–6545. [Google Scholar] [CrossRef] [PubMed]
- Sartorius, J.; Schneider, H.-J. A General Scheme Based on Empirical Increments for the Prediction of Hydrogen-Bond Associations of Nucleobases and of Synthetic Host–Guest complexes. Chem. A Eur. J. 1996, 2, 1446–1452. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, K.; Talley, S.J.; Orsino, C.M.; Moore, R.B.; Long, T.E. Quadruple hydrogen bonding containing supramolecular thermoplastic elastomers: Mechanical and morphological correlations. J. Polym. Sci. Part. A Polym. Chem. 2019, 57, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Fahs, G.B.; Aiba, M.; Moore, R.B.; Long, T.E. Nucleobase-functionalized ABC triblock copolymers: Self-assembly of supramolecular architectures. Chem. Commun. 2014, 50, 9145–9148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karikari, A.S.; Mather, B.D.; Long, T.E. Association of star-shaped poly (D,L-lactide)s containing nucleobase multiple hydrogen bonding. Biomacromolecules 2007, 8, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Mori, Y.; Lim, L.S.; Bates, F.S. Consequences of molecular bridging in lamellae-forming triblock/pentablock copolymer blends. Macromolecules 2003, 36, 9879–9888. [Google Scholar] [CrossRef]
- Zhang, K.; Talley, S.J.; Yu, Y.P.; Moore, R.B.; Murayama, M.; Long, T.E. Influence of nucleobase stoichiometry on the self-assembly of ABC triblock copolymers. Chem. Commun. 2016, 52, 7564–7567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, C.; Loveday, D.R.; Abetz, V.; Stadler, R. Morphology, dynamic mechanical properties, and phase behavior of ABC-triblock copolymers with two semicompatible elastomer blocks. Macromolecules 1998, 31, 2493–2500. [Google Scholar] [CrossRef]
- Hayashi, M.; Matsushima, S.; Noro, A.; Matsushita, Y. Mechanical Property Enhancement of ABA Block Copolymer-Based Elastomers by Incorporating Transient Cross-Links into Soft Middle Block. Macromolecules 2015, 48, 421–431. [Google Scholar] [CrossRef]
- Kawarazaki, I.; Hayashi, M. Enhancement of Mechanical Properties of ABA Triblock Copolymer-Based Elastomers by Incorporating Partial Cross-Links on the Soft Bridge Chains. ACS Appl. Polym. Mater. 2021, 2021, 1271–1275. [Google Scholar] [CrossRef]
- Kajita, T.; Tanaka, H.; Noro, A.; Matsushita, Y.; Nozawa, A.; Isobe, K.; Oda, R.; Hashimoto, S. Extremely tough block polymer-based thermoplastic elastomers with strongly associated but dynamically responsive noncovalent cross-links. Polymer 2021, 217, 123419. [Google Scholar] [CrossRef]
- Yoshida, S.; Ejima, H.; Yoshie, N. Tough Elastomers with Superior Self-Recoverability Induced by Bioinspired Multiphase Design. Adv. Funct. Mater. 2017, 27, 1701670. [Google Scholar] [CrossRef]
- Kawana, S.; Nakagawa, S.; Nakai, S.; Sakamoto, M.; Ishii, Y.; Yoshie, N. Interphase synergistic effects of dynamic bonds in multiphase thermoplastic elastomers. J. Mater. Chem. A 2019, 7, 21195–21206. [Google Scholar] [CrossRef]
- Beckingham, B.S.; Sanoja, G.E.; Lynd, N.A. Simple and Accurate Determination of Reactivity Ratios Using a Nonterminal Model of Chain Copolymerization. Macromolecules 2015, 48, 6922–6930. [Google Scholar] [CrossRef] [Green Version]
- Boulding, N.A.; Millican, J.M.; Hutchings, L.R. Understanding copolymerisation kinetics for the design of functional copolymers via free radical polymerisation. Polym. Chem. 2019, 10, 5665–5675. [Google Scholar] [CrossRef]
- Zhang, K.; Chen, M.; Drummey, K.J.; Talley, S.J.; Anderson, L.J.; Moore, R.B.; Long, T.E. Ureido cytosine and cytosine-containing acrylic copolymers. Polym. Chem. 2016, 7, 6671–6681. [Google Scholar] [CrossRef] [Green Version]
- Khandpur, A.K.; Foerster, S.; Bates, F.S.; Hamley, I.W.; Ryan, A.J.; Bras, W.; Almdal, K.; Mortensen, K. Polyisoprene-Polystyrene Diblock Copolymer Phase Diagram near the Order-Disorder Transition. Macromolecules 1995, 28, 8796–8806. [Google Scholar] [CrossRef]
- Bates, F.S.; Fredrickson, G.H. Block copolymers-designer soft materials. Phys. Today 1999, 52, 32. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | CyA/UCyA in Copolymer (mol %) | DP a | Td, 15 wt. %b (°C) | Tg1 c (°C) | Tg2 c (°C) | d d (nm) | db d (nm) |
|---|---|---|---|---|---|---|---|
| C50-b-(B480-co-C20)-b-C50 | 20 | 50–488/20–50 | 326 | −37 | 85 | 41 | N/A |
| C50-b-(B450-co-C50)-b-C50 | 25 | 51–453/50–51 | 317 | −23 | 86 | 38 | 4.2 |
| C50-b-(B420-co-C80)-b-C50 | 30 | 51–418/78–51 | 312 | 6 | 83 | 32 | 4.2 |
| U50-b-(B480-co-U20)-b-U50 | 20 | 50–488/20–50 | 280 | −35 | N/A | 41 | N/A |
| U50-b-(B450-co-U50)-b-U50 | 25 | 51–453/50–51 | 265 | −24 | N/A | 31 | 4.5 |
| U50-b-(B420-co-U80)-b-U50 | 30 | 51–418/78–51 | 262 | −9 | N/A | N/A | 4.4 |
| Sample | Young’s Modulus (MPa) | Strain at Break (%) | Stress at Break (MPa) | Toughness (MJ/m−3) |
|---|---|---|---|---|
| B450-co-U150 | 31 ± 2 | 89 ± 10 | 7.0 ± 0.5 | 4.3 ± 0.6 |
| U50-b-(B450-co-U50)-b-U50 | 23 ± 4 | 131 ± 10 | 9.1 ± 0.3 | 8.6 ± 0.7 |
| U75-b-B450-b-U75 | N/A | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, B.; Chen, X.; Spiering, G.A.; Moore, R.B.; Long, T.E. Quadruple Hydrogen Bond-Containing A-AB-A Triblock Copolymers: Probing the Influence of Hydrogen Bonding in the Central Block. Molecules 2021, 26, 4705. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26154705
Liu B, Chen X, Spiering GA, Moore RB, Long TE. Quadruple Hydrogen Bond-Containing A-AB-A Triblock Copolymers: Probing the Influence of Hydrogen Bonding in the Central Block. Molecules. 2021; 26(15):4705. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26154705
Chicago/Turabian StyleLiu, Boer, Xi Chen, Glenn A. Spiering, Robert B. Moore, and Timothy E. Long. 2021. "Quadruple Hydrogen Bond-Containing A-AB-A Triblock Copolymers: Probing the Influence of Hydrogen Bonding in the Central Block" Molecules 26, no. 15: 4705. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26154705