
A Genetically Encoded Isonitrile Lysine for Orthogonal Bioorthogonal Labeling Schemes

 , , , , and
, , , , and
Abstract
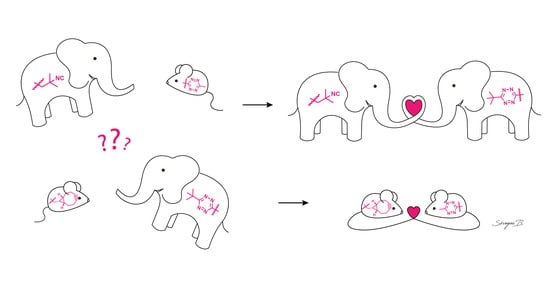
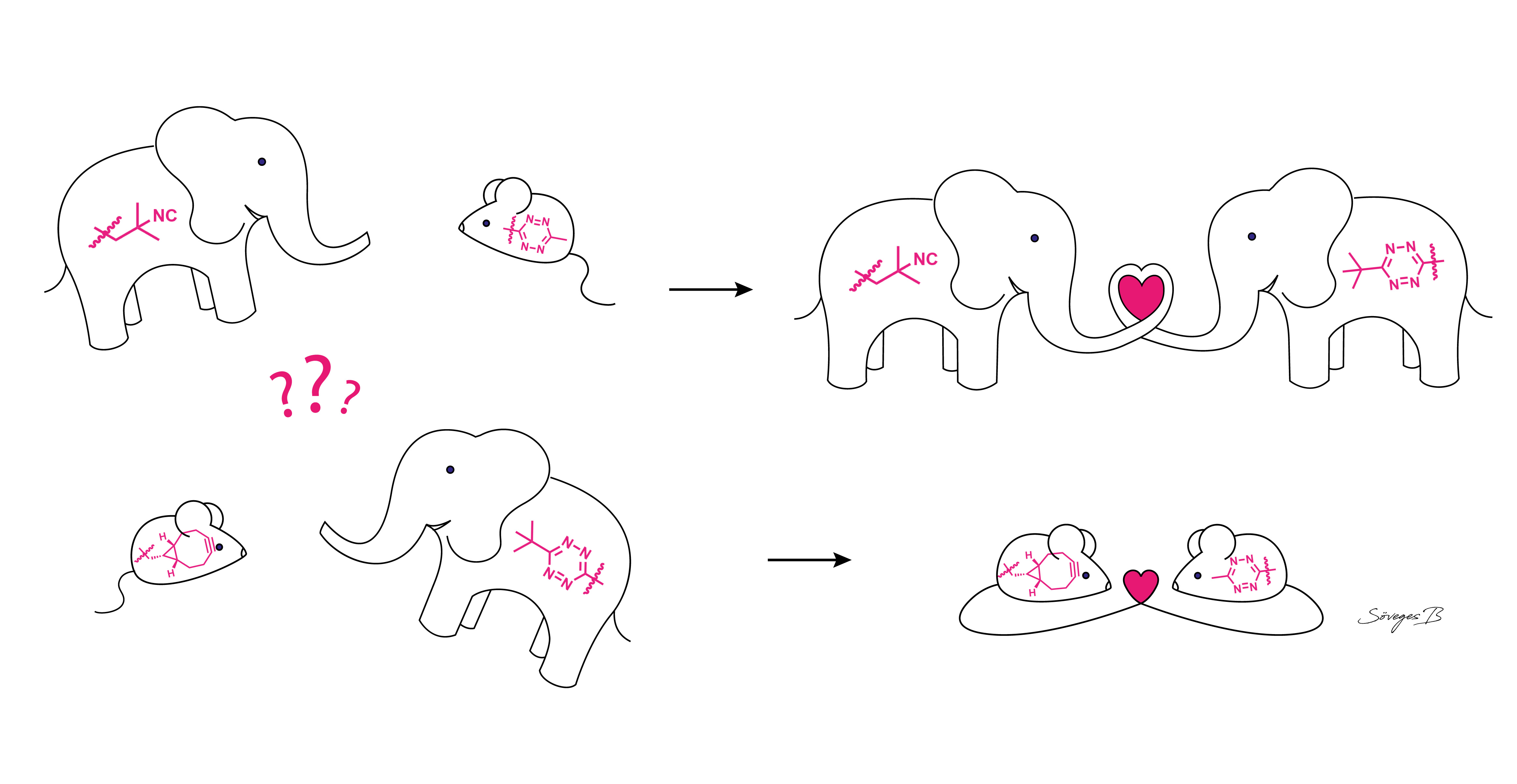
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
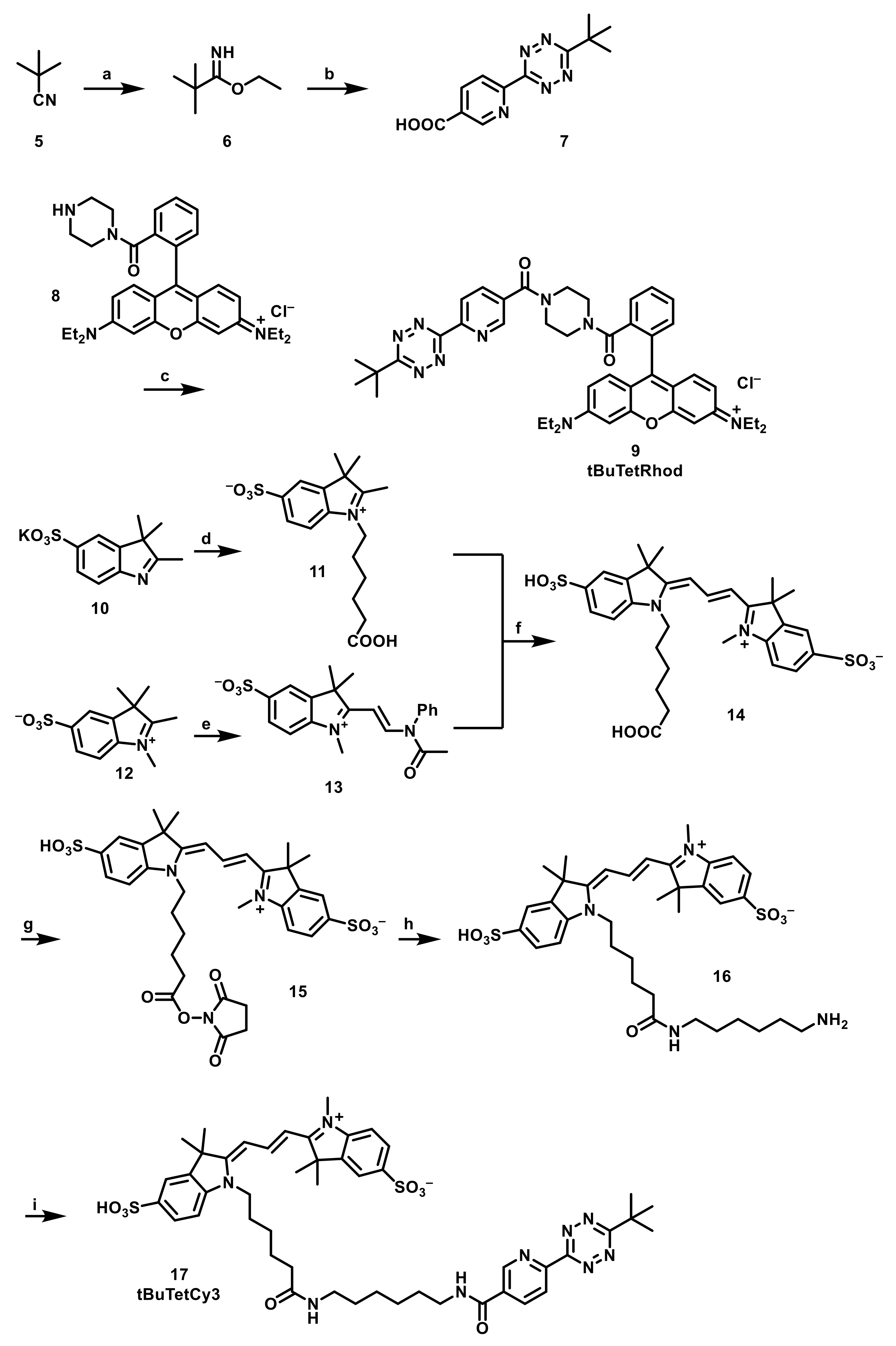
2.1. Synthesis of Bulky Isonitrile-Carbamate-Lysine (BICK), Probes and Selectivity Studies
2.2. Specificity of Protein Labeling In Vitro
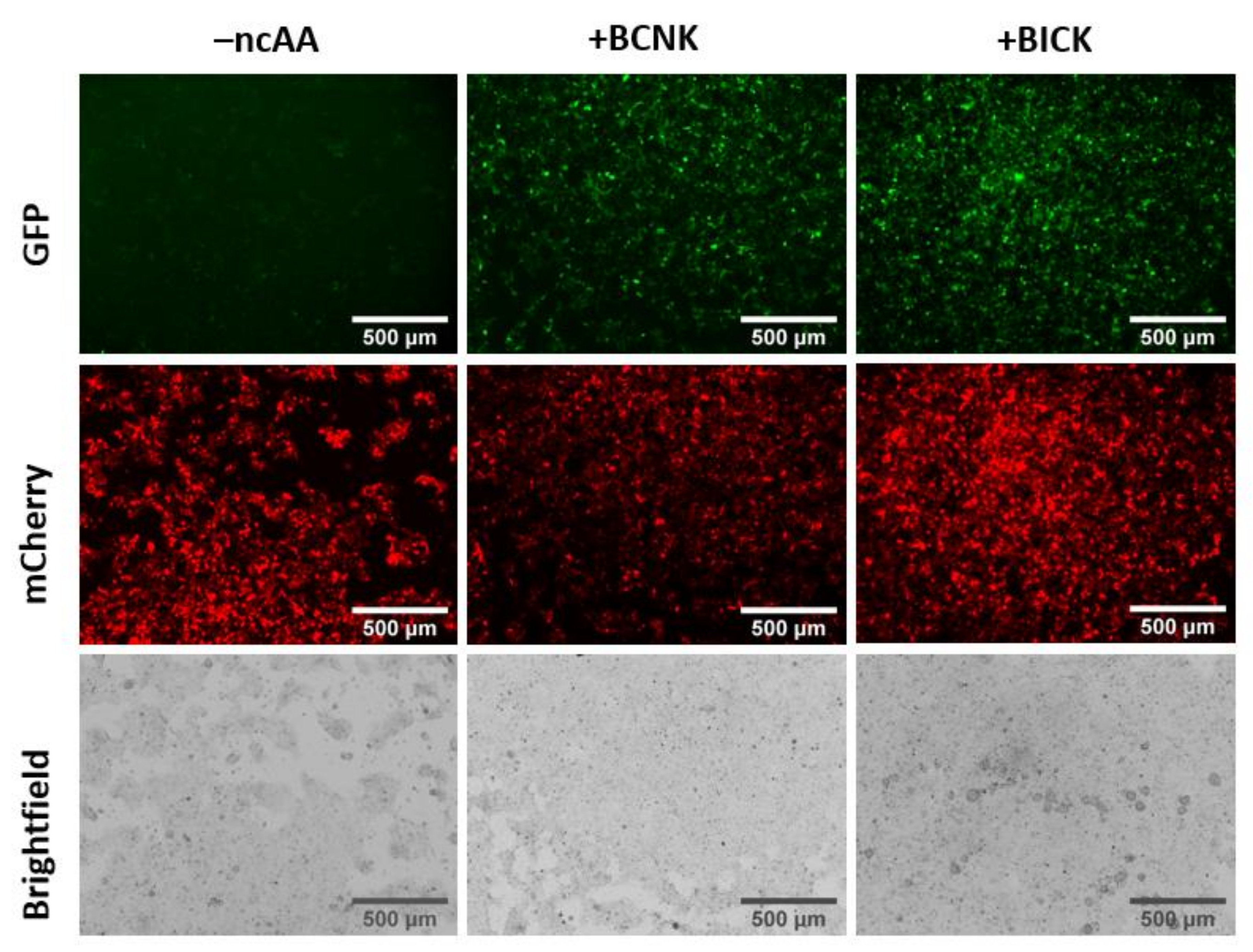
2.3. Genetic Incorporation of BICK into Mammalian Proteins in Cellulo
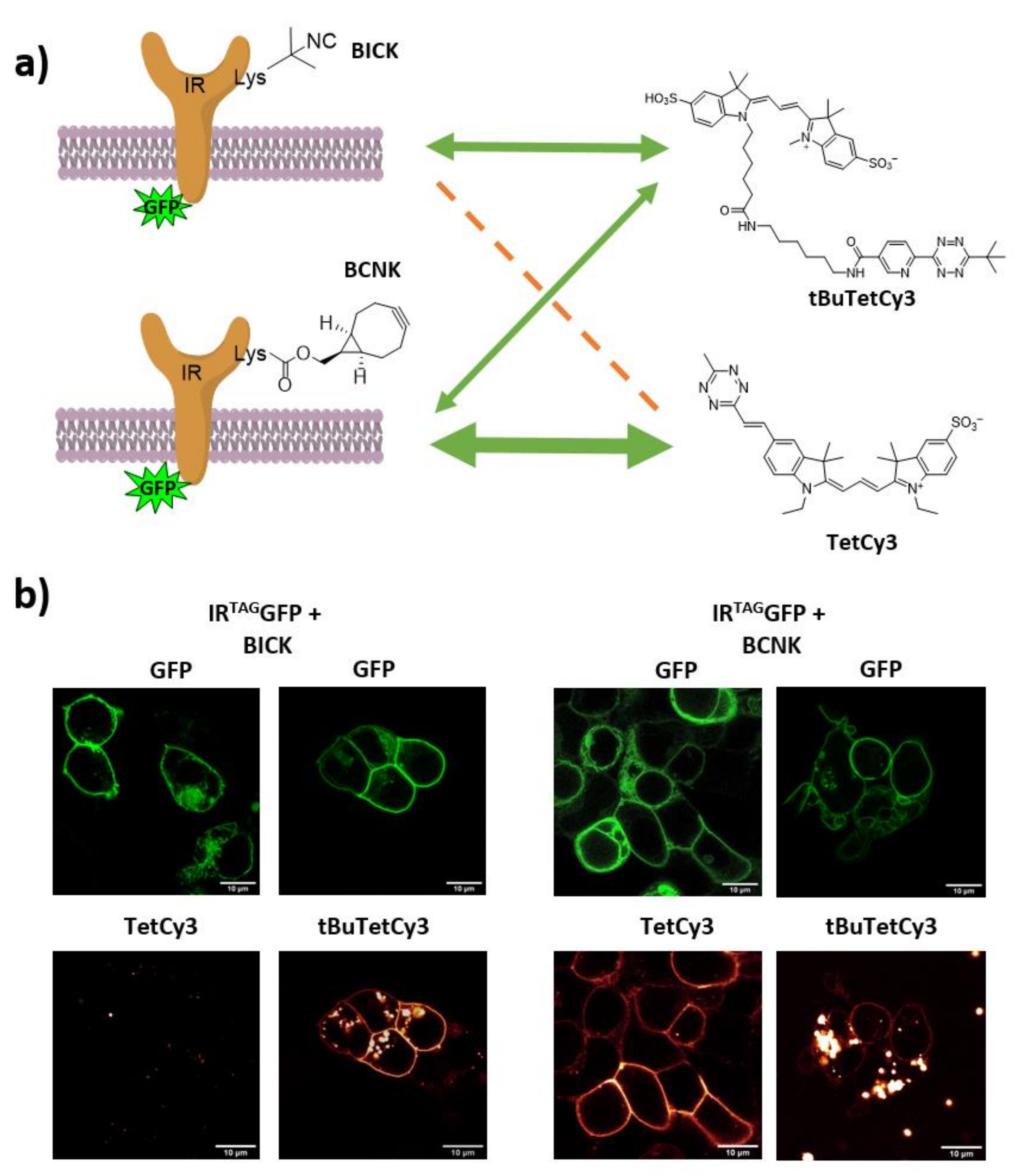
2.4. Live Cell Labeling of Insulin Receptors (IR) on the Extracellular Side of the Cell Membrane
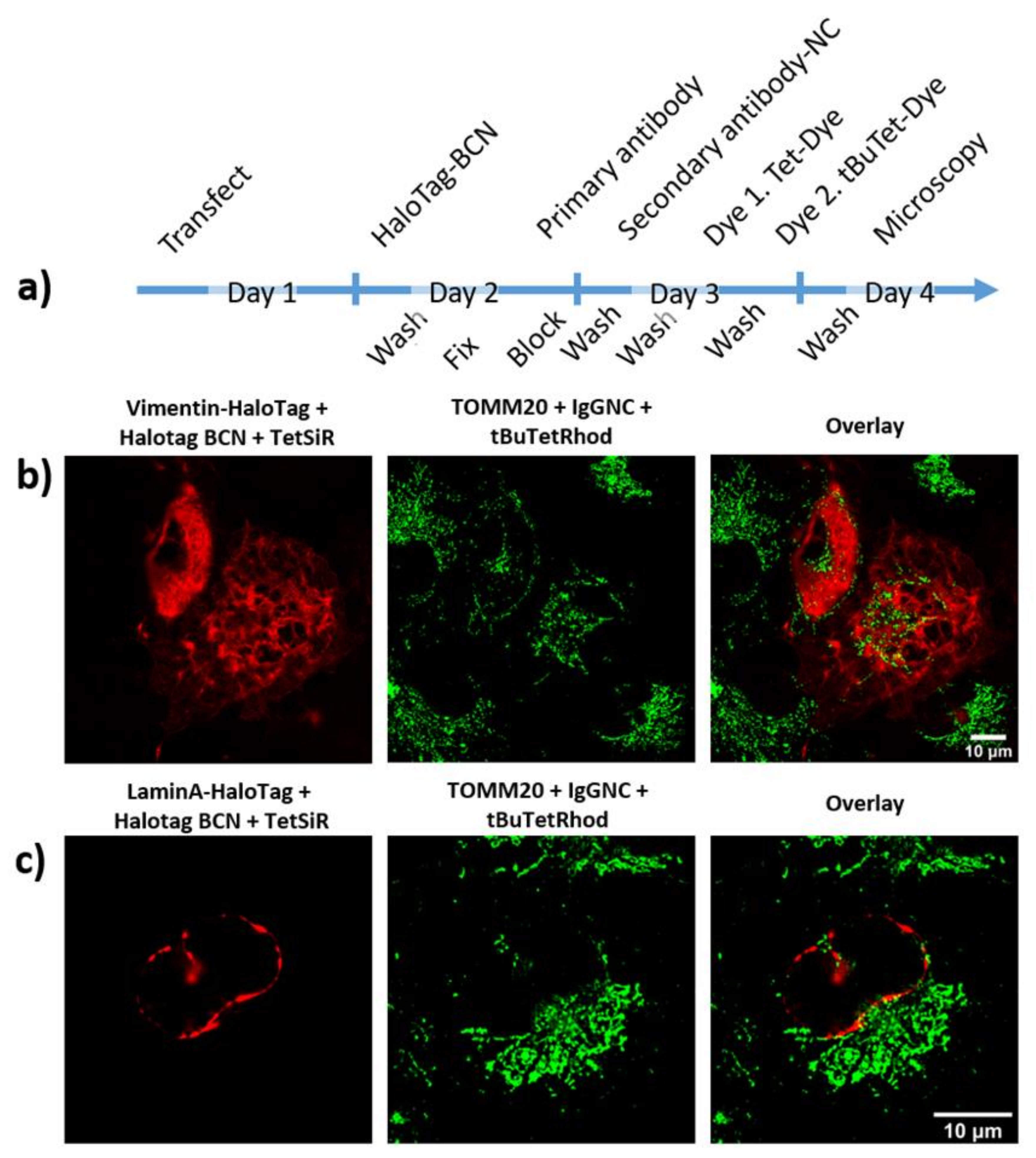
2.5. Dual Colour Labeling and Imaging of Subcellular Structures in Fixed Cells
2.6. Dual Colour Labeling and Imaging of Subcellular Structures in Live Cells
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meineke, B.; Heimgärtner, J.; Eirich, J.; Landreh, M.; Elsässer, S.J. Site-Specific Incorporation of Two ncAAs for Two-Color Bioorthogonal Labeling and Crosslinking of Proteins on Live Mammalian Cells. Cell Rep. 2020, 31, 107811. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.C.; Wu, N.; Santoro, S.W.; Lakshman, V.; King, D.S.; Schultz, P.G. An expanded genetic code with a functional quadruplet codon. Proc. Natl. Acad. Sci. USA 2004, 101, 7566–7571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunkelmann, D.L.; Willis, J.C.W.; Beattie, A.T.; Chin, J.W. Engineered triply orthogonal pyrrolysyl—tRNA synthetase/tRNA pairs enable the genetic encoding of three distinct non-canonical amino acids. Nat. Chem. 2020, 12, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Smeenk, M.L.W.J.; Agramunt, J.; Bonger, K.M. Recent developments in bioorthogonal chemistry and the orthogonality within. Curr. Opin. Chem. Biol. 2021, 60, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Kele, P.; Mezö, G.; Achatz, D.; Wolfbeis, O.S. Dual Labeling of Biomolecules by Using Click Chemistry: A Sequential Approach. Angew. Chem. Int. Ed. 2009, 48, 344–347. [Google Scholar] [CrossRef]
- Achatz, D.E.; Mező, G.; Kele, P.; Wolfbeis, O.S. Probing the Activity of Matrix Metalloproteinase II with a Sequentially Click-Labeled Silica Nanoparticle FRET Probe. ChemBioChem 2009, 10, 2316–2320. [Google Scholar] [CrossRef]
- Nikić, I.; Plass, T.; Schraidt, O.; Szymański, J.; Briggs, J.; Schultz, C.; Lemke, E.A. Minimal Tags for Rapid Dual-Color Live-Cell Labeling and Super-Resolution Microscopy. Angew. Chem. Int. Ed. 2014, 53, 2245–2249. [Google Scholar] [CrossRef]
- Tu, J.; Svatunek, D.; Parvez, S.; Liu, A.C.; Levandowski, B.J.; Eckvahl, H.J.; Peterson, R.T.; Houk, K.N.; Franzini, R.M. Stable, Reactive, and Orthogonal Tetrazines: Dispersion Forces Promote the Cycloaddition with Isonitriles. Angew. Chem. 2019, 131, 9141–9146. [Google Scholar] [CrossRef]
- Nikić, I.; Estrada Girona, G.; Kang, J.H.; Paci, G.; Mikhaleva, S.; Koehler, C.; Shymanska, N.V.; Santos, C.V.; Spitz, D.; Lemke, E.A. Debugging Eukaryotic Genetic Code Expansion for Site-Specific Click-PAINT Super-Resolution Microscopy. Angew. Chem. Int. Ed. 2016, 55, 16172–16176. [Google Scholar] [CrossRef] [Green Version]
- Ugi, I.; Fetzer, U.; Eholzer, U.; Knupfer, H.; Offermann, K. Isonitrile Syntheses. Angew. Chem. Int. Ed. Engl. 1965, 4, 472–484. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, K.-L.L.; Tang, J.; Loredo, A.; Clements, J.; Pei, J.; Peng, Z.; Gupta, R.; Fang, X.; Xiao, H. Addition of Isocyanide-Containing Amino Acids to the Genetic Code for Protein Labelling and Activation. ACS Chem. Biol. 2019, 14, 2793–2799. [Google Scholar] [CrossRef]
- Cserép, G.B.; Demeter, O.; Bätzner, E.; Kállay, M.; Wagenknecht, H.A.; Kele, P. Synthesis and Evaluation of Nicotinic Acid Derived Tetrazines for Bioorthogonal Labeling. Synthesis 2015, 47, 2738–2744. [Google Scholar] [CrossRef] [Green Version]
- Yanagisawa, T.; Ishii, R.; Fukunaga, R.; Kobayashi, T.; Sakamoto, K.; Yokoyama, S. Multistep Engineering of Pyrrolysyl-tRNA Synthetase to Genetically Encode Nε-(o-Azidobenzyloxycarbonyl) lysine for Site-Specific Protein Modification. Chem. Biol. 2008, 15, 1187–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautier, A.; Nguyen, D.P.; Lusic, H.; An, W.; Deiters, A.; Chin, J.W. Genetically encoded photocontrol of protein localization in mammalian cells. J. Am. Chem. Soc. 2010, 132, 4086–4088. [Google Scholar] [CrossRef]
- Borrmann, A.; Milles, S.; Plass, T.; Dommerholt, J.; Verkade, J.M.M.; Wießler, M.; Schultz, C.; Van Hest, J.C.M.; Van Delft, F.L.; Lemke, E.A. Genetic Encoding of a Bicyclo[6.1.0]nonyne-Charged Amino Acid Enables Fast Cellular Protein Imaging by Metal-Free Ligation. ChemBioChem 2012, 13, 2094–2099. [Google Scholar] [CrossRef]
- Ye, S.; Köhrer, C.; Huber, T.; Kazmi, M.; Sachdev, P.; Yan, E.C.; Bhagat, A.; RajBhandary, U.L.; Sakmar, T.P.; Ye, S.; et al. Site-specific incorporation of keto amino acids into functional G protein-coupled receptors using unnatural amino acid mutagenesis. J. Biol. Chem. 2008, 283, 1525–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knorr, G.; Kozma, E.; Schaart, J.M.; Németh, K.; Török, G.; Kele, P. Bioorthogonally Applicable Fluorogenic Cyanine-Tetrazines for No-Wash Super-Resolution Imaging. Bioconjug Chem. 2018, 29, 1312–1318. [Google Scholar] [CrossRef]
- Erdmann, R.S.; Baguley, S.W.; Richens, J.H.; Wissner, R.F.; Xi, Z.; Allgeyer, E.S.; Zhong, S.; Thompson, A.D.; Lowe, N.; Butler, R.; et al. Labeling Strategies Matter for Super-Resolution Microscopy: A Comparison between HaloTags and SNAP-tags. Cell Chem Biol. 2019, 26, 584–592.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murrey, H.E.; Judkins, J.C.; Am Ende, C.W.; Ballard, T.E.; Fang, Y.; Riccardi, K.; Di, L.; Guilmette, E.R.; Schwartz, J.W.; Fox, J.M.; et al. Systematic Evaluation of Bioorthogonal Reactions in Live Cells with Clickable HaloTag Ligands: Implications for Intracellular Imaging. J. Am. Chem. Soc. 2015, 137, 11461–11475. [Google Scholar] [CrossRef] [PubMed]
- Kozma, E.; Estrada Girona, G.; Paci, G.; Lemke, E.A.; Kele, P. Bioorthogonal double-fluorogenic siliconrhodamine probes for intracellular super-resolution microscopy. Chem. Commun. 2017, 53, 6696–6699. [Google Scholar] [CrossRef] [Green Version]
- Patterson, D.M.; Prescher, J.A. Orthogonal bioorthogonal chemistries. Curr. Opin. Chem. Biol. 2015, 28, 141–149. [Google Scholar] [CrossRef]
- Wan, W.; Huang, Y.; Wang, Z.; Russell, W.K.; Pai, P.-J.; Russell, D.H.; Liu, W.R. A Facile System for Genetic Incorporation of Two Different Noncanonical Amino Acids into One Protein in Escherichia coli. Angew. Chem. Int. Ed. 2010, 49, 3211–3214. [Google Scholar] [CrossRef] [PubMed]
- Wainman, Y.A.; Neves, A.A.; Stairs, S.; Stöckmann, H.; Ireland-Zecchini, H.; Brindle, K.M.; Leeper, F.J. Dual-sugar imaging using isonitrile and azido-based click chemistries. Org. Biomol. Chem. 2013, 11, 7297–7300. [Google Scholar] [CrossRef]
- Dommerholt, J.; Schmidt, S.; Temming, R.; Hendriks, L.J.A.; Rutjes, F.P.J.T.; Van Hest, J.C.M.; Lefeber, D.J.; Friedl, P.; Van Delft, F.L. Readily accessible bicyclononynes for bioorthogonal labeling and three-dimensional imaging of living cells. Angew. Chem. Int. Ed. 2010, 49, 9422–9425. [Google Scholar] [CrossRef] [PubMed]
- Sakin, V.; Hanne, J.; Dunder, J.; Anders-Össwein, M.; Laketa, V.; Nikic, I.; Kräusslich, H.-G.; Lemke, E.A.; Müller, B. A Versatile Tool for Live-Cell Imaging and Super-Resolution Nanoscopy Studies of HIV-1 Env Distribution and Mobility. Cell Chem. Biol. 2017, 24, 635–645.e5. [Google Scholar] [CrossRef] [Green Version]
- Serfling, R.; Lorenz, C.; Etzel, M.; Schicht, G.; Coin, I. NAR Breakthrough Article Designer tRNAs for efficient incorporation of non-canonical amino acids by the pyrrolysine system in mammalian cells. Nucleic Acids Res. 2017, 46, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikić-Spiegel, I. Expanding the Genetic Code for Neuronal Studies. ChemBioChem 2020, 21, 3169–3179. [Google Scholar] [CrossRef]
- Nguyen, T.; Francis, M.B. Practical synthetic route to functionalized rhodamine dyes. Org. Lett. 2003, 5, 3245–3248. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T.; Sullivan, K.F.; Wahl, G.M. Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr. Biol. 1998, 8, 377–385. [Google Scholar] [CrossRef] [Green Version]
- Ebner, M.; Lučić, I.; Leonard, T.A.; Yudushkin, I. PI(3,4,5)P3 Engagement Restricts Akt Activity to Cellular Membranes. Mol. Cell. 2017, 65, 416–431.e6. [Google Scholar] [CrossRef] [Green Version]
- Shcherbakova, D.M.; Baloban, M.; Emelyanov, A.V.; Brenowitz, M.; Guo, P.; Verkhusha, V.V. Bright monomeric near infrared fluorescent proteins as tags and biosensors for multiscale imaging. Nat. Commun. 2016, 7, 664. [Google Scholar] [CrossRef] [PubMed]
- Nikic, I.; Kang, J.H.; Girona, G.E.; Aramburu, I.V.; Lemke, E.A. Labeling proteins on live mammalian cells using click chemistry. Nat. Protoc. 2015, 10, 780–791. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szatmári, Á.; Cserép, G.B.; Molnár, T.Á.; Söveges, B.; Biró, A.; Várady, G.; Szabó, E.; Németh, K.; Kele, P. A Genetically Encoded Isonitrile Lysine for Orthogonal Bioorthogonal Labeling Schemes. Molecules 2021, 26, 4988. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26164988
Szatmári Á, Cserép GB, Molnár TÁ, Söveges B, Biró A, Várady G, Szabó E, Németh K, Kele P. A Genetically Encoded Isonitrile Lysine for Orthogonal Bioorthogonal Labeling Schemes. Molecules. 2021; 26(16):4988. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26164988
Chicago/Turabian StyleSzatmári, Ágnes, Gergely B. Cserép, Tibor Á. Molnár, Bianka Söveges, Adrienn Biró, György Várady, Edit Szabó, Krisztina Németh, and Péter Kele. 2021. "A Genetically Encoded Isonitrile Lysine for Orthogonal Bioorthogonal Labeling Schemes" Molecules 26, no. 16: 4988. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26164988