
Application of a Low Transition Temperature Mixture for the Dispersive Liquid–Liquid Microextraction of Illicit Drugs from Urine Samples

 , ,
, , 

Abstract
:1. Introduction
2. Results
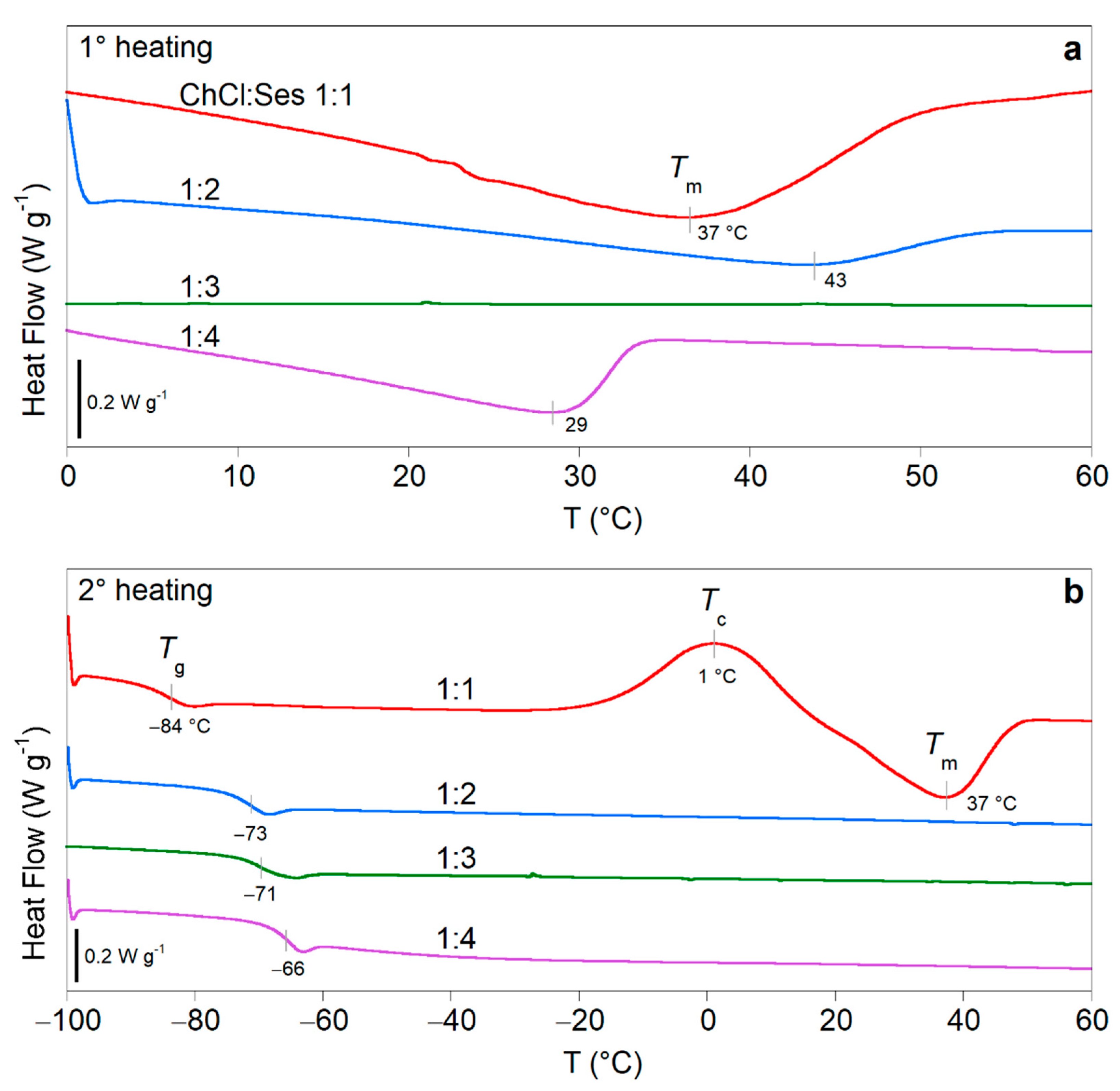
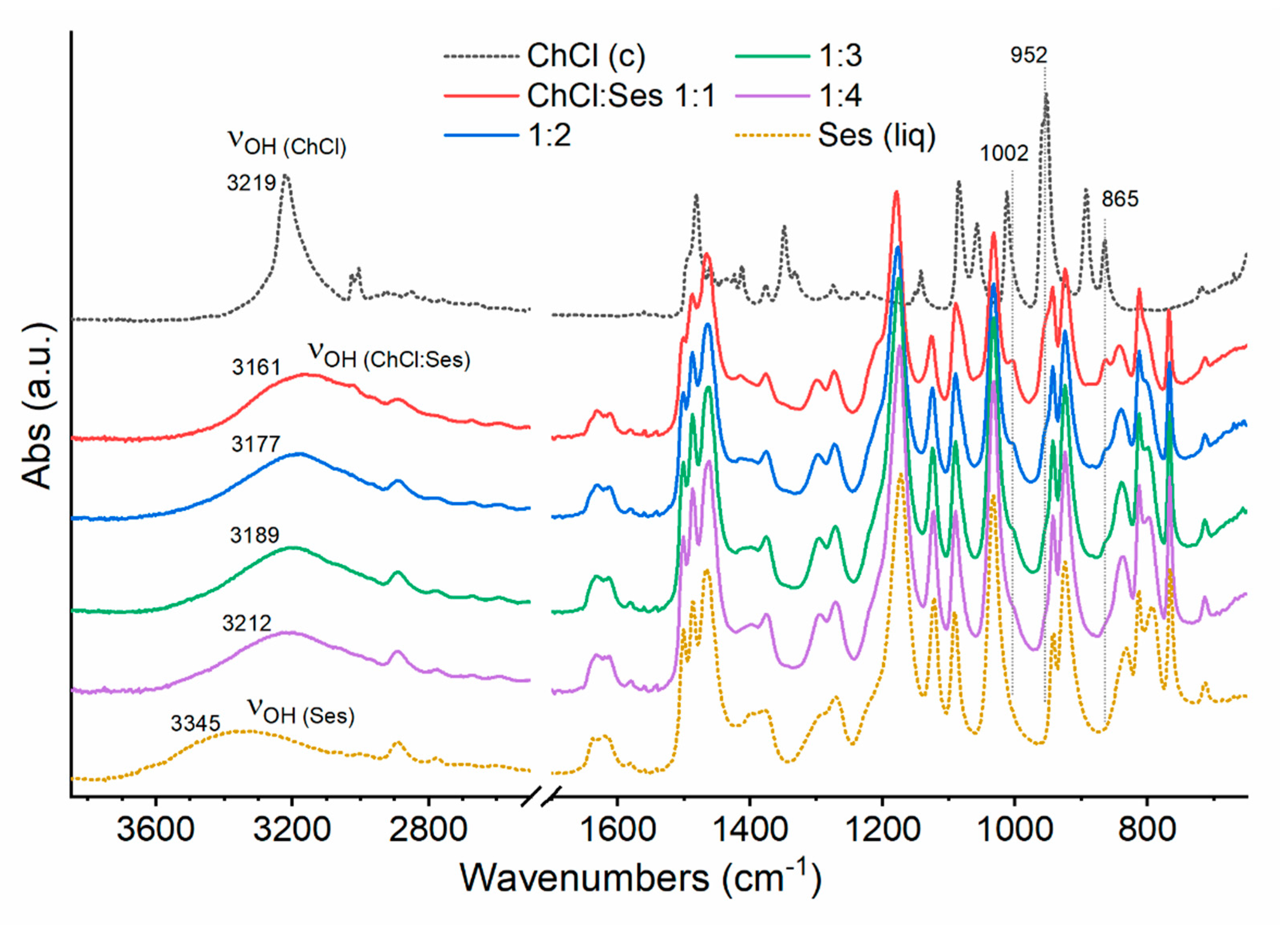
2.1. Preparation and Characterization of Some ChCl:Ses Mixtures
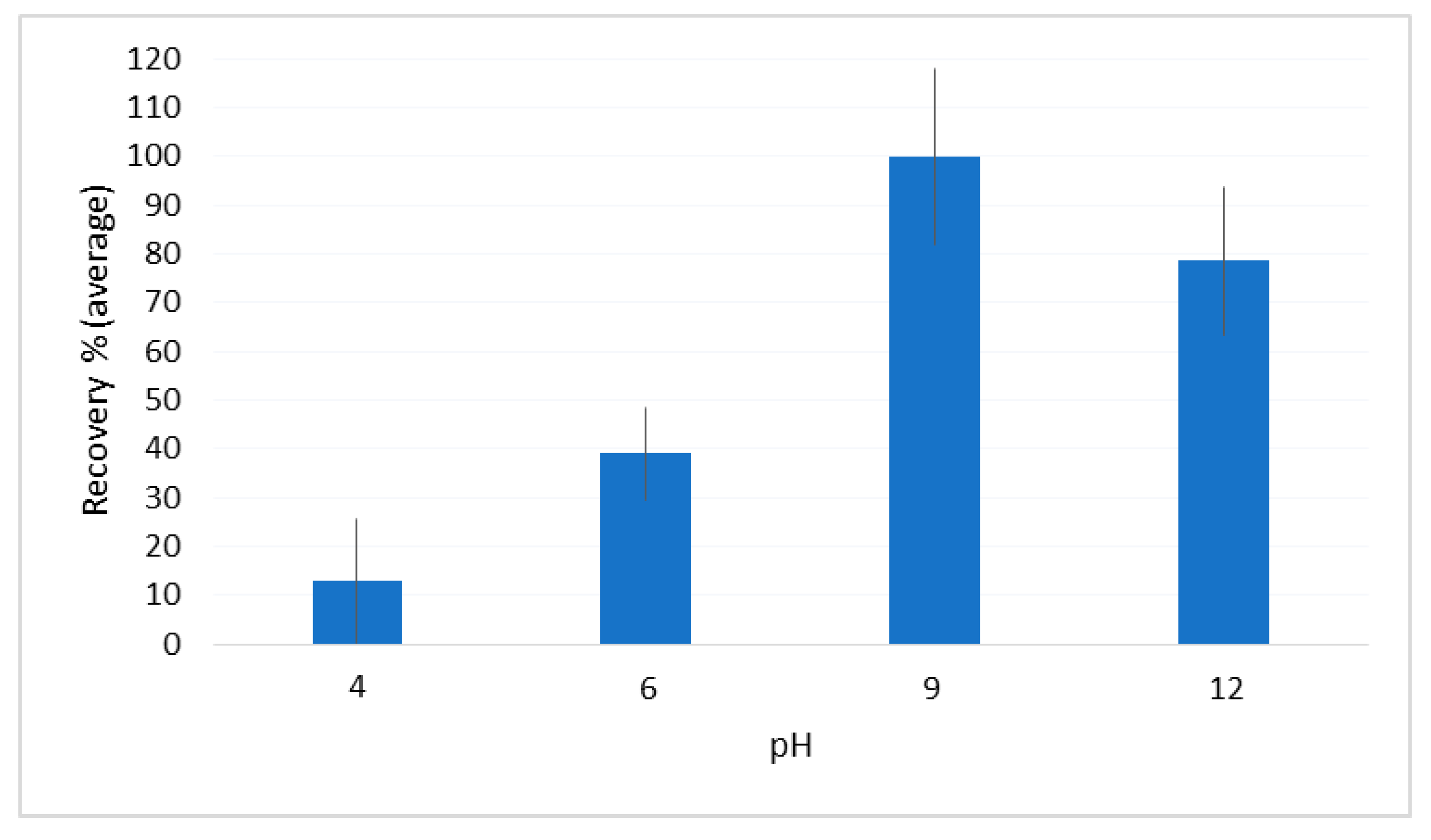
2.2. Optimization of the DLLME Procedure
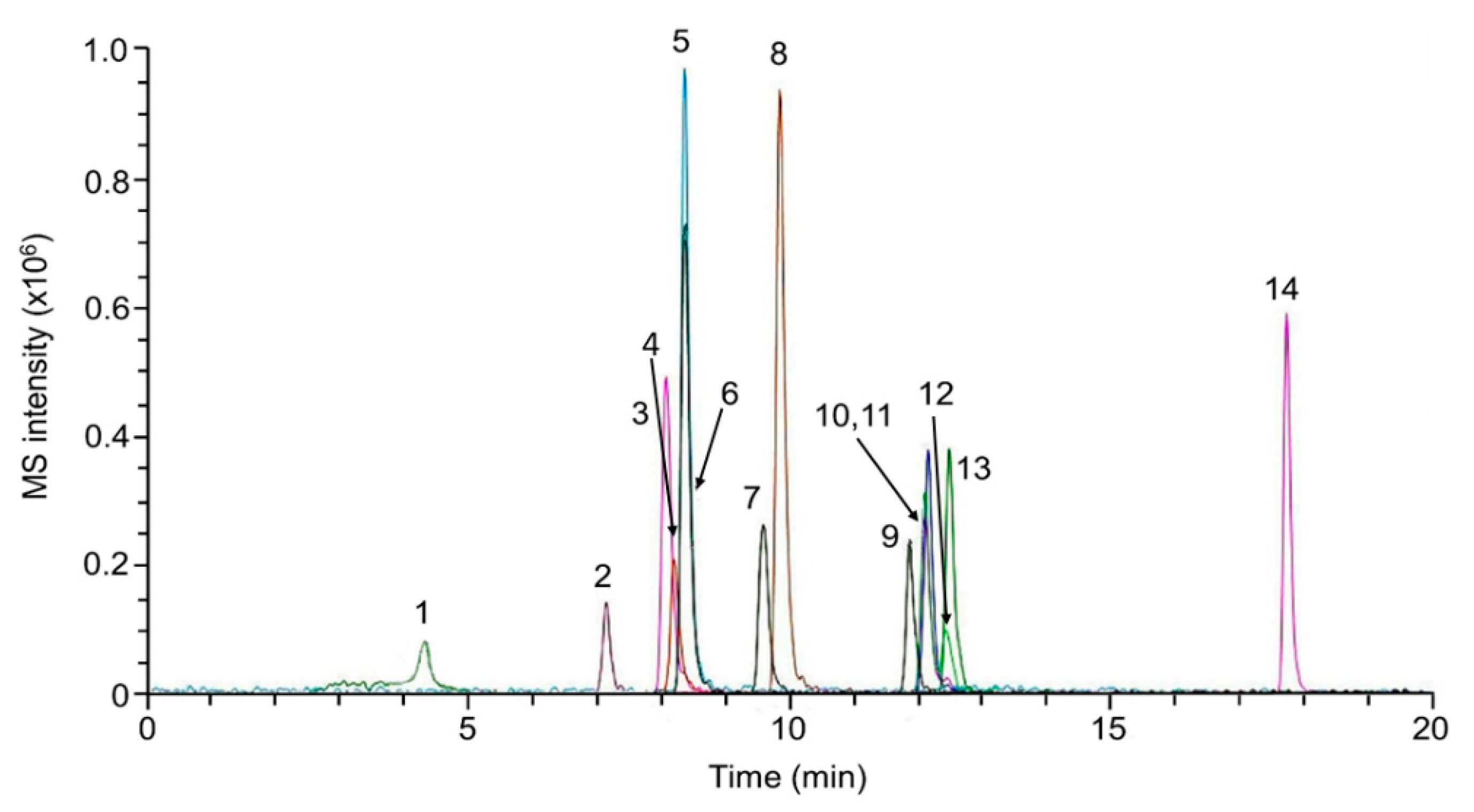
2.3. Method Validation
2.4. Comparison with Previous DLLME-Based Methods
3. Experimental
3.1. Chemicals, Materials, and Solutions
3.2. Urine Samples
3.3. Preparation of ChCl:Ses 1:3 Mixture
3.4. Differential Scanning Calorimetry
3.5. Infrared Spectroscopy
3.6. Extraction Procedure
3.7. HPLC-MS Analysis
3.8. Method Validation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Riley, A.L.; Nelson, K.H.; To, P.; López-Arnau, R.; Xu, P.; Wang, D.; Wangc, Y.; Shen, H.-W.; Kuhn, D.M.; Angoa-Perez, M.; et al. Abuse potential and toxicity of the synthetic cathinones (i.e.,“Bath salts”). Neurosci. Biobehav. Rev. 2020, 110, 150–173. [Google Scholar] [CrossRef]
- Schneir, A.; Ly, B.T.; Casagrande, K.; Darracq, M.; Offerman, S.R.; Thornton, S.; Smollin, C.; Vohra, R.; Rangun, C.; Tomaszewski, C.; et al. Comprehensive analysis of “bath salts” purchased from California stores and the internet. Clin. Toxicol. 2014, 52, 651–658. [Google Scholar] [CrossRef]
- Spiller, H.A.; Ryan, M.L.; Weston, R.G.; Jansen, J. Clinical experience with and analytical confirmation of “bath salts” and “legal highs” (synthetic cathinones) in the United States. Clin. Toxicol. 2011, 49, 499–505. [Google Scholar] [CrossRef]
- German, C.L.; Fleckenstein, A.E.; Hanson, G.R. Bath salts and synthetic cathinones: An emerging designer drug phenomenon. Life Sci. 2014, 97, 2–8. [Google Scholar] [CrossRef] [Green Version]
- Palamar, J.J.; Salomone, A.; Gerace, E.; Di Corcia, D.; Vincenti, M.; Cleland, C.M. Hair testing to assess both known and unknown use of drugs amongst ecstasy users in the electronic dance music scene. Int. J. Drug Policy 2017, 41, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Allen, K.R. Screening for drugs of abuse: Which matrix, oral fluid or urine? Ann. Clin. Biochem. 2011, 48, 531–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivier, L. Techniques for analytical testing of unconventional samples. Best Pract. Res. Clin. Endocrinol. 2000, 14, 147–165. [Google Scholar] [CrossRef]
- Hegstad, S.; Hermansson, S.; Betnér, I.; Spigset, O.; Falch, B.M.H. Screening and quantitative determination of drugs of abuse in diluted urine by UPLC–MS/MS. J. Chromatogr. B 2014, 947, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Grueninger, D.; Englert, R. Determination of the amphetamine-like designer drugs methcathinone and 4-methylmethcathinone in urine by LC-MS/MS. Annal. Toxicol. Anal. 2011, 23, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Concheiro, M.; Anizan, S.; Ellefsen, K.; Huestis, M.A. Simultaneous quantification of 28 synthetic cathinones and metabolites in urine by liquid chromatography-high resolution mass spectrometry. Anal. Bioanal. Chem. 2013, 405, 9437–9448. [Google Scholar] [CrossRef]
- Tomai, P.; Lippiello, A.; D’Angelo, P.; Persson, I.; Martinelli, A.; Di Lisio, V.; Curini, R.; Fanali, C.; Gentili, A. A low transition temperature mixture for the dispersive liquid-liquid microextraction of pesticides from surface waters. J. Chromatogr. A 2019, 1605, 360329. [Google Scholar] [CrossRef]
- Tomai, P.; Gentili, A.; Curini, R.; Gottardo, R.; Tagliaro, F.; Fanali, S. Dispersive liquid-liquid microextraction, an effective tool for the determination of synthetic cannabinoids in oral fluid by liquid chromatography–tandem mass spectrometry. J. Pharm. Anal. 2021, 11, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Li, Q.; Wei, W.; Liu, L.; Li, H. Development of a liquid–liquid microextraction method based on a switchable hydrophilicity solvent for the simultaneous determination of 11 drugs in urine by GC–MS. Chromatographia 2018, 81, 1695–1703. [Google Scholar] [CrossRef]
- Xu, F.; Liu, L. Simultaneous determination of free methamphetamine, pethidine, ketamine and tramadol in urine by dispersive liquid–liquid microextraction combined with GC–MS. Forensic Sci. Res. 2019, 4, 188–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alahyari, E.; Setareh, M.; Shekari, A.; Roozbehani, G.; Soltaninejad, K. Analysis of opioids in postmortem urine samples by dispersive liquid-liquid microextraction and high performance liquid chromatography with photo diode array detection. Egypt. J. Forensic Sci. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Xin, L.I.U.; Rao, F.U.; Min, L.I.; Li-Ping, G.U.O.; Li, Y.A.N.G. Ionic liquid-based dispersive liquid-liquid microextraction coupled with capillary electrophoresis to determine drugs of abuse in urine. Chin. J. Anal. Chem. 2013, 41, 1919–1922. [Google Scholar]
- Mercieca, G.; Odoardi, S.; Cassar, M.; Rossi, S.S. Rapid and simple procedure for the determination of cathinones, amphetamine-like stimulants and other new psychoactive substances in blood and urine by GC–MS. J Pharm. Biomed. Anal. 2018, 149, 494–501. [Google Scholar] [CrossRef]
- Kohler, I.; Schappler, J.; Sierro, T.; Rudaz, S. Dispersive liquid–liquid microextraction combined with capillary electrophoresis and time-of-flight mass spectrometry for urine analysis. J. Pharm. Biomed. Anal. 2013, 73, 82–89. [Google Scholar] [CrossRef]
- Airado-Rodríguez, D.; Cruces-Blanco, C.; García-Campaña, A.M. Dispersive liquid–liquid microextraction prior to field-amplified sample injection for the sensitive analysis of 3, 4-methylenedioxymethamphetamine, phencyclidine and lysergic acid diethylamide by capillary electrophoresis in human urine. J. Chromatogr. A 2012, 1267, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Farajzadeh, M.A.; Mogaddam, M.R.A.; Aghanassab, M. Deep eutectic solvent-based dispersive liquid–liquid microextraction. Anal. Methods 2016, 8, 2576–2583. [Google Scholar] [CrossRef]
- Shishov, A.; Bulatov, A.; Locatelli, M.; Carradori, S.; Andruch, V. Application of deep eutectic solvents in analytical chemistry. A review. Microchem. J. 2017, 135, 33–38. [Google Scholar] [CrossRef]
- Gallo, V.; Tomai, P.; Gherardi, M.; Fanali, C.; De Gara, L.; D’Orazio, G.; Gentili, A. Dispersive liquid-liquid microextraction using a low transition temperature mixture and liquid chromatography-mass spectrometry analysis of pesticides in urine samples. J. Chromatogr. A 2021, 1642, 462036. [Google Scholar] [CrossRef]
- Dal Bosco, C.; Di Lisio, V.; D’Angelo, P.; Gentili, A. Hydrophobic eutectic solvent with antioxidant properties: Application for the dispersive liquid-liquid microextraction of fatsoluble micronutrients from fruit juices. ACS Sustain. Chem. Eng. 2021, 9, 8170–8178. [Google Scholar] [CrossRef]
- Fernandez, L.; Silva, L.P.; Martins, M.A.R.; Ferreira, O.; Ortega, J.; Pinho, S.P.; Coutinho, J.A.P. Indirect assessment of the fusion properties of choline chloride from solid–liquid equilibria data. Fluid Phase Equilib 2017, 448, 9–14. [Google Scholar] [CrossRef] [Green Version]
- Omotola, E.O.; Olatunji, O.S. Quantification of selected pharmaceutical compounds in water using liquid chromatography-electrospray ionisation mass spectrometry (LC-ESI-MS). Heliyon 2020, 6, e05787. [Google Scholar] [CrossRef]
- Pearson, M.A.; Lu, C.; Schmotzer, B.J.; Waller, L.A.; Riederer, A.M. Evaluation of physiological measures for correcting variation in urinary output: Implications for assessing environmental chemical exposure in children. J. Expo. Sci. Environ. Epidemiol. 2009, 19, 336–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Araújo, W.R.; Salles, M.O.; Paixão, T.R. Development of an enzymeless electroanalytical method for the indirect detection of creatinine in urine samples. Sens. Actuators B Chem. 2012, 173, 847–851. [Google Scholar] [CrossRef]
- Guidance for Industry: Bioanalytical Method Validation; US Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research (CDER); Center for Biologics Evaluation and Research (CBER): Rockville, MD, USA, May 2001. Available online: https://www.federalregister.gov/documents/2001/05/23/01-12908/guidance-for-industry-on-bioanalytical-method-validation-availability (accessed on 25 August 2021).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Regression Equation a | R2 | |
|---|---|---|---|
| b ± sbt(0.05;6) | a ± sat(0.05;6) | ||
| BUF | 93.31 ± 0.29 | 118.81 ± 0.18 | 0.9931 |
| COD | 87.19 ± 0.72 | 273.95 ± 0.82 | 0.9624 |
| MDMA | 3241.30 ± 0.91 | −7218.30 ± 0.55 | 0.9269 |
| MDPPP | 10.91 ± 0.40 | 50.29 ± 0.36 | 0.9572 |
| PMA | 74.02 ± 0.99 | 390.06 ± 0.97 | 0.9601 |
| EPH | 2731.70 ± 0.51 | −5620.20 ± 0.63 | 0.9566 |
| bk-MMBDB | 305.42 ± 0.46 | 1237.20 ± 0.25 | 0.9802 |
| MBDB | 601.06 ± 0.98 | 1396.10 ± 3.04 | 0.9680 |
| HER | 205.63 ± 0.3 | 1046.8 ± 0.95 | 0.9611 |
| MPBP | 102.01 ± 0.6 | 816.04 ± 0.97 | 0.9597 |
| α-PVP | 38.31± 0.44 | 357.71 ± 0.20 | 0.9590 |
| KET | 465.22 ± 0.97 | 1346.40 ± 0.48 | 0.9539 |
| MDPV | 217.88 ± 0.76 | 679.06 ± 0.36 | 0.9869 |
| NAPH | 42.75 ± 0.48 | 349.17 ± 0.88 | 0.9783 |
| Creatinine | 2236.02 ± 5.03 | 77.90 ± 0.81 | 0.9988 |
| Analyte | LLOQ (µg L−1) | Enrichment Factor a | Recovery b % | Within-Run Precision b (RSD, %) | ||
|---|---|---|---|---|---|---|
| Spike Levels | Spike Levels | |||||
| LLOQ | 10xLLOQ | LLOQ | 10xLLOQ | |||
| BUF | 0.03 | 29.1 | 97 | 97 | 15 | 6 |
| COD | 0.02 | 22.5 | 71 | 79 | 14 | 11 |
| MDMA | 0.12 | 23.1 | 76 | 78 | 12 | 5 |
| MDPPP | 0.25 | 27.0 | 88 | 92 | 13 | 6 |
| PMA | 0.37 | 28.2 | 92 | 96 | 18 | 10 |
| EPH | 0.33 | 24.0 | 68 | 92 | 8 | 4 |
| bk-MMBDB | 0.01 | 28.4 | 89 | 100 | 18 | 10 |
| MBDB | 0.07 | 17.7 | 55 | 63 | 13 | 8 |
| HER | 0.07 | 21.3 | 63 | 79 | 11 | 7 |
| MPBP | 0.14 | 25.8 | 78 | 94 | 15 | 6 |
| α-PVP | 0.11 | 23.9 | 67 | 92 | 8 | 8 |
| KET | 0.09 | 28.1 | 89 | 98 | 9 | 6 |
| MDPV | 0.01 | 26.7 | 80 | 100 | 12 | 10 |
| NAPH | 0.02 | 26.6 | 80 | 97 | 13 | 6 |
| Method (Common Analytes) | Common Analytes | Enrichment Factor | Recovery % | Precision % | LOD (µg L−1) | Type and Volume of Solvents | Extraction Time (min) | Reference |
|---|---|---|---|---|---|---|---|---|
| DLLME-GC-MS | KET | Not provided | 86–93 (20, 200 µg L−1 spike level) | 5–11.7 | 1.25 | Extr a: protonated N,N-dimethylcyclohexylamine (400 µL) + NaOH 6M (400 µL) | ~15 | [13] |
| DLLME-GC/MS | KET | 204 | 91 | 3.4 | 0.91 | Pretreatment to adjust pH Extr a: carbon tetrachloride; 30 µL Disp b: EtOH; 500 µL | ~15 | [14] |
| Ionic liquid-based DLLME-capillary electrophoresis | EPH KET | 28.8 16.9 | 86–90 79–82 (1–2 mg L−1 spike level) | 6.8 6.8 | 15 30 | Pretreatment with methanol and NaOH; Extr a: [BMIM]PF6; 40 µL Disp b: acetonitrile; 350 µL | ~20 | [16] |
| DLLME-GC/MS | KET MDMA MDBD α-PVP MDPV | Not provided | 112 102 92 116 111 (50 µg L−1 spike level) | 1.6 1.0 6.7 9.7 13.5 | 5 2 5 5 10 | Pretreatment with methanol, NaOH, Na Cl; derivatization with hexyl chloroformate; Extr a: Chloroform; 100 µL Disp b: methanol; 250 µL | ~20 | [17] |
| DLLME-HPLC/MS | KET MDMA MDBD α-PVP MDPV | 28.1 23.1 17.7 23.9 26.7 | 89–98 76–78 55–63 67–92 80–100 (LLOQ spike level, see Table 2) | 6–9 5–12 8–13 8 10–12 | 0.054 0.072 0.042 0.066 0.006 | Pretreatment with NaOH to adjust pH Extr a: ChCl:Ses 1:3; 100 µL Disp b: ethyl acetate; 400 µL | ~15 | This work |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gallo, V.; Tomai, P.; Di Lisio, V.; Dal Bosco, C.; D’Angelo, P.; Fanali, C.; D’Orazio, G.; Silvestro, I.; Picó, Y.; Gentili, A. Application of a Low Transition Temperature Mixture for the Dispersive Liquid–Liquid Microextraction of Illicit Drugs from Urine Samples. Molecules 2021, 26, 5222. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26175222
Gallo V, Tomai P, Di Lisio V, Dal Bosco C, D’Angelo P, Fanali C, D’Orazio G, Silvestro I, Picó Y, Gentili A. Application of a Low Transition Temperature Mixture for the Dispersive Liquid–Liquid Microextraction of Illicit Drugs from Urine Samples. Molecules. 2021; 26(17):5222. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26175222
Chicago/Turabian StyleGallo, Valeria, Pierpaolo Tomai, Valerio Di Lisio, Chiara Dal Bosco, Paola D’Angelo, Chiara Fanali, Giovanni D’Orazio, Ilaria Silvestro, Yolanda Picó, and Alessandra Gentili. 2021. "Application of a Low Transition Temperature Mixture for the Dispersive Liquid–Liquid Microextraction of Illicit Drugs from Urine Samples" Molecules 26, no. 17: 5222. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26175222