
Oxazinethione Derivatives as a Precursor to Pyrazolone and Pyrimidine Derivatives: Synthesis, Biological Activities, Molecular Modeling, ADME, and Molecular Dynamics Studies



Abstract
:1. Introduction

2. Results and Discussion
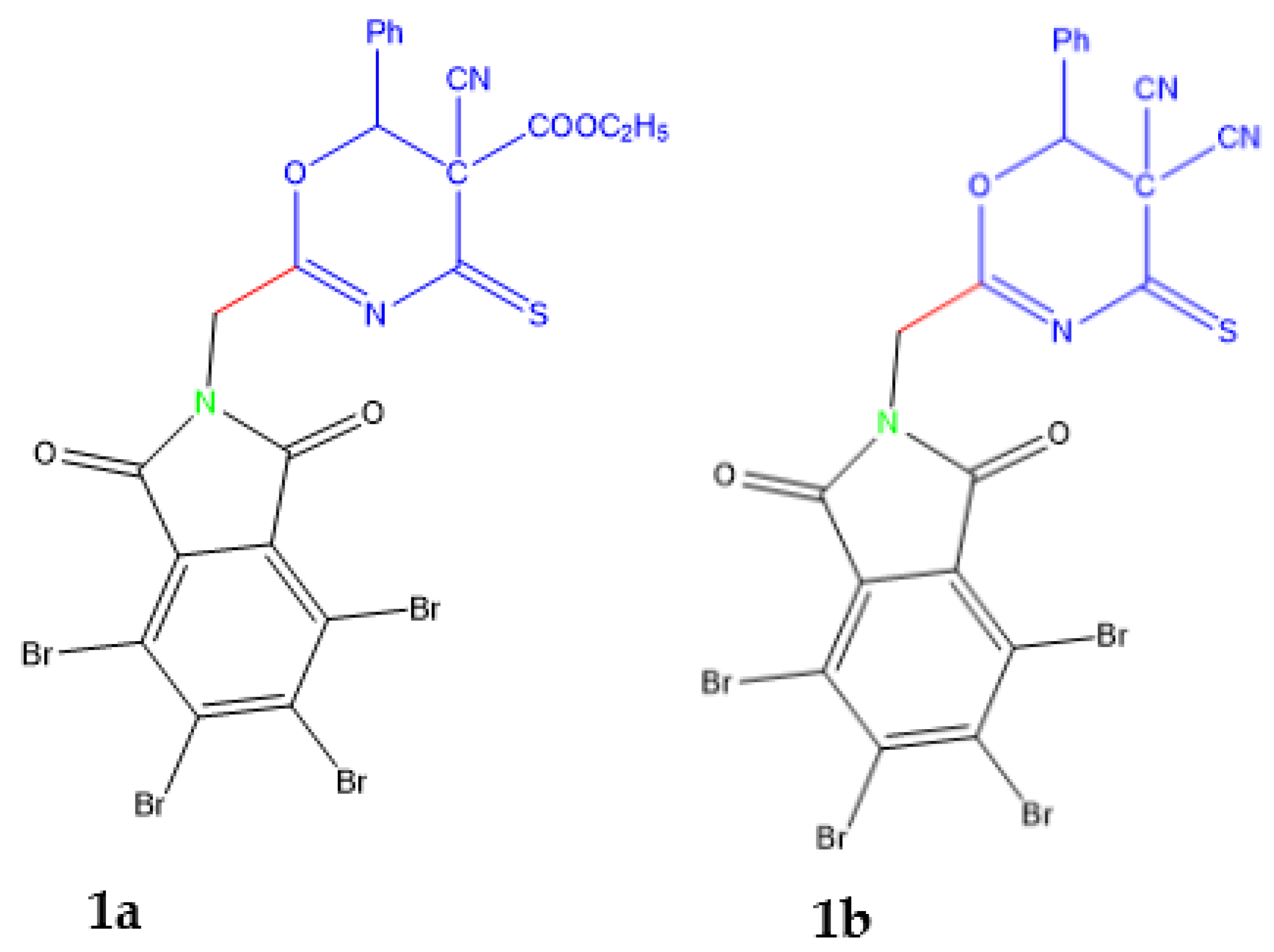
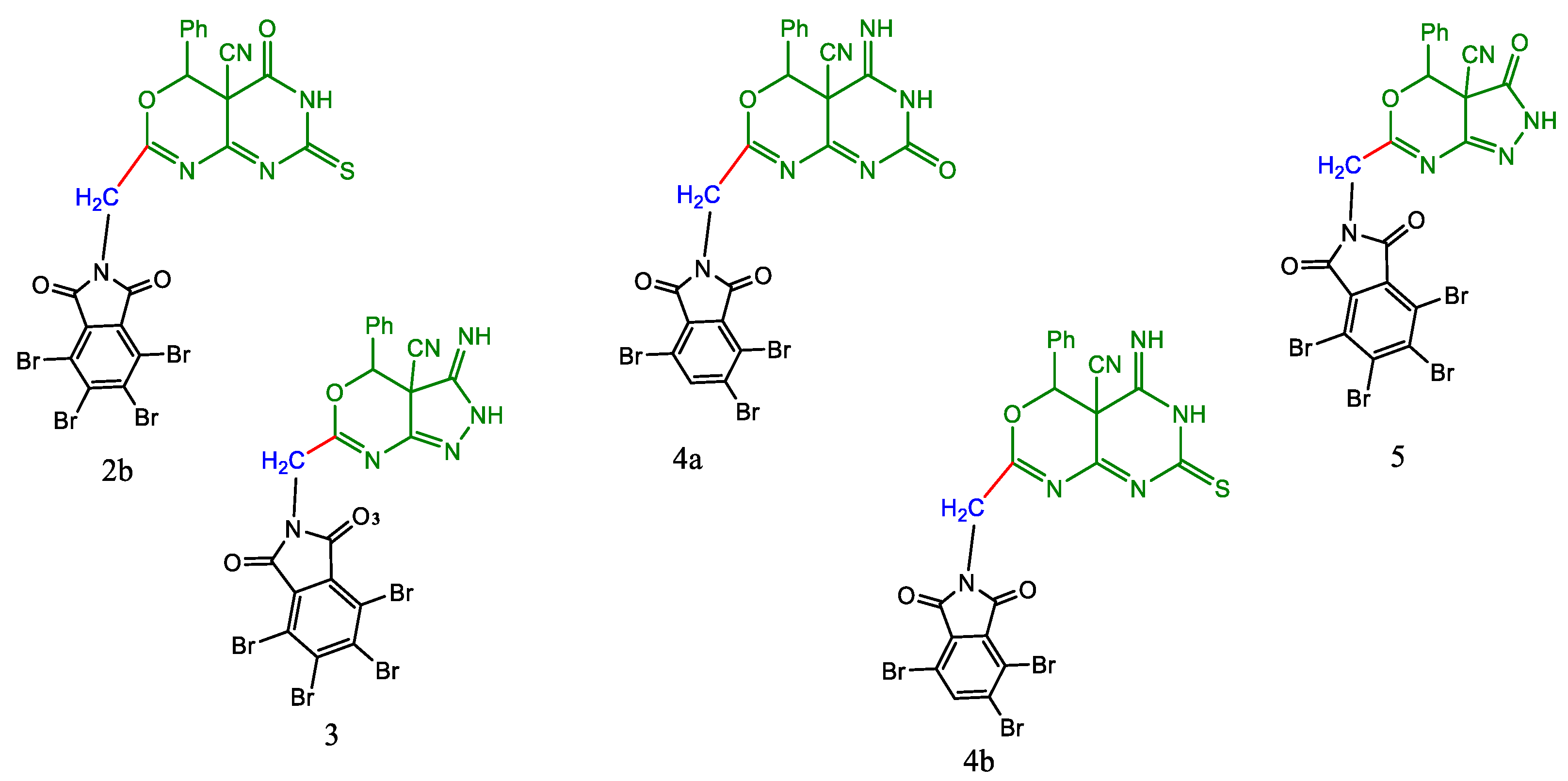
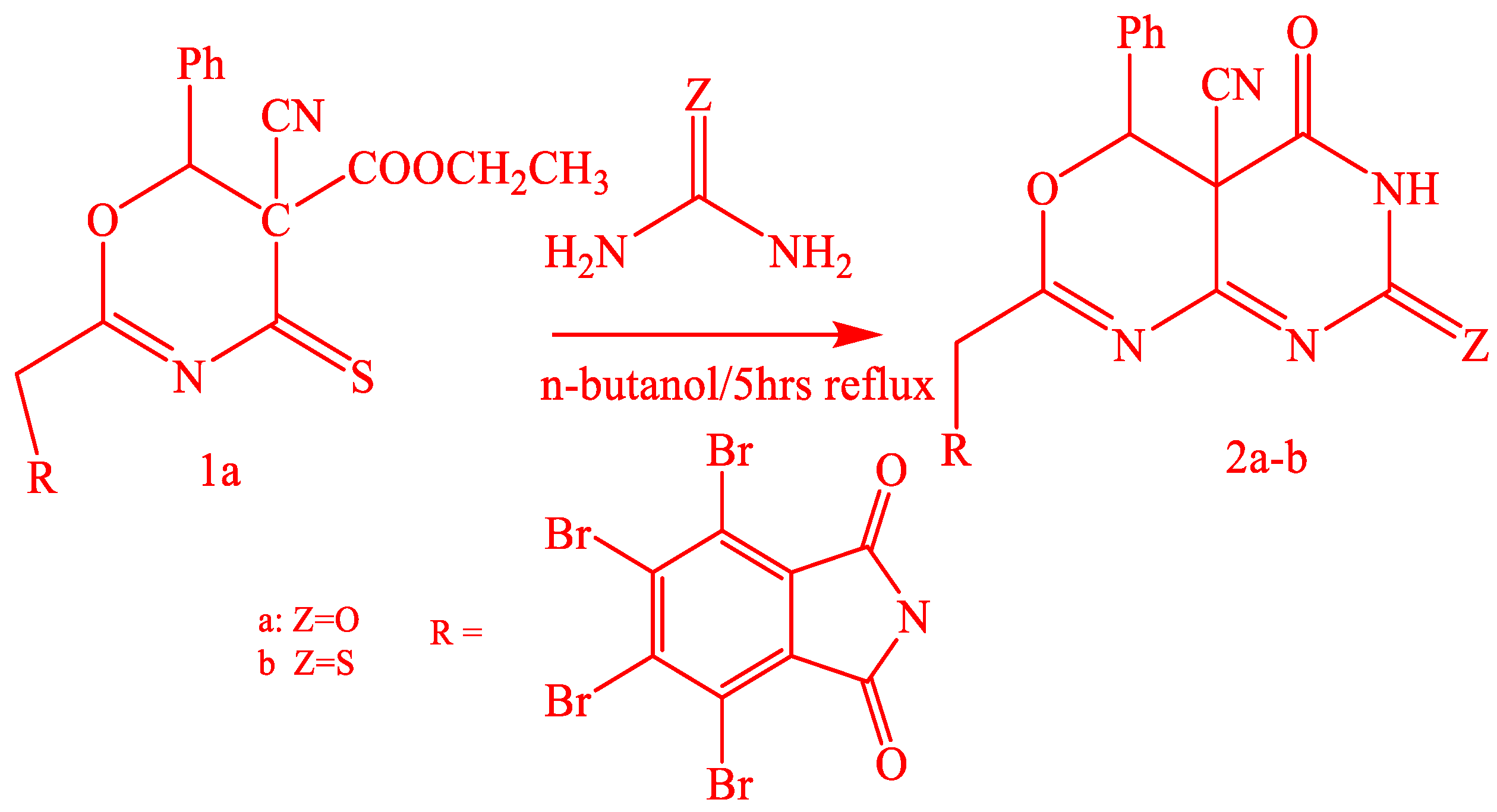
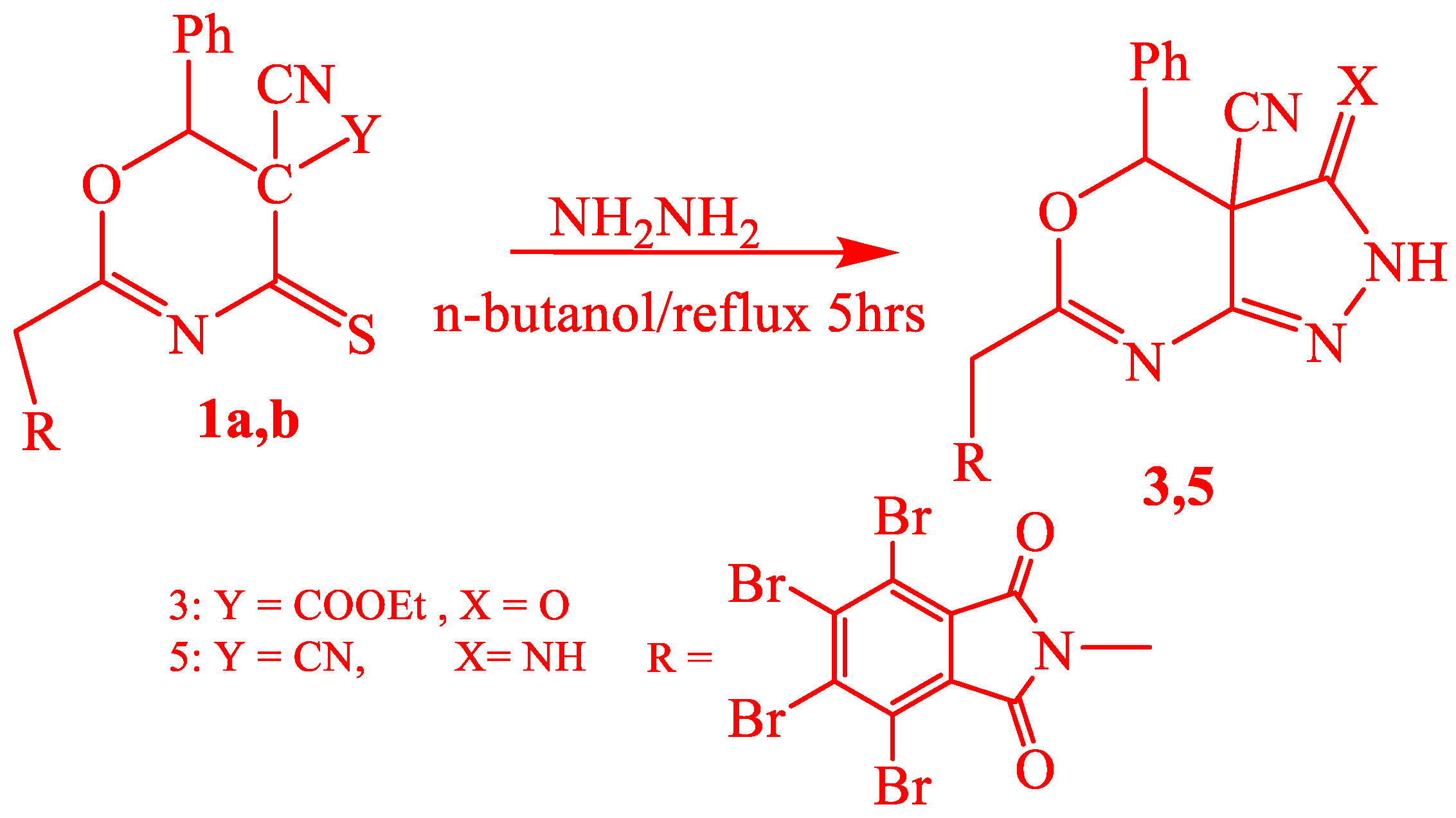
2.1. Chemistry
2.2. Biology
2.2.1. Antimicrobial Studies
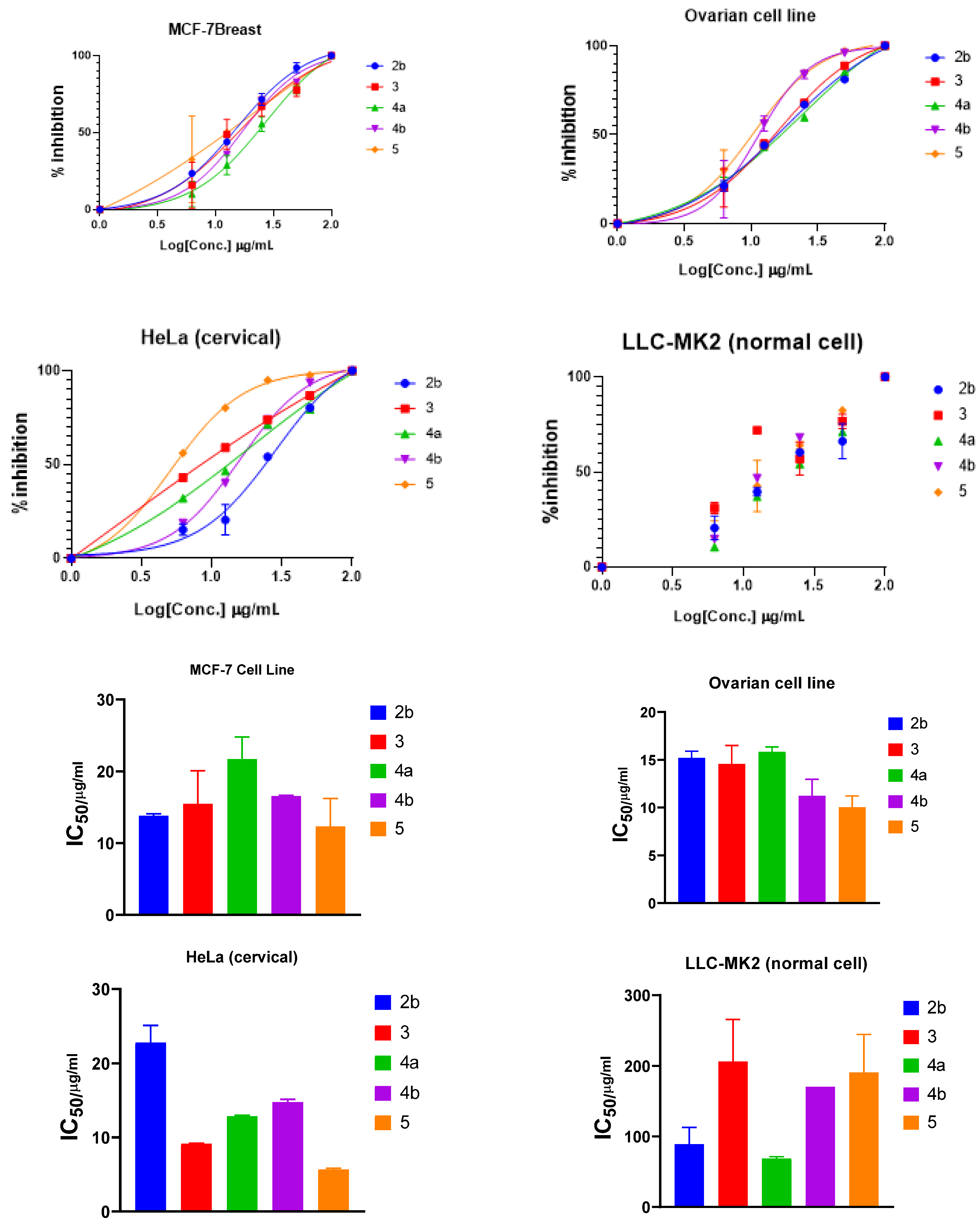
2.2.2. Anticancer Studies
2.3. Computational Studies
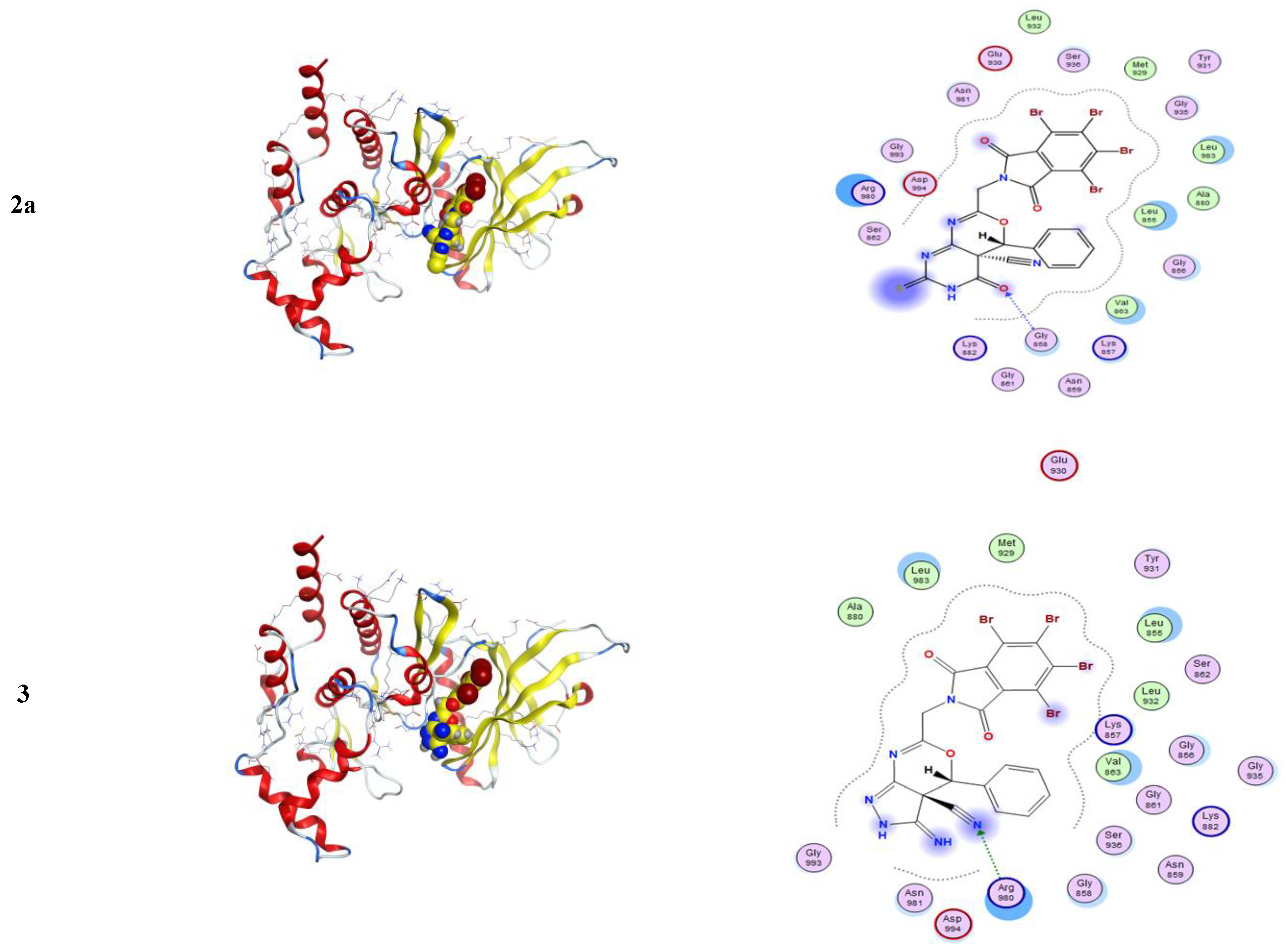
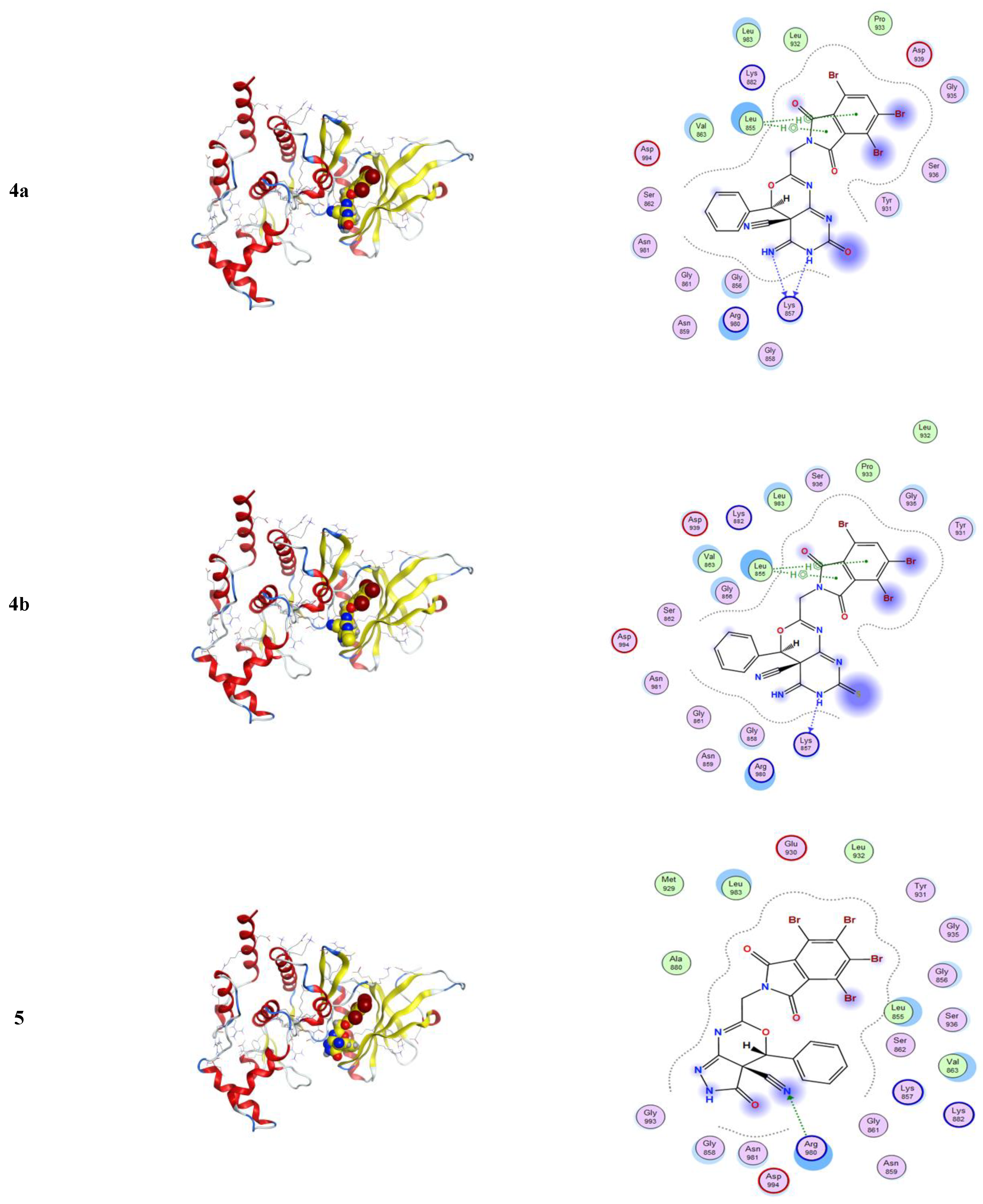
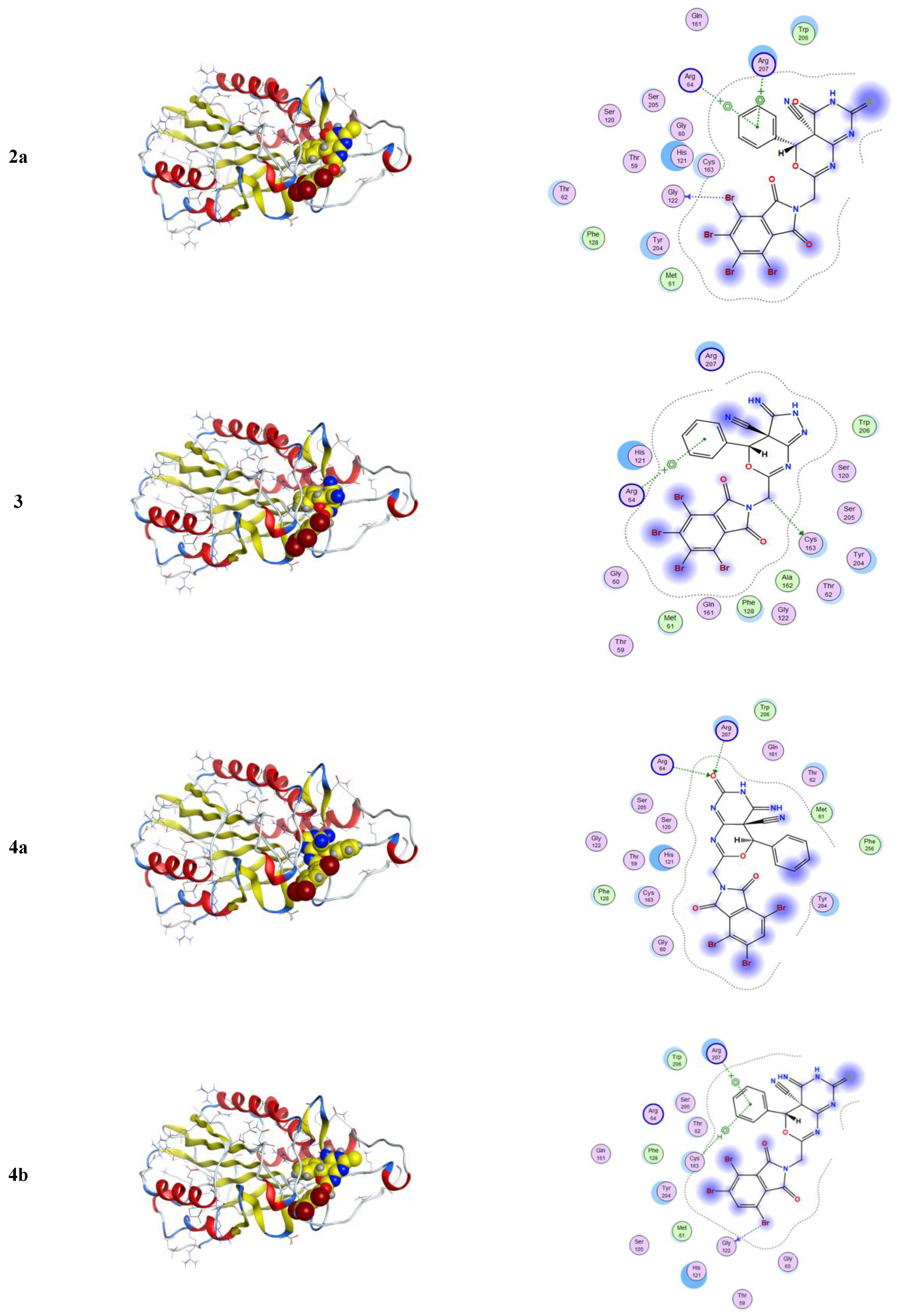
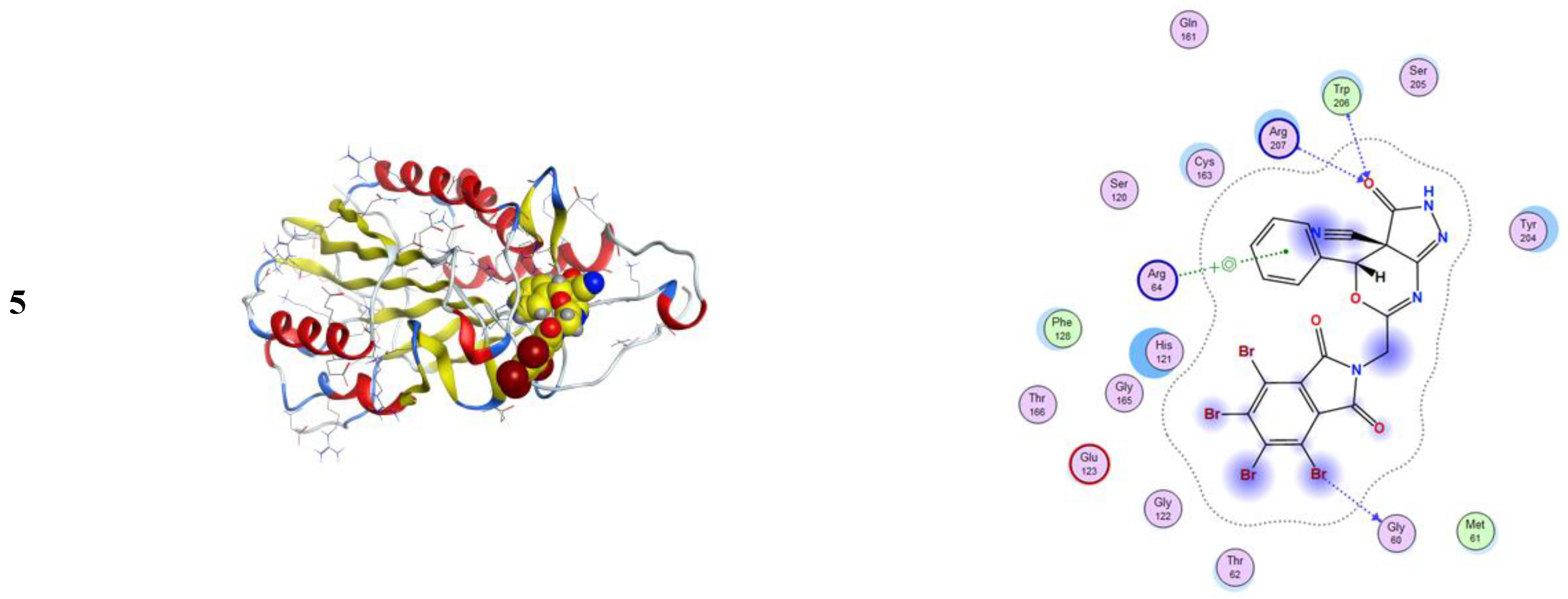
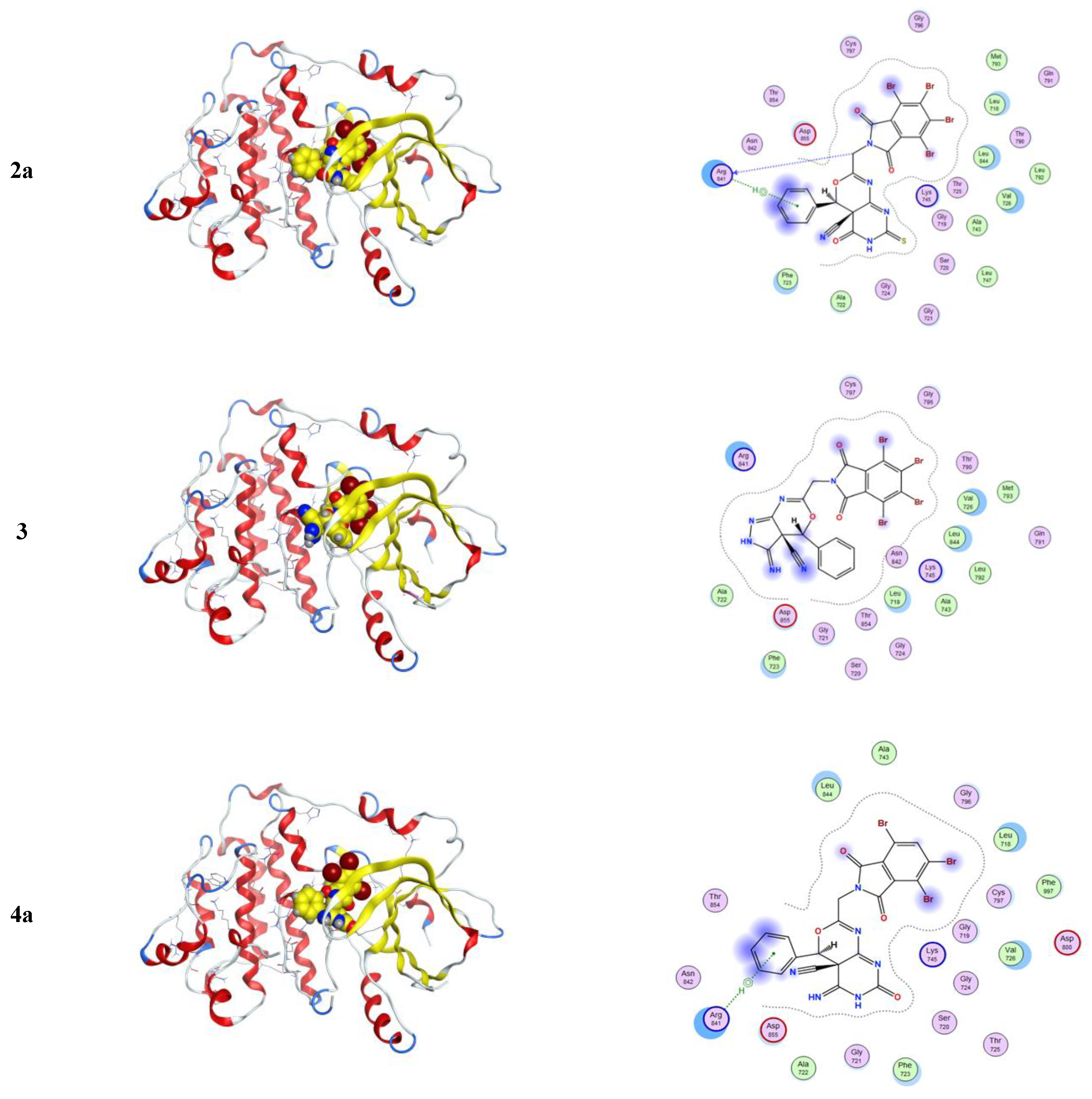
2.3.1. Molecular Modeling Studies Using MOE
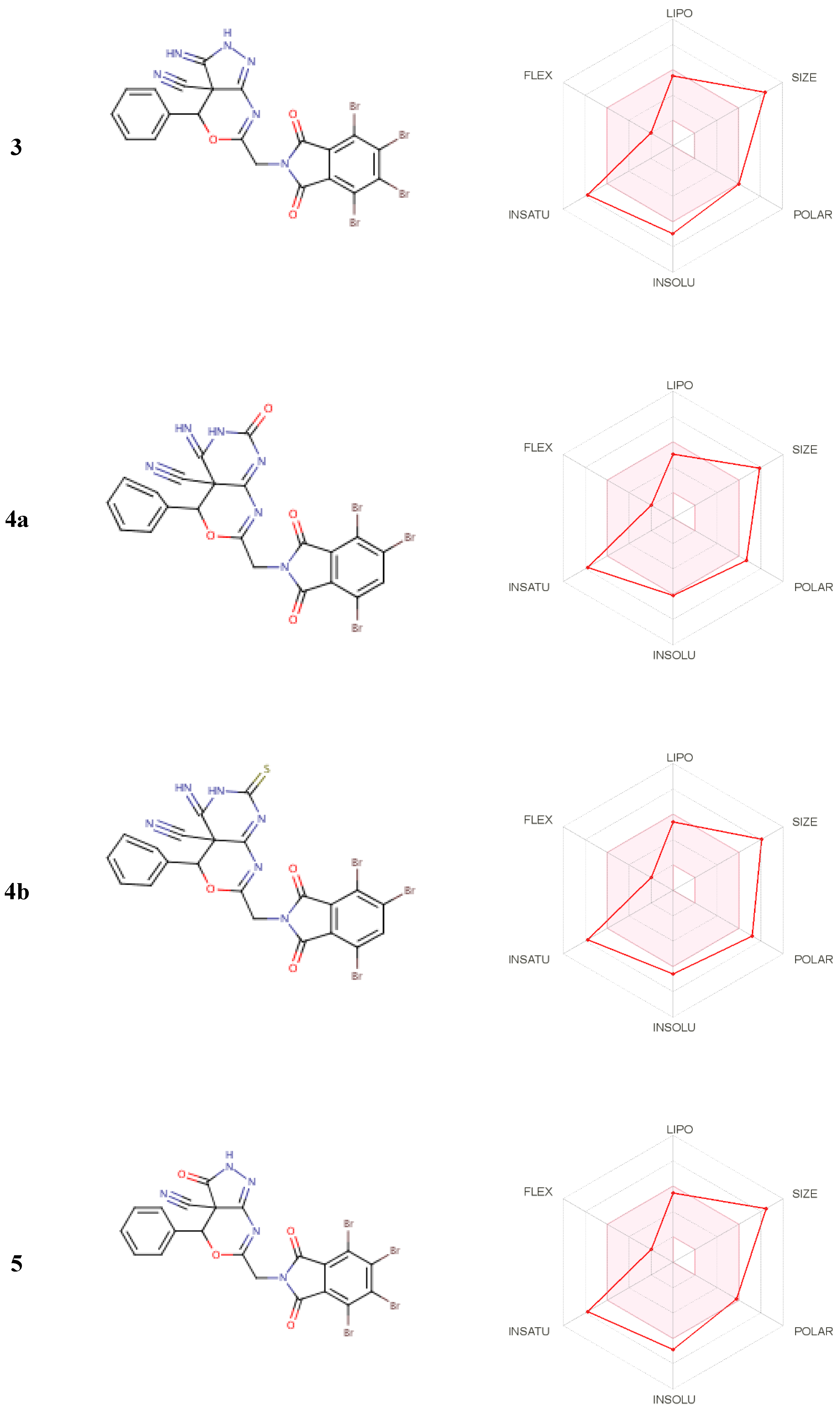
2.3.2. ADME and Pharmacophore Studies
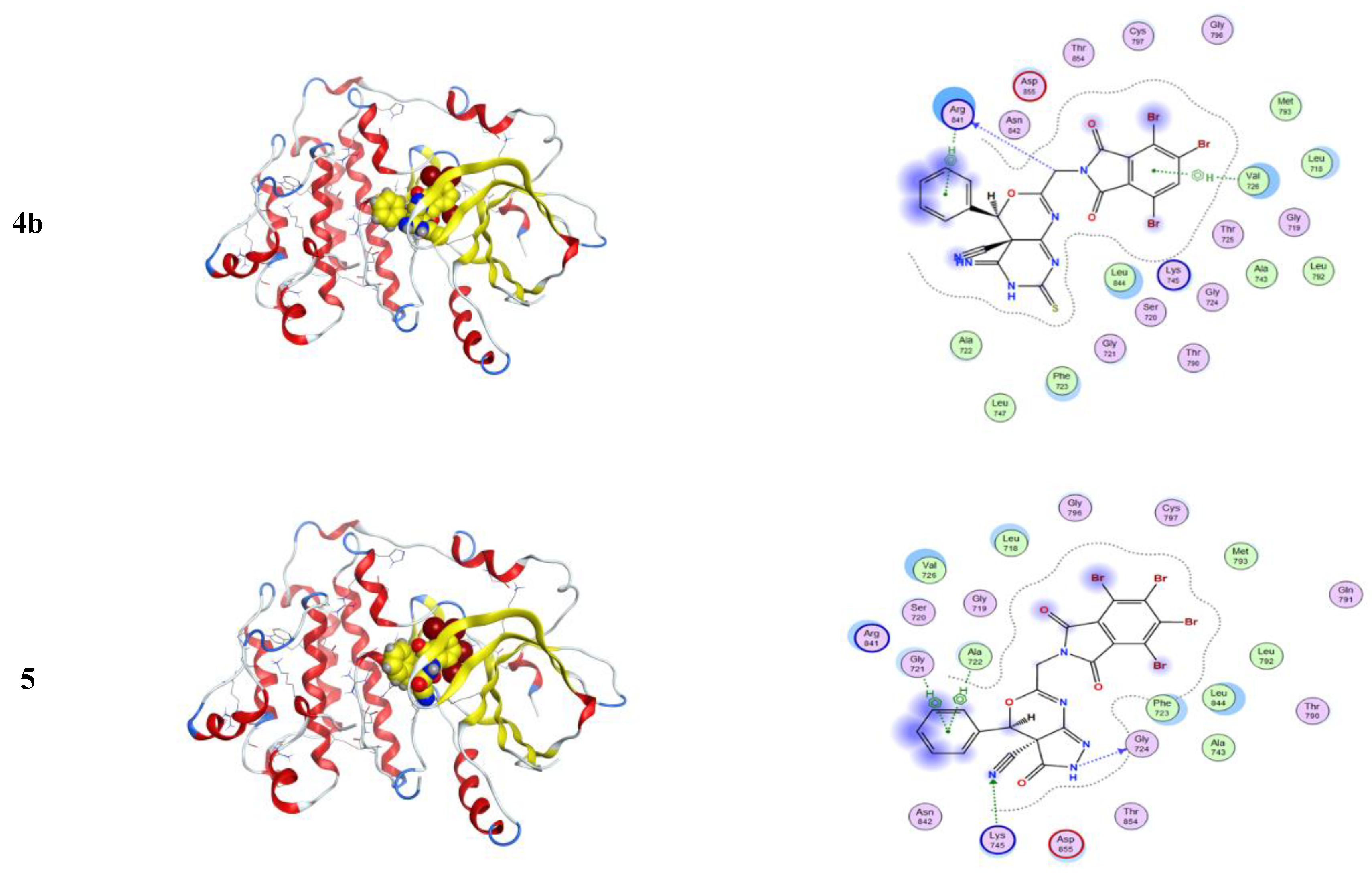
2.3.3. Molecular Dynamics Studies and Validations of the Methods Used
Analyses of the Interaction Patterns and Conformational Dynamics
3. Methodology
3.1. Chemistry
3.1.1. Synthesis of 2a-b by the Action of Urea and Thiourea on Oxazine Derivative 1a
3.1.2. 2-(Tetrabromophthalimidomethyl-6-Phenyl-5-Cyano-1,3-Oxazino[4,5-e]-1,3-Pyrimidine-3-[H]-2,4-Dione (2a)
3.1.3. 2-(Tetrabromophthalimidomethyl-6-Phenyl-5-Cyano-1,3-Oxazino[4,5-e]-1,3-Pyrimidine-3-[H]-2-Thione-4-One (2b)
3.1.4. Synthesis of 2-(Tetrabromophalimidmethyl)-6-Phenyl-5-Yano-1,3-Oxazino[4,5-d]-1,2-Pyrazole2[H]-3-One (3)
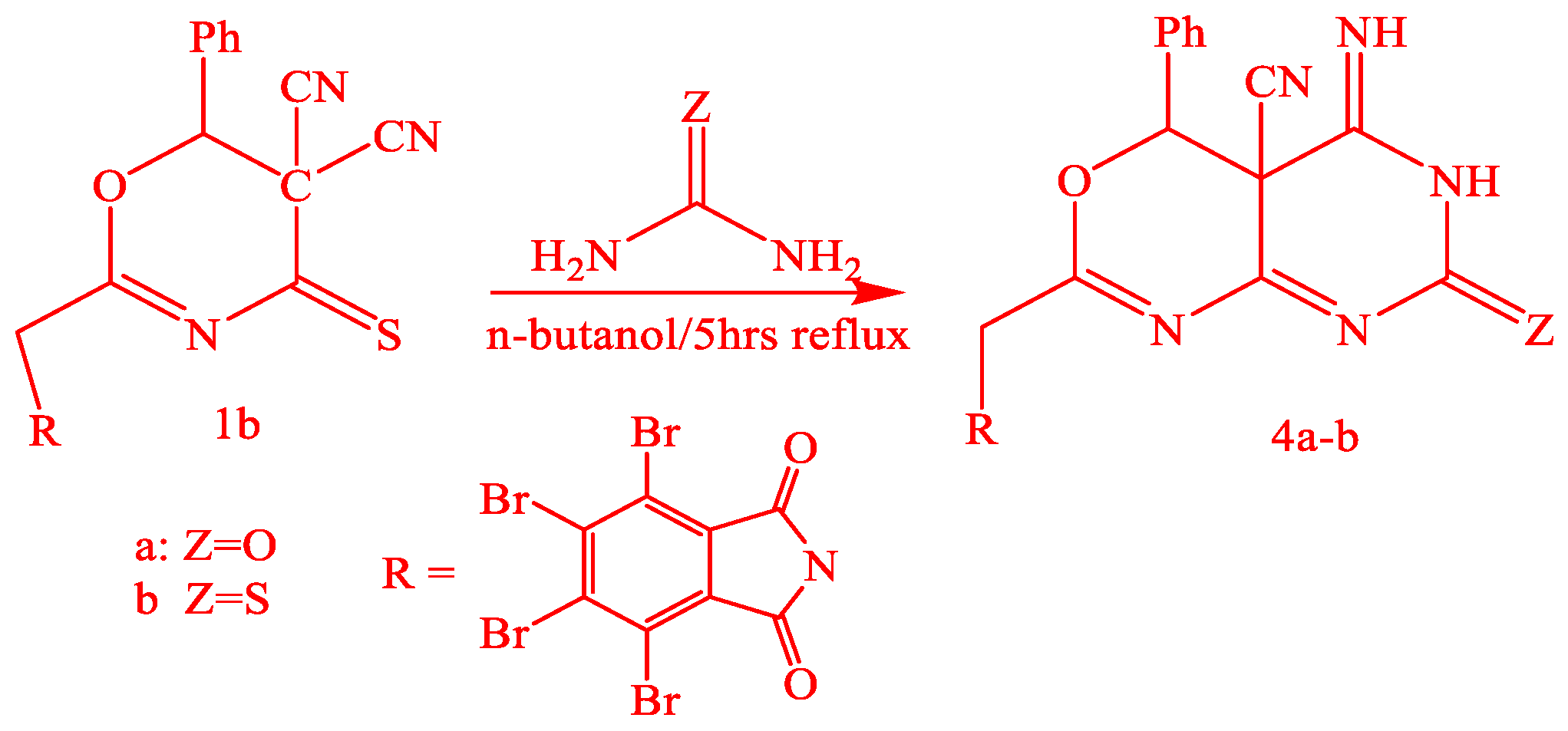
3.1.5. Synthesis of 4a-b by the Action of Urea and Thiourea on Oxazine Derivative 1b
3.1.6. Synthesis of 2-(Tetrabromophthalimidomethyl-6-Phenyl-5-Cyano-1,3-Oxazino[4,5-e]-1,3-Pyrimidine-3-[H]-2-One-5-Imide (4a)
3.1.7. 2-(Tetrabromophthalimidomethyl-6-Phenyl-5-Cyano-1,3-Oxazino[4,5-e]-1,3-Pyrimidine-3-[H]-2-Thione-4-Imide (4b)
3.1.8. Synthesis of 2-(Tetrabromophalimidmethyl)-6-Phenyl-5-Yano-1,3-Oxazino[4,5-d]-1,2-Pyrazole2[H]-3-Imide (5)
3.2. Biological Studies
3.2.1. Antimicrobial Studies
3.2.2. Anticancer
3.3. Computational Studies
3.3.1. Molecular Docking Studies with MOE
3.3.2. Virtual Screening and Validation
3.3.3. ADME and Pharmacophore Studies
3.3.4. MD Simulations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| MIC | minimum inhibitory concentration |
| MOE | Molecular Operating Environment |
| MD | Molecular Dynamics |
| A. flavus | Aspergillus flavus |
| E. coli | Escherichia coli |
| S. aureus | Staphylococcus aureus |
| F. moniliform | Fusarium moniliform |
| PDB | Protein Data Bank |
| ADME | absorption, distribution, metabolism, and excretion |
References
- Abdellattif, M.H.; Ali, O.A.; Arief, M.M.H.; Hussien, M.A. One-pot Synthesis of Novel Derivatives of Oxadiazine-4-thione, and its Antibacterial Activity, and Molecular Modeling Studies. Curr. Org. 2020, 17, 1–13. [Google Scholar] [CrossRef]
- Sondhi, S.M.; Johar, M.; Rajvanshi, S.; Dastidar, S.G.; Shukla, R.; Raghubir, R.; Lown, J.W. Anticancer, Anti-inflammatory and Analgesic Activity Evaluation of Heterocyclic Compounds Synthesized by the reaction of 4-Isothiocyanato-4-methylpentan-2-one with Substituted o-Phenylenediamines, o-Diaminopyridine and (Un)Substituted o. Aust. J. Chem. 2001, 54, 69–74. [Google Scholar] [CrossRef]
- Asif, M.; Imran, M. Pharmacological Profile of Oxazine and its Derivatives: A Mini Review. Int. J. New. Chem. 2020, 7, 60–73. [Google Scholar] [CrossRef]
- Tajima, H.; Kimoto, H.; Taketo, Y.; Taket, A. Effects of Synthetic Hydroxy Isothiocyanates on Microbial Systems. Biosci. Biotechnol. Biochem. 1998, 62, 491–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elnagdi, M.H.; Fahmy, S.M.; Elmoghayar, M.R.H.; Negm, A.M. Pyrimidine Derivatives and Related Compounds, IX/Preparation of 5-Aminopyrazolo[1,5-a]pyrimidines and of Oxazino[4,5:5,6]pyrazolo[1,5-a]pyrimidines, a New Ring System. Z. Nat. B 1977, 32, 1478–1481. [Google Scholar] [CrossRef]
- Vinita, S.; Nitin, C.; Ajay, K.A. Significance and Biological Importance of Pyrimidine in the Microbial World. Int. J. Med. Chem. 2014, 2014, 31. [Google Scholar] [CrossRef] [Green Version]
- Pyrimidine Derivatives. Available online: https://go.drugbank.com/categories/DBCAT002332 (accessed on 12 July 2021).
- Sammar, A.; Bandar, A.B.; Abdellattif, M.H.; Abdul-Hamid, E.; Mariusz, J.; Mark, G.H.; Mostafa, A.H. Effect of Net Charge on DNA-Binding, Protein-Binding and Anticancer Properties of Copper(I) Phosphine-Diimine Complexes. J. Inorg. Organomet. Polym. Mater. 2021, 1–10. [Google Scholar] [CrossRef]
- Ajmal, R.; Rajendra, B.; Dongre, S.; Aabid, H.; Gowhar, S.; Naikoo, A.; Hassan, I.U. Computational analysis for antimicrobial active pyrano[2,3-d]pyrimidine derivatives on the basis of theoretical and experimental ground. J. Taibah Univ. Sci. 2016, 20, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Bandar, A.B.; Jalal, H.A.; Bambar, D.; Abdul-Hamid, E.; Mariusz, J.; Magda, H.A.; Mostafa, A.H. Synthesis, Structural Studies, and Anticancer Properties of CuBr(PPh3)2(4,6-Dimethyl-2-Thiopyrimidine-κS. Crystals 2021, 11, 688. [Google Scholar] [CrossRef]
- Helmy, M.M.; Abdellattif, M.H.; Eldeab, H.A. New Methodology for Synthesis of Coumarin Derivatives as Potent Antimicrobial Agents. Int. J. Adv. Pharm. Biol. Chem. 2014, 3, 983–990. [Google Scholar]
- Bhatt, H.B.; Sharma, S. Synthesis and antimicrobial activity of pyrazole nucleus containing 2-thioxothiazolidin-4-one derivatives. Arab. J. Chem. 2017, 10, S1590–S1596. [Google Scholar] [CrossRef] [Green Version]
- Abdellattif, M.H.; Hussien, M.A.; Alzahrani, E. New Approaches of 4-aryl-2-hydrazinothiazole derivatives synthesis, molecular Docking and biological evaluations. Int. J. Pharm. Sci. Res. 2018, 9, 1000–1019. [Google Scholar]
- Barakat, A.; Al-Majid, A.M.; Soliman, S.M.; Lotfy, G.; Ghabbour, H.A.; Fun, H.K.; Wadood, A.; Warad, I.; Sloop, J.C. New diethyl ammonium salt of thiobarbituric acid derivative: Synthesis, molecular structure investigations and docking studies. Molecules 2015, 20, 20642–20658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNutt, M.C.; Kwon, H.J.; Chen, C.; Chen, J.R.; Horton, J.D.; Lagace, T.A. Antagonism of secreted PCSK9 increases low density lipoprotein receptor expression in HepG2 cells. J. Biol. Chem. 2009, 284, 10561–10570. [Google Scholar] [CrossRef] [Green Version]
- Mashat, K.H.; Babgi, B.A.; Hussien, M.A.; Arshad, M.N.; Abdellattif, M.H. Synthesis, structures, DNA-binding and anticancer activities of some copper(I)-phosphine complexes. Polyhedron 2019, 158, 164–172. [Google Scholar] [CrossRef]
- Subudhi, B.B.; Chattopadhyay, S.; Mishra, P.; Kumar, A. Current strategies for inhibition of chikungunya infection. Viruses 2018, 10, 235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Hashash, M.A.; Rizk, S.A.; El-Sayed, A.A. Ultrasonic and solvent free synthesis of regioselective diastereomeric adducts and heterocyclic products as antibacterial agent. J. Adv. Chem. 2017, 13, 6106–6117. [Google Scholar]
- Hegelund, F.; Larsen, R.W.; Palmer, M.H. The vibration spectrum of thiazole between (600–1400 cm−1) revisited, a combined high resolution infrared and theoretical study. J. Mol. Spectrosc. 2007, 244, 63–78. [Google Scholar] [CrossRef]
- Arikan, N.; Sumengen, D.; Dulger, B. Syntheisi and antimicrobial activity of 1,2,4 oxadiazine-5-one, 6-ones, and 5-thiones. Turk. J. Chem. 2008, 32, 147–155. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Belay, Z.S.; Sonia, K.; Pankaj, T.; Paratpar, S.; Neetu, K.T. Molecular Docking, synthesis and anticancer activity of thiosemicarbazone derivatives against MCF-7 human breast cancer cell line. Life Sci 2021, 273, 119305. [Google Scholar]
- Zhang, F.; Zhang, H.; Wang, F. EGFR inhibition studies by hybrid scaffolds for their activity against ovarian cancer. J. Balk. Union Oncol. 2016, 21, 1482–1490. [Google Scholar]
- Bochevarov, A.D.; Harder, E.; Hughes, T.F. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H. Drug-Like Properties: Concepts, Structure Design and Methods from ADME to Toxicity Optimization; Elsevier: London, UK, 2016. [Google Scholar]
- Bala, S.; Kamboj, S.; Kajal, A.; Saini, V.; Prasad, D.N. 1,3,4-oxadiazole derivatives: Synthesis, characterization, antimicrobial potential, and computational studies. BioMed Res. Int. 2014, 2014, 172791. [Google Scholar] [CrossRef] [PubMed]
- Mamoru, K.; Yusuke, Y.; Hiromune, A.; Hideharu, I. Synthesis of 1,3-Selenazetidines and 4H-1,3,5-Oxadiazines Using Acyl Isoselenocyanates. Heterocycles 2006, 68, 1267–1273. [Google Scholar]
- Helmy, M.M.; Moustafa, M.H.; Eldeab, H.A. Microwave-Assisted Synthesis of New Series Some Acetyl Coumarin Derivatives and Studying of Some their Pharmacological Activities. J. Pharm. Sci. Res. 2015, 7, 83–88. [Google Scholar]
- Magda, H.A. Synthesis of Some Novel Compounds of Saccharinyl Acetic Acid Containing Nucleus and Evaluation of Their Biological Activities as Antimicrobial. Orient. J. Chem. 2016, 32, 567–574. [Google Scholar]
- Muanza, D.N.; Kim, B.W.; Euler, K.L.; Williams, L. Antibacterial and antifungal activities of nine medicinal plants of Zaire. Int. J. Pharmacog. 1994, 32, 337–345. [Google Scholar] [CrossRef]
- Pezzuto, J.M.; Che, C.-T.; McPherson, D.D.; Zhu, J.-P.; Topcu, G.; Erdelmeier, C.A.J.; Cordell, G.A. DNA as afnity probe useful in the detection and isolation of biologically active natural products. J. Nat. Prod. 1991, 54, 1522–1530. [Google Scholar] [CrossRef]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity as anticancer drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef]
- Ekici, O.D.; Li, Z.Z.; Campbell, A.J.; James, K.E.; Asgian, J.L.; Mikolajczyk, J.; Salvesen, G.S.; Ganesan, R.; Jelakovic, S.; Grütter, M.G.; et al. Design, synthesis, and evaluation of aza-peptide Michael acceptors as selective and potent inhibitors of caspases-2, -3, -6, -7, -8, -9, and -10. J. Med. Chem. 2006, 49, 5728–5749. [Google Scholar] [CrossRef]
- Ernst, S.; Stephane, B.; Riazul, A.; Mathew, P.M.; Andreas, B.; Han, H.; Rawle, F.; Ramappa, C.; Sudhakar, J.; Aslamuzzaman, K.; et al. Development of highly potent and selective diaminothiazole inhibitors of cyclin-dependent kinases. J. Med. Chem. 2013, 56, 3768–3782. [Google Scholar]
- Edgar, R.W.; Anne, T.T.; Octerloney, B.M.D.; Derek, Y.; Anne, H.; Scott, H.D.; Byron, E.; Christopher, P.; Earnest, H. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): Relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004, 64, 6652–6659. [Google Scholar]
- Steve, W.; Li, R.; Kateri, A.A.; Ellen, R.L.; Ignacio, A.; Bruno, A.; Alex, J.B.; Edna, F.C.; Victoria, D.; Bainian, F.; et al. Pyrazolopyridine inhibitors of B-RafV600E. Part 2: Structure–activity relationships. Bioorgan. Med. Chem. Lett. 2011, 21, 5533–5537. [Google Scholar]
- Brzozowski, A.M.; Pike, A.C.W.; Dauter, Z.; Hubbard, R.E.; Bonn, T.; Engström, O.; Öhman, L.; Greene, G.L.; Gustafsson, J.; Carlquist, M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997, 389, 753–758. [Google Scholar] [CrossRef]
- Mateusz, P.C.; Adam, M.B.; Ettore, J.R.; Nikhil, R.T.; Taber, S.M.; Isabella, K.B.; Kelley, E.M.; John, H.B.; Wladek, M.; Peter, W.; et al. Structure of the Complex of an Iminopyridinedione Protein Tyrosine Phosphatase 4A3 Phosphatase Inhibitor with Human Serum Albumin. Mol. Pharmacol. 2020, 98, 648–657. [Google Scholar]
- Redinbo, M.R.; Champoux, J.J.; Hol, W.G.J. Novel Insights into Catalytic Mechanism from a Crystal Structure of Human Topoisomerase I in Complex with DNA. Biochemistry 2000, 39, 6832–6840. [Google Scholar] [CrossRef]
- Ibrahim, A.A.-S.; Naglaa, I.A.-A.; Adel, S.E.-A.; Magda, A.A.E.-S.; Amer, M.A.; Mahmoud, B.E.-A.; Alaa, A.-M.A.-A. Antitumor evaluation and molecular docking study of substituted 2-benzylidenebutane-1,3-dione, 2-hydrazonobutane-1,3-dione and trifluoromethyl-1H-pyrazole analogues. J. Enzym. Inhib. Med. Chem. 2015, 30, 679–687. [Google Scholar]
- Somaia, S.A.E.-K.; Yasmin, M.S.; Ahmed, M.E.K.; Tamer, M.A. New thiazol-hydrazono-coumarin hybrids targeting human cervical cancer cells: Synthesis, CDK2 inhibition, QSAR and molecular docking studies. Bioorganic Chem. 2019, 86, 80–96. [Google Scholar]
- Zhang, P.; Xu, S.; Zhu, Z.; Xu, J. Multi-target design strategies for the improved treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2019, 176, 228–247. [Google Scholar] [CrossRef]
- David, V.; Der, S.; Erik, L.; Berk, H.; Gerrit, G.; Alan, E.M.; Herman, J.C.B. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar]
- Robertson, M.J.; Tirado-Rives, J.; Jorgensen, W.L. Improved Peptide and Protein Torsional Energetics with the OPLSAA Force Field. J. Chem. Theory Comput. 2015, 11, 3499–3509. [Google Scholar] [CrossRef] [PubMed]
- Leela, S.D.; Israel, C.V.; Julian, T.-R.; William, L.J. LigParGen web server: An automatic OPLS-AA parameter generator for organic ligands. Nucleic Acids Res. 2017, 45, W331–W336. [Google Scholar]
- Zielkiewicz, J. Structural properties of water: Comparison of the SPC, SPCE, TIP4P, and TIP5P models of water. J. Chem. Phys. 2005, 123, 104501. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Number | A. flavus | E. coli | S. aureus | F. moniliform | ||||
|---|---|---|---|---|---|---|---|---|
| A | Mic | A | Mic | A | Mic | A | Mic | |
| 2a | - | - | - | - | + | 125 | + | 125 |
| 2b | - | - | + | 250 | ++ | 250 | + | 125 |
| 3 | + | 125 | + | 125 | + | 125 | + | 125 |
| 4a | +++ | 250 | + | 250 | - | - | + | 250 |
| 4b | - | - | + | 250 | ++ | 250 | + | 125 |
| 5 | +++ | 250 | ++ | 250 | ++ | 250 | + | 250 |
| Cpd. | MCF-7 | Hela | Ovcar-3 | LCC-MK2 | ||||
|---|---|---|---|---|---|---|---|---|
| IC50 | ±SD | IC50 | +SD | IC50 | ±SD | IC50 | ±SD | |
| 2b | 13.82 | 0.26 | 22.81 | 2.31 | 15.22 | 0.70 | 89.34 | 23.56 |
| 3 | 15.49 | 4.59 | 9.14 | 0.06 | 14.57 | 1.94 | 206.50 | 59.36 |
| 4a | 21.70 | 3.09 | 12.88 | 0.10 | 15.86 | 0.50 | 68.91 | 2.37 |
| 4b | 16.56 | 0.09 | 14.76 | 0.38 | 11.23 | 1.75 | 170.40 | 0.00 |
| 5 | 12.31 | 3.90 | 5.69 | 0.14 | 10.05 | 1.18 | 190.93 | 53.80 |
| Comp. | S | rmsd_refine | E_conf | E_place | E_score1 | E_refine | E_score2 |
|---|---|---|---|---|---|---|---|
| 2a | −8.43 | 2.30 | −212.78 | −81.54 | −12.39 | −35.32 | −8.43 |
| −8.17 | 1.29 | −203.26 | −82.90 | −12.82 | −40.22 | −8.17 | |
| −8.10 | 2.25 | −194.54 | −88.98 | −11.17 | −43.54 | −8.10 | |
| −8.09 | 1.36 | −206.26 | −72.74 | −11.47 | −33.41 | −8.09 | |
| −7.85 | 1.91 | −207.36 | −67.90 | −11.13 | −31.42 | −7.85 | |
| 3 | −8.09 | 1.54 | −65.96 | −77.00 | −11.74 | −34.65 | −8.09 |
| −8.28 | 1.97 | −65.67 | −53.76 | −11.49 | −28.68 | −8.28 | |
| −8.20 | 2.42 | −72.23 | −86.55 | −10.34 | −37.10 | −8.20 | |
| −8.15 | 1.66 | −76.70 | −119.31 | −12.66 | −48.38 | −8.15 | |
| −7.88 | 2.07 | −76.97 | −92.42 | −11.00 | −46.31 | −7.88 | |
| 4a | −8.06 | 2.27 | −215.68 | −87.53 | −10.78 | −39.39 | −8.06 |
| −7.90 | 1.10 | −212.32 | −85.86 | −10.69 | −34.31 | −7.90 | |
| −7.89 | 1.12 | −214.27 | −94.83 | −11.37 | −33.22 | −7.89 | |
| −7.83 | 1.99 | −210.89 | −55.41 | −10.83 | −38.45 | −7.83 | |
| −7.80 | 1.63 | −210.48 | −78.77 | −11.53 | −35.98 | −7.80 | |
| 4b | −8.10 | 2.64 | −192.81 | −56.79 | −9.91 | −40.36 | −8.10 |
| −7.75 | 1.64 | −193.11 | −82.44 | −11.96 | −34.88 | −7.75 | |
| −7.69 | 1.14 | −191.25 | −67.17 | −11.13 | −43.84 | −7.69 | |
| −7.67 | 2.93 | −194.16 | −84.16 | −11.16 | −44.49 | −7.67 | |
| −7.46 | 4.17 | −177.93 | −69.28 | −10.53 | −21.68 | −7.46 | |
| 5 | −8.45 | 2.08 | −76.73 | −76.05 | −11.80 | −35.78 | −8.45 |
| −8.27 | 1.65 | −76.64 | −60.46 | −10.32 | −30.34 | −8.27 | |
| −8.15 | 2.53 | −86.74 | −63.97 | −10.31 | −47.86 | −8.15 | |
| −8.09 | 2.16 | −84.21 | −75.90 | −11.73 | −45.81 | −8.09 | |
| −7.97 | 2.28 | −84.96 | −68.94 | −10.87 | −35.63 | −7.97 |
| Comp. | S | rmsd_refine | E_conf | E_place | E_score1 | E_refine | E_score2 |
|---|---|---|---|---|---|---|---|
| 2a | −6.22 | 1.41 | −215.59 | −55.68 | −10.19 | −37.97 | −6.22 |
| −7.18 | 2.38 | −215.00 | −39.36 | −10.47 | −39.33 | −7.18 | |
| −6.71 | 2.48 | −214.45 | −85.06 | −9.92 | −39.51 | −6.71 | |
| −6.67 | 1.86 | −214.32 | −48.53 | −10.00 | −31.84 | −6.67 | |
| −6.49 | 1.20 | −218.06 | −81.95 | −11.05 | −28.54 | −6.49 | |
| 3 | −7.45 | 1.46 | −64.61 | −63.24 | −9.74 | −37.05 | −7.45 |
| −6.42 | 1.66 | −71.66 | −58.22 | −10.24 | −29.76 | −6.42 | |
| −6.40 | 1.50 | −73.19 | −82.55 | −11.59 | −36.54 | −6.40 | |
| −6.39 | 2.08 | −74.87 | −67.51 | −10.52 | −35.25 | −6.39 | |
| −6.38 | 1.98 | −67.73 | −73.49 | −11.36 | −28.50 | −6.38 | |
| 4a | −6.74 | 1.48 | −224.62 | −96.40 | −10.43 | −30.40 | −6.74 |
| −6.59 | 4.54 | −223.62 | −41.68 | −11.62 | −34.24 | −6.59 | |
| −6.51 | 1.69 | −222.06 | −46.45 | −10.35 | −31.55 | −6.51 | |
| −6.45 | 1.28 | −217.10 | −42.55 | −9.81 | −27.20 | −6.45 | |
| −6.43 | 3.94 | −223.58 | −74.74 | −10.32 | −33.29 | −6.43 | |
| 4a | −7.15 | 2.11 | −193.36 | −72.65 | −10.43 | −38.75 | −7.15 |
| −6.80 | 1.21 | −201.45 | −92.81 | −10.69 | −34.58 | −6.80 | |
| −6.61 | 4.02 | −201.54 | −68.46 | −9.64 | −36.48 | −6.61 | |
| −6.61 | 2.94 | −199.69 | −45.26 | −9.68 | −34.36 | −6.61 | |
| −6.44 | 1.98 | −194.51 | −61.61 | −10.37 | −31.49 | −6.44 | |
| 5 | −7.91 | 2.58 | −81.90 | −45.97 | −9.62 | −43.31 | −7.91 |
| −6.49 | 1.61 | −83.08 | −50.78 | −9.51 | −42.46 | −6.49 | |
| −6.39 | 1.52 | −82.96 | −67.06 | −10.80 | −34.79 | −6.39 | |
| −6.37 | 2.03 | −85.56 | −80.15 | −9.61 | −34.14 | −6.37 | |
| −6.26 | 2.93 | −86.09 | −61.42 | −9.86 | −31.70 | −6.26 |
| Comp. | S | rmsd_refine | E_conf | E_place | E_score1 | E_refine | E_score2 |
|---|---|---|---|---|---|---|---|
| 2a | −7.70 | 1.60 | −219.01 | −77.08 | −10.27 | −44.05 | −7.70 |
| −7.19 | 2.26 | −218.05 | −78.25 | −9.69 | −39.51 | −7.19 | |
| −7.14 | 2.42 | −215.81 | −77.27 | −10.93 | −40.10 | −7.14 | |
| −7.00 | 1.80 | −200.88 | −62.91 | −9.93 | −25.82 | −7.00 | |
| −6.96 | 1.70 | −224.02 | −50.21 | −9.71 | −36.41 | −6.96 | |
| 3 | −7.39 | 1.59 | −70.23 | −91.55 | −9.50 | −43.12 | −7.39 |
| −7.54 | 2.85 | −73.44 | −84.32 | −10.71 | −45.10 | −7.54 | |
| −7.46 | 3.09 | −78.20 | −44.13 | −10.22 | −45.39 | −7.46 | |
| −7.21 | 2.26 | −78.10 | −57.97 | −9.53 | −45.34 | −7.21 | |
| −7.12 | 1.23 | −70.06 | −60.35 | −9.94 | −34.02 | −7.12 | |
| 4a | −7.66 | 1.10 | −228.66 | −99.53 | −10.21 | −44.05 | −7.66 |
| −7.66 | 1.75 | −214.85 | −88.63 | −10.97 | −41.02 | −7.66 | |
| −7.06 | 1.13 | −223.12 | −92.78 | −10.98 | −34.17 | −7.06 | |
| −7.06 | 2.36 | −205.71 | −78.95 | −9.73 | −33.29 | −7.06 | |
| −6.74 | 1.14 | −221.63 | −46.92 | −9.61 | −32.15 | −6.74 | |
| 4b | −8.38 | 2.98 | −201.86 | −89.52 | −10.94 | −38.96 | −8.38 |
| −7.36 | 1.18 | −199.20 | −93.88 | −11.48 | −39.58 | −7.36 | |
| −7.29 | 1.10 | −205.78 | −110.63 | −11.67 | −40.47 | −7.29 | |
| −7.07 | 2.36 | −192.78 | −64.95 | −10.54 | −39.50 | −7.07 | |
| −6.96 | 1.94 | −196.57 | −78.63 | −10.34 | −33.65 | −6.96 | |
| 5 | −8.77 | 2.24 | −86.58 | −74.44 | −12.15 | −50.19 | −8.77 |
| −7.69 | 1.78 | −87.46 | −83.46 | −9.87 | −39.94 | −7.69 | |
| −7.54 | 2.64 | −73.58 | −50.75 | −10.56 | −40.73 | −7.54 | |
| −7.46 | 1.71 | −81.62 | −70.61 | −10.04 | −40.33 | −7.46 | |
| −7.45 | 1.65 | −88.79 | −67.97 | −10.21 | −42.42 | −7.45 |
| Compound | Ligand | Receptor | Interaction | Distance E | (kcal/mol) |
|---|---|---|---|---|---|
| 2a | O 13 | CA GLY 858 | H-acceptor | 3.31 | −1.6 |
| 3 | N 44 | NH1 ARG 980 | H-acceptor | 3.22 | −1.1 |
| 4a | N 10 | O LYS 857 | H-donor | 2.95 | −1.2 |
| N 13 | O LYS 857 | H-donor | 2.84 | −0.5 | |
| 5-ring | CB LEU 855 | pi-H | 4.28 | −0.7 | |
| 6-ring | CB LEU 855 | pi-H | 4.32 | −0.7 | |
| 4b | N 10 | O LYS 857 | H-donor | 2.94 | −0.3 |
| 5-ring | CB LEU 855 | pi-H | 4.28 | −0.4 | |
| 6-ring | CB LEU 855 | pi-H | 4.32 | −0.4 | |
| 5 | N 43 | NH1 ARG 980 | H-acceptor | 3.30 | −1.9 |
| Compound | Ligand | Receptor | Interaction | Distance E | (kcal/mol) |
|---|---|---|---|---|---|
| 2a | Br 26 | O GLY 122 (A) | H-donor | 3.37 | −2.5 |
| 6-ring | NE ARG 64 (A) | pi-cation | 4.79 | −0.9 | |
| 6-ring | NE ARG 207 (A) | pi-cation | 3.98 | −0.9 | |
| 6-ring | NH1 ARG 207 (A) | pi-cation | 3.28 | −0.6 | |
| 3 | C 27 | SG CYS 163 (A) | H-donor | 3.85 | −0.5 |
| 6-ring | NE ARG 64 (A) | pi-cation | 4.43 | −1.9 | |
| 4a | O 15 | NE ARG 64 (A) | H-acceptor | 3.36 | −1.0 |
| O 15 | NH2 ARG 64 (A) | H-acceptor | 3.33 | −2.4 | |
| O 15 | NE ARG 207 (A) | H-acceptor | 3.10 | −1.6 | |
| 4b | Br 28 | O GLY 122 (A) | H-donor | 3.44 | −0.5 |
| 6-ring | SG CYS 163 (A) | pi-H | 4.18 | −0.7 | |
| 6-ring | NE ARG 207 (A) | pi-cation | 3.71 | −1.1 | |
| 5 | Br 15 | O GLY 60 (A) | H-donor | 3.50 | −0.8 |
| O 30 | CA TRP 206 (A) | H-acceptor | 3.42 | −0.5 | |
| O 30 | N ARG 207 (A) | H-acceptor | 3.14 | −2.4 | |
| 6-ring | NE ARG 64 (A) | pi-cation | 4.89 | −0.9 |
| Compound | Ligand | Receptor | Interaction | Distance E | (kcal/mol) |
|---|---|---|---|---|---|
| 2a | C 30 | O ARG 841 (A) | H-donor | 3.24 | −0.5 |
| 6-ring | CD ARG 841 (A) | pi-H | 3.75 | −1.0 | |
| 3 | No measurable interactions | ||||
| 4a | 6-ring | CD ARG 841 (A) | pi-H | 3.92 | −0.7 |
| 4b | C 31 | O ARG 841 (A) | H-donor | 3.38 | −0.8 |
| 6-ring | CG1 VAL 726 (A) | pi-H | 4.50 | −0.5 | |
| 6-ring | CD ARG 841 (A) | pi-H | 3.74 | −0.7 | |
| 5 | N 24 | O GLY 724 (A) | H-donor | 2.85 | −2.8 |
| N 43 | NZ LYS 745 (A) | H-acceptor | 3.00 | −10.2 | |
| 6-ring | CA GLY 721 (A) | pi-H | 4.24 | −0.9 | |
| 6-ring | N ALA 722 (A) | pi-H | 4.79 | −0.7 | |
| Comp. | 2b | 3 | 4a | 4b | 5 |
|---|---|---|---|---|---|
| Formula | C22H9Br4N5O4S | C21H10Br4N6O3 | C22H11Br3N6O4 | C22H11Br3N6O3S | C21H9Br4N5O4 |
| Molecular weight | 759.02 g/mol | 713.96 g/mol | 663.07 g/mol | 679.14 g/mol | 714.94 g/mol |
| Num. heavy atoms | 36 | 34 | 35 | 35 | 34 |
| Num. arom. heavy atoms | 12 | 12 | 12 | 12 | 12 |
| Fraction Csp3 | 0.14 | 0.14 | 0.14 | 0.14 | 0.14 |
| Num. rotatable bonds | 3 | 3 | 3 | 3 | 3 |
| Num. H-bond acceptors | 7 | 7 | 8 | 7 | 7 |
| Num. H-bond donors | 1 | 2 | 2 | 2 | 1 |
| Molar Refractivity | 160.73 | 151.53 | 149.22 | 156.42 | 148.13 |
| TPSA | 156.31 Å2 | 131.00 Å2 | 148.07 Å2 | 163.09 Å2 | 124.22 Å2 |
| Lipophilicity | |||||
| Log Po/w (iLOGP) | 2.81 | 2.19 | 2.09 | 2.50 | 2.40 |
| Log Po/w (XLOGP3) | 4.53 | 4.14 | 3.33 | 3.93 | 4.05 |
| Log Po/w (WLOGP) | 2.98 | 3.06 | 2.50 | 2.67 | 2.61 |
| Log Po/w (MLOGP) | 3.88 | 4.56 | 3.76 | 3.72 | 4.16 |
| Log Po/w (SILICOS-IT) | 6.82 | 5.57 | 4.81 | 6.27 | 5.44 |
| Consensus Log Po/w | 4.20 | 3.91 | 3.30 | 3.82 | 3.73 |
| Water Solubility | |||||
| Log S (ESOL) | −7.45 | −6.94 | −6.10 | −6.58 | –6.89 |
| Solubility | 2.70 × 10−5 mg/mL; 3.56 × 10−8 mol/L | 8.24 × 10−5 mg/mL; 1.15 × 10−7 mol/L | 5.21 × 10−5 mg/mL; 7.86 × 10−7 mol/L | 1.78 × 10−5 mg/mL; 2.62 × 10−7 mol/L | 9.27 × 10−5 mg/mL; 1.30 × 10−7 mol/L |
| Class | Poorly soluble | Poorly soluble | Poorly soluble | Poorly soluble | Poorly soluble |
| Log S (Ali) | −7.53 | −6.60 | −6.12 | −7.05 | −6.36 |
| Solubility | 2.22 × 10−5 mg/mL; 2.92 × 10−8 mol/L | 1.80 × 10−4 mg/mL; 2.52 × 10−7 mol/L | 5.07 × 10−4 mg/mL; 7.65 × 10−7 mol/L | 5.99 × 10−5 mg/mL; 8.83 × 10−8 mol/L | 3.10 × 10−4 mg/mL; 4.34 × 10−7 mol/L |
| Class | Poorly soluble | Poorly soluble | Poorly soluble | Poorly soluble | Poorly soluble |
| Log S (SILICOS-IT) | −9.20 | −9.04 | −8.44 | −8.62 | −8.90 |
| Solubility | 4.76 × 10−7 mg/mL; 6.27 × 10−10 mol/L | 6.44 × 10−7 mg/mL; 9.02 × 10−10 mol/L | 2.43 × 10−6 mg/mL; 3.66 × 10−9 mol/L | 1.64 × 10−6 mg/mL; 2.42 × 10−9 mol/L | 9.02 × 10−7 mg/mL; 1.26 × 10−9 mol/L |
| Class | Poorly soluble | Poorly soluble | Poorly soluble | Poorly soluble | Poorly soluble |
| Pharmacokinetics | |||||
| G.I. absorption | Low | High | Low | Low | High |
| BBB permeant | No | No | No | No | No |
| P-gp substrate | No | No | No | No | No |
| CYP1A2 inhibitor | Yes | Yes | No | Yes | Yes |
| CYP2C19 inhibitor | No | No | No | No | No |
| CYP2C9 inhibitor | No | No | No | No | No |
| CYP2D6 inhibitor | No | No | No | No | No |
| CYP3A4 inhibitor | No | No | No | No | No |
| Log Kp (skin permeation) | −7.71 cm/s | −7.72 cm/s | −7.98 cm/s | −7.65 cm/s | −7.79 cm/s |
| Drug-likeness | |||||
| Lipinski | Yes; 1 violation: MW > 500 | No; 2 violations: MW > 500, MLOGP > 4.15 | Yes; 1 violation: MW > 500 | Yes; 1 violation: MW > 500 | No; 2 violations: MW > 500, MLOGP > 4.15 |
| Ghose | No; 2 violations: MW > 480, MR > 130 | No; 2 violations: MW > 480, MR > 130 | No; 2 violations: MW > 480, MR > 130 | No; 2 violations: MW > 480, MR > 130 | No; 2 violations: MW > 480, MR > 130 |
| Veber | No; 1 violation: TPSA > 140 | Yes | No; 1 violation: TPSA > 140 | No; 1 violation: TPSA > 140 | Yes |
| Egan | No; 1 violation: TPSA > 131.6 | Yes | No; 1 violation: TPSA > 131.6 | No; 1 violation: TPSA > 131.6 | Yes |
| Muegge | No; 2 violations: MW > 600, TPSA > 150 | No; 1 violation: MW > 600 | No; 1 violation: MW > 600 | No; 2 violations: MW > 600, TPSA > 150 | No; 1 violation: MW > 600 |
| Bioavailability Score | 0.55 | 0.17 | 0.55 | 0.55 | 0.17 |
| Medicinal Chemistry | |||||
| PAINS | 0 alert | 0 alert | 0 alert | 0 alert | 0 alert |
| Brenk | 4 alerts: halogenated_ring_1, halogenated_ring_2, phthalimide, thiocarbonyl_group | 4 alerts: halogenated_ring_1, halogenated_ring_2, imine_1, phthalimide | 3 alerts: halogenated_ring_1, imine_1, phthalimide | 4 alerts: halogenated_ring_1, imine_1, phthalimide, thiocarbonyl_group | 3 alerts: halogenated_ring_1, halogenated_ring_2, phthalimide |
| Lead-likeness | No; 2 violations: MW > 350, XLOGP3 > 3.5 | No; 2 violations: MW > 350, XLOGP3 > 3.5 | No; 1 violation: MW > 350 | No; 2 violations: MW > 350, XLOGP3 > 3.5 | No; 2 violations: MW > 350, XLOGP3 > 3.5 |
| Synthetic accessibility | 4.61 | 4.58 | 4.74 | 4.66 | 4.52 |
| Model Name | Pharmacokinetic Properties | ||||
|---|---|---|---|---|---|
| Water solubility (log mol/L) | −5.621 | −4.823 | −4.608 | −4.749 | −5.483 |
| Caco2 permeability (log Papp in 10−6cm/s) | 0.69 | 0.621 | 0.001 | 0.58 | 0.665 |
| Intestinal absorption (human) (% Absorbed) | 83.586 | 84.559 | 80.732 | 82.507 | 84.596 |
| Skin Permeability (log Kp) | −2.826 | −2.8 | −2.779 | −2.782 | −2.863 |
| P-glycoprotein substrate | Yes | Yes | Yes | Yes | Yes |
| P-glycoprotein I inhibitor | Yes | Yes | Yes | Yes | Yes |
| P-glycoprotein II inhibitor | Yes | Yes | No | No | Yes |
| Compound | 2b | 3 | 4a | 4b | 5 |
|---|---|---|---|---|---|
| Predicted LD50 | 10,000 mg/kg | 1000 mg/kg | 1000 mg/kg | 1168 mg/kg | 300 mg/kg |
| Predicted ToxicityClass * | 6 | 4 | 4 | 4 | 3 |
| Average similarity | 34.04% | 31.94% | 35.06% | 33.99% | 32.14% |
| Prediction accuracy: | 23% | 23% | 23% | 23% | 23% |
 * * | |||||
| S. No | Inhibitors | Free Energy of Binding (kcal/mol) | ||||||
|---|---|---|---|---|---|---|---|---|
| Caspase-3 | Human Cyclin-Dependent Kinase 2 (CDK2) | Epidermal Growth Factor Receptor (EGFR) | Human B-Raf Kinase | Human Estrogen Receptor Ligand-Binding Domain | Human Serum Albumin | Human Topoisomerase I | ||
| 1 | 2a | −6.7 | −8.7 | −9.1 | −8.5 | −8.1 | −8.6 | −8.1 |
| 2 | 2b | −6.7 | −7.5 | −8.2 | −8.2 | −8.2 | −8.2 | −7.9 |
| 3 | 3 | −6.4 | −8.0 | −7.9 | −9.2 | −7.6 | −9.8 | −8.2 |
| 4 | 4a | −6.6 | −9.0 | −8.1 | −8.3 | −8.5 | −9.9 | −8.1 |
| 5 | 4b | −6.9 | −7.4 | −8.5 | −8.5 | −7.8 | −8.4 | −8.2 |
| 6 | 5 | −6.7 | −8.8 | −8.9 | −9.2 | −7.7 | −9.3 | −8.2 |
| S. No | Docked Complex | MM-PBSA-Based Calculated Energies (kJ/mol) | |||
|---|---|---|---|---|---|
| Van Der Waals | Electrostatic | SASA | Binding e Energy | ||
| 1 | 3 | −308.232 | −22.682 | −22.504 | −375.922 |
| 2 | 4a | −244.065 | −29.981 | −23.239 | −320.525 |
| 3 | 5 | −259.583 | −41.077 | −23.113 | −346.887 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdellattif, M.H.; Shahbaaz, M.; Arief, M.M.H.; Hussien, M.A. Oxazinethione Derivatives as a Precursor to Pyrazolone and Pyrimidine Derivatives: Synthesis, Biological Activities, Molecular Modeling, ADME, and Molecular Dynamics Studies. Molecules 2021, 26, 5482. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26185482
Abdellattif MH, Shahbaaz M, Arief MMH, Hussien MA. Oxazinethione Derivatives as a Precursor to Pyrazolone and Pyrimidine Derivatives: Synthesis, Biological Activities, Molecular Modeling, ADME, and Molecular Dynamics Studies. Molecules. 2021; 26(18):5482. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26185482
Chicago/Turabian StyleAbdellattif, Magda H., Mohd Shahbaaz, M. M. H. Arief, and Mostafa A. Hussien. 2021. "Oxazinethione Derivatives as a Precursor to Pyrazolone and Pyrimidine Derivatives: Synthesis, Biological Activities, Molecular Modeling, ADME, and Molecular Dynamics Studies" Molecules 26, no. 18: 5482. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26185482