
Selective Esterification of Phosphonic Acids †


Institute of Organic Chemistry, Polish Academy of Science, Kasprzaka 44/52, 01-224 Warsaw, Poland
*
Author to whom correspondence should be addressed.
†
This work is dedicated to Professor Janusz Jurczak on the occasion of his 80th birthday.
Molecules 2021, 26(18), 5637; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26185637
Submission received: 30 July 2021
/
Revised: 1 September 2021
/
Accepted: 13 September 2021
/
Published: 17 September 2021
(This article belongs to the Special Issue In Honor of the 80th Birthday of Professor Janusz Jurczak)
Abstract
:Here, we report straightforward and selective synthetic procedures for mono- and diesterification of phosphonic acids. A series of alkoxy group donors were studied and triethyl orthoacetate was found to be the best reagent as well as a solvent for the performed transformations. An important temperature effect on the reaction course was discovered. Depending on the reaction temperature, mono- or diethyl esters of phosphonic acid were obtained exclusively with decent yields. The substrate scope of the proposed methodology was verified on aromatic as well as aliphatic phosphonic acids. The designed method can be successfully applied for small- and large-scale experiments without significant loss of selectivity or reaction yield. Several devoted experiments were performed to give insight into the reaction mechanism. At 30 °C, monoesters are formed via an intermediate (1,1-diethoxyethyl ester of phosphonic acid). At higher temperatures, similar intermediate forms give diesters or stable and detectable pyrophosphonates which were also consumed to give diesters. 31P NMR spectroscopy was used to assign the structure of pyrophosphonate as well as to monitor the reaction course. No need for additional reagents and good accessibility and straightforward purification are the important aspects of the developed protocols.
1. Introduction
Organophosphonates are a relatively diverse and common group of compounds, which feature the presence of a stable C–P bond. In nature, they are present in microorganisms as well as in metazoans. Furthermore, they constitute a considerable part of phosphorus compounds found in oceans [1]. It is also considered that organophosphonates could play an important role at the early stages of the development of life on Earth when the oxygen concentration in the atmosphere was negligible [2]. Currently, phosphonates have a large number of applications in agriculture [3] and medicine [4,5], as well as markers of chemical warfare agents (CWAs), and many others [6].
The synthesis of organophosphorus esters from related phosphites in reaction with organic halides (Michaelis–Arbuzov reaction) is well-established chemistry [7,8]. The corresponding transformations leading to phosphonates from hydrogenphosphonates (Michaelis–Becker [9,10], Atherton–Todd [11,12] reactions), phosphonic dichlorides [13,14] and phosphonochloridates [15,16] (alcoholysis) or even simpler substrates such as phosphoryl chloride [17] are also broadly employed as well. However, the abovementioned methods are connected with the generation of corrosive and hazardous halide waste [18]. Furthermore, most of the described methods are not suitable for application in a standard laboratory or require harsh reaction conditions, toxic solvents [10] and laborious preparation [19,20].
An alternative route for target compounds is direct esterification or transesterification of the phosphonic acids. The esterification can be achieved in the presence of chlorinated silica gel [21], by exploitation of Garegg–Samuelsson conditions [22], dicyclohexylcarbodiimide mediated esterification [23] or O-alkylation [24], whereas alkyl hydrogen phosphonates can be received from the diesters by partial hydrolysis [20,25] or via transesterification with silyl halides and selective cleavage by protic solvents [26]. Monoesters can also be obtained directly from phosphonic acids upon selective monoesterification. For successful syntheses, phenylarsonic acid [27,28], carbodiimides [29,30], solid-phase synthesis [31] and ionic liquids [24] were used. Unfortunately, none of the mentioned methods can be performed under mild conditions, with high yields and good selectivity towards monoesters.
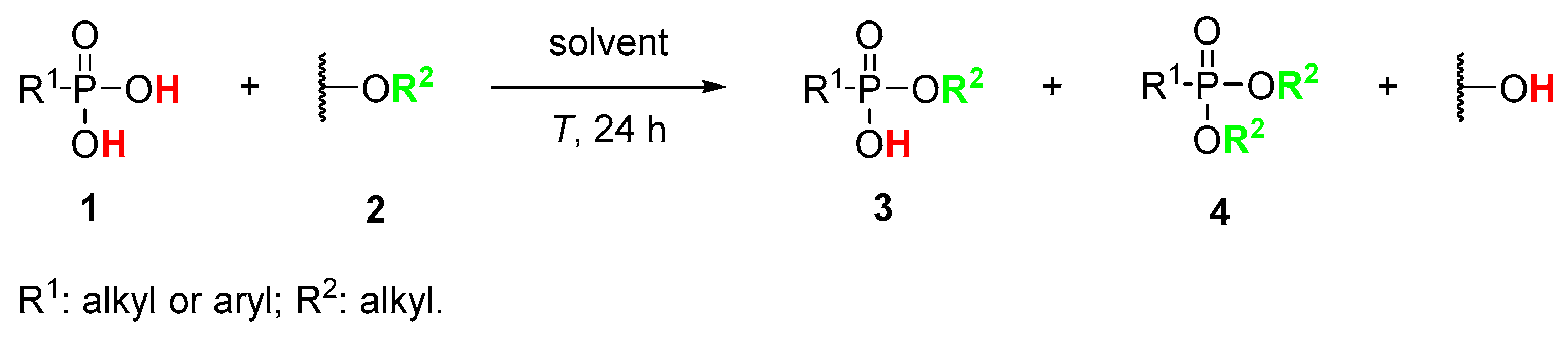
Recently, we have found that esterification of carboxylic acid proceeds efficiently upon the application of orthoesters [32], carbonates [33], orthocarbonates [34], acetals and ketals [35] as alkoxy group donors. The studies on the reaction mechanism proved that dialkoxy carboxylate is a key intermediate that is subsequently and irreversibly transformed into ethyl acetate which is a driving force for the reaction [32]. Since phosphonic acids are the isosteres of carboxylic acids, the same principle is proposed for the esterification of phosphonic acids (1) using corresponding alkoxy group donors (2). The esterification of phosphonic acids is more complicated since during the reaction two possible products, mono- (3) and diesters (4), can be formed simultaneously (Scheme 1). The straightforward synthesis of diester 4 is obvious [36,37,38,39] while the selective formation of monoesters is very tricky. The application of alkoxy group donors in monoesterification of phosphonic acids has never been reported before.
2. Results and Discussion
For preliminary experiments, both butylphosphonic (1a) and phenylphosphonic (1b) acids were selected as model substrates. Initially, various potential alkoxy group donors were investigated in the monoesterification reaction (Table 1). Their structures are presented in Figure 1. The reactions were conducted in methyl tert-butyl ether (MTBE) at 30 °C for 24 h. The conversion of substrates and selectivity toward products were determined by 1H and 31P NMR analyses.
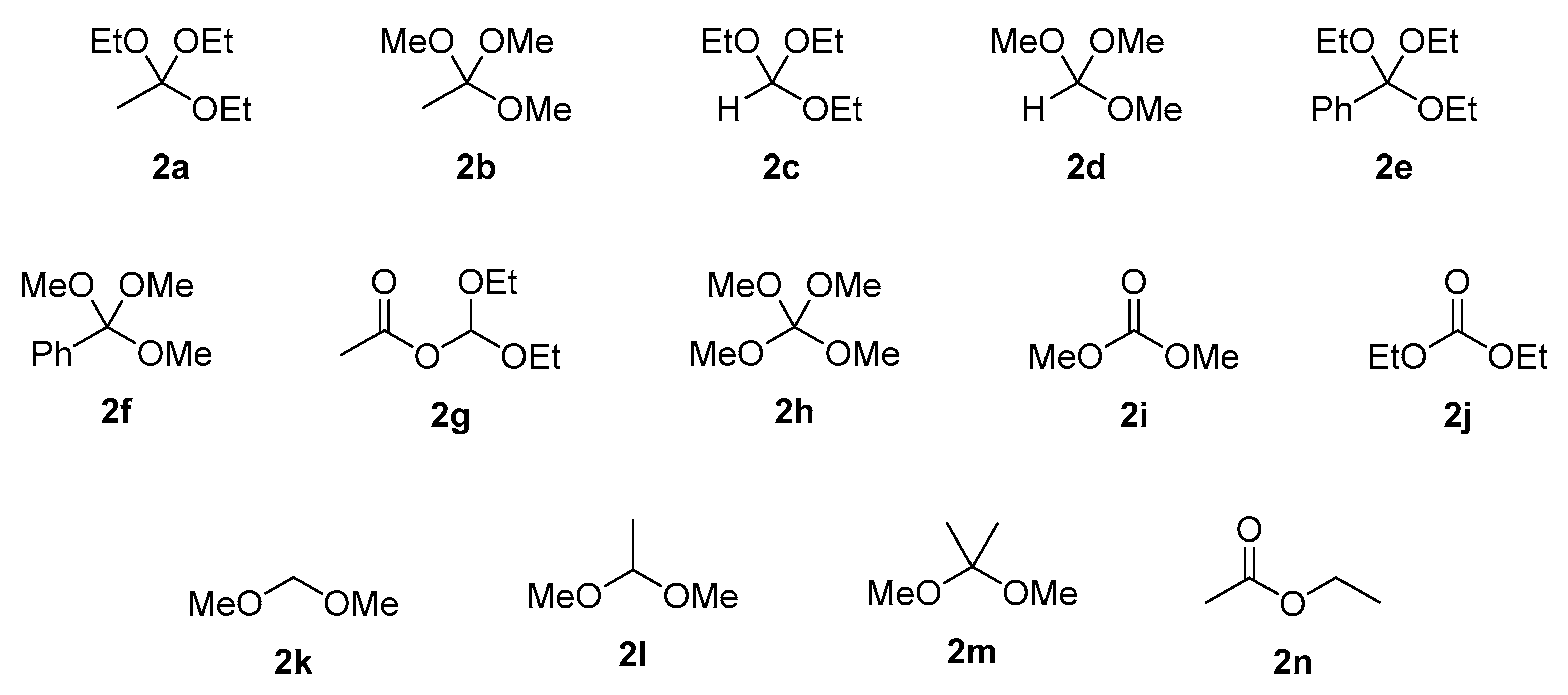
The application of triethyl orthoacetate (2a) as an ethoxy group donor resulting in high conversion, yielding 84% of 3a and 83% of 3b (Table 1, entries 1 and 15). In both cases, excellent selectivity towards monoesters 3a and 3b was observed (yields of diesters 4a and 4b were below 3%). Trimethyl orthoacetate (2b) was found to be a less efficient donor of an alkoxy group, the conversion of acid 1a reached 25% and that of acid 2b was 50% (Table 1, entries 2 and 16). In the case of triethyl orthoformate (2c), trimethyl orthoformate (2d), triethyl orthobenzoate (2e) and trimethyl orthobenzoate (2f), no substrate conversion was observed (Table 1, entries 3–6, 17–20). Along with 2a, diethoxymethyl acetate (2g) was also found to be a selective ethoxy group donor. In reaction with butylphosphonic acid (1a), the conversion of substrate was 39% (Table 1, entry 7) and for phenylphosphonic acid (1b) it was 89% (Table 1, entry 21). The studied transformation was very selective towards monoesters 3. The application of donor 2g provides a higher conversion of phenylphosphonic acid with respect to butylphosphonic acid. Tetramethyl orthocarbonate (2h) was also active in the esterification reaction, providing ester 3a in 23% and ester 3b in 44% yields, respectively (Table 1, entries 8, 22). When other donors such as dimethyl carbonate (2i), diethyl carbonate (2j), formaldehyde dimethyl acetal (2k), acetaldehyde dimethyl acetal (2l), 2,2-dimethoxypropane (2m) and ethyl acetate (2n) were applied, no reaction occurred (Table 1, entries 9–14 and 23–28).
In the next step of our research, we tested the impact of the reaction medium using 2a as a donor (Table 2), because the preliminary used MTBE was selected arbitrarily. For butylphosphonic acid 1a, when the reaction was conducted in tetrahydrofuran, a slight drop in reaction yield was observed with respect to MTBE while in the case of phenylphosphonic acid 1b, the result was similar (Table 2, entries 1 and 2). In dichloromethane, conversion was high for acid 1a and almost quantitative for acid 1b, with good selectivity towards monoesters 3a and 3b (Table 2, entry 3). In ethyl acetate and acetonitrile, the conversions were lower for both substrates (Table 2, entries 4 and 5). Acetone was found to be an inappropriate solvent with very low substrate conversion (Table 2, entry 6). The reaction proceeds effectively in toluene (Table 2, entry 7) but the highest conversion (95%) was observed when 2a was used as a reagent and solvent (Table 2, entry 8). No reaction progress was observed in experiments conducted in dimethyl sulfoxide and dimethylformamide (Table 2, entries 9 and 10).
The analysis of data from Table 2 concludes that the best medium for the studied transformation is triethyl orthoacetate 2a when used in large excess with respect to substrate (over 5 equiv.). After selection of the best solvent for reaction, we studied the impact of temperature on the esterification (Table 3). Reactions of butylphosphonic acid 1a with orthoacetate 2a were performed at temperatures varying from 30°C to 100 °C. The increase in the reaction temperature from 30 to 40 °C resulted in a higher conversion of the substrate (over 99%) and slightly influenced the yield of the product (Table 3, entries 1 and 2). At elevated temperatures, the conversion of the substrate became quantitative (Table 3, entries 2–8) and the selectivity towards diester increased gradually. The amount of monoester 3a in the reaction mixture dropped to 1% at 90 °C (Table 3, entry 7). At 100 °C, partial decomposition of the product was observed (Table 3, entry 8).
In conclusion, the considered data indicate that at higher temperatures, diester 4a is formed as a single product (Table 3, entries 7 and 8). The best conditions for diesterification of butylphosphonic acid 1a involved reaction in excess of orthoester 2a at 90 °C. An analogous esterification performed at 30 °C led to the selective formation of monoethyl butylphosphonate (3a).
With the optimized reaction conditions in hand, we proceeded to assess the generality and scope of the developed procedures. Table 4 summarizes the results of the preparation of several mono- and diesters from various phosphonic acids. It should be noted that for the experiments performed on a laboratory scale, the favourable amount of orthoacetate 2a was 15 equiv. for monoesterification and 30 equiv. for diesterification.
The preparative esterification of model butylphosphonic acid (1a) proceeded efficiently and monoester 3a and diester 4a were obtained in 98% yields (Table 4, entries 1 and 2). For effective monoesterification of benzylphosphonic acid, a longer reaction time, 48 h, was required and respective monoester 3b was obtained in a 76% isolated yield (Table 4, entry 3). Diester 4b was obtained in an almost quantitative 98% yield (Table 4, entry 4). Monoesterification proceeded smoothly for the other alkylphosphonic acids (Table 4, entries 5, 7, 9 and 11) and yields of monoesters exceeded 83%. The selective monoesterification of aryl and benzylphosphonic acids was also successful. Presumably due to poor solubility of 4-hydroxybenzylphosphonic acid (1i) and [1,4-phenylenebis(methylene)]-bis(phosphonic acid) (1m), no product formation (3i and 3m) was observed (Table 4, entries 17, 25).
Exhaustive esterification of other aliphatic acids in established conditions gave respective diesters 4c, 4e and 4f in 89–97% yields (Table 4, entries 6, 10, and 12). For vinylphosphonic acid (2d), product 4d was not received due to its rapid polymerization at elevated temperatures (Table 4, entry 8). Diesterification for arylphosphonic acids 1g and 1h proceeded smoothly and esters 4g and 4h were isolated in quantitative yield (Table 4, entries 14–16). Substituted derivatives of benzylphosphonic acid 1i–l underwent double esterification with good to excellent yields (Table 4, entries 18, 20, 22 and 24). For 24 h, at 90 °C, reaction with substrate 1m (p-xylene diphosphonic acid) proceeded with low conversion. Therefore, to accelerate the transformation, the temperature was raised to 145 °C, and the reaction mixture was refluxed for 24 h which led to product 4m with quantitative yield (Table 4, entry 26).
It is important to note that using a single reagent—triethyl orthoacetate—two types of products, mono- (3) and diethylesters (4), were obtained under mild reaction conditions in a reasonable time without the use of any toxic or polluting reagents. In almost all cases, products were obtained as single compounds upon evaporation of the reaction mixture to dryness. The developed procedure partially fulfills green chemistry principles and can be applied to a fair range of phosphonic acids.
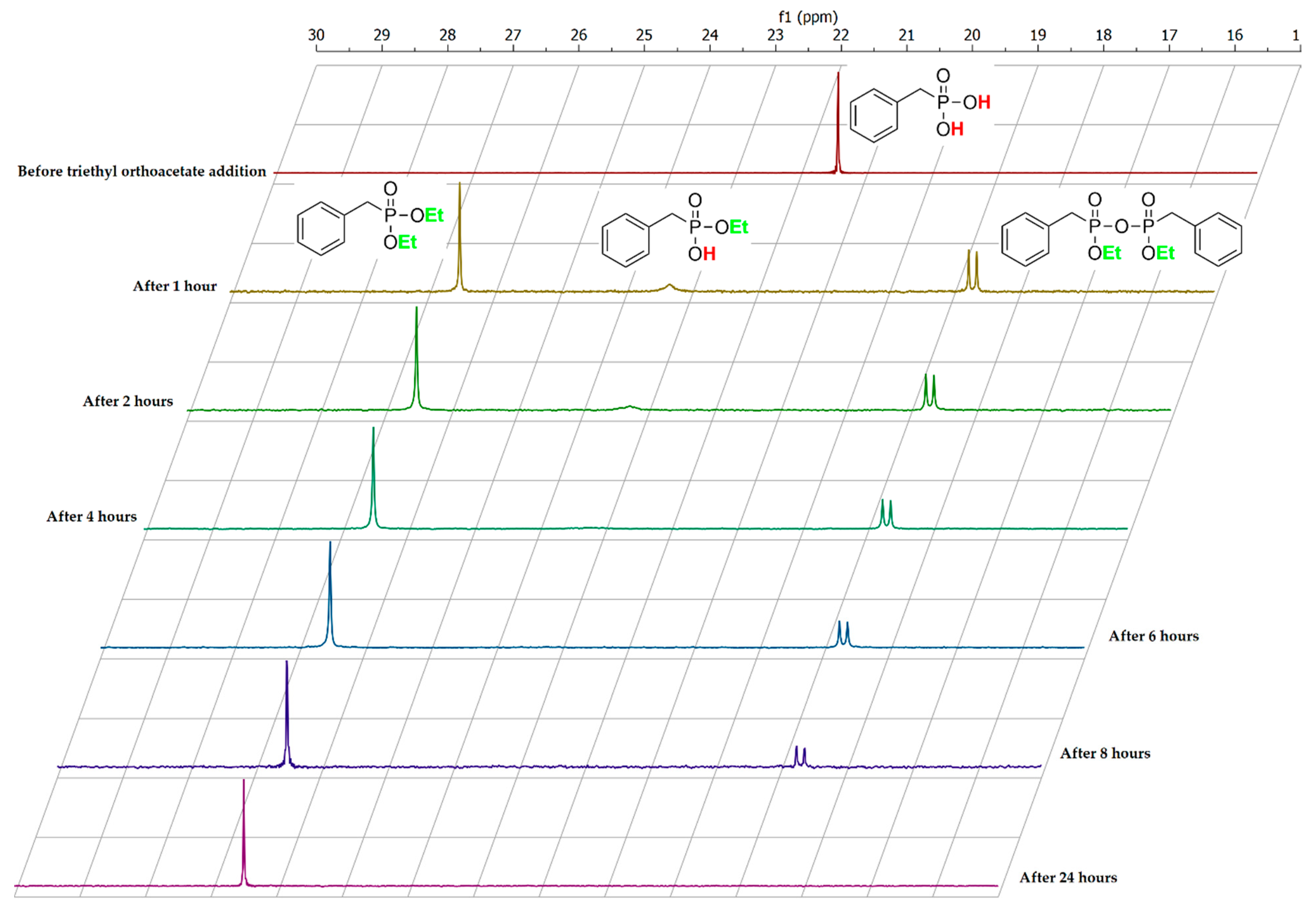
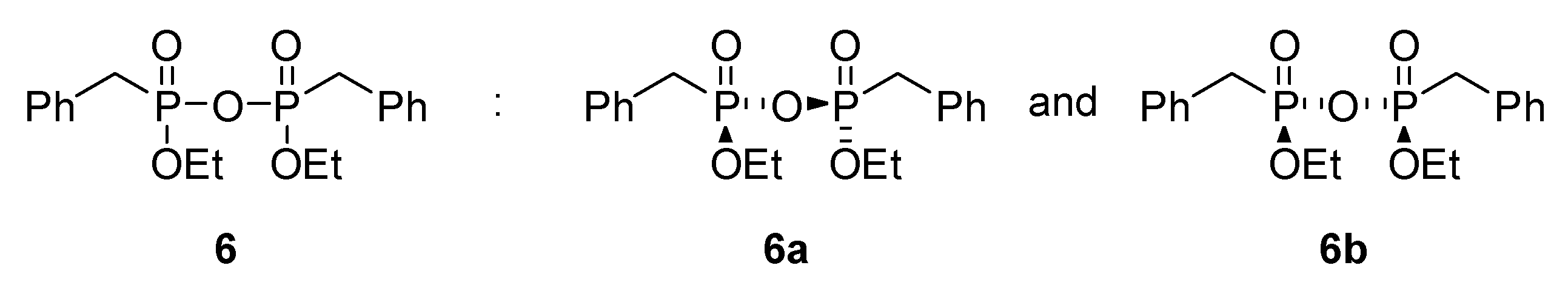
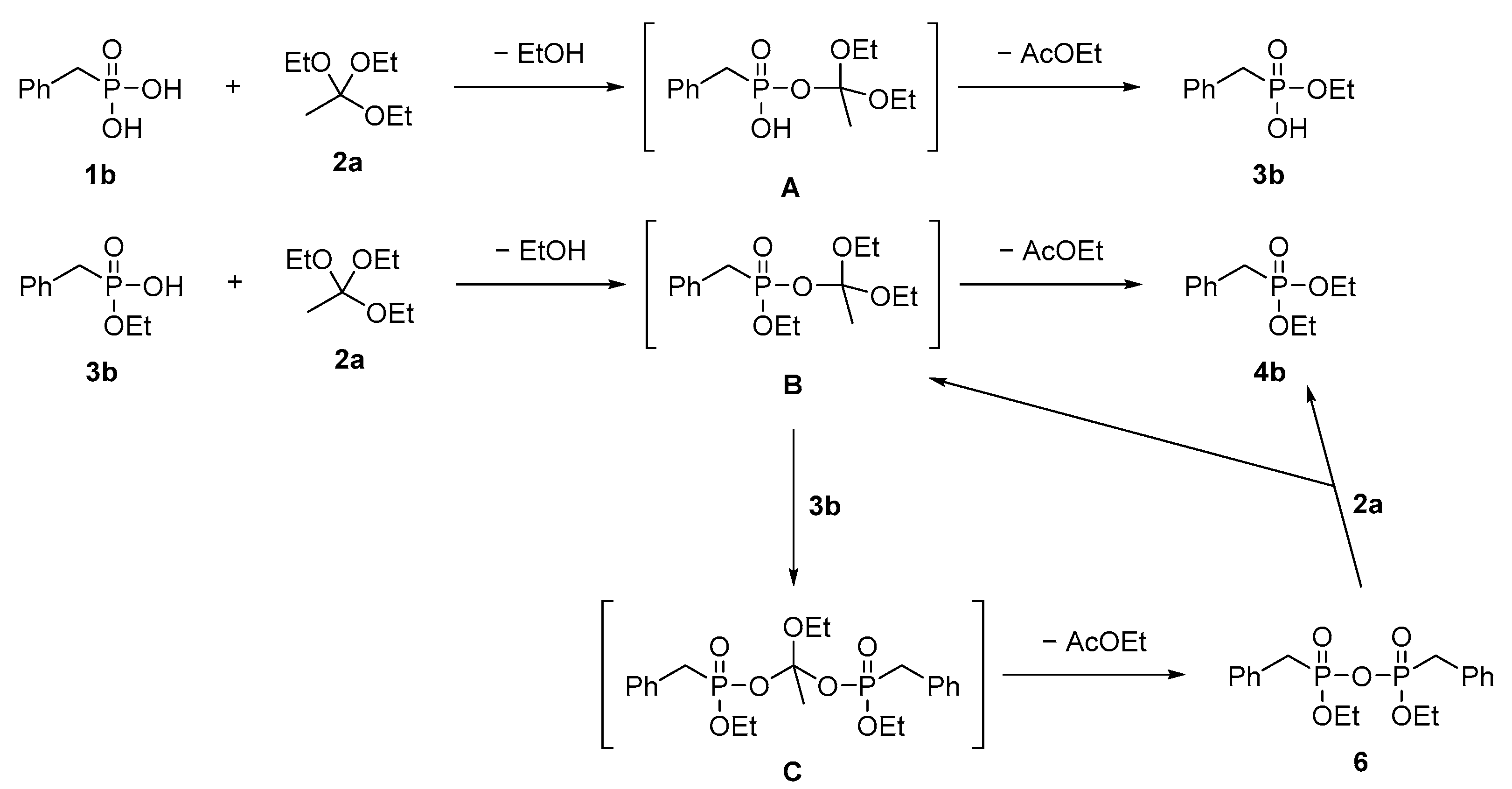
The gain insight into the reaction mechanism, additional experiments were performed on one of the model substrates—benzylphosphonic acid 1b—with selected donor 2a at 30° and 75 °C. Close inspection of 31P NMR spectra of crude 3b product obtained in reaction at 30 °C did not show any additional signals from possible reaction intermediates (see Supplementary Materials Section S2.3). However, in the reaction performed at the higher temperature, an interesting phenomenon was observed. Formally, before the reaction had started, the spectrum of the substrate 1b was measured. It is at the top of Figure 2 and shows a single peak at 21.3 ppm. After the addition of triethyl orthoacetate and heating the reaction mixture for 1 h at 75 °C, the NMR spectra changed substantially. The signal associated with substrate 1b diminished almost to zero and four new signals appeared on the spectra. The main peak at 26.5 ppm comes from diester 4b. The amount of product 4b increased rapidly during this first hour and reached a maximum after 24 h. The small and broad signal at 23.3 ppm represents monoester 3b. The most significant peak of the 3b product appeared in the first spectrum (after an hour). Further measurements demonstrated that the amount of 3b dropped close to zero after 8 h. Surprisingly, two new signals at 18.65 and 18.77 ppm of similar intensity appeared. Close inspection of literature data [40] allowed us to assign those two signals to diastereomers 6a and 6b of pyrophosphonate 6 (Figure 3). The appearance of compound 6 as a reaction intermediate is a valuable clue to the reaction mechanism.
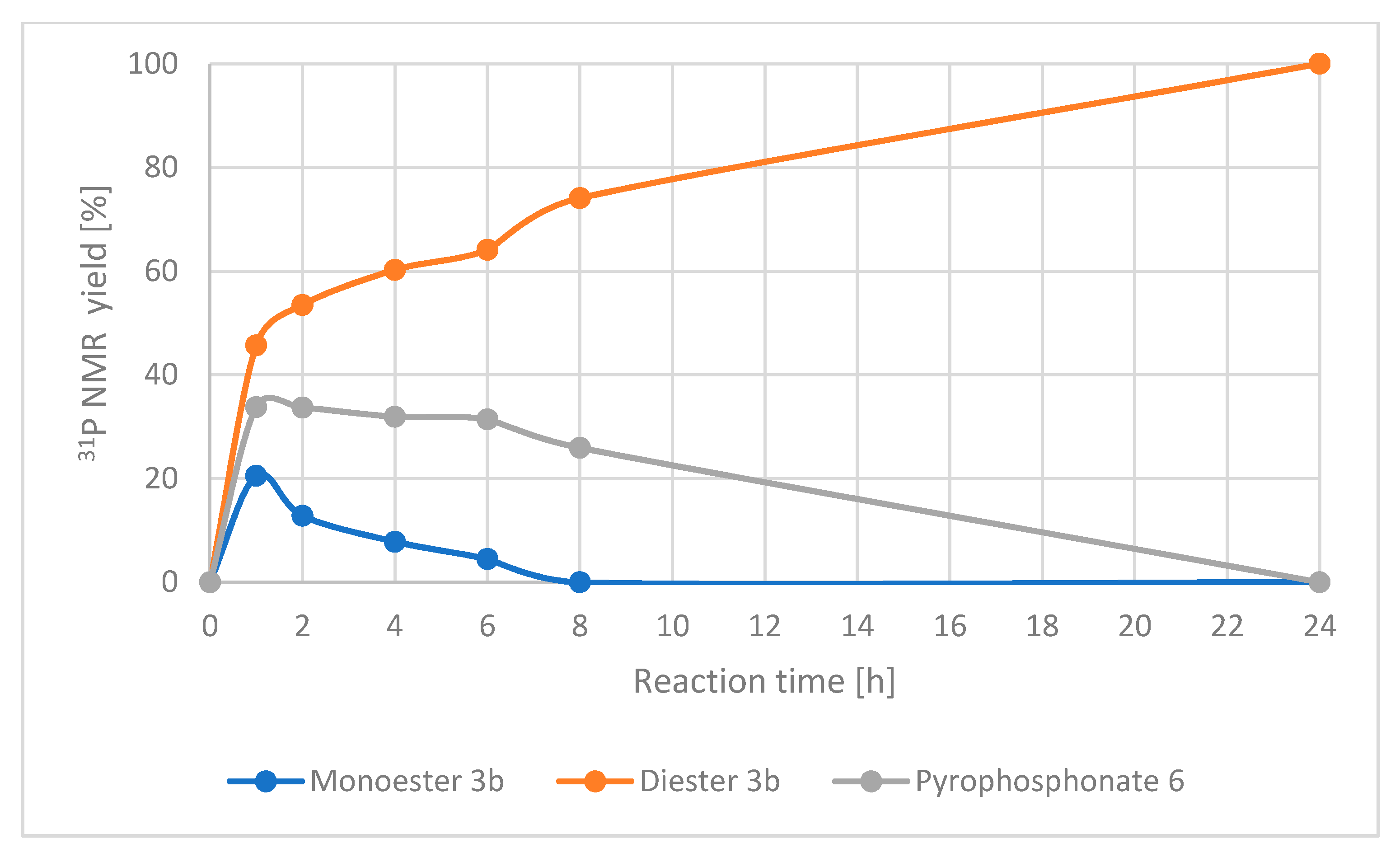
The calculated 31P NMR yield of pyrophosphonate 6 reached 34% after 1 h and its amount remained at a similar level for the next 5 h and then slowly decreased to zero. The detailed data from the 31P NMR spectra analysis are summarized in Table S1 (SI). To sum up, the NMR spectra recorded during the reaction course showed that the amount of diester 4b was continuously increasing with time while amounts of 3b and 6 reached a kind of plateau and then were diminished to zero after 24 h. As mentioned above, at a lower reaction temperature (30 °C) signals from pyrophosphonate 6 were not observed and a higher reaction temperature was required to transform monoester 3b into pyrophosphonate 6 and finally into diester 4b. The graphical representation of the results described above is shown in Figure 4.
The analysis of obtained data provided some insight into the reaction mechanism (Scheme 2). The first step is the reaction between triethyl orthoacetate (2a) and benzylphosphonic acid (1b) which leads to the formation of an unstable intermediate A. It readily decomposes to monoester 3b by releasing one molecule of ethanol and ethyl acetate in a two-step process, at room temperature. The intermediate A can be formed only from selected substrates used as alkoxy group donors, depicted in Figure 1. This mechanism allowed us to explain why only monophosphonates were formed from orthoesters 2a and 2b, diethoxymethyl acetate 2g and tetramethyl orthocarbonate 2h. At higher temperatures, monoester 3b reacts with a second molecule of triethyl orthoacetate to give intermediate B, similar to A. This intermediate can react with the second molecule of monoester 3b to give compound C which is readily transformed into pyrophosphonate 6 and its structure was ambiguously assigned by kinetic experiments. Compound 6 reacts with the third molecule of orthoester 2a to give the final product diester 4b and intermediate B. According to this mechanism, for the formation of one molecule of diester 4, three molecules of orthoester 2a are required. This readily explains why the larger amount of triethyl orthoacetate favors double esterification with respect to monoesterification.
3. Materials and Methods
3.1. General
Commercially available reagents were used without additional purification. Phosphonic acids were purchased or prepared according to reported literature procedures. The water and hexane mixtures were previously distilled. Other solvents (analytical grade) were used without extra drying and purification. Reactions were performed in dry laboratory glassware under an air atmosphere (otherwise noted) using magnetic stirrers. Solvents and volatile reagents were evaporated under reduced pressure. Merck (Darmstadt, Germany) silica gel plates 60 F254 were used for TLC analysis.
The majority of obtained compounds did not need additional purification after reaction workup. Those requiring isolation were separated via distillation under reduced pressure on Kugelrohr apparatus. Melting points were measured on a Boetius hot plate microscope (Nagema, Dresden, Germany). The phosphorous nuclear magnetic resonance (31P{1H} NMR) spectra of analyzed compounds, dissolved in DMSO-d6, D2O or CDCl3, were recorded with a Bruker spectrometer (162.0 MHz) at 30 °C and using 85% H3PO4 as external calibration. 1H and 13C NMR spectra were also recorded in DMSO-d6, D2O or CDCl3 (30 °C) on the same Bruker spectrometer at 400 and 101 MHz, respectively. Chemical shifts were reported in ppm and referred to residual deuterated solvent signal; coupling constants (J) were noted in Hz. Low-resolution mass spectra were recorded on the API365i API 3000 spectrometer, and the ESI technique was used to analyte ionization. High-resolution mass spectra were recorded on the Maldi SYNAPT G2-S HDMS (Waters, Milford, MA, USA) apparatus with a QqTOF analyzer.
3.2. General Procedure for Esterification of Phosphonic Acid Using Various Alkoxy Group Donors
To a solution of butylphosphonic acid 1a (1 equiv., 7 mg, 0.05 mmol) or benzylphosphonic acid 1b (1 equiv., 9 mg, 0.05 mmol) in MTBE (1 mL), the respective alkoxy group donor 2 (3 equiv.) was added and stirred at 30 °C for 24 h. The solvent and other volatile compounds were evaporated under reduced pressure. The crude product was dissolved in 0.6 mL DMSO-d6 and the 31P NMR spectrum was recorded. Subsequently, the conversion of substrate 1 and yields of products 3 and 4 were determined on the basis of relative peak integrals.
3.3. General Procedure for Esterification of Phosphonic Acids Using Triethyl Orthoacetate in Different Solvents
To a solution of butylphosphonic acid 1a (1 equiv., 7 mg, 0.05 mmol) or benzylphosphonic acid 1b (1 equiv., 9 mg, 0.05 mmol), 1 mL of respective solvent triethyl orthoacetate 2a (3 equiv., 25 mg, 0.15 mmol) was added and stirred at 30 °C for 24 h. The solvent and other volatile substances were evaporated under reduced pressure. Obtained crude was dissolved in 0.6 mL DMSO-d6 and the spectrum was recorded. The conversion of substrate 1 and yield of products 3 and 4 were determined based on 31P NMR spectrum analysis.
3.4. The Influence of Temperature on the Selectivity of Esterification Reaction of Butylphosphonic acid with Triethyl Orthoacetate
Butylphosphonic 1a (1 equiv., 14 mg, 0.1 mmol)) in 1 mL orthoacetate 2a was heated at the temperature indicated in Table 3 for 24 h. After that, the reaction mixture was cooled to room temperature and the excess 2a, as well as volatile byproducts, was evaporated to dryness. Received crude was dissolved in DMSO-d6 and the prepared sample was used for the 31P NMR measurements, based on which the conversion of 1a and product 3a and 4a yields were determined.
3.5. General Procedure for Selective Monoesterification of Phosphonic Acids 1 Using Triethyl Orthoacetate 2a Leading to Ethyl Hydrogen Phosphonates 3
Phosphonic acid 1 (1 mmol) and triethyl orthoacetate (15 mmol, 2.75 mL) were stirred at 30 °C and the substrate conversion was monitored by 31P NMR. After the competition of the reaction, the orthoacetate excess and volatile by-product were evaporated off under vacuum. In several cases, the remaining product required no further purification. If necessary, the extraction procedure proposed by Campbell [41] was applied: the crude was diluted with diethyl ether (10 mL) and extracted twice with NaHCO3 (5 mL, 5% w/v solution in water). The aqueous fractions were combined and washed with two portions of ethyl ether (2 × 5 mL). The aqueous phase was acidified with hydrogen chloride solution (1 M) to reach pH 2 and extracted three times with ethyl acetate (3 × 5 mL). Combined ethyl acetate fractions were dried over magnesium sulfate and pure products were received after solvent evaporation to dryness.
3.6. General Procedure for Esterification of Phosphonic Acids 1 Using Triethyl Orthoacetate Leading to Diethyl Phosphonic Esters 4
Phosphonic acid 1 (1 mmol) and triethyl orthoacetate (5.5 mL, 30 mmol) were stirred at 90 °C (or, for sparingly soluble substrates, at a higher temperature) for 24 h under a reflux condenser. The progress of the reaction was monitored by 31P NMR. The excess of orthoester was evaporated under reduced pressure and the product—dialkyl phosphonate 4—was received in pure form. In some cases, the purity of diester 4 was not satisfactory and additional purification by bulb-to-bulb vacuum distillation with Kugelrohr apparatus or column chromatography was performed.
3.6.1. Ethyl Hydrogen Butylphosphonate (3a)
The synthesis of 3a was conducted according to Section 3.5.
Yield 98%; 1H NMR data in accordance with the previous report [42]; colorless oil; 1H NMR (400 MHz, DMSO-d6) 3.90 (dq, JH-P = 8.0, JH-H = 7.0 Hz, 2H), 1.63–1.50 (m, 2H), 1.50–1.29 (m, 4H), 1.20 (t, JH-H = 7.1 Hz, 3H), 0.86 (t, JH-H = 7.2 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 59.9 (d, JC-P = 6.0 Hz), 25.5 (d, JC-P = 137.6 Hz), 24.5 (d, JC-P = 4.9 Hz), 23.1 (d, JC-P = 16.2 Hz), 16.4 (d, JC-P = 5.9 Hz), 13.6; 31P NMR (162 MHz, DMSO-d6) δ 29.0.
3.6.2. Ethyl Hydrogen Benzylphosphonate (3b)
The synthesis of 3b was conducted according to Section 3.5, with the only difference being that the reaction time was extended to 48 h. Yield: 76%; 1H NMR and 31P NMR spectra were in accordance with previous literature reports [43]; pale yellow oil; 1H NMR (400 MHz, DMSO-d6) δ 7.51–7.15 (m, 5H; broad -OH, 1H), 3.93–3.84 (m, 2H), 3.06 (d, JH-P = 21.4 Hz, 2H), 1.15 (t, JH-H = 7.1 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 133.4 (d, JC-P = 8.9 Hz), 129.7 (d, JC-P = 6.4 Hz), 128.1 (d, JC-P = 2.7 Hz), 126.1 (d, JC-P = 3.4 Hz), 60.5 (d, JC-P = 6.0 Hz), 33.6 (d, JC-P = 133.6 Hz), 16.3 (d, JC-P = 6.0 Hz); 31P NMR (162 MHz, DMSO-d6) δ 23.3.
3.6.3. Ethyl Hydrogen Ethylphosphonate (3c)
The synthesis of 3c was conducted according to Section 3.5.
Yield 89%; NMR data consistent with the previous report [44]; colorless oil; 1H NMR (400 MHz, DMSO-d6) δ 3.95–3.86 (m, 2H), 1.57 (dq, JH-P = 17.8, JH-H = 7.7 Hz, 2H), 1.20 (t, J = 7.1 Hz, 3H), 1.00 (dt, JH-P = 19.2, JH-H = 7.7 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 59.9 (d, JC-P = 6.0 Hz), 18.9 (d, JC-P = 139.3 Hz), 16.4 (d, JC-P = 6.1 Hz), 6.8 (d, JC-P = 6.4 Hz); 31P NMR (162 MHz, DMSO-d6) δ 30.2.
3.6.4. Ethyl Hydrogen Vinylphosphonate (3d)
The synthesis of 3d was conducted according to Section 3.5.
Yield 83%; NMR data in accordance with the previous report [45]; colorless oil. 1H NMR (400 MHz, DMSO-d6) δ 9.05 (broad s, 1H), 6.21–5.91 (m, 3H), 3.88 (dq, JH-P = 8.2, JH-H = 7.1 Hz, 2H), 1.20 (t, JH-H = 7.1 Hz, 4H); 13C NMR (101 MHz, DMSO-d6) δ 132.9, 129.8, 128.1, 60.5 (d, JC-P = 5.2 Hz), 16.32 (d, JC-P = 6.2 Hz); 31P NMR (162 MHz, DMSO-d6) δ 14.0; HRMS (ESI−, m/z) calcd for C4H9O3P; [M − H]−: 135.0211; found: 135.0206.
3.6.5. Ethyl Hydrogen Hexylphosphonate (3e)
The synthesis of 3e was conducted according to Section 3.5, with the only difference being that the reaction time was extended to 48 h.
Yield 93%; colorless oil. 1H NMR (400 MHz, DMSO-d6) δ 7.08 (broad s, 1H), 3.94–3.85 (m, 2H), 1.62–1.51 (m, 2H), 1.51–1.38 (m, 2H), 1.38–1.22 (m, 6H), 1.20 (t, J = 7.1 Hz, 3H), 0.86 (t, J = 6.9 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 59.8 (d, JC-P = 5.9 Hz), 30.8, 29.6 (d, JC-P = 15.7 Hz), 25.8 (d, JC-P = 137.4 Hz), 22.4 (d, JC-P = 4.9 Hz), 21.9, 16.4 (d, JC-P = 6.1 Hz), 13.9; 31P NMR (162 MHz, DMSO-d6) δ 28.9; HRMS (ESI+, m/z) calcd for C8H19O3P; [M + Na]+: 217.0970; found: 217.0966.
3.6.6. Ethyl Hydrogen Dodecylphosphonate (3f)
The synthesis of 3f was conducted according to Section 3.5, with the only difference being that the reaction time was extended to 72 h.
Yield 89%; NMR data in accordance with the previous report [46]; colorless thick oil; 1H NMR (400 MHz, DMSO-d6) δ 3.94–3.84 (m, 2H), 1.62–1.50 (m, 2H), 1.50–1.38 (m, 2H), 1.38–1.22 (m, 18H), 1.20 (t, J = 7.1 Hz, 3H), 0.92–0.78 (t, J = 7.0 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 59.7 (d, JC-P = 6.0 Hz), 31.3, 29.9 (d, JC-P = 15.8 Hz), 28.99, 28.96, 28.94, 28.8, 28.7, 28.6, 25.8 (d, JC-P = 137.2 Hz), 22.6 (d, JC-P = 4.9 Hz), 22.0, 16.4 (d, JC-P = 5.9 Hz), 13.9; 31P NMR (162 MHz, DMSO-d6) δ 28.9.
3.6.7. Ethyl Hydrogen Phenylphosphonate (3g)
The synthesis of 3g was conducted according to Section 3.5.
Yield 56%; NMR data consistent with reported spectra [47]; yellowish oil. 1H NMR (400 MHz, DMSO-d6) δ 7.79–7.62 (m, 2H), 7.63–7.53 (m, 1H), 7.52–7.39 (m, 2H), 7.02 (broad s, 1H), 3.88 (dq, JH-P = 8.0 Hz, JH-H = 7.0 Hz, 2H), 1.17 (t, JH-H = 7.1 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 131.6 (d, JC-P = 3.0 Hz), 131.3 (d, JC-P = 183.0 Hz), 130.9 (d, JC-P = 9.6 Hz), 128.4 (d, JC-P = 14.3 Hz), 60.6 (d, JC-P = 5.2 Hz), 16.2 (d, JC-P = 6.4 Hz); 31P NMR (162 MHz, DMSO-d6) δ 15.0.
3.6.8. Ethyl Hydrogen (4-Methoxyphenyl)phosphonate (3h)
The synthesis of 3h was conducted according to Section 3.5, with the only difference being that the reaction time was extended to 96 h.
Yield 65%; NMR data consistent with reported spectra [48]; pale yellow oil. 1H NMR (400 MHz, DMSO-d6) δ 7.88–7.40 (m, 2H), 7.26–6.59 (m, 2H), 6.15 (broad s, -OH and H2O), 3.97–3.81 (m, 2H), 3.80 (s, 3H), 1.15 (t, J = 7.0 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 161.7 (d, JC-P = 2.8 Hz), 132.9 (d, JC-P = 11.0 Hz), 122.7 (d, JC-P = 190.1 Hz), 113.8 (d, JC-P = 15.3 Hz), 60.4 (d, JC-P = 4.8 Hz), 55.3, 16.2 (d, JC-P = 6.2 Hz); 31P NMR (162 MHz, DMSO-d6) δ 15.5.
3.6.9. Ethyl Hydrogen [(4-Nitrophenyl)methyl]phosphonate (3j)
Due to the poor solubility of the substrate 1j in triethyl orthoacetate and its low conversion, the reaction was carried out according to the modified Section 3.5. The reaction was conducted at 40 °C for 24 h. The excess of orthoester was then evaporated off, and 83% conversion was ascertained by 31P NMR analysis, and the product was isolated via extraction.
Yield: 29%; 1H NMR is in accordance with foregoing literature data [49]; milky white solid; m.p. 144.1–144.7 °C; 1H NMR (400 MHz, DMSO-d6) δ 8.22–7.88 (m, 2H), 7.73–7.20 (m, 2H), 4.02 (broad s, -OH and H2O), 3.91 (dq, JH-P ≈ JH-H = 7.1 Hz, 2H), 3.27 (d, JH-P = 22.0 Hz, 2H), 1.16 (t, JH-H = 7.1 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 146.1 (d, JC-P = 3.4 Hz), 142.4, 130.9 (d, JC-P = 6.2 Hz), 123.1 (d, JC-P = 2.8 Hz), 60.7 (d, JC-P = 6.0 Hz), 33.8 (d, JC-P = 131.2 Hz), 16.3 (d, JC-P = 6.0 Hz).31P NMR (162 MHz, DMSO-d6) δ 21.4.
3.6.10. Ethyl Hydrogen [(4-Bromophenyl)methyl]phosphonate (3k)
The synthesis of 3k was conducted according to Section 3.5.
Yield 78%; NMR spectra are in accordance with literature data [50]; white solid; m.p. 84.0–85.0 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.62–7.33 (m, 2H), 7.33–7.05 (m, 2H), 4.46 (broad s, -OH and H2O), 3.88 (dq, JH-H = 7.0 Hz, JH-P = 6.6 Hz, 2H), 3.05 (d, JH-P = 21.3 Hz, 2H), 1.15 (t, JH-H = 7.0 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 133.0 (d, JC-P = 8.9 Hz), 131.9 (d, JC-P = 6.5 Hz), 130.9 (d, JC-P = 2.8 Hz), 119.3 (d, JC-P = 4.3 Hz), 60.5 (d, JC-P = 5.9 Hz), 33.0 (d, JC-P = 133.1 Hz), 16.3 (d, JC-P = 6.1 Hz); 31P NMR (162 MHz, DMSO-d6) δ 22.4.
3.6.11. Ethyl Hydrogen [(3-Bromophenyl)methyl]phosphonate (3l)
The synthesis of 3l was conducted according to Section 3.5, with the only difference being that the reaction time was extended to 48 h.
Yield 49%; white solid; m.p. 85.1–85.6 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.57–7.45 (m, 1H), 7.45–7.31 (m, 1H), 7.31–7.14 (m, 2H), 6.96 (broad s, OH), 3.90 (dq, JH-P ≈ JH-H = 7.1 Hz, 2H), 3.10 (d, JH-P = 21.4 Hz, 1H), 1.16 (t, JH-H = 7.1 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 136.4 (d, JC-P = 8.7 Hz), 132.3 (d, JC-P = 6.4 Hz), 130.2 (d, JC-P = 2.9 Hz), 129.0 (d, JC-P = 3.3 Hz), 128.8 (d, JC-P = 6.4 Hz), 121.3 (d, JC-P = 3.4 Hz), 60.6 (d, JC-P = 6.1 Hz), 33.0 (d, JC-P = 133.3 Hz), 16.3 (d, JC-P = 5.9 Hz); 31P NMR (162 MHz, DMSO-d6) δ 22.7; HRMS (ESI−, m/z) calcd for C9H11O3PBr; [M-H]−: 276.9637; found: 276.9629.
3.6.12. Diethyl Butylphosphonate (4a)
The synthesis of 4a was conducted according to Section 3.6.
Yield: 98%; 1H NMR data consistent with the previous report [42]; colorless oil; 1H NMR (400 MHz, DMSO-d6) δ 3.97 (m, 4H), 1.74–1.62 (m, 2H), 1.54–1.31 (m, 4H), 1.22 (t, JH-H = 7.1 Hz, 6H), 0.87 (t, JH-H = 7.2 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 60.7 (d, JC-P = 6.3 Hz), 24.2 (d, JC-P = 138.7 Hz), 24.1 (d, JC-P = 5.2 Hz), 22.9 (d, JC-P = 16.3 Hz), 16.2 (d, JC-P = 5.7 Hz), 13.4; 31P NMR (162 MHz, DMSO-d6) δ 32.0.
3.6.13. Diethyl Benzylphosphonate (4b)
The synthesis of 4b was conducted according to Section 3.6.
Yield 98%; 1H and 31P NMR spectra were in accordance with a previous report [43]; transparent oil. 1H NMR (400 MHz, DMSO-d6) δ 7.39–7.14 (m, 5H), 3.94 (dq, JH-P = 7.9, JH-H = 7.0, 4H), 3.21 (d, JH-P = 21.6 Hz, 2H), 1.16 (t, JH-H = 7.0 Hz, 6H); 13C NMR (101 MHz, DMSO-d6) δ 132.3 (d, JC-P = 9.0 Hz), 129.7 (d, JC-P = 6.6 Hz), 128.2 (d, JC-P = 3.0 Hz), 126.4 (d, JC-P = 3.4 Hz), 61.3 (d, JC-P = 6.5 Hz), 32.3 (d, JC-P = 135.1 Hz), 16.1 (d, JC-P = 5.8 Hz); 31P NMR (162 MHz, DMSO-d6) δ 26.5.
3.6.14. Diethyl Ethylphosphonate (4c)
The synthesis of 4c was conducted according to Section 3.6.
Yield 97%; NMR data consistent with the previous report [44]; colorless oil. 1H NMR (400 MHz, DMSO-d6) δ 4.02–3.92 (m, 4H), 1.68 (dq, JH-P = 17.8, JH-H = 7.6 Hz, 2H), 1.22 (t, JH-H = 7.0 Hz, 6H), 1.01 (dt, JH-P = 19.7, JH-H = 7.7 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 60.8 (d, JC-P = 6.3 Hz), 17.8 (d, JC-P = 140.5 Hz), 16.3 (d, JC-P = 5.7 Hz), 6.4 (d, JC-P = 6.6 Hz); 31P NMR (162 MHz, DMSO-d6) δ 33.1.
3.6.15. Diethyl Hexylphosphonate (4e)
The synthesis of 4e was conducted according to Section 3.6.
Yield 91%; NMR data in accordance with the previous report [51]; colorless oil; 1H NMR (400 MHz, DMSO-d6) δ 4.02–3.91 (m, 4H), 1.76–1.57 (m, 2H), 1.52–1.39 (m, 2H), 1.39–1.29 (m, 2H), 1.29–1.18 (m, 10H), 0.86 (t, J = 6.8 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 60.7 (d, JH-P = 6.4 Hz), 30.7, 29.4 (d, JH-P = 15.7 Hz), 24.5 (d, JH-P = 138.4 Hz), 22.0 (d, JH-P = 5.0 Hz), 21.8, 16.2 (d, JH-P = 5.5 Hz), 13.8; 31P NMR (162 MHz, DMSO-d6) δ 31.9.
3.6.16. Diethyl Dodecylphosphonate (4f)
The synthesis of 4e was conducted according to Section 3.6.
Yield 89%; NMR data in accordance with the previous report [48]; transparent oil; 1H NMR (400 MHz, DMSO-d6) δ 4.02–3.90 (m, 4H), 1.75–1.61 (m, 2H), 1.51–1.38 (m, 2H), 1.37–1.18 (m, 18H and t, J = 7.1 Hz, 6H), 0.93–0.73 (m, 3H); 13C NMR (101 MHz, DMSO-d6) δ 60.7 (d, JC-P = 6.3 Hz), 31.3, 29.7 (d, JC-P = 15.7 Hz), 29.0, 28.9, 28.9, 28.8, 28.7, 28.5, 24.5 (d, JC-P = 138.4 Hz), 22.0, 22.0 (d, JC-P = 5.2 Hz), 16.2 (d, JC-P = 5.6 Hz), 13.9; 31P NMR (162 MHz, DMSO-d6) δ 31.9.
3.6.17. Diethyl Phenylphosphonate (4g)
The synthesis of 4g was conducted according to Section 3.6.
Yield 98%; NMR data consistent with a previous report [52]; transparent oil; 1H NMR (400 MHz, DMSO-d6) δ 7.78–7.68 (m, 2H), 7.67–7.59 (m, 1H), 7.58–7.43 (m, 2H), 4.16–3.85 (m, 4H), 1.23 (t, J = 7.0 Hz, 6H); 13C NMR (101 MHz, DMSO-d6) δ 132.4 (d, JC-P = 3.1 Hz), 131.2 (d, JC-P = 9.8 Hz), 128.7 (d, JC-P = 14.7 Hz), 128.5 (d, JC-P = 186.3 Hz), 61.6 (d, JC-P = 5.6 Hz), 16.1 (d, JC-P = 6.0 Hz); 31P NMR (162 MHz, DMSO-d6) δ 17.8.
3.6.18. Diethyl (4-Methoxyphenyl)phosphonate (4h)
The synthesis of 4h was conducted according to Section 3.6.
Yield 99%; NMR data in accordance with the literature [53]; colorless oil. 1H NMR (400 MHz, DMSO-d6) δ 7.68–7.59 (m, 2H), 7.11–7.03 (m, 2H), 4.03–3.91 (m, 4H), 3.82 (s, 3H), 1.21 (t, J = 7.1 Hz, 6H); 13C NMR (101 MHz, DMSO-d6) δ 162.4 (d, JC-P = 3.5 Hz), 133.3 (d, JC-P = 11.2 Hz), 119.6 (d, JC-P = 193.2 Hz), 114.2 (d, JC-P = 15.7 Hz), 61.3 (d, JC-P = 5.4 Hz), 55.3, 16.1 (d, JC-P = 6.1 Hz); 31P NMR (162 MHz, DMSO-d6) δ 18.8.
3.6.19. Diethyl [(4-Hydroxyphenyl)methyl]phosphonate (4i)
The synthesis of 4i was conducted according to Section 3.6.
Yield 79%; NMR spectra were in accordance with a previous report [54]; colorless oil; 1H NMR (400 MHz, DMSO-d6) δ 9.25 (s, -OH), 7.21–6.87 (m, 2H), 6.76–6.54 (m, 2H), 3.91 (dq, JH-P ≈ JH-H ≈ 7.1 Hz, 4H), 3.06 (d, JH-P = 20.9 Hz, 1H), 1.16 (t, JH-H = 7.1 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 156.0 (d, JC-P = 3.2 Hz), 130.6 (d, JC-P = 6.6 Hz), 121.9 (d, JC-P = 9.1 Hz), 115.1 (d, JC-P = 2.8 Hz), 61.2 (d, JC-P = 6.6 Hz), 31.3 (d, JC-P = 136.2 Hz), 16.2 (d, JC-P = 5.7 Hz); 31P NMR (162 MHz, DMSO-d6) δ 27.1.
3.6.20. Diethyl [(4-Nitrophenyl)methyl]phosphonate (4j)
The synthesis of 4j was conducted according to Section 3.6.
Yield 99%; 1H and 13C NMR spectra were in accordance with reported data [55]; pinkish oil; 1H NMR (400 MHz, DMSO-d6) δ 8.31–8.04 (m, 2H), 7.56 (m, 2H), 3.98 (dq, JH-P = 8.4 Hz, JH-H = 7.0 Hz, 4H), 3.45 (d, JH-P = 22.4 Hz, 2H), 1.18 (t, JH-H = 7.1 Hz, 6H); 13C NMR (101 MHz, DMSO-d6) δ 146.3 (d, JC-P = 4.1 Hz), 141.0 (d, JC-P = 9.0 Hz), 131.0 (d, JC-P = 6.4 Hz), 123.3 (d, JC-P = 2.9 Hz), 61.6 (d, JC-P = 6.5 Hz), 32.3 (d, JC-P = 133.3 Hz), 16.1 (d, JC-P = 5.6 Hz); 31P NMR (162 MHz, DMSO-d6) δ 24.9.
3.6.21. Diethyl [(4-Bromophenyl)methyl]phosphonate (4k)
The synthesis of 4k was conducted according to Section 3.6.
Yield 75%; NMR data in accordance with the literature [56]; colorless oil; 1H NMR (400 MHz, DMSO-d6) δ 7.64–7.39 (m, 2H), 7.39–7.12 (m, 2H), 3.95 (dq, JH-P = 8.3, JH-H = 7.1 Hz, 4H), 3.22 (d, JH-P = 21.6 Hz, 2H), 1.17 (t, JH-H = 7.1 Hz, 6H); 13C NMR (101 MHz, DMSO-d6) δ 131.9 (d, JC-P = 9.0 Hz), 131.9 (d, JC-P = 6.6 Hz), 131.1 (d, JC-P = 3.0 Hz), 119.7 (d, JC-P = 4.5 Hz), 61.4 (d, JC-P = 6.5 Hz), 31.6 (d, JC-P = 134.7 Hz), 16.1 (d, JC-P = 5.7 Hz), 31P NMR (162 MHz, DMSO-d6) δ 25.8.
3.6.22. Diethyl [(3-Bromophenyl)methyl]phosphonate (4l)
The synthesis of 4l was conducted according to Section 3.6. The product was purified on a silica gel column (hexanes/ethyl acetate 1:1).
Yield 74%; NMR data in accordance with the literature [57]; colorless liquid. 1H NMR (400 MHz, DMSO-d6) δ 7.54–7.46 (m, 1H), 7.47–7.37 (m, 1H), 7.33–7.18 (m, 2H), 3.96 (dq, JH-P = 8.3 Hz, JH-H = 7.0 Hz, 4H), 3.30 (d, JH-P = 10.3 Hz, 2H), 1.17 (t, JH-H = 7.1 Hz, 6H); 13C NMR (101 MHz, DMSO-d6) δ 135.2 (d, JC-P = 9.0 Hz), 132.4 (d, JC-P = 6.5 Hz), 130.3 (d, JC-P = 2.8 Hz), 129.3 (d, JC-P = 3.4 Hz), 128.8 (d, JC-P = 6.5 Hz), 121.3 (d, JC-P = 3.4 Hz), 61.4 (d, JC-P = 6.4 Hz), 31.6 (d, JC-P = 134.8 Hz), 16.1 (d, JC-P = 5.8 Hz); 31P NMR (162 MHz, DMSO-d6) δ 25.9.
3.6.23. Tetraethyl [1,4-Phenylenebis(methylene)]bis(phosphonate) (4m)
The synthesis of 4m was conducted according to Section 3.6, with the only difference being that the reaction temperature was set to 145 °C.
Yield 99%; NMR data in accordance with the literature report [58]; colorless solid; m.p. 75–77 °C; 1H NMR (400 MHz, DMSO-d6) δ 7.21 (s, 4H), 4.66–3.63 (m, 8H), 3.18 (d, JH-P = 20.2 Hz, 2H), 1.16 (t, JH-H = 7.1 Hz, 12H); 13C{ NMR (101 MHz, DMSO-d6) δ 130.4, 130.4, 130.4, 129.6, 129.6, 129.6, 129.5, 129.5, 61.3, 61.3, 61.2, 31.9 (d, JC-P = 135.8 Hz), 16.16, 16.13, 16.11; 31P NMR (162 MHz, DMSO-d6) δ 26.5.
4. Conclusions
Systematic studies were undertaken to design green and sustainable synthesis of monoethyl and diethyl esters of phosphonic acids. Several alkoxy group donors were utilized for direct esterification of substrates and only for triethyl or trimethyl orthoacetate, diethoxymethyl acetate and tetramethyl orthocarbonate were the expected products obtained. Triethyl orthoacetate (2a) was found to be the best reagent as well as a solvent for esterification reactions. In the present studies, we discovered the effect of temperature on selective esterification of phosphonic acids. For the reactions conducted, at 30 °C the dominant formation of monoethyl esters was observed, while at 90 °C diesters 4 were obtained in good to excellent yields. The generality and applicability of the new method were verified on thirteen aromatic and aliphatic phosphonic acids. For benzylphosphonic acid (1b), mono- and diesterification procedures were performed in 1 and 10 mmol scales without significant loss of selectivity or reaction yield.
Additional experiments were performed to give insight into the reaction mechanism. The first step in the reaction led to the formation of highly unstable intermediate A which readily transforms into monoester 3 at room temperature. At a higher temperature, a subsequent reaction starts giving diesters 4 via an important pyrophosphonate 6 intermediate which was characterised by 31P NMR spectroscopy.
It is important to note that for both reactions, no additional reagents are required and crude products obtained after reaction are sufficiently pure for most applications.
Supplementary Materials
The following are available online. Table S1: Summarized data from the 31P NMR measurements during the course of the esterification at elevated temperature, Table S2: Recorded 31P NMR shifts for substrate phosphonic acids, Table S3: Recorded 31P NMR shifts for obtained mono- and diesters, as well as 1H, 13C and 31P NMR spectra for all isolated products.
Author Contributions
Conception and design of the work, R.O.; methodology, D.T.; synthesis of all compounds, D.T., D.K.; project administration, R.O.; funding acquisition, R.O., formal analysis, A.B., writing—original draft preparation, D.T.; writing—review and editing, R.O., A.B. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by National Science Center, Poland project OPUS 2019/33/B/ST4/01118.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
On request to those interested.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are available from the authors.
References
- Kafarski, P. Phosphonates: Their natural occurrence and physiological role. In Contemporary Topics about Phosphorus in Biology and Materials; IntechOpen: London, UK, 2019. [Google Scholar]
- Jia, Y.; Lu, Z.; Huang, K.; Herzberg, O.; Dunaway-Mariano, D. Insight into the mechanism of phosphoenolpyruvate mutase catalysis derived from site-directed mutagenesis studies of active site residues. Biochemistry 1999, 38, 14165. [Google Scholar] [CrossRef]
- Maier, L. What are the requirements in the glyphosate molecule in order for it to be herbicidally active? Heteroat. Chem. Int. J. Main Group Elem. 2000, 11, 454. [Google Scholar] [CrossRef]
- Krise, J.P.; Stella, V.J. Prodrugs of phosphates, phosphonates, and phosphinates. Adv. Drug Deliv. Rev. 1996, 19, 287. [Google Scholar] [CrossRef]
- Pradere, U.; Garnier-Amblard, E.C.; Coats, S.J.; Amblard, F.; Schinazi, R.F. Synthesis of nucleoside phosphate and phosphonate prodrugs. Chem. Rev. 2014, 114, 9154. [Google Scholar] [CrossRef] [Green Version]
- Engel, R. Handbook of Organophosphorus Chemistry; CRC Press: Boca Raton, FL, USA, 1992. [Google Scholar]
- Bhattacharya, A.K.; Thyagarajan, G. Michaelis-arbuzov rearrangement. Chem. Rev. 1981, 81, 415. [Google Scholar] [CrossRef]
- Rajeshwaran, G.G.; Nandakumar, M.; Sureshbabu, R.; Mohanakrishnan, A.K. Lewis acid-mediated michaelis–arbuzov reaction at room temperature: A facile preparation of arylmethyl/heteroarylmethyl phosphonates. Org. Lett. 2011, 13, 1270. [Google Scholar] [CrossRef]
- Kem, K.M.; Nguyen, N.V.; Cross, D.J. Phase-transfer-catalyzed Michaelis-Becker reaction. J. Org. Chem. 1981, 46, 5188. [Google Scholar] [CrossRef]
- Cohen, R.J.; Fox, D.L.; Eubank, J.F.; Salvatore, R.N. Mild and efficient Cs2CO3-promoted synthesis of phosphonates. Tetrahedron Lett. 2003, 44, 8617. [Google Scholar] [CrossRef]
- Ilia, G.; Petric, M.; Bálint, E.; Keglevich, G. Synthesis of the mixed alkyl esters of phenylphosphonic acid by two variations of the Atherton–Todd protocol. Heteroat. Chem. 2015, 26, 29. [Google Scholar] [CrossRef]
- Wang, G.; Shen, R.; Xu, Q.; Goto, M.; Zhao, Y.; Han, L.-B. Stereospecific coupling of H-phosphinates and secondary phosphine oxides with amines and alcohols: A general method for the preparation of optically active organophosphorus acid derivatives. J. Org. Chem. 2010, 75, 3890. [Google Scholar] [CrossRef]
- Norlin, R.; Juhlin, L.; Lind, P.; Trogen, L. A-haloenamines as reagents for the conversion of phosphorus oxyacids to their halogenated analogues. Synthesis 2005, 2005, 1765. [Google Scholar] [CrossRef]
- Nowlan, C.; Li, Y.; Hermann, J.C.; Evans, T.; Carpenter, J.; Ghanem, E.; Shoichet, B.K.; Raushel, F.M. Resolution of chiral phosphate, phosphonate, and phosphinate esters by an enantioselective enzyme library. J. Am. Chem. Soc. 2006, 128, 15892. [Google Scholar] [CrossRef]
- Van Helden, H.P.; Benschop, H.P.; Wolthuis, O.L. New simulators in the prophylaxis against soman poisoning: Structural specificity for the depot site (s). J. Pharm. Pharmacol. 1984, 36, 305. [Google Scholar] [CrossRef] [PubMed]
- Roussis, V.; Wiemer, D.F. Synthesis of phosphonates from alpha-hydroxy carbonyl compounds and dialkyl phosphorochloridites. J. Org. Chem. 1989, 54, 627. [Google Scholar] [CrossRef]
- Verbelen, B.; Dehaen, W.; Binnemans, K. Selective substitution of POCl3 with organometallic reagents: Synthesis of phosphinates and phosphonates. Synthesis 2018, 50, 2019. [Google Scholar] [CrossRef] [Green Version]
- Cui, L.; Yin, C.; Chen, T.; Quan, G.; Ippolito, J.A.; Liu, B.; Yan, J.; Ding, C.; Hussain, Q.; Umer, M. Remediation of organic halogen-contaminated wetland soils using biochar. Sci. Total Environ. 2019, 696, 134087. [Google Scholar] [CrossRef]
- Poshkus, A.; Herweh, J. The reaction of neutral esters of trivalent phosphorus acids with inorganic acid chlorides. III. The reaction of trialkyl phosphites with thionyl chloride. J. Am. Chem. Soc. 1962, 84, 555. [Google Scholar] [CrossRef]
- Rabinowitz, R. Synthesis of monoesters of phosphonic acids. J. Am. Chem. Soc. 1960, 82, 4564. [Google Scholar] [CrossRef]
- Sathe, M.; Gupta, A.K.; Kaushik, M. An efficient method for the esterification of phosphonic and phosphoric acids using silica chloride. Tetrahedron Lett. 2006, 47, 3107. [Google Scholar] [CrossRef]
- Purohit, A.K.; Pardasani, D.; Tak, V.; Kumar, A.; Jain, R.; Dubey, D. Mild and efficient esterification of alkylphosphonic acids using polymer-bound triphenylphosphine. Tetrahedron Lett. 2012, 53, 3795. [Google Scholar] [CrossRef]
- Kumar, R.; Gupta, A.; Kaushik, M. Surface-mediated synthesis of O-Alkyl 2-methoxyethyl alkylphosphonates under solvent free conditions: Potential marker of nerve agents. Phosphorus Sulfur Silicon 2010, 185, 2064. [Google Scholar] [CrossRef]
- Henyecz, R.; Kiss, A.; Mórocz, V.; Kiss, N.Z.; Keglevich, G. Synthesis of phosphonates from phenylphosphonic acid and its monoesters. Synth. Commun. 2019, 49, 2642. [Google Scholar] [CrossRef] [Green Version]
- Pardasani, D.; Purohit, A.; Kumar, A.; Tak, V.; Goud, D.R.; Gupta, A.K.; Dubey, D.K. Synthesis of O-Alkyl Alkylphosphonates via hydrazine mediated partial dealkylation of phosphonate diesters. ChemistrySelect 2018, 3, 253. [Google Scholar] [CrossRef] [Green Version]
- Timperley, C.M.; Bird, M.; Holden, I.; Black, R.M. Organophosphorus chemistry. Part 1. The synthesis of alkyl methylphosphonic acids. J. Chem. Soc. Perkin Trans. 1 2001, 1, 26–30. [Google Scholar] [CrossRef]
- Crenshaw, M.D.; Cummings, D.B. Preparation, derivatization with trimethylsilyldiazomethane, and GC/MS analysis of a “pool” of alkyl methylphosphonic acids for use as qualitative standards in support of counterterrorism and the chemical weapons convention. Phosphorus Sulfur Silicon 2004, 179, 1009. [Google Scholar] [CrossRef]
- Crenshaw, M.D. Synthesis of alkyl-and arylphosphonic acid monoesters by direct esterification of dibasic phosphonic acids in the presence of an arsonic acid catalyst. Phosphorus Sulfur Silicon 2004, 179, 1509. [Google Scholar] [CrossRef]
- Gupta, A.; Kumar, R.; Gupta, H.; Tak, V.; Dubey, D. DCC—Celite hybrid immobilized solid support as a new, highly efficient reagent for the synthesis of O-alkyl hydrogen alkylphosphonates under solvent-free conditions. Tetrahedron Lett. 2008, 49, 1656. [Google Scholar] [CrossRef]
- Leypold, M.; Wallace, P.W.; Kljajic, M.; Schittmayer, M.; Pletz, J.; Illaszewicz-Trattner, C.; Guebitz, G.M.; Birner-Gruenberger, R.; Breinbauer, R. A robust and simple protocol for the synthesis of arylfluorophosphonates. Tetrahedron Lett. 2015, 56, 5619. [Google Scholar] [CrossRef]
- Barucki, H.; Black, R.M.; Kinnear, K.I.; Holden, I.; Read, R.W.; Timperley, C.M. Solid-phase synthesis of some alkyl hydrogen methylphosphonates. Phosphorus Sulfur Silicon Relat. Elem. 2003, 178, 2279. [Google Scholar] [CrossRef]
- Brodzka, A.; Koszelewski, D.; Zysk, M.; Ostaszewski, R. The mechanistic promiscuity of the enzymatic esterification of chiral carboxylic acids. Catal. Commun. 2018, 106, 82. [Google Scholar] [CrossRef]
- Zysk, M.; Zadlo, A.; Brodzka, A.; Wisniewska, C.; Ostaszewski, R. The unexpected kinetic effect of enzyme mixture: The case of enzymatic esterification. J. Mol. Cat. B Enzym. 2014, 102, 225–229. [Google Scholar] [CrossRef]
- Koszelewski, D.; Brodzka, A.; Zadlo, A.; Paprocki, D.; Trzepizur, D.; Zysk, M.; Ostaszewski, R. Dynamic kinetic resolution of 3-aryl-4-pentenoic acids. ACS Catal. 2016, 6, 3287–3292. [Google Scholar] [CrossRef]
- Brodzka, A.; Koszelewski, D.; Cwiklak, M.; Ostaszewski, R. Studies on the chemoenzymatic synthesis of 3-phenyl-GABA and 4-phenyl-pyrrolid-2-one: The influence of donor of the alkoxy group on enantioselective esterification. Tetrahedron Asymmetry 2013, 24, 427–433. [Google Scholar] [CrossRef]
- Kafarski, P.; Lejczak, B. A facile conversion of aminoalkanephosphonic acids into their diethyl esters. The use of unblocked aminoalkanephosphonic acids in phosphono peptide synthesis. Synthesis 1988, 4, 307–310. [Google Scholar] [CrossRef]
- Lagadic, E.; Bruyneel, F.; Demeyer, N.; Herent, M.-F.; Garcia, Y.; Marchand-Brynaert, J. Phosphonated benzoxazole derivatives: Synthesis and metal-complexing properties. Synlett 2013, 24, 817–822. [Google Scholar]
- Yoshino, T.; Imori, S.; Togo, H. Efficient esterification of carboxylic acids and phosphonic acids with trialkyl orthoacetate in ionic liquid. Tetrahedron 2006, 62, 1309–1317. [Google Scholar] [CrossRef]
- Lagadic, E.; Garcia, Y.; Marchand-Brynaert, J. Selective protection and deprotection of ortho-functionalized arylphosphonates. Synthesis 2012, 44, 93–98. [Google Scholar]
- Fredriksen, K.A.; Amedjkouh, M. Investigation of reactive intermediates and reaction pathways in the coupling agent mediated phos-phonamidation reaction. Eur. J. Org. Chem. 2016, 2016, 474–482. [Google Scholar] [CrossRef]
- Campbell, D.A. The synthesis of phosphonate esters; an extension of the Mitsunobu reaction. J. Org. Chem. 1992, 57, 6331–6335. [Google Scholar] [CrossRef]
- Gobec, S.; Kikelj, D. Synthesis of ethyl 3-(hydroxyphenoxy) benzyl butylphosphonates as potential antigen 85C inhibitors. Tetrahedron 2007, 63, 10698–10708. [Google Scholar]
- Schrader, T. Strong binding of alkylguanidinium ions by molecular tweezers: An artificial selective arginine receptor molecule with a biomimetic recognition pattern. Chem. Eur. J. 1997, 3, 1537–1541. [Google Scholar] [CrossRef]
- Olagnon-Bourgeot, S.; Chastrette, F.; Wilhelm, D. 31P NMR—Structure correlations for phosphonocarboxylic acids and esters. Magn. Reson. Chem. 1995, 33, 971–976. [Google Scholar] [CrossRef]
- Boutevin, B.; Hervaud, Y.; Jeanmaire, T.; Boulahna, A.; Elasri, M. Monodealkylation des esters phosphoniques synthese de monosels et de monoacides phosphoniques. Phosphorus Sulfur Silicon Relat. Elem. 2001, 174, 1–14. [Google Scholar] [CrossRef]
- Meziane, D.; Hardouin, J.; Elias, A.; Guenin, E.; Lecouvey, M. Microwave Michaelis–Becker synthesis of diethyl phosphonates, tetraethyl diphosphonates, and their total or partial dealkylation. Heteroat. Chem. Int. J. Main Group Elem. 2009, 20, 369–377. [Google Scholar] [CrossRef]
- Ryu, T.; Kim, J.; Park, Y.; Kim, S.; Lee, P.H. Rhodium-catalyzed oxidative cyclization of arylphosphonic acid monoethyl esters with alkenes: Efficient synthesis of benzoxaphosphole 1-oxides. Org. Lett. 2013, 15, 3986–3989. [Google Scholar] [CrossRef]
- Seo, J.; Park, Y.; Jeon, I.; Ryu, T.; Park, S.; Lee, P.H. Synthesis of phosphaisocoumarins through rhodium-catalyzed cyclization using alkynes and arylphosphonic acid monoesters. Org. Lett. 2013, 15, 3358–3361. [Google Scholar] [CrossRef]
- Yu, J.; Li, M.; Yu, Y.; Gao, Y.; Liu, J.; Sun, F. Synthetic strategy and performances of a UV-curable poly acryloyl phosphinate flame retardant by carbene polymerization. Phosphorus Sulfur Silicon Relat. Elem. 2015, 190, 1958–1970. [Google Scholar] [CrossRef]
- Fu, Z.; Sun, S.; Yang, A.; Sun, F.; Xu, J. Transition metal-free access to 3,4-dihydro-1,2-oxaphosphinine-2-oxides from phosphonochloridates and chalcones through tandem Michael addition and nucleophilic substitution. Chem. Commun. 2019, 55, 13124–13127. [Google Scholar] [CrossRef] [PubMed]
- Baumann, A.L.; Schwagerus, S.; Broi, K.; Kemnitz-Hassanin, K.; Stieger, C.E.; Trieloff, N.; Schmieder, P.; Hackenberger, C.P. Chemically induced vinylphosphonothiolate electrophiles for thiol–thiol bioconjugations. J. Am. Chem. Soc. 2020, 142, 9544–9552. [Google Scholar] [CrossRef]
- Besse, V.; Le Pluart, L.; Cook, W.D.; Pham, T.N.; Madec, P.J. Synthesis and polymerization kinetics of acrylamide phosphonic acids and esters as new dentine adhesives. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 149–157. [Google Scholar] [CrossRef]
- Hu, G.; Chen, W.; Fu, T.; Peng, Z.; Qiao, H.; Gao, Y.; Zhao, Y. Nickel-catalyzed C–P cross-coupling of arylboronic acids with P (O) H compounds. Org. Lett. 2013, 15, 5362–5365. [Google Scholar] [CrossRef]
- Luo, H.; Liu, H.; Chen, X.; Wang, K.; Luo, X.; Wang, K. Ar–P bond construction by the Pd-catalyzed oxidative cross-coupling of arylsilanes with H-phosphonates via C–Si bond cleavage. Chem. Commun. 2017, 53, 956–958. [Google Scholar] [CrossRef]
- Ma, X.; Xu, Q.; Li, H.; Su, C.; Yu, L.; Zhang, X.; Cao, H.; Han, L.-B. Alcohol-based Michaelis–Arbuzov reaction: An efficient and environmentally-benign method for C–P (O) bond formation. Green Chem. 2018, 20, 3408–3413. [Google Scholar] [CrossRef]
- Al-Riyami, L.; Pineda, M.A.; Rzepecka, J.; Huggan, J.K.; Khalaf, A.I.; Suckling, C.J.; Scott, F.J.; Rodgers, D.T.; Harnett, M.M.; Harnett, W. Designing anti-inflammatory drugs from parasitic worms: A synthetic small molecule analogue of the Acanthocheilonema viteae product ES-62 prevents development of collagen-induced arthritis. J. Med. Chem. 2013, 56, 9982–10002. [Google Scholar] [CrossRef]
- Huang, H.; Denne, J.; Yang, C.H.; Wang, H.; Kang, J.Y. Direct aryloxylation/alkyloxylation of dialkyl phosphonates for the synthesis of mixed phosphonates. Angew. Chem. 2018, 130, 6734–6738. [Google Scholar] [CrossRef]
- Huang, T.; Chen, T.; Han, L.-B. Oxidative dephosphorylation of benzylic phosphonates with dioxygen generating symmetrical trans-stilbenes. J. Org. Chem. 2018, 83, 2959–2965. [Google Scholar] [CrossRef]
Scheme 1.
The general concept of esterification of phosphonic acids.

Figure 1.
Structures of alkoxy group donors used in experiments.

Figure 2.
31P NMR spectrum of substrate 1b stacked with spectra of reaction mixture recorded during the course of the reaction.
Figure 2.
31P NMR spectrum of substrate 1b stacked with spectra of reaction mixture recorded during the course of the reaction.

Figure 3.
Structures of diastereomers 6a and 6b of diethyl dibenzylpyrophosphonate 6 which was identified in the reaction mixture.
Figure 3.
Structures of diastereomers 6a and 6b of diethyl dibenzylpyrophosphonate 6 which was identified in the reaction mixture.

Figure 4.
Changes in concentration of products with time.

Scheme 2.
A plausible mechanism for esterification of phosphonic acid 1b with orthoester 2a.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
The influence of alkoxy group donors on esterification of butylphosphonic (1a) and benzylphosphonic (1b) acids; 1 Section 3.2.
Table 1.
The influence of alkoxy group donors on esterification of butylphosphonic (1a) and benzylphosphonic (1b) acids; 1 Section 3.2.
| Entry | Substrate | Donor | Conversion of 1 [%] 2 | Product Yield [%] 2 | |
|---|---|---|---|---|---|
| 3 | 4 | ||||
| 1 | 1a | 2a | 98 | 84 | 3 |
| 2 | 2b | 25 | 25 | 0 | |
| 3 | 2c | 0 | 0 | 0 | |
| 4 | 2d | 0 | 0 | 0 | |
| 5 | 2e | 0 | 0 | 0 | |
| 6 | 2f | 0 | 0 | 0 | |
| 7 | 2g | 39 | 34 | 0 | |
| 8 | 2h | 24 | 23 | 1 | |
| 9 | 2i | 0 | 0 | 0 | |
| 10 | 2j | 0 | 0 | 0 | |
| 11 | 2k | 0 | 0 | 0 | |
| 12 | 2l | 0 | 0 | 0 | |
| 13 | 2m | 0 | 0 | 0 | |
| 14 | 2n | 0 | 0 | 0 | |
| 15 | 1b | 2a | 89 | 83 | 1 |
| 16 | 2b | 50 | 49 | 1 | |
| 17 | 2c | 0 | 0 | 0 | |
| 18 | 2d | 0 | 0 | 0 | |
| 19 | 2e | 0 | 0 | 0 | |
| 20 | 2f | 0 | 0 | 0 | |
| 21 | 2g | 89 | 83 | 6 | |
| 22 | 2h | 52 | 44 | 8 | |
| 23 | 2i | 0 | 0 | 0 | |
| 24 | 2j | 0 | 0 | 0 | |
| 25 | 2k | 0 | 0 | 0 | |
| 26 | 2l | 0 | 0 | 0 | |
| 27 | 2m | 0 | 0 | 0 | |
| 28 | 2n | 0 | 0 | 0 | |
1 Reaction conditions: phosphonic acid 1a–b (1 equiv., 0.05 mmol), respective donor 2a–n (3 equiv., 0.15 mmol) in 1 mL of MTBE at 30 °C for 24 h. 2 The substrate conversion and product yield determined by 31P NMR.
Table 2.
The influence of a solvent on the substrate conversion and the selectivity of esterification reaction with triethyl orthoacetate (2a); 1 Section 3.3.
Table 2.
The influence of a solvent on the substrate conversion and the selectivity of esterification reaction with triethyl orthoacetate (2a); 1 Section 3.3.
| Entry | Solvent | Substrate 1a Conversion [%] 2 | Product Yield [%] 2 | Substrate 1b Conversion [%] 2 | Product Yield [%] 2 | ||
|---|---|---|---|---|---|---|---|
| 3a | 4a | 3b | 4b | ||||
| 1 | MTBE | 89 | 87 | 2 | 90 | 85 | 5 |
| 2 | THF | 66 | 62 | 1 | 89 | 80 | 1 |
| 3 | DCM | 88 | 78 | 1 | 98 | 84 | 3 |
| 4 | EtOAc | 76 | 75 | 1 | 68 | 66 | 2 |
| 5 | MeCN | 71 | 65 | 2 | 50 | 41 | 2 |
| 6 | Me2CO | 15 | 10 | 0 | 3 | 2 | 1 |
| 7 | PhMe | 76 | 74 | 2 | 89 | 81 | 3 |
| 8 | Neat 3 | 95 | 89 | 5 | 95 | 88 | 7 |
| 9 | DMSO | 0 | 0 | 0 | 0 | 0 | 0 |
| 10 | DMF | 0 | 0 | 0 | 0 | 0 | 0 |
1 Reaction conditions: phosphonic acid 1a–b (1 equiv., 0.05 mmol), triethyl orthoacetate (3 equiv., 0.15 mmol) in 1 mL of solvent at 30 °C for 24 h. 2 The substrate conversion and product yield determined by 31P NMR. 3 One milliliter of triethyl orthoacetate.
Table 3.
Temperature influence on the selectivity of esterification of butylphosphonic acid (1a) with triethyl orthoacetate (2a); 1 Section 3.4.
Table 3.
Temperature influence on the selectivity of esterification of butylphosphonic acid (1a) with triethyl orthoacetate (2a); 1 Section 3.4.
| Entry | Temperature [°C] | Substrate 1a Conversion [%] 2 | Product Yield [%] 2 | |
|---|---|---|---|---|
| 3a | 4a | |||
| 1 | 30 | 97 | 92 | 5 |
| 2 | 40 | >99 | 94 | 6 |
| 3 | 50 | 87 | 13 | |
| 4 | 60 | 73 | 27 | |
| 5 | 70 | 29 | 66 | |
| 6 | 80 | 21 | 79 | |
| 7 | 90 | 1 | 96 | |
| 8 | 100 | 0 | 95 | |
1 Reaction conditions: butylphosphonic acid 1a (1 equiv., 0.1 mmol), triethyl orthoacetate (1 mL). 2 The substrate conversion and product yield determined by 31P NMR.
Table 4.
The yields of mono- (3) and diesters (4) obtained from respective phosphonic acids (1) with triethyl orthoacetate 2a as ethoxy group donor and solvent; 1 Section 3.5 and Section 3.6.
Table 4.
The yields of mono- (3) and diesters (4) obtained from respective phosphonic acids (1) with triethyl orthoacetate 2a as ethoxy group donor and solvent; 1 Section 3.5 and Section 3.6.
| Entry | R | Product | Conditions | Yield [%] 2 | 31P NMR [ppm] |
|---|---|---|---|---|---|
| 1 | n-Butyl | 3a | 30 °C, 24 h | 98 | 29.1 |
| 2 | 4a | 90 °C, 24 h | 98 | 32.0 | |
| 3 | Benzyl | 3b | 30 °C, 48 h | 76 | 23.3 |
| 4 | 4b | 90 °C, 24 h | 98 | 26.5 | |
| 5 | Ethyl | 3c | 30 °C, 96 h | 89 | 30.2 |
| 6 | 4c | 90 °C, 24 h | 97 | 33.1 | |
| 7 | Vinyl | 3d | 25 °C, 24 h | 83 | 14.0 |
| 8 3 | 4d | 90 °C, 24 h | <1 | 16.9 | |
| 9 | n-Hexyl | 3e | 30 °C, 48 h | 93 | 28.9 |
| 10 | 4e | 90 °C, 24 h | 91 | 31.9 | |
| 11 | n-Dodecyl | 3f | 30 °C, 72 h | 89 | 28.9 |
| 12 | 4f | 90 °C, 24 h | 89 | 31.9 | |
| 13 | Phenyl | 3g | 30 °C, 24 h | 56 | 15.0 |
| 14 | 4g | 90 °C, 24 h | 98 | 17.8 | |
| 15 | 4-methoxyphenyl | 3h | 30 °C, 96 h | 65 | 15.5 |
| 16 | 4h | 90 °C, 24 h | 99 | 18.8 | |
| 17 4 | 4-hydroxybenzyl | 3i | 30 °C, 96 h | <1 | - |
| 18 | 4i | 90 °C, 24 h | 79 | 27.1 | |
| 19 | 4-nitrobenzyl | 3j | 40 °C, 24 h | 29 | 21.4 |
| 20 | 4j | 90 °C, 24 h | 99 | 24.9 | |
| 21 | 4-bromobenzyl | 3k | 30 °C, 24 h | 78 | 22.4 |
| 22 | 4k | 90 °C, 24 h | 75 | 25.8 | |
| 23 | 3-bromobenzyl | 3l | 30 °C, 48 h | 49 | 22.7 |
| 24 | 4l | 90 °C, 24 h | 74 | 25.9 | |
| 25 4 | p-xylylene | 3m | 30 °C, 48 h | <1 | - |
| 26 | 4m | 145 °C, 24 h | 99 | 26.5 |
1 Monoesterification and diesterification were conducted according to Section 3.5 and Section 3.6, respectively. Reactions were monitored by 31P NMR and terminated after complete conversion of substrate. 2 The yield of isolated product. 3 The substrate readily polymerized at elevated temperatures. 4 No reaction, due to insolubility of substrate.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Trzepizur, D.; Brodzka, A.; Koszelewski, D.; Ostaszewski, R. Selective Esterification of Phosphonic Acids. Molecules 2021, 26, 5637. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26185637
AMA Style
Trzepizur D, Brodzka A, Koszelewski D, Ostaszewski R. Selective Esterification of Phosphonic Acids. Molecules. 2021; 26(18):5637. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26185637
Chicago/Turabian StyleTrzepizur, Damian, Anna Brodzka, Dominik Koszelewski, and Ryszard Ostaszewski. 2021. "Selective Esterification of Phosphonic Acids" Molecules 26, no. 18: 5637. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26185637