
Antimalarial Inhibitors Targeting Epigenetics or Mitochondria in Plasmodium falciparum: Recent Survey upon Synthesis and Biological Evaluation of Potential Drugs against Malaria



Abstract
:1. Introduction
2. Epigenetics: A New Antimalarial Biological Target
2.1. DNMT Antimalarial Inhibitors
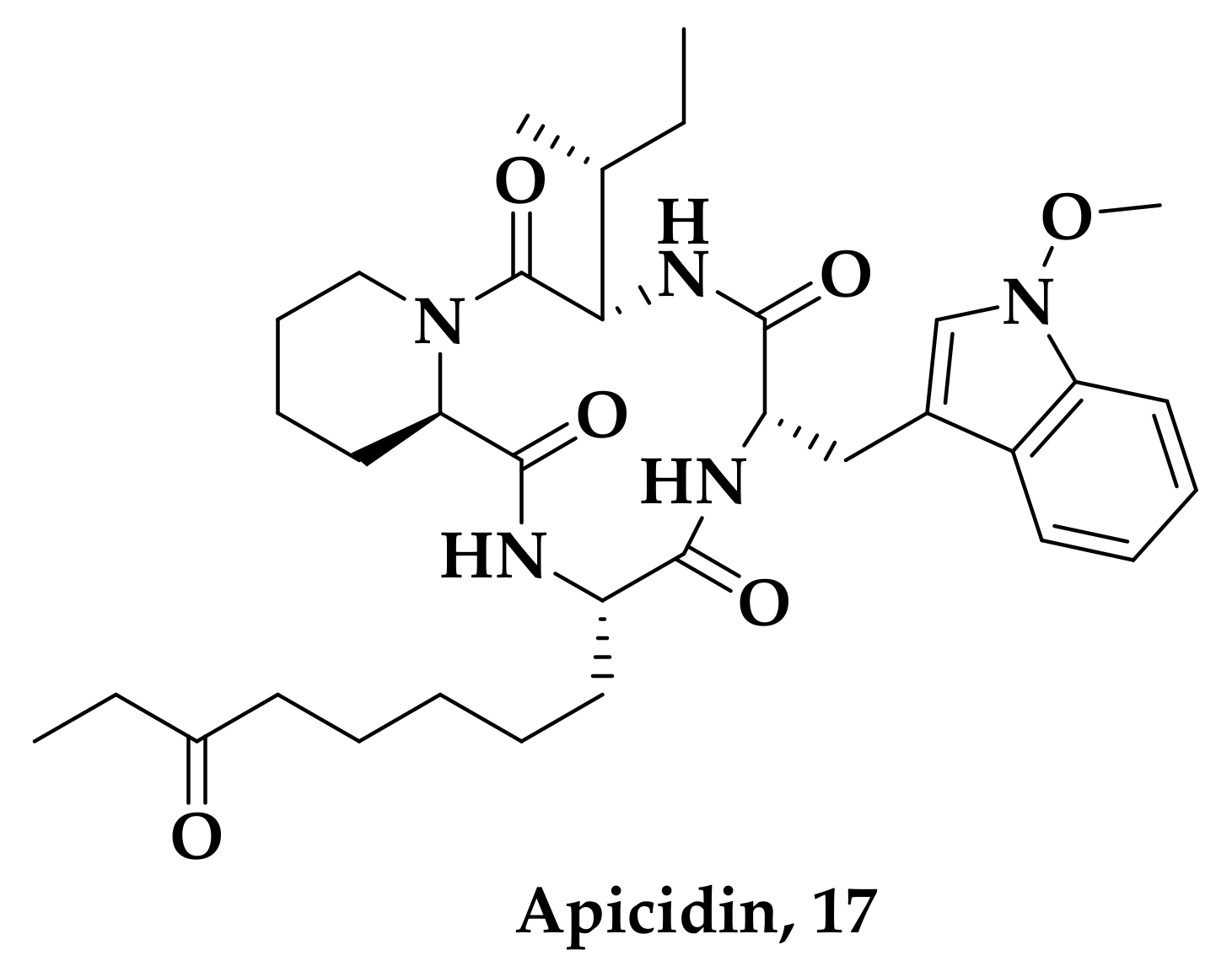
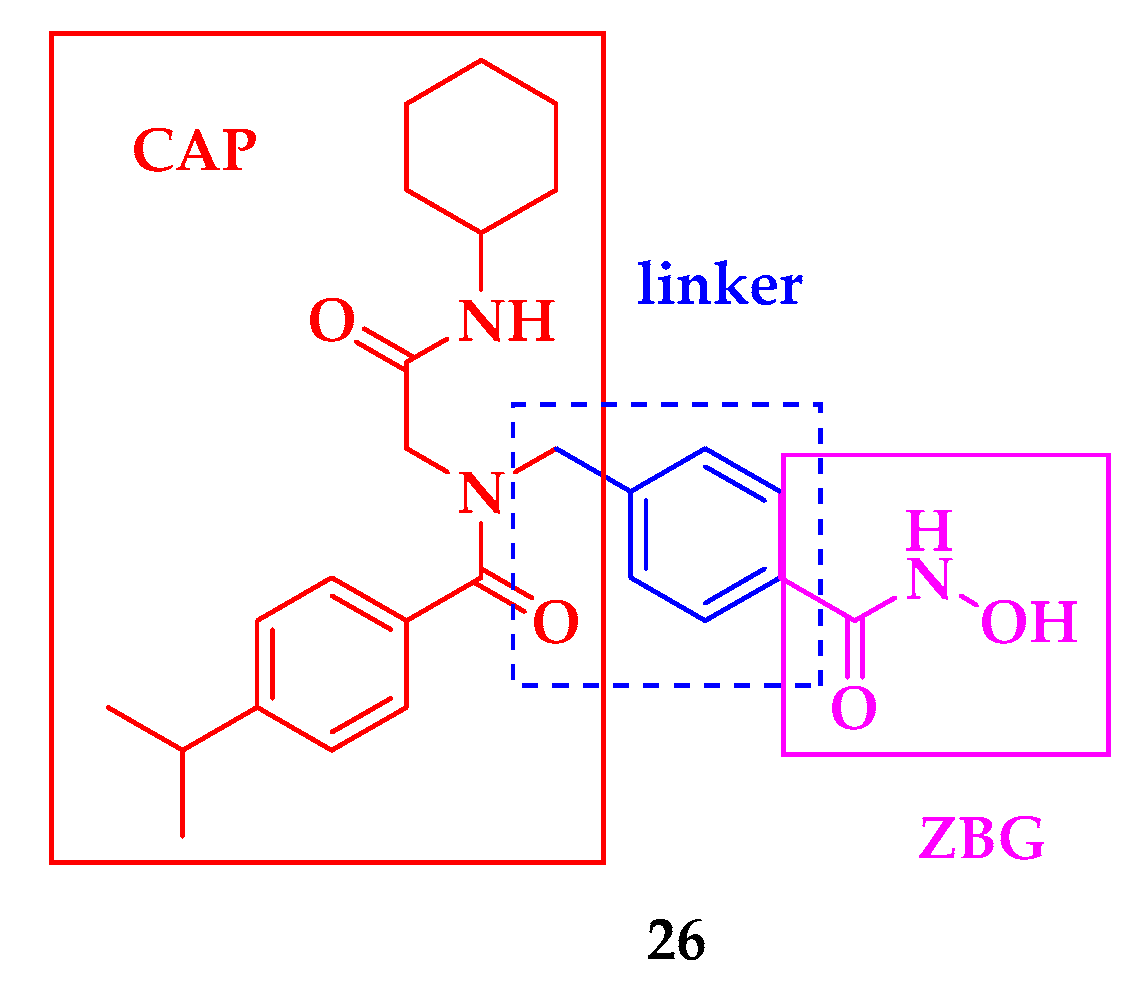
2.2. Histone Deacetylase (HDAC) Inhibition
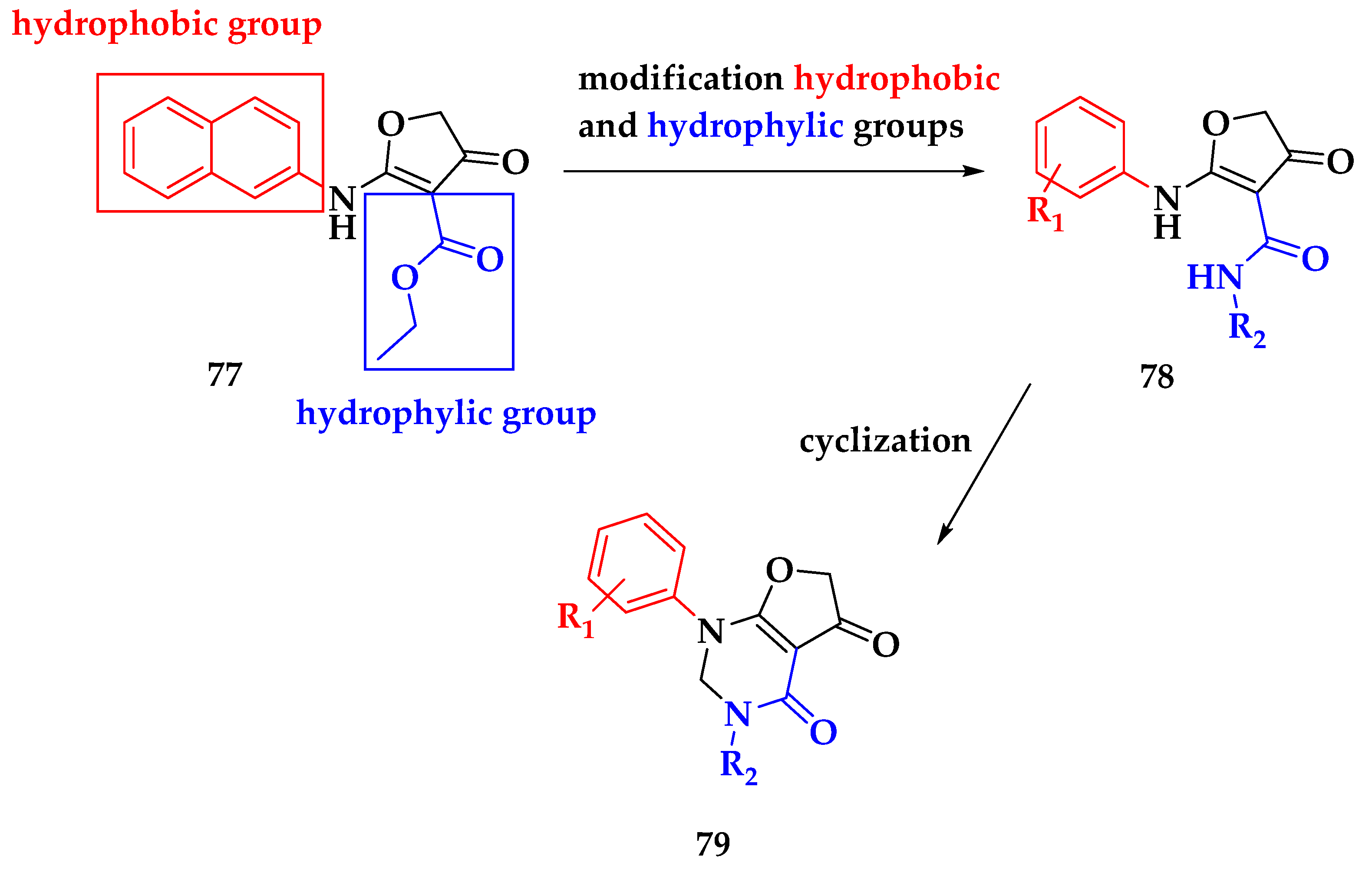
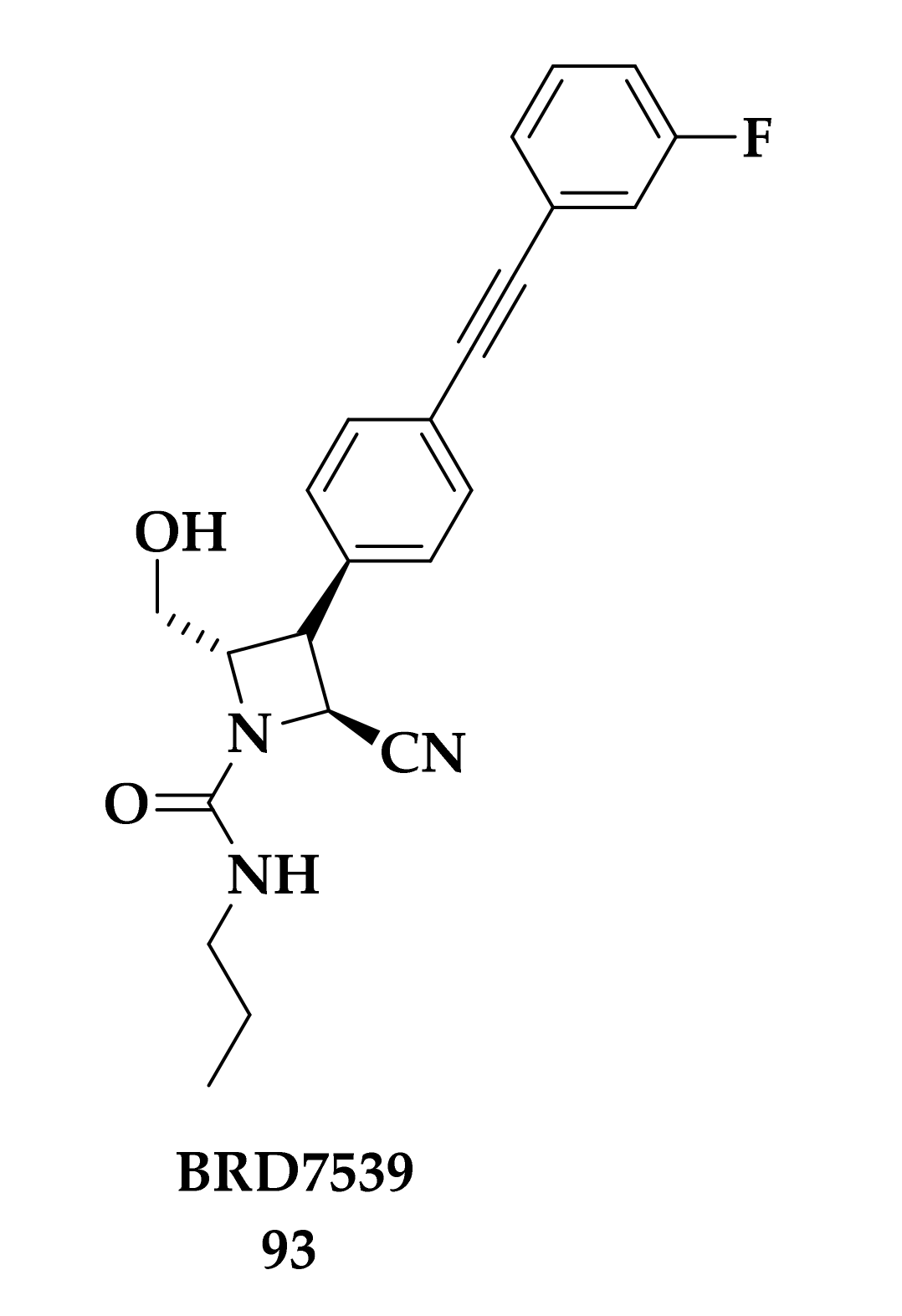
3. Malaria Focused Mitochondrial Targets
3.1. Atovaquone: A bc1 Inhibitor
3.2. bc1 and/or DHODH Antimalarial Inhibitors
3.3. Developing bc1 Antimalarial Inhibitors
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. World Malaria Report 2020; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Centers for Disease Control and Prevention. Available online: https://www.cdc.gov/ (accessed on 3 June 2021).
- Ledford, H. Malaria vaccine shows promise—Now come tougher trials. Nature 2021, 593, 17. [Google Scholar] [CrossRef]
- World Health Organization. Guidelines for the treatment of Malaria, 3rd ed.; World Health Organization: Geneva, Switzerland, 2015. [Google Scholar]
- Blasco, B.; Leroy, D.; Fidock, D.A. Antimalarial drug resistance: Linking Plasmodium falciparum parasite biology to the clinic. Nat. Med. 2017, 23, 917–928. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Status Report on Artemisinin Resistance and ACT Efficacy; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Cortés, A.; Crowley, V.M.; Vaquero, A.; Voss, T.S.; Rall, G.F. A View on the Role of Epigenetics in the Biology of Malaria Parasites. PLoS Pathog. 2012, 8, e1002943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krungkrai, J. The multiple roles of the mitochondrion of the malarial parasite. Parasitology 2004, 129, 511–524. [Google Scholar] [CrossRef]
- Vaidya, A.B.; Mather, M.W. Mitochondrial Evolution and Functions in Malaria Parasites. Annu. Rev. Microbiol. 2009, 63, 249–267. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.P.; Chin, W.H.; Zhu, L.; Mok, S.; Luah, Y.H.; Lim, E.H.; Bozdech, Z. Dynamic Epigenetic Regulation of Gene Expression during the Life Cycle of Malaria Parasite Plasmodium falciparum. PLoS Pathog. 2013, 9, e1003170. [Google Scholar] [CrossRef] [PubMed]
- Malmquist, N.A.; Sundriyal, S.; Caron, J.; Chen, P.; Witkowski, B.; Menard, D.; Suwanarusk, R.; Renia, L.; Nosten, F.; Jiménez-Díaz, M.B.; et al. Histone Methyltransferase Inhibitors Are Orally Bioavailable, Fast-Acting Molecules with Activity against Different Species Causing Malaria in Humans. Antimicrob. Agents Chemother. 2015, 59, 950–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponts, N.; Fu, L.; Harris, E.Y.; Zhang, J.; Chung, D.W.D.; Cervantes, M.C.; Prudhomme, J.; Atanasova-Penichon, V.; Zehraoui, E.; Bunnik, E.M.; et al. Genome-wide mapping of DNA methylation in the human malaria parasite Plasmodium falciparum. Cell Host Microbe. 2013, 14, 696–706. [Google Scholar] [CrossRef] [Green Version]
- Hammam, E.; Ananda, G.; Sinha, A.; Scheidig-Benatar, C.; Bohec, M.; Preiser, P.R.; Dedon, P.C.; Scherf, A.; Vembar, S.S. Discovery of a new predominant cytosine DNA modification that is linked to gene expression in malaria parasites. Nucleic Acids Res. 2020, 48, 184–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coetzee, N.; von Grüning, H.; Opperman, D.; van der Watt, M.; Reader, J.; Birkholtz, L.R. Epigenetic inhibitors target multiple stages of Plasmodium falciparum parasites. Sci. Rep. 2020, 10, 2355. [Google Scholar] [CrossRef] [Green Version]
- Ngwa, C.J.; Gross, M.R.; Musabyimana, J.P.; Pradel, G.; Deitsch, K.W. The Role of The Histone Methyltransferase PfSET10 in Antigenic Variation by Malaria Parasites: A Cautionary Tale. mSphere 2021, 6, e01217-20. [Google Scholar] [CrossRef]
- Chan, A.; Dziedziech, A.; Kirkman, L.A.; Deitsch, K.W.; Ankarklev, J. A histone methyltransferase inhibitor can reverse epigenetically acquired drug resistance in the malaria parasite Plasmodium falciparum. Antimicrob. Agents Chemother. 2020, 64, e02021-19. [Google Scholar] [CrossRef]
- Fioravanti, R.; Mautone, N.; Rovere, A.; Rotili, D.; Mai, A. Targeting histone acetylation/deacetylation in parasites: An update (2017–2020). Curr. Opin. Chem. Biol. 2020, 57, 65–74. [Google Scholar] [CrossRef]
- Bouchut, A.; Rotili, D.; Pierrot, C.; Valente, S.; Lafitte, S.; Schultz, J.; Hoglund, U.; Mazzone, R.; Lucidi, A.; Fabrizi, G.; et al. Identification of novel quinazoline derivatives as potent antiplasmodial agents. Eur. J. Med. Chem. 2019, 161, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Halby, L.; Menon, Y.; Rilova, E.; Pechalrieu, D.; Masson, V.; Faux, C.; Bouhlel, M.A.; David-Cordonnier, M.H.; Novosad, N.; Aussagues, Y.; et al. Rational Design of Bisubstrate-Type Analogs as Inhibitors of DNA Methyltransferases in Cancer Cells. J. Med. Chem. 2017, 60, 4665–4679. [Google Scholar] [CrossRef] [PubMed]
- Nardella, F.; Halby, L.; Hammam, E.; Erdmann, D.; Cadet-Daniel, V.; Peronet, R.; Meńard, D.; Witkowski, B.; Mecheri, S.; Scherf, A.; et al. DNA Methylation Bisubstrate Inhibitors Are Fast-Acting Drugs Active against Artemisinin-Resistant Plasmodium falciparum Parasites. ACS Cent. Sci. 2020, 6, 16–21. [Google Scholar] [CrossRef] [Green Version]
- Buchini, S.; Leumann, C. 2-O’-Aminoethyl Oligoribonucleotides Containing Novel Base Analogs: Synthesis and Triple-Helix Formation at Pyrimidine/Purine Inversion Sites. Eur. J. Org. Chem. 2006, 3152–3168. [Google Scholar] [CrossRef]
- Philips, D.M. The presence of acetyl groups of histones. Biochem. J. 1963, 87, 258–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. Acetylation and Methylation of Histones and Their Possible Role in the Regulation of RNA Synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef] [Green Version]
- Benedetti, R.; Conte, M.; Altucci, L. Targeting histone deacetylases in diseases: Where are we? Antioxid. Redox. Signal. 2015, 23, 99–126. [Google Scholar] [CrossRef]
- Mann, B.S.; Johnson, J.R.; Cohen, M.H.; Justice, R.; Pazdur, R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist 2007, 12, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Moore, D. Panobinostat (Farydak): A novel option for the treatment of relapsed or relapsed and refractory multiple myeloma. Pharm. Ther. 2016, 41, 296–300. [Google Scholar]
- Lee, H.Z.; Kwitkowski, V.E.; Del Valle, P.L.; Ricci, M.S.; Saber, H.; Habtemariam, B.A.; Bullock, J.; Bloomquist, E.; Li Shen, Y.; Chen, X.H.; et al. FDA approval: Belinostat for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma. Clin. Cancer. Res. 2015, 21, 2666–2670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbarotta, L.; Hurley, K. Romidepsin for the treatment of peripheral T-cell lymphoma. J. Adv. Pract. Oncol. 2015, 6, 22–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganesan, A.; Arimondo, P.B.; Rots, M.G.; Jeronimo, C.; Berdasco, M. The Timeline of Epigenetic Drug Discovery: From Reality to Dreams. Clin. Epigenet. 2019, 11, 174–190. [Google Scholar] [CrossRef] [Green Version]
- Morales Torres, C.; Wu, M.Y.; Hobor, S.; Wainwright, E.N.; Martin, M.J.; Patel, H.; Grey, W.; Grönroos, E.; Howell, S.; Carvalho, J.; et al. Selective inhibition of cancer cell self-renewal through a Quisinostat-histone H1.0 axis. Nat. Commun. 2020, 11, 1792–1807. [Google Scholar] [CrossRef] [Green Version]
- Engel, J.A.; Jones, A.J.; Avery, V.M.; Sumanadasa, S.D.M.; Ng, S.S.; Fairlie, D.P.; Adams, T.S.; Skinner-Adams, K.T. Profiling the anti-protozoal activity of anti-cancer HDAC inhibitors against Plasmodium and Trypanosoma parasites. Int. J. Parasitol. Drug. Resist. 2015, 5, 117–126. [Google Scholar] [CrossRef] [Green Version]
- Andrews, K.T.; Gupta, A.P.; Tran, T.N.; Fairlie, D.P.; Gobert, G.N.; Bozdhek, Z. Comparative gene expression profiling of P. falciparum malaria parasites exposed to three different histone deacetylase inhibitors. PLoS ONE 2012, 7, e31847. [Google Scholar] [CrossRef] [Green Version]
- Andrews, K.T.; Tran, T.N.; Wheatley, N.C.; Fairlie, D.P. Targeting Histone Deacetylase Inhibitors for Anti-Malarial Therapy. Curr. Top. Med. Chem. 2009, 9, 292–308. [Google Scholar] [CrossRef]
- Darkin-Rattray, S.J.; Gurnett, A.M.; Myers, R.W.; Dulski, P.M.; Crumley, T.M.; Allocco, J.J.; Cannova, C.; Meinke, P.T.; Colletti, S.L.; Bednarek, M.A.; et al. Apicidin: A Novel Antiprotozoal Agent that Inhibits Parasite Histone Deacetylase. Proc. Natl. Acad. Sci. USA 1996, 93, 13143–13147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, K.; Tran, T.N.; Fairlie, D.P. Towards Histone Deacetylase Inhibitors as New Antimalarial Drugs. Curr. Pharm. Des. 2012, 18, 3467–3479. [Google Scholar] [CrossRef] [PubMed]
- Ontoria, J.M.; Paonessa, G.; Ponzi, S.; Ferrigno, F.; Nizi, E.; Biancofiore, I.; Malancona, S.; Graziani, R.; Roberts, D.; Willis, P.; et al. Discovery of a Selective Series of Inhibitors of Plasmodium falciparum HDACs. ACS Med. Chem. Lett. 2016, 7, 454–459. [Google Scholar] [CrossRef] [PubMed]
- Atenni, B.; Ferrigno, F.; Jones, P.; Ingenito, R.; Kinzel, O.; Llauger-Bufi, L.; Ontoria, J.M.; Pescatore, G.; Rowley, M.; Scarpelli, R.; et al. Heterocycles Derivatives as Histone Deacetylase (HDAC) Inhibitors. WO/2006/061638, 15 June 2006. [Google Scholar]
- Deziel, R.; Rahil, J.; Wahhab, A.; Allan, M.; Nguyen, N. Sirtuin Inhibitors. WO/2009/026701, 3 March 2009. [Google Scholar]
- Mackwitz, M.K.W.; Hesping, E.; Antonova-Koch, Y.; Diedrich, D.; Woldearegai, T.G.; Skinner-Adams, T.; Clarke, M.; Schöler, A.; Limbach, L.; Kurz, T.; et al. Structure-Activity and Structure-Toxicity Relationships of Peptoid-Based Histone Deacetylase Inhibitors with Dual-Stage Antiplasmodial Activity. Chem. Med. Chem. 2019, 14, 912–926. [Google Scholar] [CrossRef]
- Andrews, K.T.; Tran, T.N.; Lucke, A.J.; Kahnberg, P.; Le, G.T.; Boyle, G.M.; Gardiner, D.L.; Skinner-Adams, T.S.; Fairlie, D.P. Potent antimalarial activity of histone deacetylase inhibitor analogs. Antimicrob. Agents Chemother. 2008, 52, 1454–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chua, M.J.; Arnold, M.S.J.; Xu, W.; Lancelot, J.; Lamotte, S.; Späth, G.F.; Prina, E.; Pierce, R.J.; Fairlie, D.P.; Skinner-Adams, T.S.; et al. Effect of clinically approved HDAC inhibitors on Plasmodium, Leishmania and Schistosoma parasite growth. Int. J. Parasitol. Drugs Drug Resist. 2017, 7, 42–50. [Google Scholar] [CrossRef]
- Melesina, J.; Robaa, D.; Pierce, R.J.; Romier, C.; Sippl, W. Homology modeling of parasite histone deacetylases to guide the structure-based design of selective inhibitors. J. Mol. Graphics Modell. 2015, 62, 342–361. [Google Scholar] [CrossRef]
- Hailu, G.S.; Robaa, D.; Forgione, M.; Sippl, W.; Rotili, D.; Mai, A. Lysine Deacetylase Inhibitors in Parasites: Past, Present, and Future Perspectives. J. Med. Chem. 2017, 60, 4780–4804. [Google Scholar] [CrossRef]
- Diedrich, D.; Stenzel, K.; Hesping, E.; Antonova-Koch, Y.; Gebru, T.; Duffy, S.; Fisher, G.; Schçler, A.; Meister, S.; Kurz, T.; et al. One-pot, multi-component synthesis and structure-activity relationships of peptoid-based histone deacetylase (HDAC) inhibitors targeting malaria parasites. Eur. J. Med. Chem. 2018, 158, 801–813. [Google Scholar] [CrossRef]
- Li, R.; Ling, D.; Tang, T.; Huang, Z.; Wang, M.; Ding, Y.; Liu, T.; Wei, H.; Xu, W.; Mao, F.; et al. Discovery of Novel Plasmodium falciparum HDAC1 Inhibitors with Dual-Stage Antimalarial Potency and Improved Safety Based on the Clinical Anticancer Drug Candidate Quisinostat. J. Med. Chem. 2021, 64, 2254–2271. [Google Scholar] [CrossRef]
- Jiang, L.; Haung, Z. Novel High-Efficiency Antimalarial Drug, Quisinostat. WO/2017/143964, 31 August 2017. [Google Scholar]
- Zheng, Y.; Tice, C.M.; Singh, S.B. The Use of Spirocyclic Scaffolds in Drug Discovery. Bioorg. Med. Chem. Lett. 2014, 24, 3673–3682. [Google Scholar] [CrossRef] [Green Version]
- Dickens, J.W.J.; Houpis, I.N.; Lang, Y.L.; Leys, C.; Stokbroekx, S.C.M.; Weerts, J.E.E. Mono-Hydrochloric Salts of an Inhibitor of Histone Deacetylase. WO2008138918A1, 20 November 2008. [Google Scholar]
- Nardella, F.; Halby, L.; Dobrescu, I.; Viluma, J.; Bon, J.; Claes, A.; Cadet-Daniel, V.; Tafit, A.; Roesch, C.; Hammam, E.; et al. Procainamide–SAHA Fused Inhibitors of hHDAC6 Tackle Multidrug-Resistant Malaria Parasites. J. Med. Chem. 2021, 64, 10403–10417. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.H.; Yegnasubramanian, S.; Lin, X.; Nelson, W.G. Procainamide Is a Specific Inhibitor of DNA Methyltransferase 1. J. Biol. Chem. 2005, 280, 40749–40756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Dhar, S.K.; Subbarao, N. In silico identification of inhibitors against Plasmodium falciparum histone deacetylase 1 (PfHDAC-1). J. Mol. Model. 2018, 24, 232. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Protein structure modeling with MODELLER. Methods Mol. Biol. 2014, 1137, 1–15. [Google Scholar] [CrossRef]
- Singh, A.; Maqbool, M.; Mobashir, M.; Hoda, N. Dihydroorotate dehydrogenase: An inevitable drug target for the development of antimalarials. Eur. J. Med. Chem. 2017, 125, 640–651. [Google Scholar] [CrossRef]
- Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 2018, 12, 865–886. [Google Scholar] [CrossRef] [Green Version]
- Berry, E.A.; Huang, L.S.; Saechao, L.K.; Pon, N.G.; Valkova-Valchanova, M.; Daldal, F. X-ray structure of Rhodobacter capsulatus cytochrome bc1: Comparison with its mitochondrial and chloroplast counterparts. Photosynth. Res. 2004, 81, 251–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunte, C.; Koepke, J.; Lange, C.; Rossmanith, T.; Michel, H. Structure at 2.3 A resolution of the cytochrome bc1 complex from the yeast Saccharomyces cerevisiae co-crystallized with an antibody Fv fragment. Structure 2000, 8, 669–684. [Google Scholar] [CrossRef] [Green Version]
- Xia, D.; Yu, C.A.; Kim, H.; Xia, J.Z.; Kachurin, A.M.; Zhang, L.; Yu, L.; Deisenhofer, J. Crystal Structure of the Cytochrome bc1 Complex from Bovine Heart Mitochondria. Science 1997, 277, 60–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Huang, L.; Shulmeister, V.M.; Chi, Y.I.; Kim, K.K.; Hung, L.W.; Crofts, A.R.; Berry, E.A.; Kim, S.H. Electron transfer by domain movement in cytochrome bc1. Nature 1998, 392, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Trumpower, B.L. Purification of a three-subunit ubiquinol-cytochrome c oxidoreductase complex from Paracoccus denitrificans. J. Biol. Chem. 1986, 261, 12282–12289. [Google Scholar] [CrossRef]
- Mitchell, P. Possible molecular mechanisms of the protonmotive function of cytochrome systems. J. Theor. Biol. 1976, 62, 327–367. [Google Scholar] [CrossRef]
- Erecińska, M.; Chance, B.; Wilson, D.F.; Dutton, P.L. Aerobic Reduction of Cytochrome b566 in Pigeon-Heart Mitochondria. Proc. Natl. Acad. Sci. USA 1972, 69, 50–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.S.; Cobessi, D.; Tung, E.Y.; Berry, E.A. Binding of the Respiratory Chain Inhibitor Antimycin to the Mitochondrial bc1 Complex: A New Crystal Structure Reveals an Altered Intramolecular Hydrogen-bonding Pattern. J. Mol. Biol. 2005, 351, 573–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, G.F.; Wang, F.; Li, H.; Zhu, X.L.; Yang, W.C.; Huang, L.S.; Wu, J.W.; Berry, E.A.; Yang, G.F. Computational Discovery of Picomolar Qo Site Inhibitors of Cytochrome bc1 Complex. J. Am. Chem. Soc. 2012, 134, 11168–11176. [Google Scholar] [CrossRef]
- Solmaz, S.R.N.; Hunte, C. Structure of complex III with bound cytochrome c in reduced state and definition of a minimal core interface for electron transfer. J. Biol. Chem. 2008, 283, 17542–17549. [Google Scholar] [CrossRef] [Green Version]
- Esser, L.; Elberry, M.; Zhou, F.; Yu, C.A.; Yu, L.; Xia, D. Inhibitor-complexed Structures of the Cytochrome bc1 from the Photosynthetic Bacterium Rhodobacter sphaeroides. J. Biol. Chem. 2008, 283, 2846–2857. [Google Scholar] [CrossRef] [Green Version]
- Painter, H.J.; Morrisey, J.M.; Mather, M.W.; Vaidya, A.B. Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature 2007, 446, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Fagan, R.L.; Nelson, M.N.; Pagano, P.M.; Palfey, B.A. Mechanism of Flavin Reduction in Class 2 Dihydroorotate Dehydrogenases. Biochemistry 2006, 45, 14926–14932. [Google Scholar] [CrossRef]
- Phillips, M.A.; Rathod, P.K. Plasmodium dihydroorotate dehydrogenase: A promising target for novel anti-malarial chemotherapy. Infect. Disord. Drug Targets 2010, 10, 226–239. [Google Scholar] [CrossRef] [Green Version]
- Coteron, J.M.; Marco, M.A.; Esquivias, J.; Deng, X.; White, K.L.; White, J.; Koltun, M.; El Mazouni, F.; Kokkonda, S.; Katneni, K.; et al. Structure-Guided Lead Optimization of Triazolopyrimidine-Ring Substituents Identifies Potent Plasmodium falciparum Dihydroorotate Dehydrogenase Inhibitors with Clinical Candidate Potential. J. Med. Chem. 2011, 54, 5540–5561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
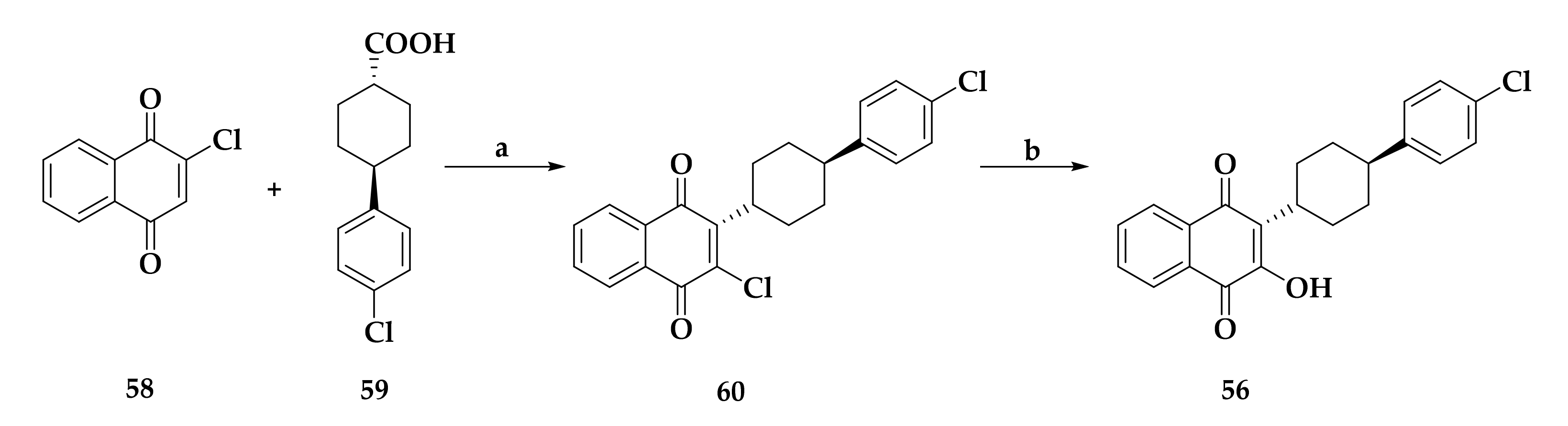
- Fry, M.; Pudney, M. Site of action of the antimalarial hydroxynaphthoquinone, 2-[trans-4-(4′-chlorophenyl)cyclohexyl]-3-hydroxy-1,4-naphthoquinone (566C80). Biochem. Pharmacol. 1992, 43, 1545–1553. [Google Scholar] [CrossRef]
- Shanks, G.D.; Gordon, D.M.; Klotz, F.W.; Aleman, G.M.; Oloo, A.J.; Sadie, D.; Scott, T.R. Efficacy and Safety of Atovaquone/Proguanil as Suppressive Prophylaxis for Plasmodium falciparum Malaria. Clin. Infect. Dis. 1998, 27, 494–499. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, I.K.; Morrisey, J.M.; Darrouzet, E.; Daldal, F.; Vaidya, A.B. Resistance mutations reveal the atovaquone-binding domain of cytochrome b in malaria parasites. Mol. Microbiol. 1999, 33, 704–711. [Google Scholar] [CrossRef]
- Srivastava, I.K.; Vaidya, A.B. A mechanism for the Synergistic Antimalarial Action of Atovaquone and Proguanil. Antimicrob. Agents Chemother. 1999, 43, 1334–1339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birth, D.; Kao, W.C.; Hunte, C. Structural analysis of atovaquone-inhibited cytochrome bc1 complex reveals the molecular basis of antimalarial drug action. Nat. Commun. 2014, 5, 4029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudson, A.T.; Randall, A.W. Naphthoquinone derivatives. U.S. Patent 5053432A, 1 October 1991. [Google Scholar]
- Hudson, A.T.; Latter, V. Medicaments. EP0580185A1, 26 January 1994. [Google Scholar]
- Antonio, N.; Mara, S.; Annibale, S.; Stefan, M. Process for the preparation of trans-2,3-disubstituted naphthoquinones. U.S. Patent 7,842,840 B2. 2010. Chem. Abstr. 2008, 149, 576277. [Google Scholar]
- Saralya, S.S.; Shasikumar, S.H.; Shashipraba, S.; Kanakamajalu, S.; Koottungalamadhom, R.R.; Ananathalakshmi, V.; Govindarajalu, J.; Rao, K.S.; Nagarajan, K. Preparation of naphthoquinone compounds using 2,3-dihalonaphthoquinone. WO 2009122432A2, 8 October 2009. [Google Scholar]
- Williams, D.R.; Clark, M.P. Synthesis of atovaquone. Tetrahedron Lett. 1998, 39, 7629–7632. [Google Scholar] [CrossRef]
- Dwyer, A.N.; Gordon, A.; Urquhart, M. Novel process. WO/2012/080243 A2, 21 June 2012. [Google Scholar]
- Britton, H.; Catterick, D.; Dwyer, A.N.; Gordon, A.H.; Leach, S.G.; McCormick, C.; Mountain, C.E.; Simpson, A.; Stevens, D.R.; Urquhart, M.W.J.; et al. Discovery and Development of an Efficient Process to Atovaquone. Org. Process Res. Dev. 2012, 16, 1607–1617. [Google Scholar] [CrossRef]
- Dike, S.Y.; Singh, D.; Thankachen, B.N.; Sharma, B.; Mathur, P.K.; Kore, S.; Kumar, A. A Single-Pot Synthesis of Atovaquone: An Antiparasitic Drug of Choice. Org. Process Res. Dev. 2014, 18, 618–625. [Google Scholar] [CrossRef]
- Borgati, T.F.; Alves do Nascimento, M.F.; Bernardino, J.F.; Oliveira Martins, L.C.; Gutterres Taranto, A.; Braga de Oliveira, A. Synthesis, SAR, and Docking Studies Disclose 2-Arylfuran-1,4-naphthoquinones as In Vitro Antiplasmodial Hits. J. Trop. Med. 2017, 2017, 7496934. [Google Scholar] [CrossRef] [Green Version]
- Nogueira, F.; Rosário, V.E. Methods for assessment of antimalarial activity in the different phases of the Plasmodium life cycle. Rev. Pan-Amaz. Saude 2010, 3, 109–124. [Google Scholar] [CrossRef]
- Makler, M.T.; Piper, R.C.; Milhous, W.K. Lactate dehydrogenase and the diagnosis of malaria. Parasitol. Today 1998, 9, 376–377. [Google Scholar] [CrossRef]
- Kapadia, G.J.; Azuine, M.A.; Balasubramanian, V.; Sridhar, R. Amino-naphthoquinones—a novel class of compounds with potent antimalarial activity against Plasmodium falciparum. Pharmacol. Res. 2001, 4, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Oramas-Royo, S.; López-Rojas, P.; Amesty, A.; Gutiérrez, D.; Flores, N.; Martín-Rodríguez, P.; Fernández-Pérez, L.; Estévez-Braun, A. Synthesis and Antiplasmodial Activity of 1,2,3-Triazole-Naphthoquinone Conjugates. Molecules 2019, 24, 3917. [Google Scholar] [CrossRef] [Green Version]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Hurt, D.E.; Widom, J.; Clardy, J. Structure of Plasmodium falciparum dihydroorotate dehydrogenase with a bound inhibitor. Acta Cryst. 2006, 62, 312–323. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Matthews, D.; Rathod, P.K.; Phillips, M.A. The X-ray structure of Plasmodium falciparum dihydroorotate dehydrogenase bound to a potent and selective N-phenylbenzamide inhibitor reveals novel binding-site interactions. Acta Cryst. Sect. F Struct. Biol. Commun. 2015, 71, 553–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Li, W.; Diao, Y.; Sun, H.; Li, H.; Zhu, L.; Zhou, H.; Zhao, Z. Synthesis, Design, and Structure–Activity Relationship of the Pyrimidone Derivatives as Novel Selective Inhibitors of Plasmodium falciparum Dihydroorotate Dehydrogenase. Molecules 2018, 23, 1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Zhu, J.; Diao, Y.; Zhou, H.; Ren, X.; Sun, D.; Huang, J.; Han, D.; Zhao, Z.; Zhu, L.; et al. Novel selective and potent inhibitors of malaria parasite dihydroorotate dehydrogenase: Discovery and optimization of dihydrothiophenone derivatives. J. Med. Chem. 2013, 56, 7911–7924. [Google Scholar] [CrossRef]
- Knecht, W.; Henseling, J.; Löffler, M. Kinetics of Inhibition of Human and Rat Dihydroorotate Dehydrogenase by Atovaquone, Lawsone Derivatives, Brequinar Sodium and Polyporic Acid. Chem. Biol. Interact. 2000, 124, 61–76. [Google Scholar] [CrossRef]
- Woody, S.; Tyler, D.; Matthew, P.J.; Richard, A.F.; Ramy, F. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Azeredo, L.F.S.P.; Coutinho, J.P.; Jabor, V.A.P.; Feliciano, P.R.; Nonato, M.R.; Kaiser, C.R.; Menezes, C.M.S.; Hammes, A.S.O.; Caffarena, E.R.; Hoelz, L.V.B.; et al. Evaluation of 7-arylaminopyrazolo[1,5-a]pyrimidines as anti-Plasmodium falciparum, antimalarial, and Pf-dihydroorotate dehydrogenase inhibitors. Eur. J. Med. Chem. 2017, 126, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Boechat, N.; Pinheiro, L.C.S.; Silva, T.S.; Aguiar, A.C.C.; Carvalho, A.S.; Bastos, M.M.; Costa, C.C.P.; Pinheiro, S.; Pinto, A.C.; Mendonça, J.S.; et al. New trifluoromethyl triazolopyrimidines as anti-Plasmodium falciparum agents. Molecules 2012, 17, 8285–8302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senga, K.; Novinson, T.; Wilson, H.R. Synthesis and Antischistosomal activity of certain Pyrazolo[1,5-a]pyrimidines. J. Med. Chem. 1981, 24, 610–613. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Maetani, M.; Kato, N.; Jabor, V.A.P.; Calil, F.A.; Nonato, M.C.; Scherer, C.A.; Schreiber, S.L. Discovery of Antimalarial Azetidine-2-carbonitriles That Inhibit P. falciparum Dihydroorotate Dehydrogenase. ACS Med. Chem. Lett. 2017, 8, 438–442. [Google Scholar] [CrossRef] [Green Version]
- Kato, N.; Comer, E.; Sakata-Kato, T.; Sharma, A.; Sharma, M.; Maetani, M.; Bastien, J.; Brancucci, N.M.; Bittker, J.A.; Corey, V.; et al. Diversity-oriented synthesis yields novel multistage antimalarial inhibitors. Nature 2016, 538, 344–349. [Google Scholar] [CrossRef]
- Dancík, V.; Seiler, K.P.; Young, D.W.; Schreiber, S.L.; Clemons, P.A. Distinct biological network properties between the targets of natural products and disease genes. J. Am. Chem. Soc. 2010, 132, 9259–9261. [Google Scholar] [CrossRef]
- Okada-Junior, C.Y.; Monteiro, G.C.; Campos Aguiar, A.N.; Batista, V.S.; Oliveira de Souza, J.; Souza, G.E.; Bueno, R.V.; Oliva, G.; Nascimento-Juńior, N.M.; Victorio, R.; et al. Phthalimide Derivatives with Bioactivity against Plasmodium falciparum: Synthesis, Evaluation, and Computational Studies Involving bc1 Cytochrome Inhibition. ACS Omega 2018, 3, 9424–9430. [Google Scholar] [CrossRef] [Green Version]
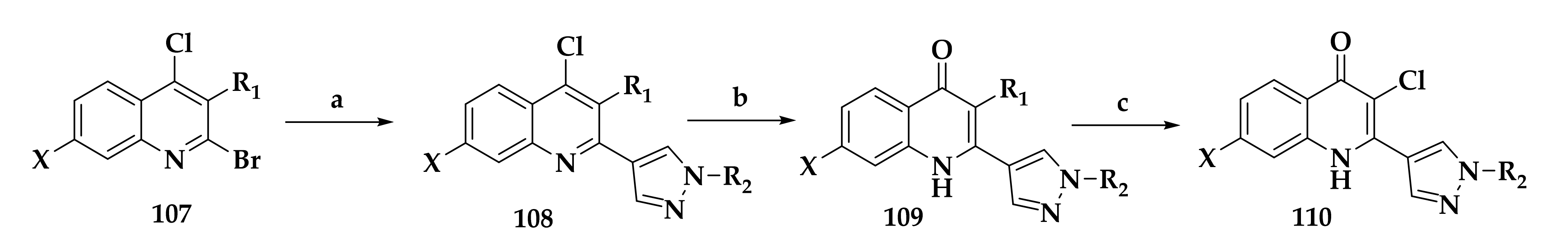
- Hong, W.D.; Leung, S.C.; Amporndanai, K.; Davies, J.; Priestley, R.S.; Nixon, G.L.; Berry, N.G.; Hasnain, S.S.; Antonyuk, S.; Ward, S.A.; et al. Potent Antimalarial 2-Pyrazolyl Quinolone bc1 (Qi) Inhibitors with Improved Drug-like Properties. ACS Med. Chem. Lett. 2018, 9, 1205–1210. [Google Scholar] [CrossRef] [PubMed]
- Pidathala, C.; Amewu, R.; Pacorel, B.; Nixon, G.L.; Gibbons, P.; Hong, W.D.; Leung, S.C.; Berry, N.G.; Sharma, R.; Stocks, P.A.; et al. Identification, Design and Biological Evaluation of Bisaryl Quinolones Targeting Plasmodium falciparum Type II NADH: Quinone Oxidoreductase (PfNDH2). J. Med. Chem. 2012, 55, 1831–1843. [Google Scholar] [CrossRef] [PubMed]
- Biagini, G.A.; Fisher, N.; Berry, N.; Stocks, P.A.; Meunier, B.; Williams, D.P.; Bonar-Law, R.; Bray, P.G.; Owen, A.; O’Neill, P.M.; et al. Acridinediones: Selective and potent inhibitors of the malaria parasite mitochondrial bc(1) complex. Mol. Pharmacol. 2008, 73, 1347–1355. [Google Scholar] [CrossRef] [Green Version]
- Capper, M.J.; O’Neill, P.M.; Fisher, N.; Strange, R.W.; Moss, D.; Ward, S.A.; Berry, N.G.; Lawrenson, A.S.; Hasnain, S.S.; Biagini, G.A.; et al. Antimalarial 4(1H)-pyridones bind to the Qi site of cytochrome bc1. Proc. Natl. Acad. Sci. USA 2015, 112, 755–760. [Google Scholar] [CrossRef] [Green Version]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Gallo Cassarino, T.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, 252–258. [Google Scholar] [CrossRef] [PubMed]

































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | P. falciparum (nM) | HepG2 Cells (nM) |
|---|---|---|
| 2 | 71 ± 23 | 1400 ± 200 |
| 3 | 513 ± 63 | ND |
| 4 | 60 ± 14 | 2500 ± 500 |
| Dihydroartemisin (DHA) | 5 ± 1 | ND |

| Compound | Ar | Pf Growth EC50 (μΜ) | HeLa Class I HDAC IC50 (μΜ) | hHDAC1 IC50 (μΜ) | SI |
|---|---|---|---|---|---|
| 25a |  | 0.27 ± 0.12 | 4.75 ± 1.44 | 0.60 ± 0.15 | 18 |
| 25b |  | 0.10 ± 0.02 | 2.04 ± 0.71 | 0.29 ± 0.04 | 20 |
| 25c |  | 0.10 ± 0.05 | 1.37 ± 0.38 | 0.39 ± 0.02 | 14 |
| 25d |  | 0.45 ± 0.08 | 16.63 ± 3.26 | 1.85 ± 0.31 | 37 |
| 25e |  | 0.5 ± 0.17 | >25 | >2.5 | 54 |
| 19 (Figure 5) | used for comparison | 0.21 ± 0.07 | 0.07 ± 0.02 | 0.002 ± 0.001 | 0.3 |

| Compound | R1 | R2 | Pf3D7 IC50 (μM) |
|---|---|---|---|
| 31c | c-Hex | 4–CH3)2N | 0.095 ± 0.015 |
| 31e | t Bu | 2–CH3 | 0.11 ± 0.080 |
| 31h | Bn | 4–iPr | 0.0052 ± 0.0036 |
| 31s | CH3 | 3,5–CH3 | 0.054 ± 0.0029 |
| 31t | CH3 | 2–CH3 | 0.071 ± 0.015 |
| SAHA (12) | - | - | 0.17 ± 0.035 |
| Chloroquine | - | - | 0.0068 ± 0.0023 |
| Compound | CAP | Erythrocytic IC50 (nM)a | SI | |
|---|---|---|---|---|
| 3D7 | Dd2 | |||
| 36a |  | 2.8 ± 0.2 | 2.5 ± 0.1 | 22 |
| 36b |  | 3.19 ± 0.07 | 1.9 ± 0.1 | 75 |
| 36c |  | 3.0 ± 0.2 | 3.4 ± 0.3 | 132 |
| 36d |  | 3.0 ± 0.5 | 5.6 ± 0.1 | 180 |
| 36e |  | 4.0 ± 0.1 | 4.9 ± 1.2 | 370 |
| 36f |  | 3.22 ± 0.02 | 4.3 ± 0.3 | 235 |
| DHA | - | 2.7 ± 0.2 | 2.68 ± 0.02 | ND |

| Compound | R | Pf Mean IC50 (nM) | hHDAC Extracts IC50 (μM) | PfHDAC Extracts IC50 (μM) |
|---|---|---|---|---|
| 53a |  | 41 ± 7 | 0.06 ± 0.05 | 0.38 ± 0.07 |
| 53b |  | 64 ± 2 | 0.16 ± 0.01 | ND |
| 53c |  | 52 ± 12 | 0.14 ± 0.07 | 0.48 ± 0.07 |
| SAHA | - | 175 ± 33 | 0.18 ± 0.07 | 0.46 ± 0.11 |
| DHA | - | 3 ± 7 | - | - |

| Compound ID | Group | R | Reported Activity (Target) a |
|---|---|---|---|
| CHEMBL152862 | I |  | IC50: 289.2 ± 25.8 nM (A) IC50: 34 nM (B) |
| CHEMBL244454 | I |  | IC50: 78 nM (C) IC50: 13 nM (D) |
| CHEMBL152635 | I |  | IC50: 159 nM (C) IC50: 19 nM (D) |
| CID: 11730425 | I |  | IC50: 34.7 nM (A) |
| CHEMBL3103569 | II |  | IC50: 110 nM (E) IC50: 80 nM (F) |
| CHEMBL211750 | III | - | IC50: 3.3 nM (E) IC50: 1.9 nM (F) |

| Compound ID | Group | R | Docking Score (kcal/mol) | Predicted Binding Affinity (X Score) (kcal/mol) |
|---|---|---|---|---|
| CHEMBL152862 | I | | −10.715 | −8.52 |
| CHEMBL244454 | I | | −10.529 | −8.80 |
| CHEMBL152635 | I | | −10.116 | −8.42 |
| CHEMBL3103569 | II | | −9.723 | −10.52 |
| CHEMBL211750 | III | - | −9.117 | −8.13 |
| CID: 11730425 | I | | -10.315 | −8.64 |
| Vorinostat | - | - | −8.019 | −7.74 |
| Pracinostat | - | - | −7.336 | −8.39 |
| Panobinostat | - | - | −3.897 | −7.69 |
| Belinostat | - | - | −2.432 | −8.43 |

| Compound | R | % Reduction at 25 μg/mL | IC50 (μM) |
|---|---|---|---|
| 67a | −CH2CH2CH3 | 50% | 52.65 ± 3.71 |
| 67b |  | 58% | 39.55 ± 8.98 |
| 66b | −CH3 | 75% | 26.57 ± 3.25 |
| 66a | −CH2CH2CH3 | 86% | 11.65 ± 2.50 |
| 66c |  | 85% | 18.78 ± 0.83 |
| 66d |  | 78% | 21.80 ± 2.64 |
| Chloroquine | - | - | 0.280 ± 0.01 |

| Compound | R | P. falciparum IC50 (μM) | SkBr3 IC50 (μM) |
|---|---|---|---|
| 71a |  | 1.9 ± 0.09 | >10 |
| 71b |  | 1.6 ± 0.1 | >10 |
| 71c |  | 1.2 ± 0.2 | >10 |
| 71d |  | 1.7 ± 0.1 | >10 |
| 71e |  | 1.7 ± 0.09 | >10 |
| 71f |  | 0.8 ± 0.1 | >10 |
| Chloroquine | - | 0.15 ± 0.02 | - |
| Adriamycin | - | - | 0.1 ± 0.06 |
| Compound | Structure | IC50 (μM) PfDHODH | IC50 (μM) hDHODH |
|---|---|---|---|
| 86a |  | 0.070 ± 0.01 | >10 |
| 79a |  | 0.023 ± 0.001 | >10 |
| DSM1 | - | 0.042 ± 0.004 | - |
| Compound | Substituents R1/R2 | Cytotoxicity (MLD50) (μΜ) | IC50 against PfDHODH (μΜ) |
|---|---|---|---|
| 88a | 2-CF3/5-CH3 | 561.4 ± 49.7 | 6 ± 1 |
| 88b | 2-CH3/5-CH3 | 406 ± 216 | 4 ± 1 |
| 88c | 2-CH3/5-CF3 | 2341 ± 105 | 0.16 ± 0.01 |
| 87 | - | 425 | 0.70 ± 0.08 |

| Compound | R1 | R2 | R3 | EC50 (μΜ) |
|---|---|---|---|---|
| BRD7539 |  |  |  | 0.010 |
| 97 |  | | | 0.019 |
| 98 |  | | | 0.010 |
| 99 |  | | | 0.039 |
| 100 |  | | | 0.016 |
| 101 |  | | | 0.015 |

| Compound | R/X | % Inhibition at 10 μΜ | IC50 (μΜ) | SI |
|---|---|---|---|---|
| 102a | R = H, X = OMe | 75 ± 4 | 4.2 (3.2–5.2) | >59 |
| 102b | R = Bz, X = OMe | 65 ± 5 | 6.8 (5.6–8.0) | >36 |
| artesunate | - | - | 0.006 (0.005–0.008) | 50.833 |

| Compound | R1/X | R2 | IC50 W2 (nM) |
|---|---|---|---|
| 116a | R1 = Me, X = H |  | 26 |
| 116b | R1 = Me, X = H |  | 33 |
| 116c | R1 = Cl, X = H | | 14 |
| 116d | R1 = Cl, X = 7-OMe | | 11 |
| 116e | R1 = Me, X = 7-OMe | | 15 |
| Chloroquine | - | - | 12.3 |
| Atovaquone | - | - | 0.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koumpoura, C.L.; Robert, A.; Athanassopoulos, C.M.; Baltas, M. Antimalarial Inhibitors Targeting Epigenetics or Mitochondria in Plasmodium falciparum: Recent Survey upon Synthesis and Biological Evaluation of Potential Drugs against Malaria. Molecules 2021, 26, 5711. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26185711
Koumpoura CL, Robert A, Athanassopoulos CM, Baltas M. Antimalarial Inhibitors Targeting Epigenetics or Mitochondria in Plasmodium falciparum: Recent Survey upon Synthesis and Biological Evaluation of Potential Drugs against Malaria. Molecules. 2021; 26(18):5711. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26185711
Chicago/Turabian StyleKoumpoura, Christina L., Anne Robert, Constantinos M. Athanassopoulos, and Michel Baltas. 2021. "Antimalarial Inhibitors Targeting Epigenetics or Mitochondria in Plasmodium falciparum: Recent Survey upon Synthesis and Biological Evaluation of Potential Drugs against Malaria" Molecules 26, no. 18: 5711. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26185711