1. Introduction
Protein degradation in living cells is indispensable for preserving homeostasis and is a crucial component of the regulatory, immune and stress response systems [
1]. Removing aberrant or denatured proteins prevents the formation of toxic aggregates and allows recycling of amino acids [
2]. On the other hand, specific regulation of protein half-lives can adapt cellular processes in response to the changing environment [
3]. Bacteria have several pathways for specific protein degradation in which major proteolytic complexes are members of the ATPases Associated with diverse cellular Activities (AAA+) family [
4,
5]. A common element of protein degradation pathways is recognition of the target proteins by specific elements known as degrons [
6]. Several proteases might recognize the same degrons [
7,
8,
9] and further specificity can be conferred by the action of specialized adaptor proteins that cooperate with chosen substrate-protease pairs [
10,
11].
Modification of endogenous protein stability became a popular trend in protein research and drug discovery known as targeted protein degradation. Multiple protein degradation techniques were developed in eukaryotic cells including Proteolysis Targeting Chimeras (PROTACs), auxin-inducible degradation (AID), and hydrophobic tagging [
12,
13]. In bacteria, all directed protein degradation methods developed so far are based on the use of degrons [
14]. Knowledge of degron properties, such as the strength of binding to the components of the degradation machinery or the impact on the efficiency of degradation is necessary for their informed use in fusion proteins.
One of the best described and most exploited bacterial degrons is the ssrA tag sequence [
14,
15]. In the bacterial quality control pathway, ssrA tag is added in a process called trans-translation. During trans-translation, tmRNA (transfer-messenger RNA) enters stalled ribosomes, replaces the mRNA and promotes the translation of a peptide tag (ssrA) onto the C terminus of the nascent polypeptide [
16]. The ssrA tag is conserved across bacteria and in
Escherichia coli is recognized mainly by the ClpXP protease, a member of the AAA+ family. To a lesser extent, also other AAA+ enzymes such as the ClpAP complex can degrade ssrA-tagged substrates [
7,
17,
18]. ClpX is the unfoldase subunit comprising a hexameric ring decorated with dimers of the ClpX N-terminal Zinc Binding Domain (ZBD), while the peptidase ClpP forms a double ring of heptamers that can bind ClpX hexamers on one or both faces of the ClpP barrel (
Figure 1a). The ssrA tag consists of 11 amino acids (AANDENYALAA) in which the AANDENY motif is responsible for binding of the ClpX adaptor, i.e., SspB protein, and the ALAA motif binds to the ClpX unfoldase central pore itself [
19]. This modularity of ssrA is frequently exploited in biotechnology for diverse applications [
15].
The biochemical parameters for various variants of the ssrA tag have already been described with the use of multiple experimental setups and methods [
20,
21,
22,
23]. However, typically only a few selected variants have been characterized per study, making it difficult to make proper variant comparison across studies due to different experimental conditions. Moreover, simultaneous evaluation of properties such as SspB binding and degradation efficiency is available only for a limited set of ssrA variants.
Herein, we strive to establish a benchmark that will facilitate the comparison of the ssrA degron and its variants under uniform conditions. We included methods for the expression and purification of the ClpX, ClpP, SspB, and eGFP-degron proteins (
Figure 1b), as well as tests for the ATPase activity of ClpX, measurements of eGFP-degrons binding to the SspB protein and methods for measuring the degradation of eGFP-degrons in vitro and in vivo. Using uniform, precise and sensitive methods under the same conditions on a range of eGFP-degrons allowed us to determine subtle differences in their properties and to indicate their potential applications. Our findings can be a valuable point of reference and a resource for researchers using targeted protein degradation approaches.
3. Discussion
For the purpose of comparing and characterizing the properties of ssrA variants, it was necessary to set up expression and purification protocols for all the eGFP-degrons and the components of the ClpX-ClpP-SspB complex, as well as methods required to check their activity and interactions. The expression of proteins at the beginning in Top10 for eGFP-degrons and in BL21 (DE3) for ClpX, ClpP and SspB unfortunately resulted in a noticeable degradation of eGFP-degrons and ClpX. Difficulties in ClpX expression, which in our hands was generally the most challenging protein to purify, could be attributed to the high mobility of the ZBD domain, which can be easily digested by various proteases. Similar problems were encountered previously in structural studies of ClpX. Typically, ClpX structures were obtained without the ZBD domain [
25], and if ZBD was present, the resolution for ZBD was much lower than for the rest of the ClpX structure [
26]. For eGFP-degrons the observed degradation likely took place during their expression in bacteria, as might be expected for efficiently working degradation signals. Only by switching to expression in the
ΔclpP strain could we obtain proteins that did not show significant degradation.
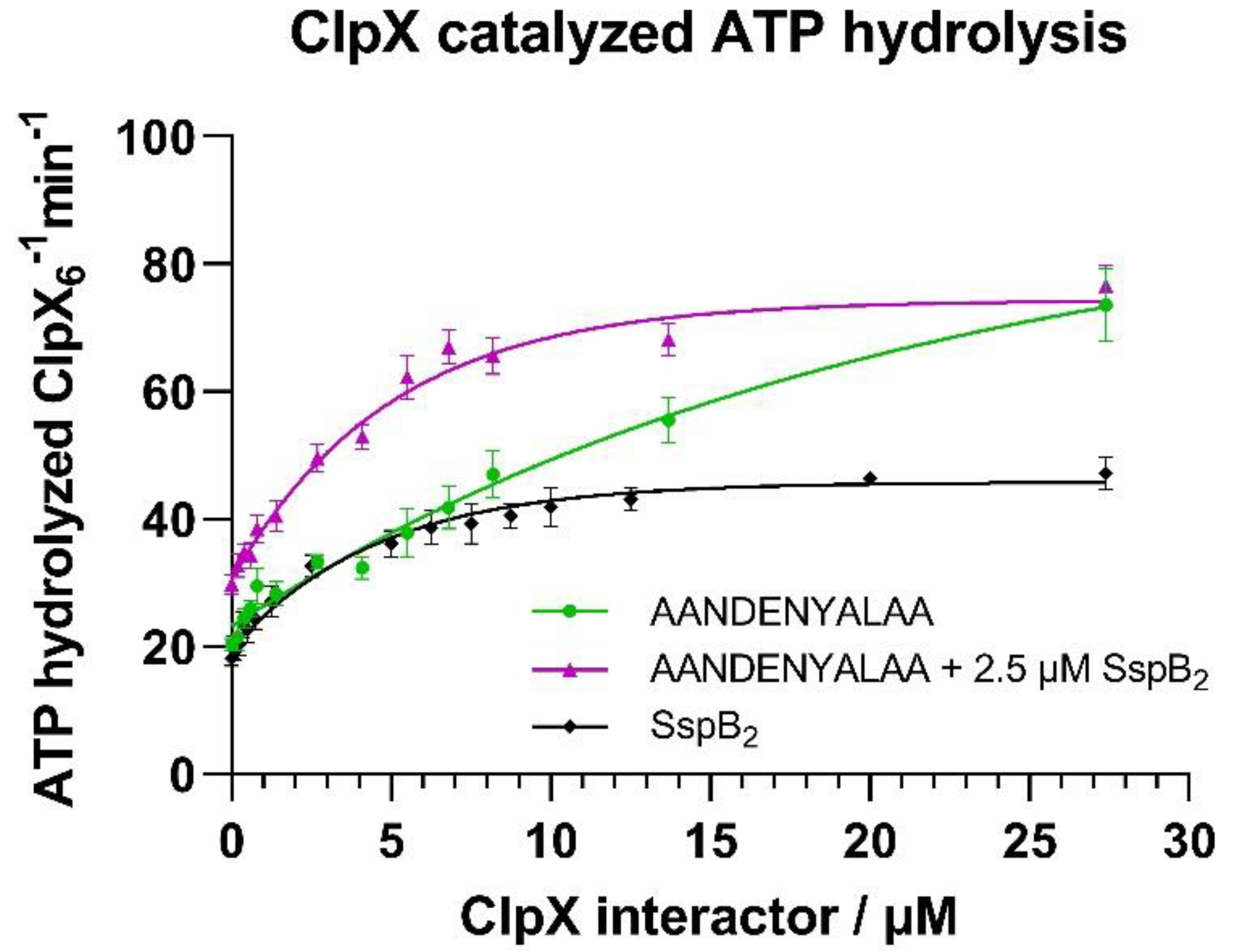
In the next step we ascertained that ClpX was active in an ATPase assay. Our results confirmed the previously reported [
27,
28] positive effect of the SspB adaptor on the ATPase activity of ClpX. Since ZBD is explicitly necessary for the interaction with SspB [
29], this strongly indicated the presence of a functional ZBD domain within the ClpX protein, with the downstream consequences on degradation by improving substrate engagement by ClpX.
The analysis of binding of eGFP-degrons to SspB protein studied on native gels confirmed the previously reported results of a mutational analysis of the GFP-ssrA tag [
30] that AANDENY sequence was necessary for the eGFP-degrons to bind SspB. Our MST measurements allowed for a more detailed binding analysis and the determination of the K
D for eGFP-ssrA and SspB at ~80 nM. Similar K
D values were observed previously in ITC measurements for GFP-ssrA and SspB, where the binding constant was determined to be 75 ± 30nM [
22] or even 16 ± 4 nM [
27]. The application of the eGFP-degron fusions allowed us not only to evaluate the interactions using MST but also to reduce the K
D value by one order of magnitude compared to the isolated ssrA tag, which is known to bind SspB more weakly [
27]. Additionally, MST is a relatively high-throughput and precise method that allowed us to determine even minor differences in the binding of the SspB protein to the eGFP-degrons. We noted that the ADAS sequence caused a slight decrease in the eGFP-degron binding to SspB in contrast to the WT ALAA end, despite the expected independence of the AANDENY and ALAA modules in binding to SspB and ClpX, respectively. This difference may be due to steric requirements or electrostatic interactions related to the presence of the negatively charged aspartate in the DAS fragment. To our knowledge, this is the first time that the DAS fragment has been reported to negatively influence SspB binding since, typically, the C-terminal amino acids of ssrA have been tested mostly for recognition by ClpX [
20]. It is not clear whether the disruptive effect of DAS would have any physiological relevance since it was observed under isolated conditions. However it could be of importance in the design of AANDENY-containing modules used to mediate interactions with the SspB protein. Our results suggest that the sequence that follows the AANDENY fragment should also meet specific requirements for the most efficient binding of the substrate protein by SspB. Overall degron length did not matter in SspB binding, even in the case of the eGFP-AANDENYAANDENY variant which seemed to bind better to SspB but without a significant change of the K
D. This may suggest the impossibility of SspB occupying more than one binding site on such a short peptide regardless of the presence of two SspB binding sites. This result may also indicate relatively slow dissociation of the complex between SspB and the AANDENY motif, which would render the second binding motif present in AANDENYAANDENY less relevant for improving SspB binding despite the increased local concentration of binding sites.
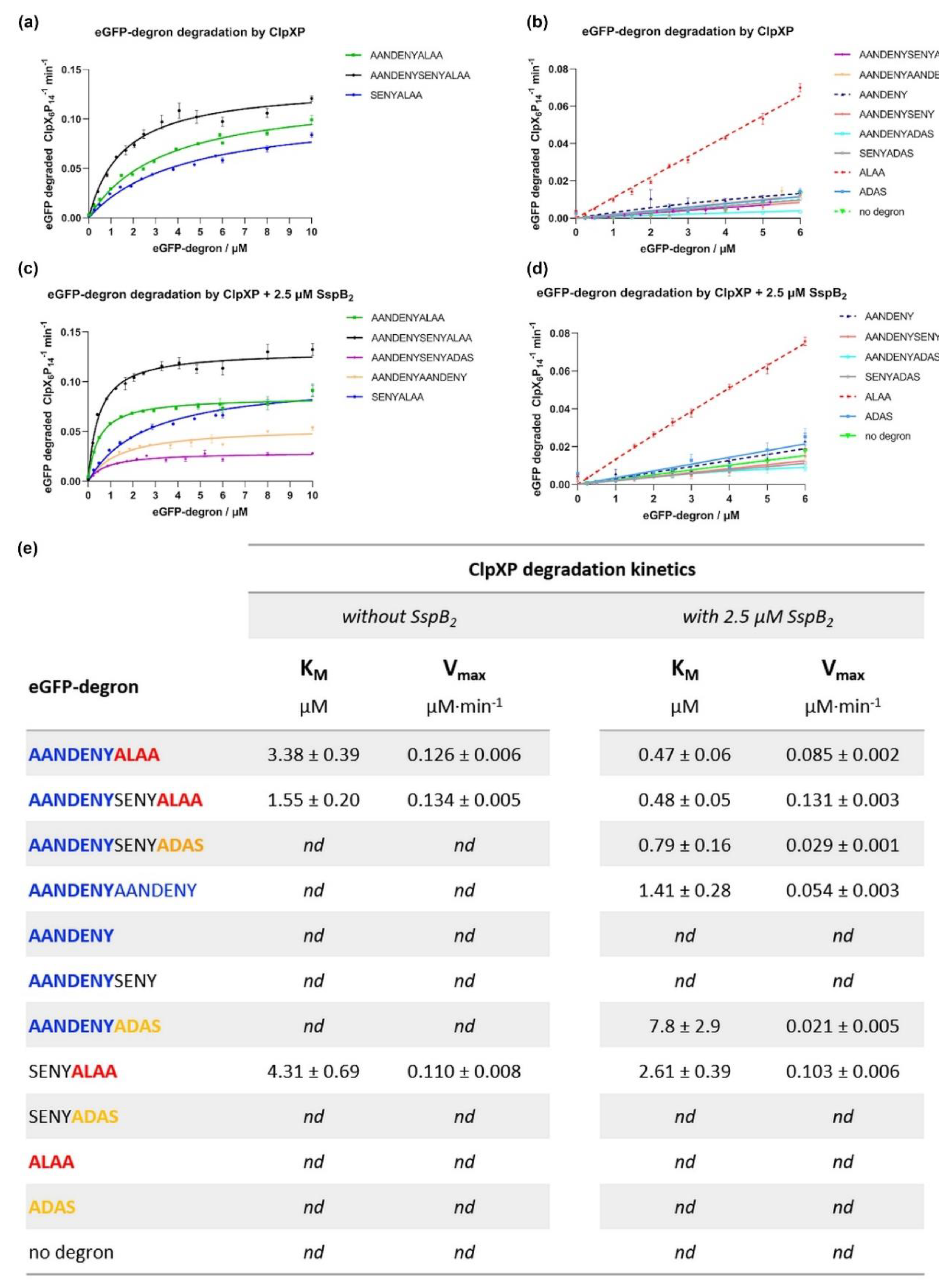
The key part of our work was comparing the degradation effectiveness of eGFP-degrons by the ClpXP complex and the effect of SspB on it. The application of our standardized reaction conditions throughout our assays to a whole set of ssrA tag variants made it easier to compare their K
M values and draw conclusions which parts of degrons are necessary for degradation, which can be replaced, and which are largely negligible. In the absence of SspB, degradation was observed only for eGFP-degrons containing the ALAA end preceded with an extending fragment such as the SENY linker. The longer the preceding fragment, the more efficient the degradation was observed. The ALAA fragment alone was insufficient for degradation. The ADAS fragment which is known to interact weaker with ClpX [
20], rendered eGFP fusions retardant to degradation regardless of the extension preceding ADAS, even for the longest AANDENYSENYADAS degron variant.
In the presence of SspB the K
M values for eGFP-degrons which were degraded without SspB were further lowered. Additionally, degradation was induced for 3 more eGFP-degrons (AANDENYADAS, AANDENYSENYADAS, AANDENYAANDENY), proving the positive influence of the AANDENY motif on the degradation efficiency and even compensating for the weaker binding of the mutated ADAS equivalent of ALAA. eGFP-AANDENYAANDENY was degraded efficiently despite the lack of the ClpX binding motif (ALAA or ADAS), suggesting that the terminal repeat of the AANDENY fragment provided sufficient reach to bind nonspecifically to ClpX pore entrance for degradation. The decrease in K
M was also observed for the eGFP-SENYALAA, which does not have the AANDENY motif and did not bind to SspB. This improvement in degradation efficiency could be attributed to the increased ClpX activity in the presence of SspB, as observed in our ATPase activity assays. Such a boost could result in the increased degradation of eGFP-SENYALAA by ClpX without direct SspB binding to the degron. However, the increased ATPase activity was not sufficient to efficiently degrade short degrons or degrons lacking the ALAA fragment. In summary, the eGFP-degron must be able to engage ClpX and be at least slightly degradable in the absence of SspB for SspB to positively influence its degradation through stimulation of ClpX ATPase activity. The positive effect of SspB on degradation efficiency was consistent with the literature data [
20,
21,
27]. The slight differences in the intensity of its effect may be attributed to the differences in the experimental conditions. Despite these differences, our results and the literature data both indicated [
21] that the highest impact of SspB on the eGFP-degron degradation efficiency was observed for the eGFP-AANDENYSENYADAS variant.
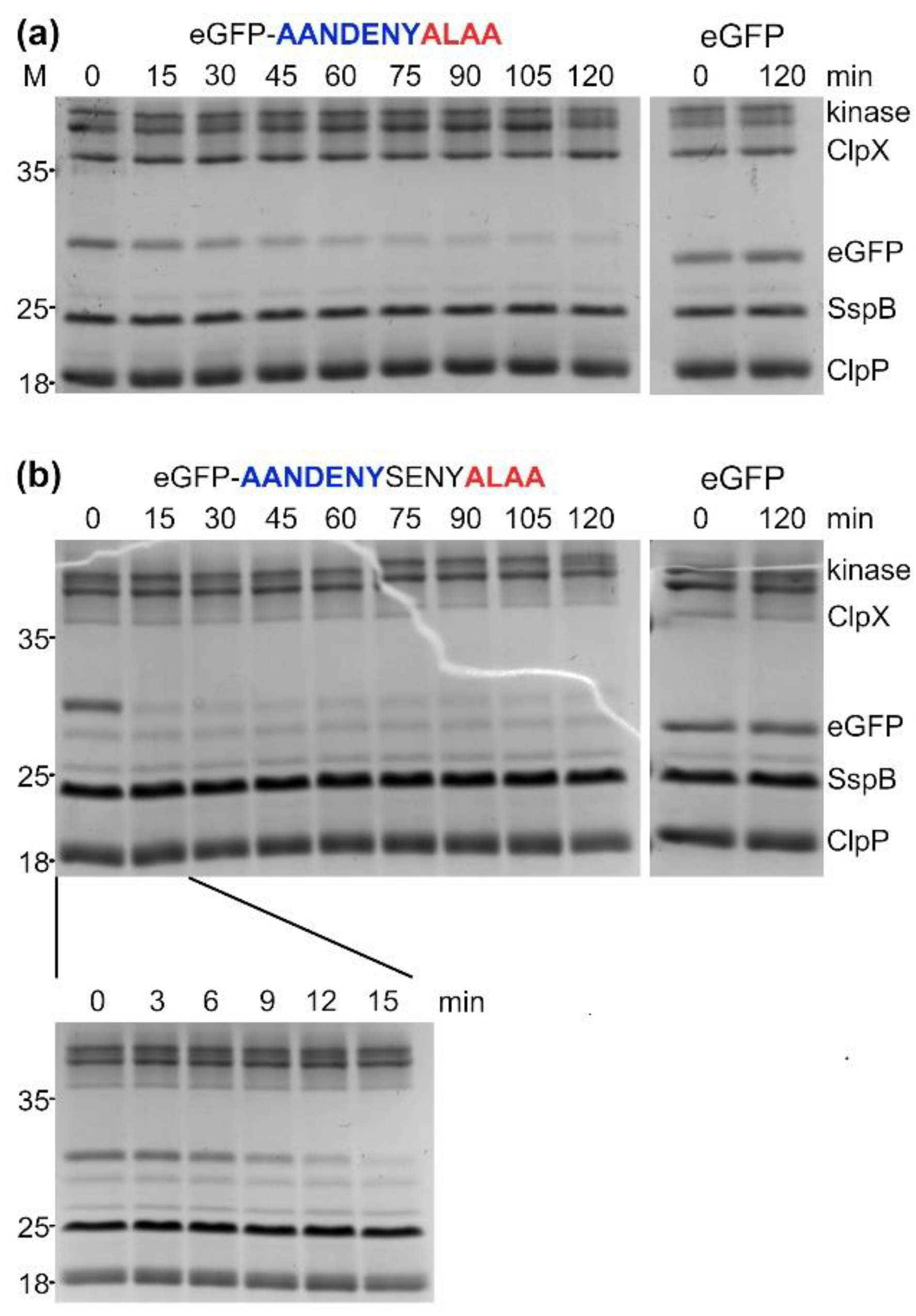
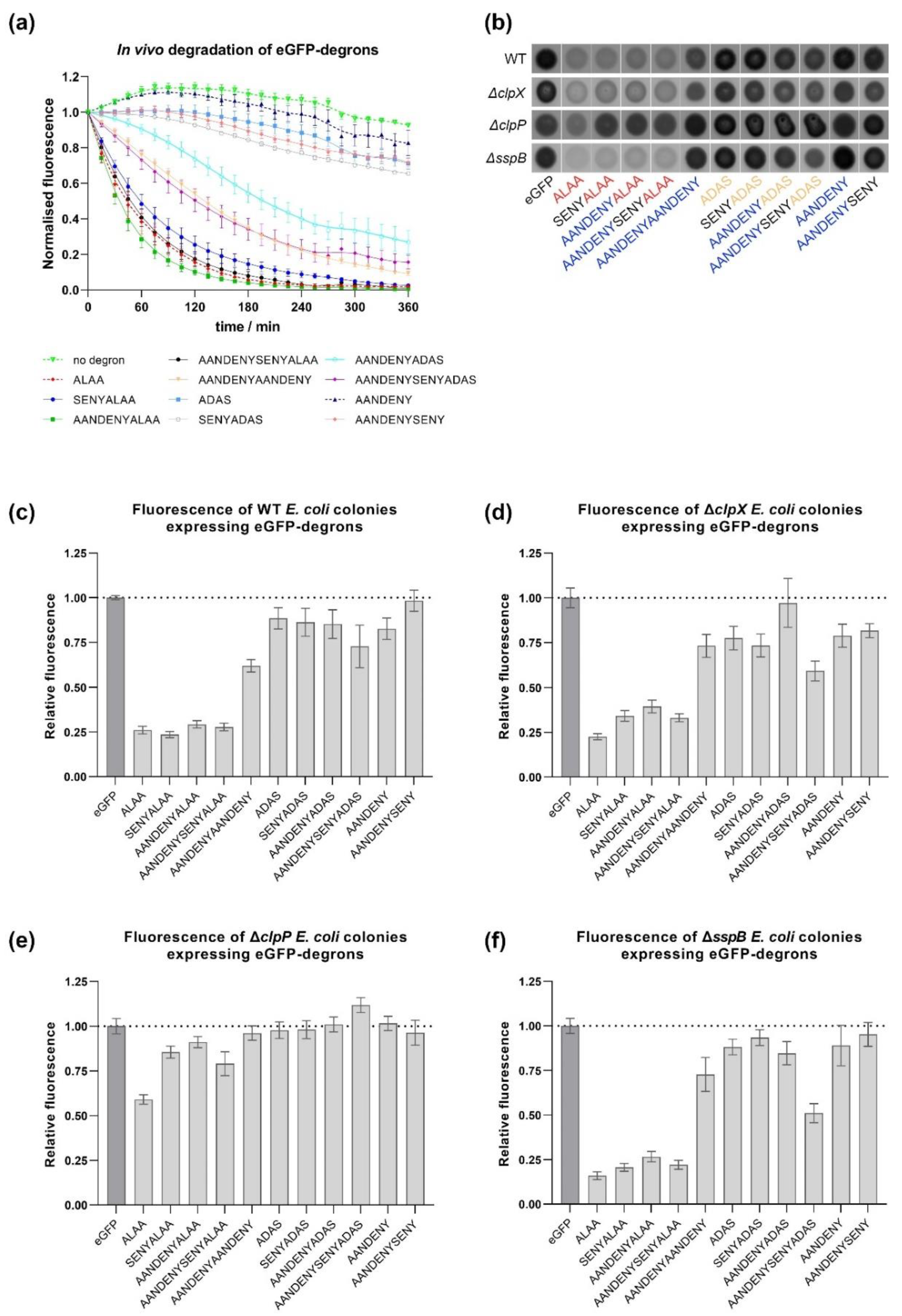
The results obtained in vivo were consistent with our in vitro results showing that eGFP-degrons containing the ALAA sequence were degraded the most efficiently. Significantly reduced fluorescence was observed for those variants (AANDENYALAA, AANDENYSENYALAA and SENYALAA), for which eGFP-degrons were also degraded in vitro even in the absence of SspB. For the remaining eGFP-degrons, the decrease of fluorescence was not pronounced, confirming again in vitro degradation requirements for the proper degron length and the ALAA fragment. An additional common element for our in vitro and in vivo results was the atypical behavior of the eGFP-ALAA fusion, which was degraded in vitro with an intermediate efficiency among our set of degrons even in the absence of SspB, but with a linear dependence on the tested substrate concentration that prevented determination of K
M values. eGFP-ALAA was degraded very efficiently in vivo independently of all the factors we addressed. This was not due to the low level of eGFP-ALAA expression which was deemed sufficiently high by our time-course experiments, but rather that it could be degraded by other proteases outside the ClpXP pathway. This could result from a cross-talk of the ssrA system with other pathways in which hydrophobic or alanine-rich degrons are recognized as signals for degradation. The involvement of other proteases is likely if we take into account that in the
ΔclpX strain some eGFP-degrons were still degraded, whereas in the
ΔclpP strain only the aforementioned eGFP-ALAA was depleted. Our results indicate that in BW25113 cells, ClpX is redundant for ssrA-tagged substrates and could be successfully replaced by another unfoldase such as ClpA [
7,
18]. Degradation observed in the
ΔsspB strain was similar to that observed for WT and
ΔclpX strains, corroborating the redundancy of the ClpX pathway or the SspB itself. Recently, it was shown that the
E. coli ribosome-associated trigger factor (TF), a chaperonin which is highly abundant in the cell and acts early during the folding process, enhances degradation of various ClpXP complex substrates including ssrA-tagged proteins both in vitro and in vivo [
31]. The role of TF as an additional adaptor could contribute to the lack of an effect of
sspB deletion on our tested eGFP-degrons.
The results and protocols presented here can serve as a benchmark for designing new degrons as well as providing guidelines on their comparison. However, the degron research strategies we used also have their limitations. Firstly, the complicated stoichiometry of oligomeric components may pose a problem. Changes in stoichiometry can cause changes in degradation kinetics (especially if the substrate is also an oligomer) or in the selectivity of the degradation system. Secondly, it is crucially important to test degron performance in the case of individual substrates/targeted proteins, as the substrate itself may alter degron availability and thus affect the efficiency of degron binding or its degradation. With these difficulties in mind, we aimed at establishing the most robust and straightforward strategies and methods, which allowed us to discover subtle differences between degrons in terms of their binding strength or degradation efficiency.
Building on this type of knowledge, degron variants can be fine-tuned for two basic types of applications. We ranked the propensity of the tested ssrA variants for SspB binding and for eGFP-degron degradation (
Figure 7), which should facilitate the choice of the degron appropriate to the desired task. If efficient degradation of the fusion protein is desired, the best binding to SspB is provided by the WT ssrA sequence, but in the absence of SspB, AANDENYSENYALAA would be an even better choice. Our results confirm that AANDENYSENYADAS and AANDENYADAS enable degradation which might be regulated by SspB presence [
32,
33,
34]. The SsrA degron is often used simply as an interaction module, for example in photoswitches [
35]. In this case, if only promoted interaction with SspB is desired, then AANDENY or AANDENYSENY degrons would be the most suitable choice for efficient tag binding without degradation.
Degrons can also be exploited as protease recruiting ligands in the design of degrader molecules in the targeted protein degradation field [
14]. For this purpose, it is essential to distinguish the effects of degron sequence and length on adaptor and protease binding from those parameters that lead to degradation and depletion of the degron-based molecular tools. Our benchmark of methods and protocols may serve the budding targeted protein degradation studies in bacteria in establishing more stable and efficient bacterial degraders.
4. Materials and Methods
4.1. Cloning of clpP, clpX and sspB Genes to pET28a Expression Vector by Sequence and Ligation Independent Cloning (SLIC)
DNA encoding full-length
clpP,
clpX and
sspB genes of
Escherichia coli (Top10 strain) was obtained by colony PCR amplification using specific primers (
Supplementary Materials, Table S1) containing sequences complementary to pET28a plasmid with His tag and SUMO protease cleavage site.
The SLIC reaction was performed by mixing 100 ng of pET28a linearized empty vector, 100 ng of PCR-amplified DNA fragment (with clpP, clpX or sspB gene) and 1U of T4 DNA polymerase (Thermo Fisher Scientific, Waltham, MA, USA) in 1X FastDigest Green Buffer (Thermo Fisher Scientific, Waltham, MA, USA) in a volume of 30 µL. After 30 min incubation in RT, reactions were stopped by adding dCTP (EURx, Poland) to a final concentration of 1 mM, and the tubes were incubated for 30 min at 37 °C for annealing. Finally, reactions were transformed to E. coli Top10 for selection on kanamycin.
For the preparation of pBAD-eGFP-degron constructs (
Supplementary Materials, Table S2), we modified eGFP-pBAD plasmid (later referred to as pBAD-eGFP) containing His-TEV-tagged eGFP gene, obtained from Michael Davidson (Addgene plasmid # 54762;
http://n2t.net/addgene:54762 (accessed on 26 July 2021); RRID:Addgene_54762). The C-terminal degrons were appended to the eGFP gene by site-directed mutagenesis. Primers used for PCR (
Supplementary Materials, Table S1) were designed to amplify an entire pBAD-eGFP plasmid while simultaneously adding degrons. The PCR products were then ligated and phosphorylated, and the obtained constructs were transformed into
E. coli Top10 for selection on ampicillin.
Successfully transformed bacteria were used for plasmid isolation, and all constructs were verified by sequencing. The sequences of clpP, clpX and sspB were identical to the corresponding genes from strain K-12 substrain MG1655 which is widely used as a model E. coli strain (GenBank: U00096.3).
4.2. Protein Expression and Purification
For in vitro studies protein expression was carried out in BL21 DE3 bacteria (for ClpX, ClpP, SspB), in
E. coli Top10 strain (for eGFP-degrons) and in
ΔclpP Keio collection strain [
36] (JW0427) (for ClpX, eGFP-degrons) using LB media with induction at OD
600 ~ 0.5 with 0.5 mM IPTG for pET28a constructs in BL21 DE3 strain or 0.02% arabinose (VWR International, USA) for pBAD constructs in Top10 and
ΔclpP strains. Overexpression of ClpP, SspB and eGFP-degrons was carried out at 30 °C for 3 h, ClpX at 18 °C overnight, all at 140 rpm. Bacterial pellet was collected by centrifugation (5000 rpm [5180×
g], 30 min, at 4 °C) and frozen at −20 °C for later use.
ClpX, ClpP, SspB and eGFP-degrons were purified on HisTrap HP 5 mL column (GE Healthcare, North Richland Hills, TX, USA) following the manufacturer’s guidelines. The lysis/loading buffer contained 50 mM Tris pH 8.0, 20 mM imidazole, 500 mM NaCl (for ClpX and eGFP-degrons) or alternatively 1 M NaCl (for ClpP and SspB) and with addition of 1 mM DTT for ClpX. Lysis by incubation for 30 min (on a rotator at 4 °C) with lysozyme (100 mg) and DNase (20 U) was followed by sonication (15 min, 45 on/15 off s, 40% amplitude at 4 °C) (Sonics & Materials Inc., Newtown, CT, USA) and centrifugation (20,000 rpm [48,380× g], 30 min, 4 °C). Lysates were cleared by filtration (0.45 μm) and applied on the HisTrap column. ClpX, ClpP and SspB elutions (using lysis/loading buffer supplemented to 1 M imidazole) were visualized on SDS-PAGE (Bio-Rad Laboratories, Hercules, CA, USA) and proper fractions were selected, then combined and treated with SUMO protease for His-SUMO tag removal (2 h on a rotator at 4 °C). After cleavage, samples were concentrated in 10K MWCO centricons (Pall Corporation, Washington, DC, USA) (5000 rpm [3214× g] at 4 °C) and applied on a Superdex200 HiLoad 16/600 column (GE Healthcare, North Richland Hills, TX, USA) using 25 mM Hepes pH 7.6, 200 mM KCl, 5 mM MgCl2, 10% glycerol buffer with the addition of 0.5 mM of TCEP in case of ClpX. The eGFP-degrons were purified on HisTrap and Superdex200 columns in a tandem configuration without purification tag (His-TEV) removal in between. Based on SDS-PAGE, selected SEC fractions were combined, and protein concentration was measured using the Bradford method (for ClpX) (Tecan, Männedorf, Switzerland) or based on the UV absorbance profile (for ClpP, SspB, His-TEV-eGFP-degrons, further denoted as eGFP-degrons) (DeNovix Inc., Wilmington, NC, USA). All proteins were flash frozen in liquid nitrogen and stored at −80°C for later experiments.
4.3. ATP Activity Assay
In vitro ATP hydrolysis by ClpX was measured using an NADH-coupled assay as previously described [
37]. A mixture of ClpX interacting protein (0–27.4 μM SspB
2, eGFP-SsrA or eGFP-SsrA with constant concentration of 2.5 μM SspB
2), 2.5 mM ATP, 2.5 mM phosphoenolpyruvate, 50 µg/mL pyruvate kinase, 1 mM NADH, and 50 µg/mL lactate dehydrogenase (Merck, Darmstadt, Germany) in the reaction buffer (50 mM Tris-HCl pH 7.5, 200 mM NaCl, 5 mM MgCl
2) was incubated at 30 °C for 30 min. As a negative control for the study of the eGFP-ssrA interaction, an eGFP protein not fused to ssrA degron was used. The assay was started by the addition of ClpX
6 to a final concentration of 0.08 µM in the 100 µL final reaction volume. Changes in NADH concentration were monitored by measuring absorbance at 340 nm for 1.5 h (in 1 min intervals) at 30 °C in a 96 well plate (Greiner Bio-One, Kremsmünster, Austria) in a Tecan Infinite M200 Pro plate reader instrument (Tecan, Männedorf, Switzerland). All measurements were performed at least in a triplicate. The rate of ADP formation was calculated from the linear loss of fluorescence assuming a 1:1 correspondence between ATP regeneration and NADH oxidation and a Δε
340 of 6.23 µM
−1cm
−1. Curve fitting and statistical analysis was performed via Graphpad Prism 9 software (Graphpad Software, LLC, San Diego, CA, USA).
4.4. Native Electrophoresis
In vitro protein-protein interaction studies were performed by means of non-denaturing polyacrylamide gel electrophoresis. Individual proteins (8 μM for eGFP-degron and 8 μM for SspB2) or protein mixtures (8 μM each) were incubated prior to electrophoresis for approximately 10 min at RT in the reaction buffer (50 mM Tris-HCl pH 7.5, 200 mM NaCl, 5 mM MgCl2). Protein samples were separated on 15% native polyacrylamide gels in Tris/borate buffer and the results visualized by staining the gels with SimplyBlue Safe Stain (Invitrogen, LC6060, Thermo Fisher Scientific).
4.5. Microscale Thermophoresis (MST)
For measurements of binding between SspB and eGFP-degrons, 100 nM eGFP-degrons and a series of 16 two-fold dilutions of SspB ranging from 7.45 nM to 244 μM in MST buffer (25 mM Hepes pH 7.6, 200 mM KCl, 5 mM MgCl2, 10% glycerol) supplemented with 0.05% Tween 20 were prepared. Then each ligand dilution was mixed with one volume of eGFP-degron, which led to the final eGFP-degron concentration of 50 nM and final SspB concentrations ranging from 3.72 nM to 122 μM. After 15 min incubation at RT, the samples were loaded into Monolith NT.115 Premium Capillaries (NanoTemper Technologies, München, Germany), and measurements were carried out using the Monolith NT.115 instrument (NanoTemper Technologies, München, Germany) at 20 °C. Instrument parameters were adjusted to 20% LED power and medium MST power.
All measurements were performed at least in triplicates. Protein binding parameters were calculated using the signal from an MST-on time of 10 s. Curve fitting and Scatchard plots, and statistical analysis of the obtained constants were performed via Graphpad Prism 9 software (Graphpad Software, LLC, San Diego, CA, USA).
4.6. Fluorescence-Based Measurements of In Vitro Degradation
In vitro degradation of eGFP-degron by ClpXP analysis was performed in the 100 µL final volume in the reaction buffer (50 mM Tris-HCl pH 7.5, 200 mM NaCl, 5 mM MgCl2). A mixture of 0.08 µM ClpX6, eGFP-degron (0–10 μM), SspB (2.5 μM SspB2, if added), 4 mM ATP, 2.5 mM creatine phosphate, and 50 µg/mL creatine kinase was incubated at 30 °C for 30 min. As a negative control, the eGFP protein not fused with any degron was used. The degradation assay was started by the addition of 0.21 µM ClpP14 for the final protease machinery assembly in ClpX6:ClpP14 = 3:8 ratio. Changes in eGFP fluorescence (at excitation λ = 489 nm and emission λ = 550 nm) were monitored for 1.5 h at 30 °C in a Tecan Infinite M200 Pro plate reader instrument. All measurements were performed at least in triplicates. Degradation rates were calculated from the initial linear loss of eGFP fluorescence. Curve fitting using the Michaelis-Menten equation and statistical analysis was performed via Graphpad Prism 9 software (Graphpad Software, LLC, San Diego, CA, USA).
4.7. SDS-PAGE Analysis of In Vitro Degradation
Reactions for SDS-PAGE analysis of in vitro eGFP-degron degradation by ClpXP were prepared the same way as for fluorescence readouts, with eGFP-degron or eGFP concentration set at 1 μM. 10 µL samples were collected in 15 min intervals up to 120 min (with additional time points for AANDENYSENYALAA of 3, 6, 9 and 12 min). The reaction was stopped by adding the SDS-PAGE loading buffer followed by 5 min incubation at 95 °C. Prepared samples were applied on 12% SDS-PAGE gels, which were developed at 200 V. Resolved proteins were visualized using the SafeStain method.
4.8. Time-Course Measurements of In Vivo Degradation
Precultures were prepared by inoculating a small amount of a glycerol stock of E. coli BW25113 (previously transformed with pBAD-eGFP constructs) in liquid LB medium with ampicillin (50 μg/mL) and incubating overnight at 37 °C in an orbital shaker. The following day, the precultures were diluted 50 times in fresh medium and the cultures were grown to mid-exponential phase. The expression of eGFP-degron fusions was induced by addition of l-arabinose to the final concentration of 0.001%. The induced cultures were incubated overnight in 18 °C. Bacteria were then diluted 10 times by adding 20 μL of the cultures to 180 μL of M9 medium supplemented with glucose (5%), thiamine (10 μg/mL), biotin (10 μg/mL), trace elements, and additionally spectinomycin (100 μg/mL) to stop protein translation. The measurements of OD600 and fluorescence (excitation at λ = 489 nm and emission at λ = 520 nm) were made every 15 min for 6 h using Tecan Infinite M200 Pro plate reader (Tecan, Männedorf, Switzerland) at 30 °C. The fluorescence results were normalized to the optical density. The measurements were further normalized by setting the values at zero time points as 100%.
4.9. Plate Spot Assay of In Vivo Degradation
2–3µ. l drops of 100x diluted overnight cultures (prepared as described above) were placed in triplicates or quadruplicates on LB agar plates supplemented with 0.001%
l-arabinose and antibiotics: ampicillin (50 μg/mL) for WT strain (BW25113), and ampicillin (50 μg/mL) with kanamycin (15 μg/mL) for the deletant strains (
ΔclpX (JW0428-KC),
ΔclpP (JW0427-KC) and
ΔsspB (JW0866-KC)). The plates were incubated overnight at 37 °C. Images of the plates were taken with the Bio-Rad Chemidoc Imager (Bio-Rad, Philadelphia, PA, USA) using a fluorescein filter and analyzed with Fiji software [
38].


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






