
Chemistry of Spontaneous Alkylation of Methimazole with 1,2-Dichloroethane

 ,
,  , and
, and 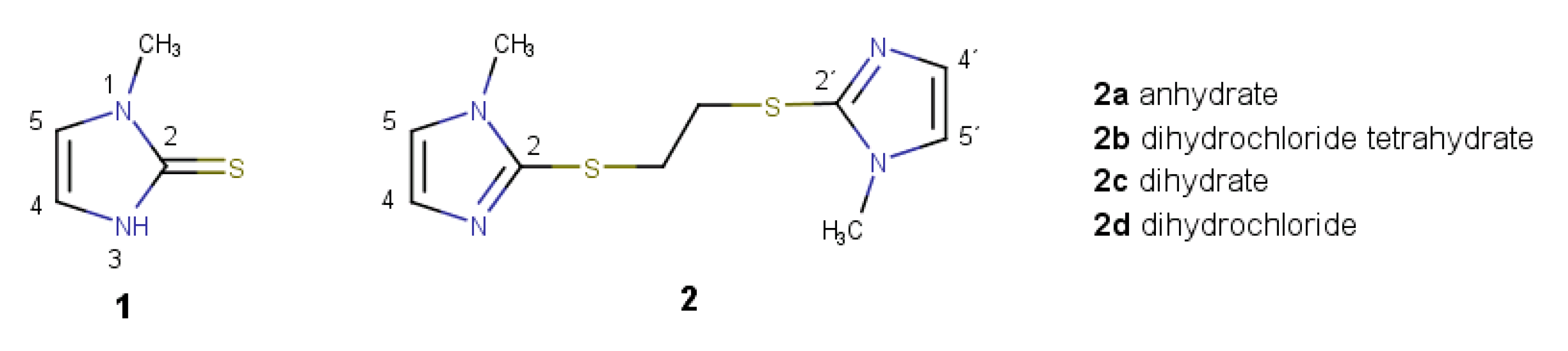{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
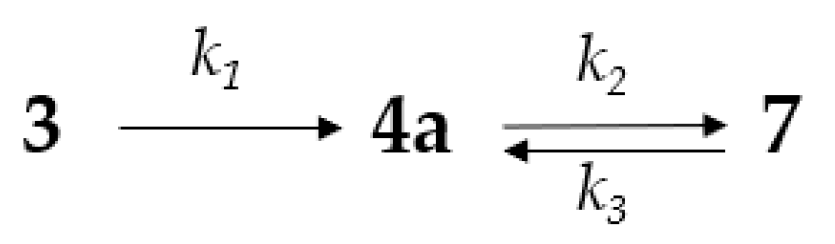{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
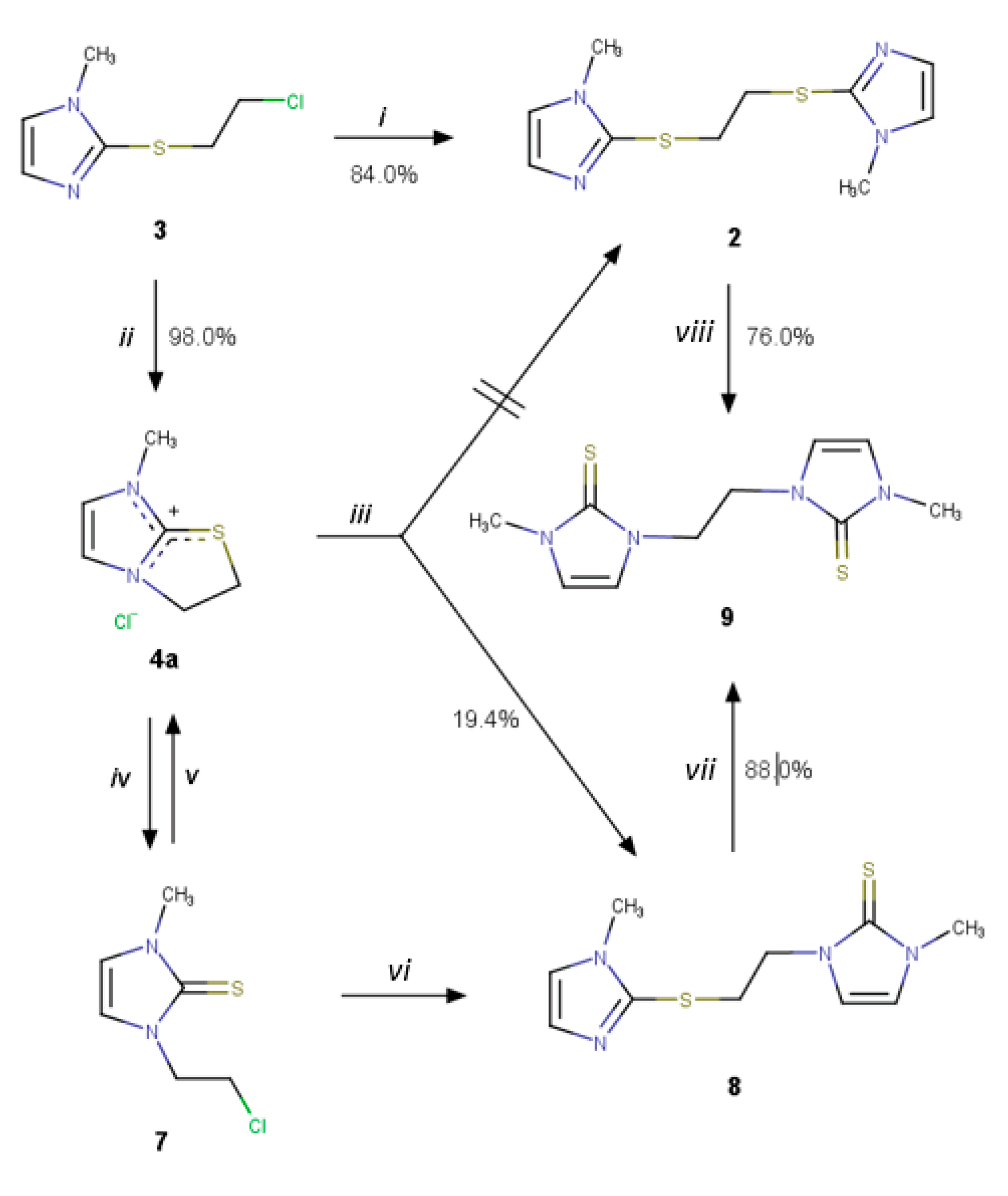
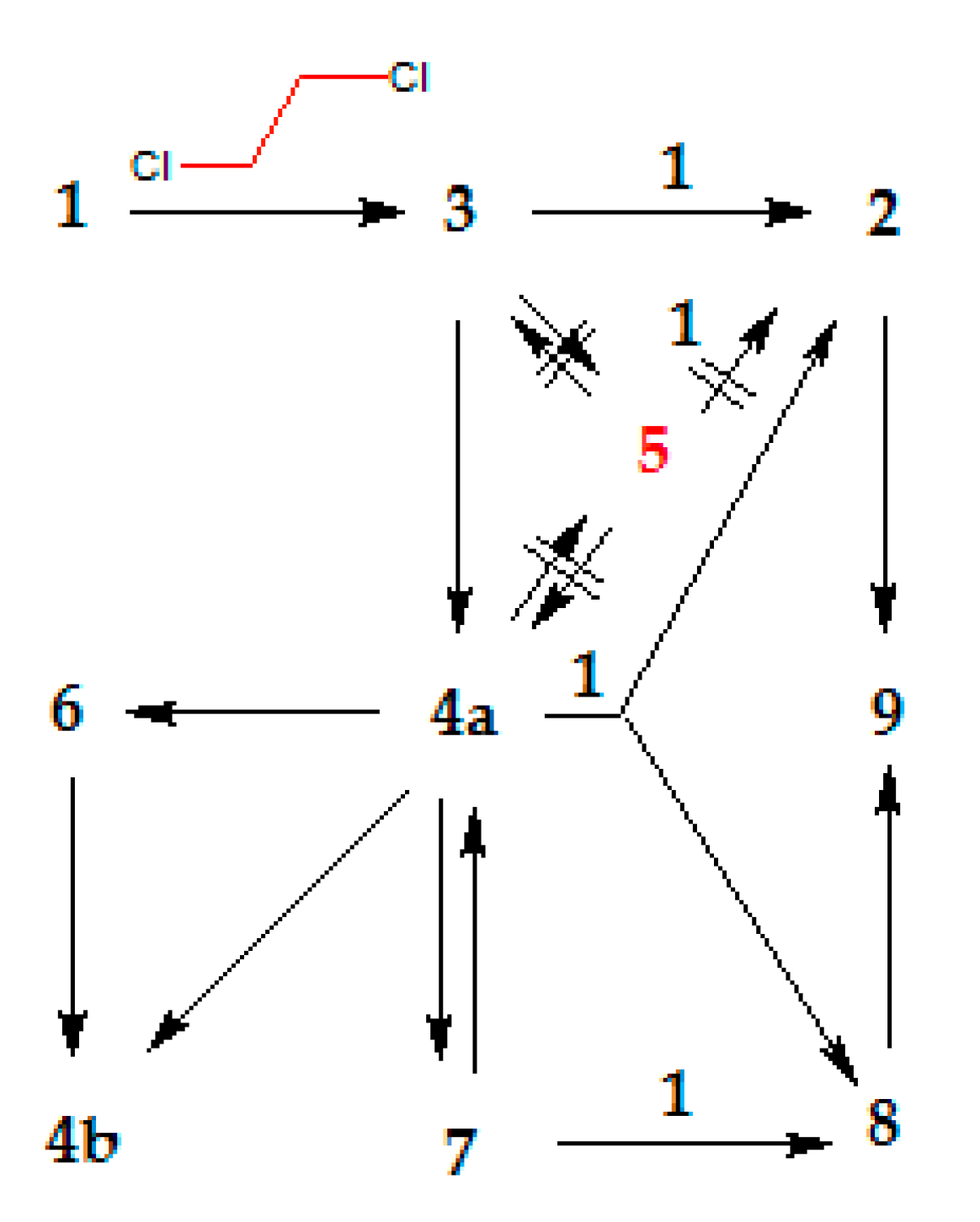
2. Results and Discussion
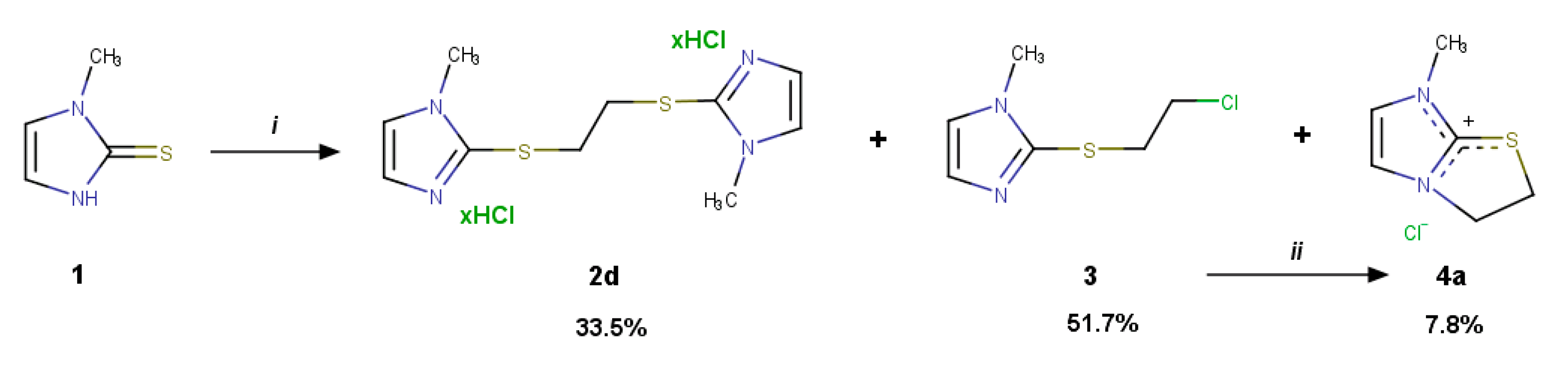
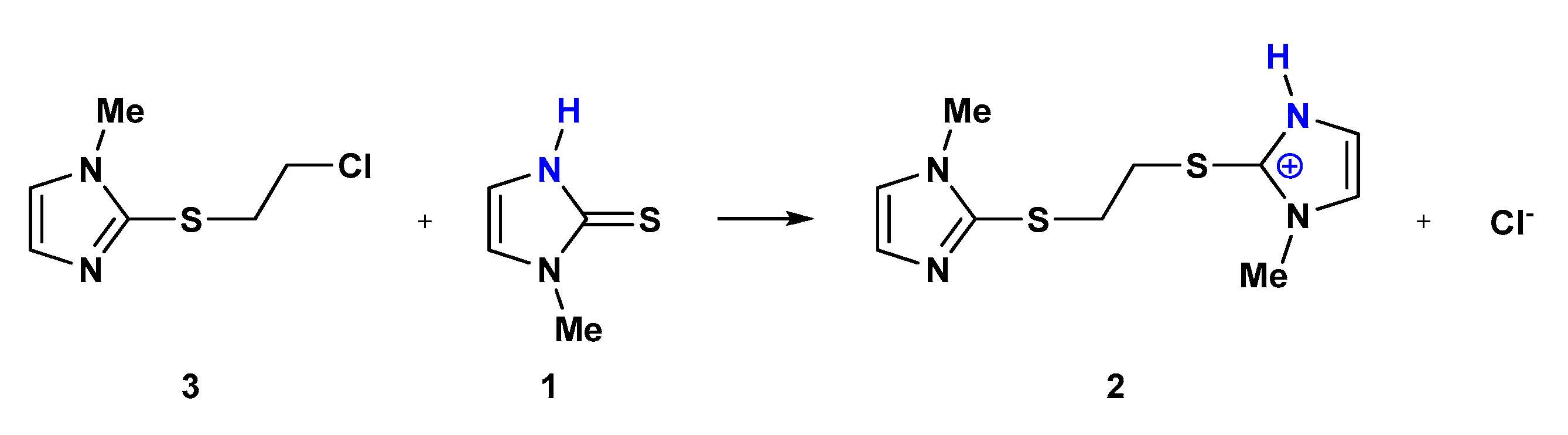
2.1. Reaction of Methimazole (1) with 1,2-Dichoroethane
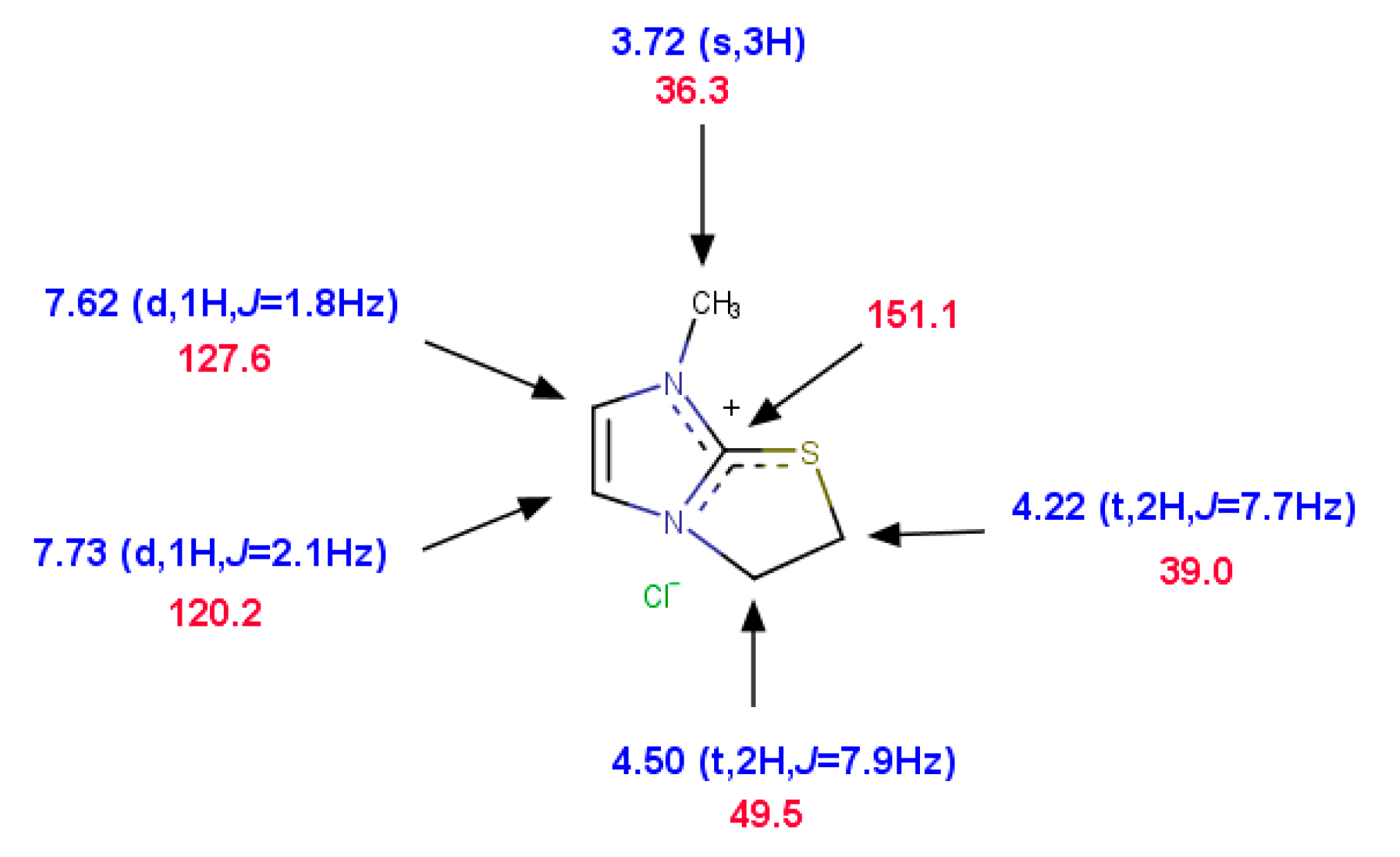
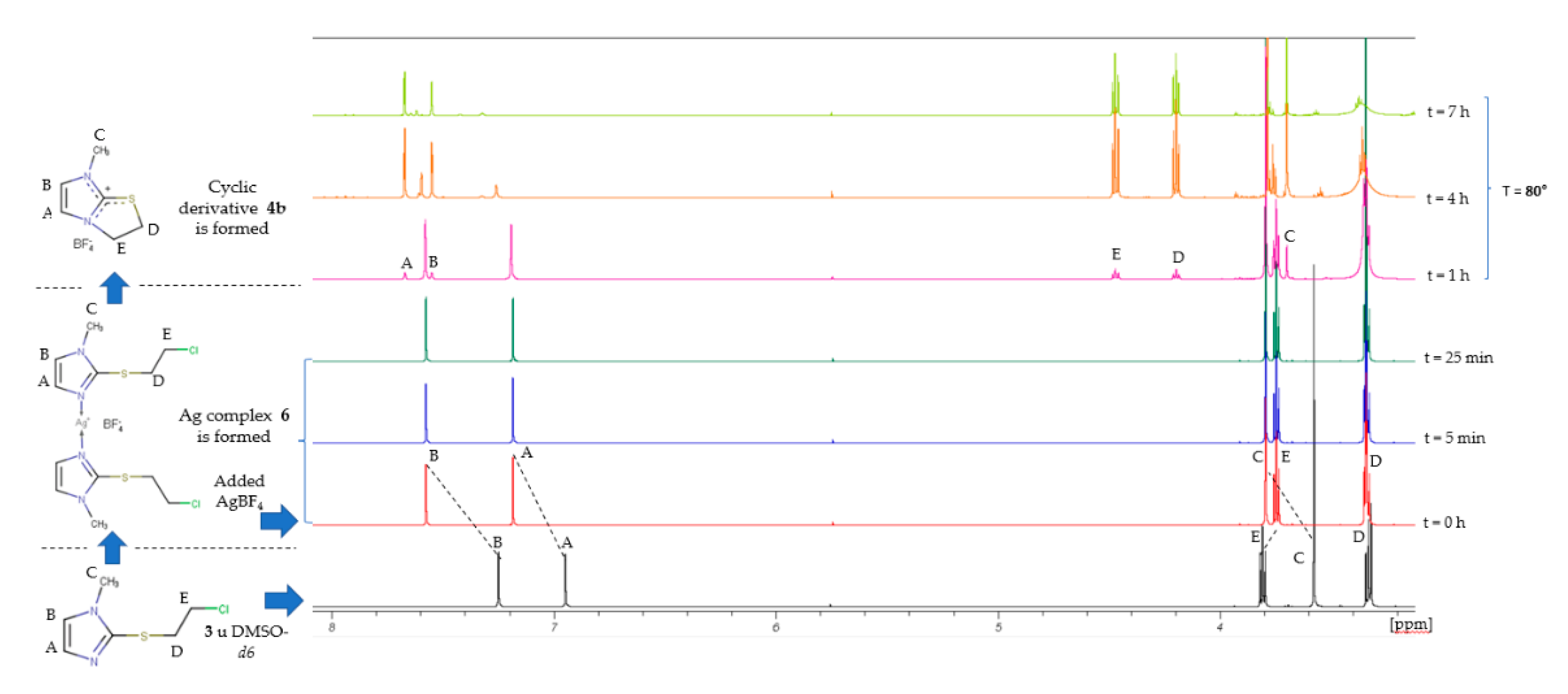
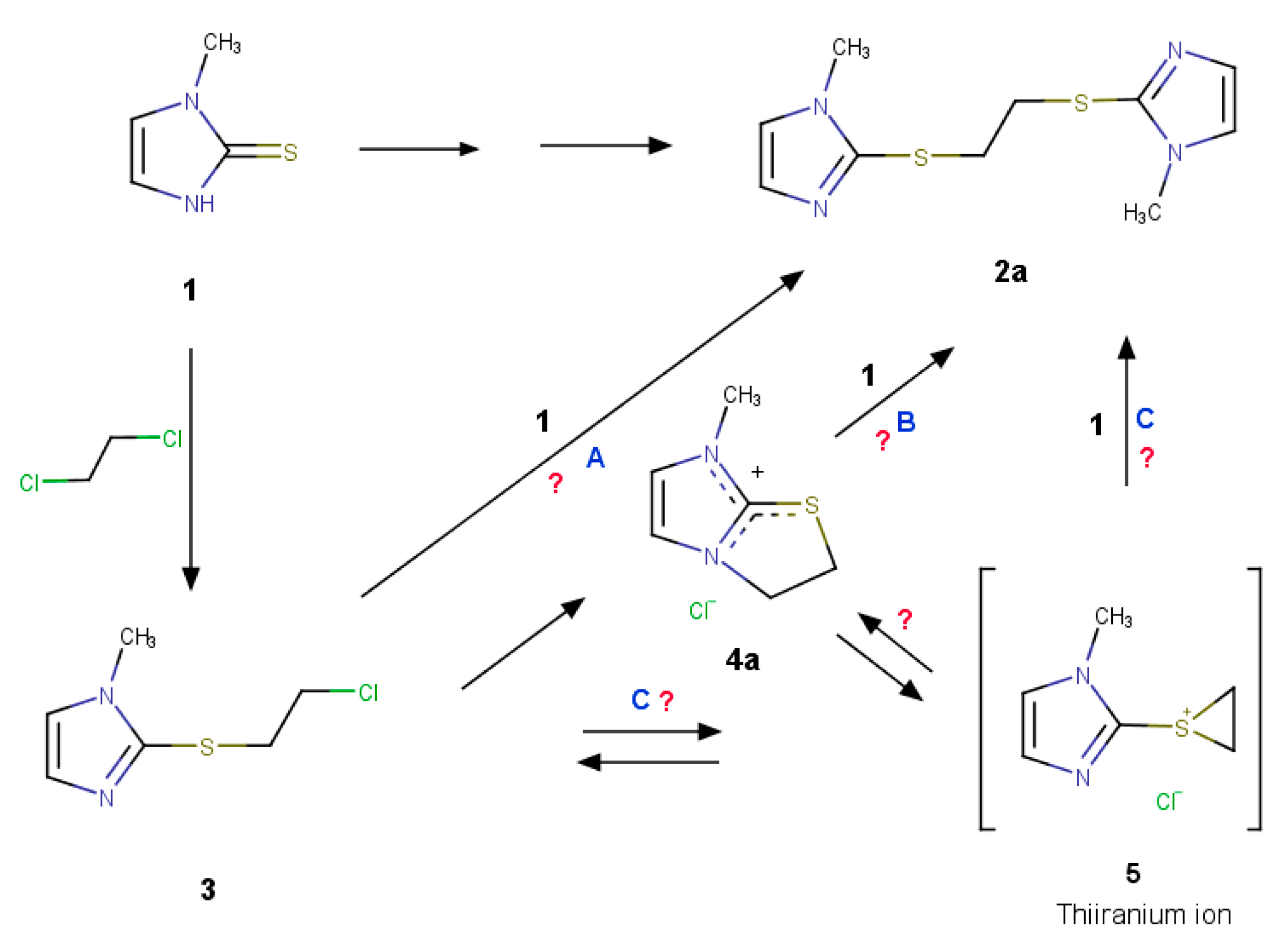
2.2. Search for Thiiranium Ion Intermediate 5 by NMR Spectroscopy
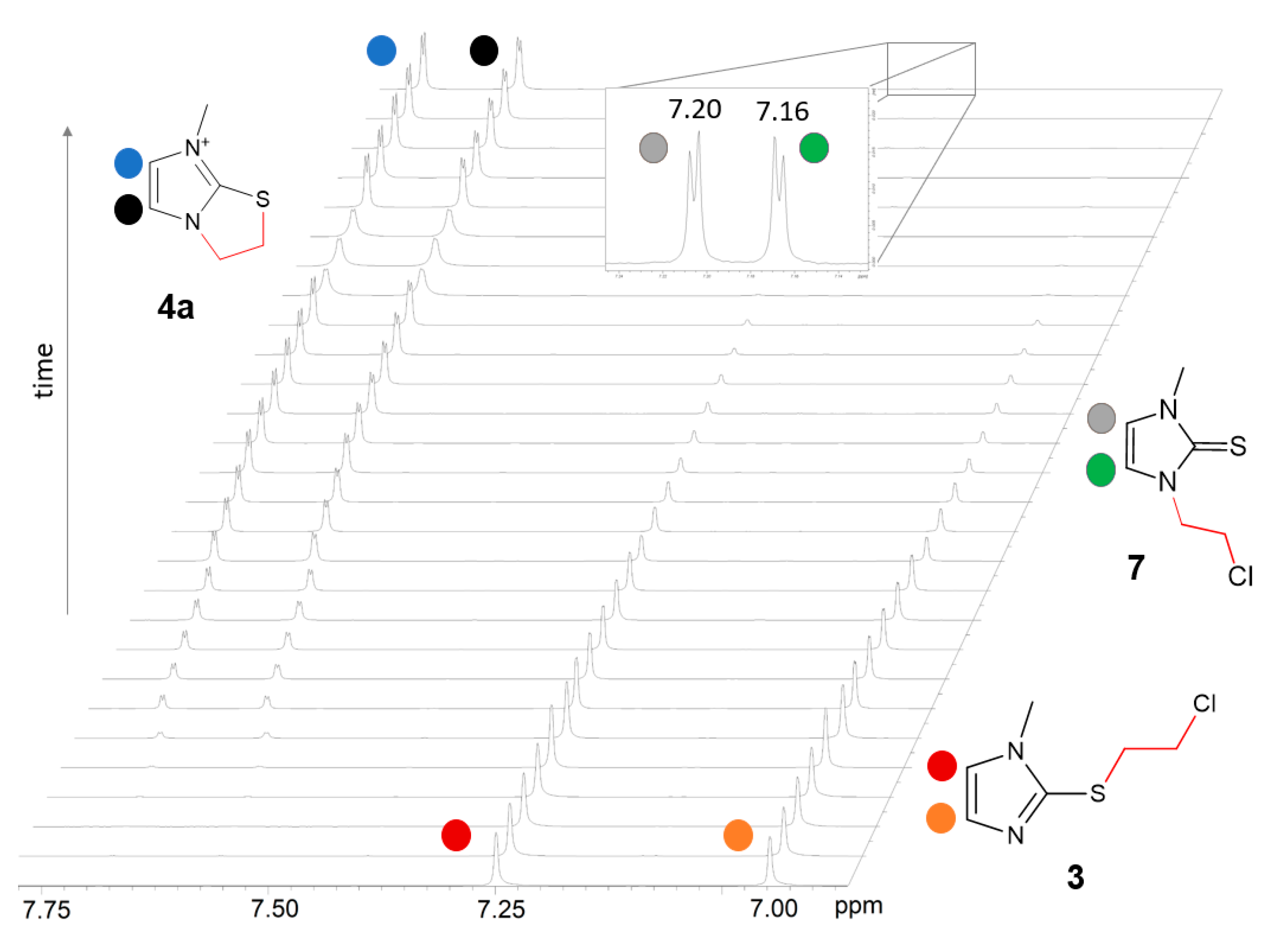
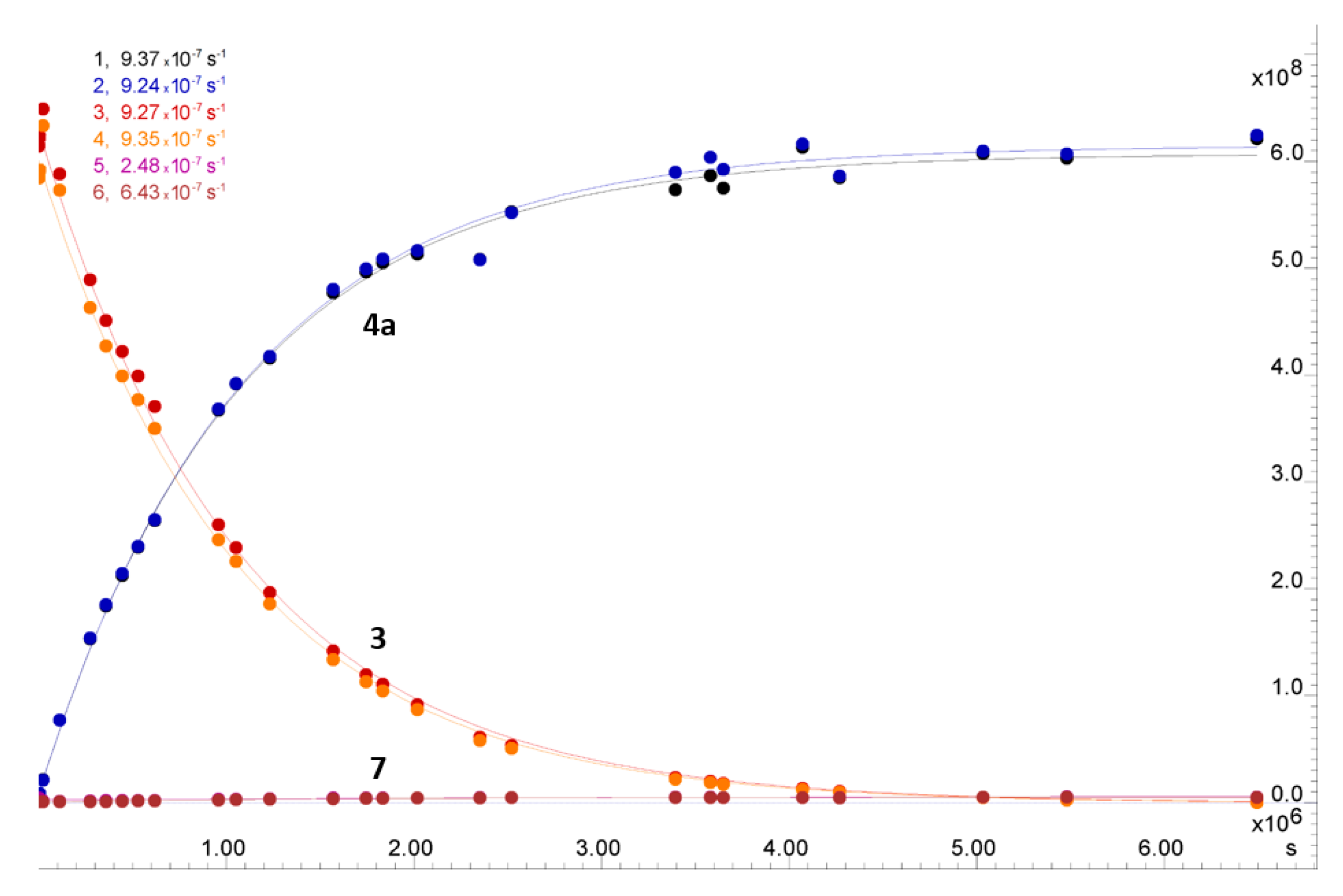
2.2.1. Kinetics of the S-Chloroethyl Derivative 3 Degradation
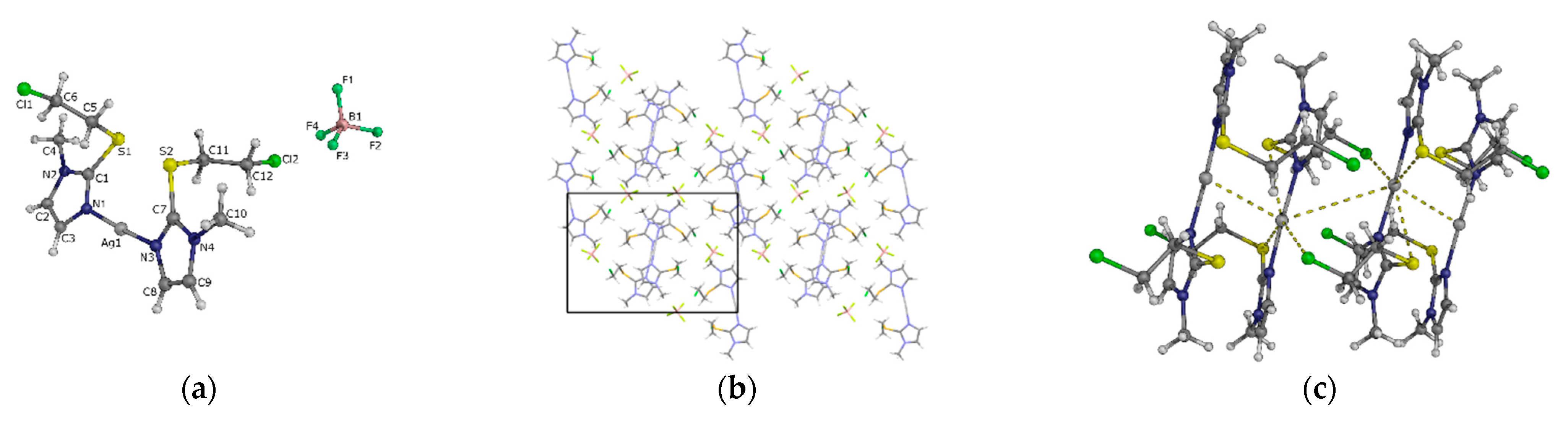
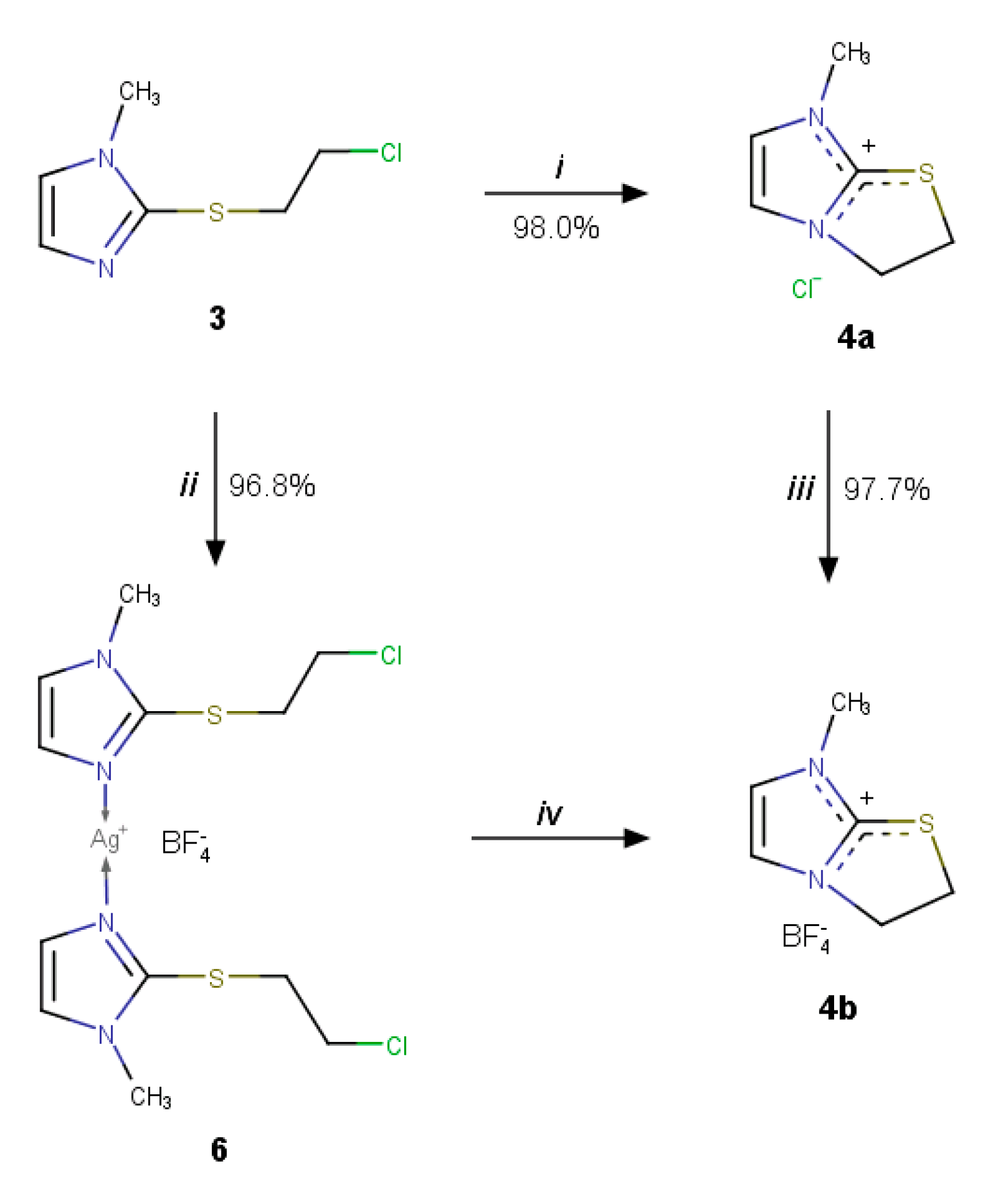
2.2.2. Trapping the Thiiranium Intermediate 5 with AgBF4
2.3. Further Synthetic Transformation
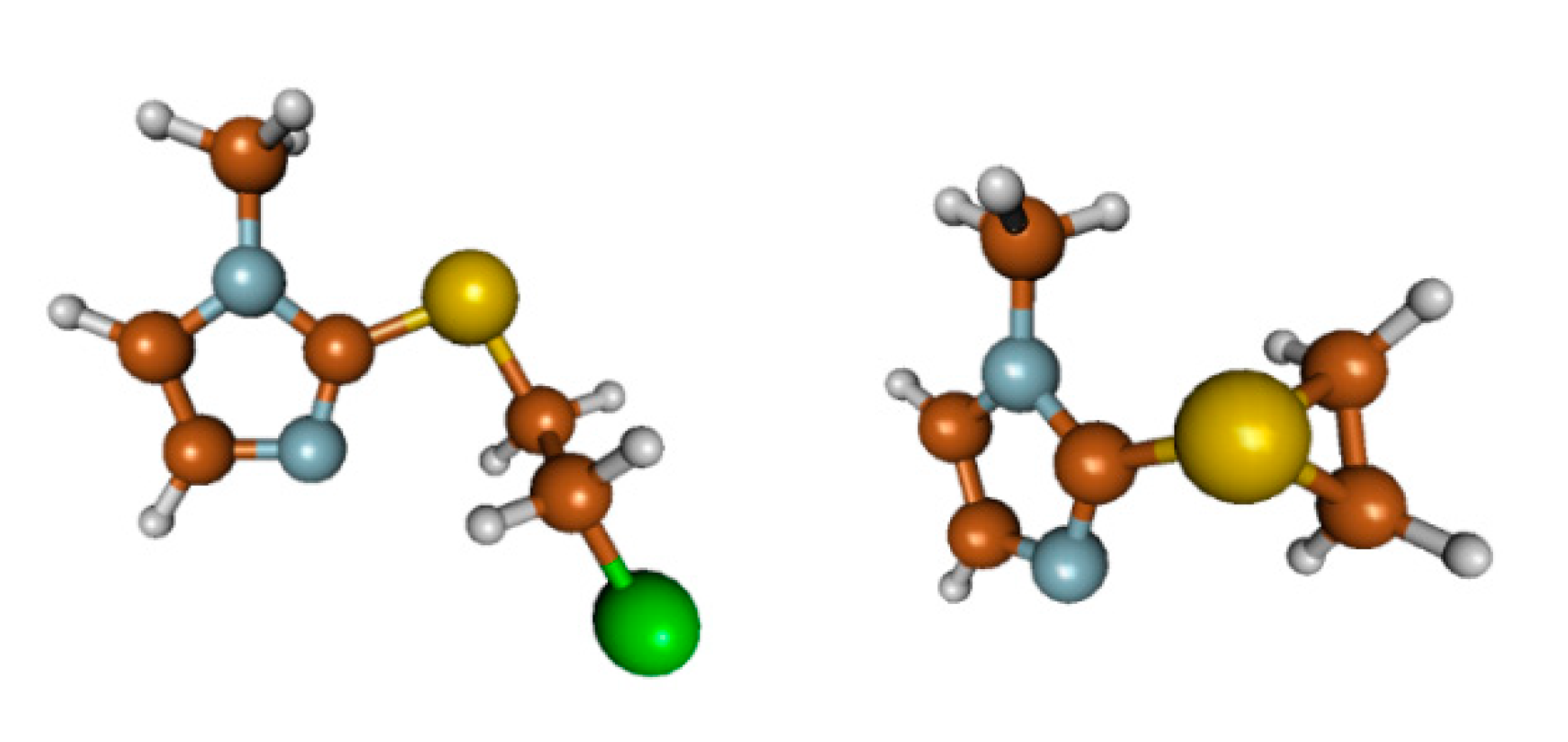
2.4. Computational Search for the Thiiranium Ion Intermediate 5
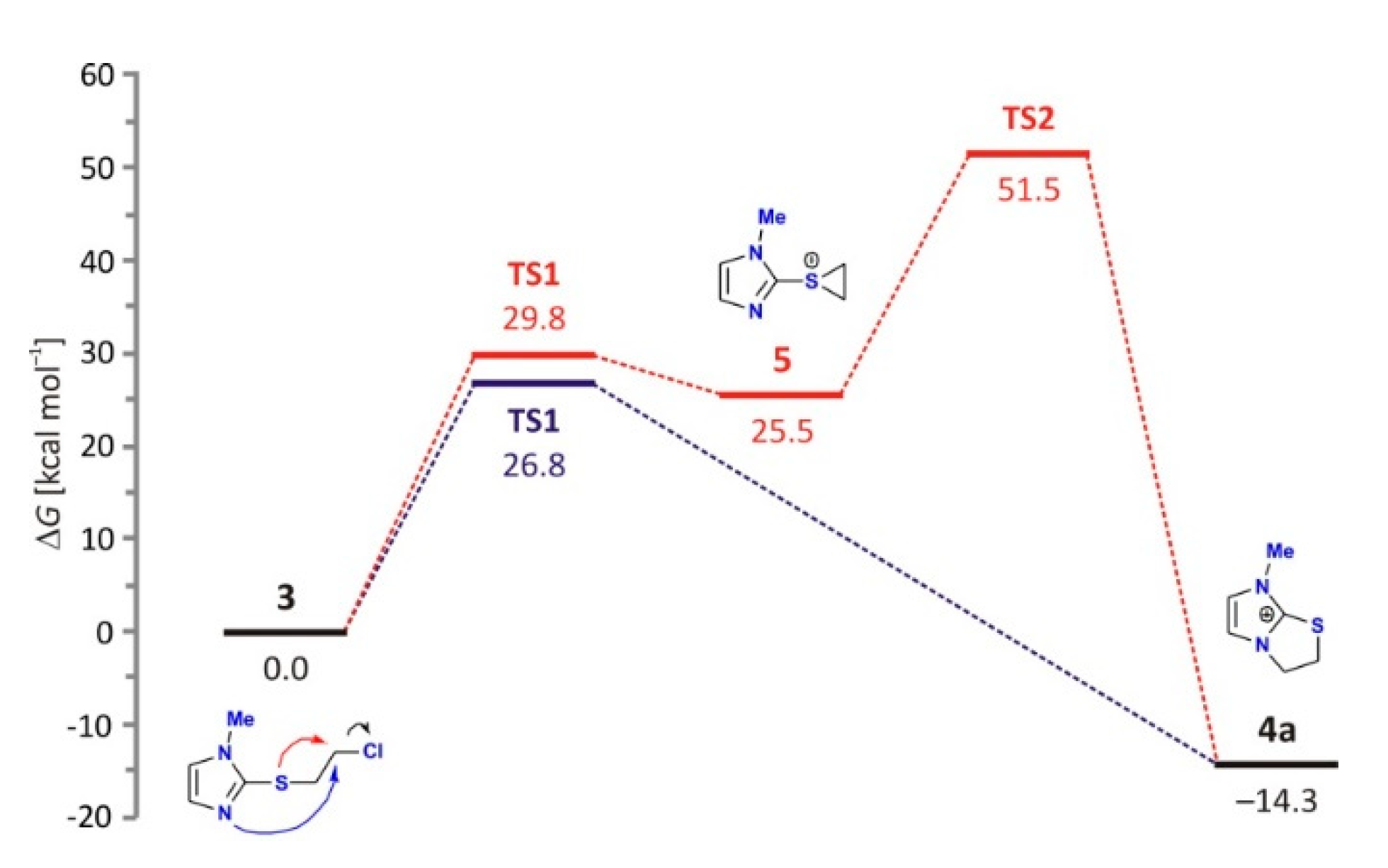
2.4.1. Reaction without External Nucleophiles
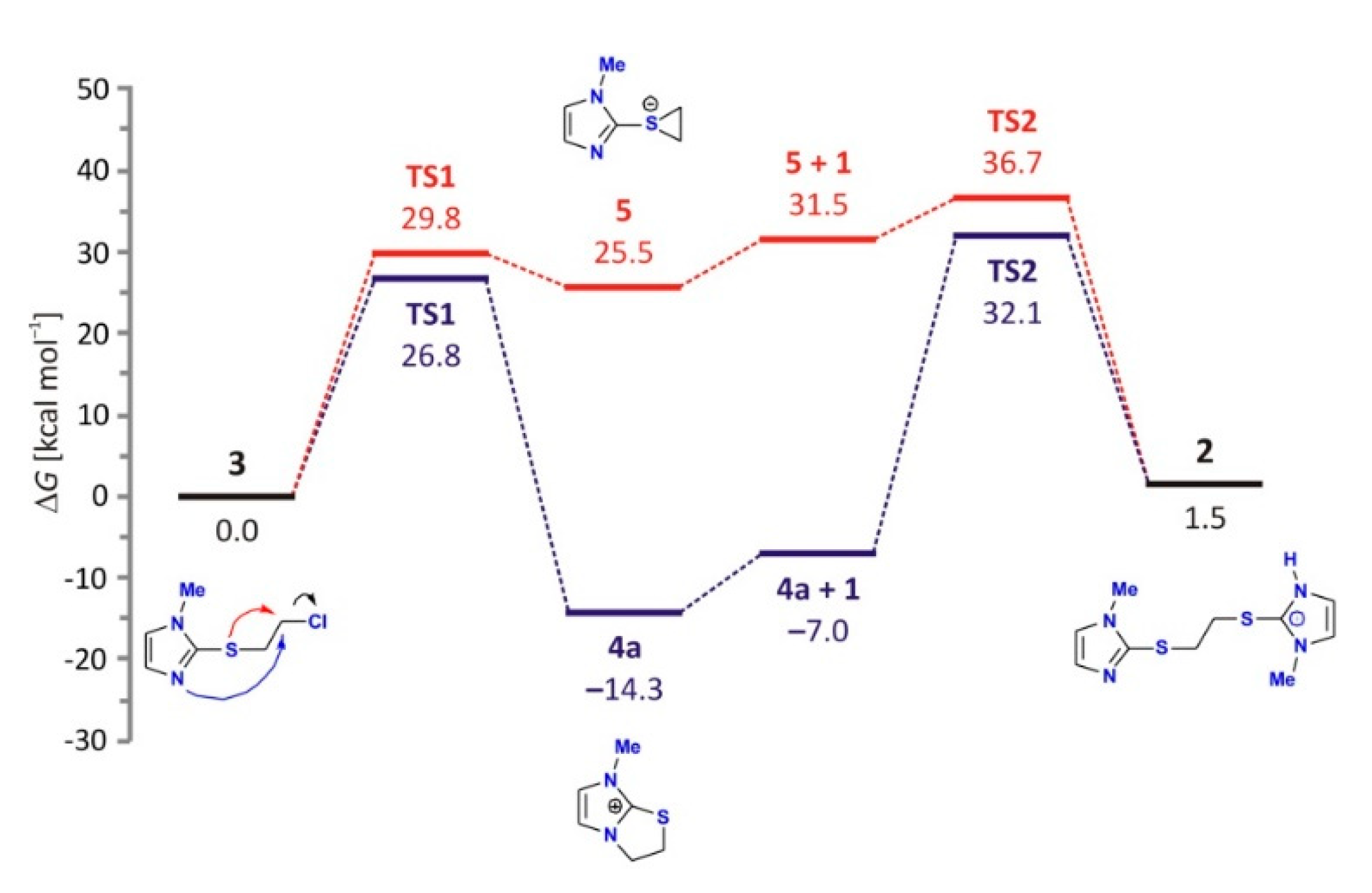
2.4.2. Reaction with the Introduction of External Nucleophile
3. Materials and Methods
3.1. General
3.2. NMR Spectroscopy
3.3. Synthesis
3.3.1. Reaction of Methimazole in 1,2-Dichloroethane at Room Temperature
3.3.2. 2-[(Chloroethyl)thio]-1-methyl-1H-imidazole (3)
3.3.3. Reaction of Methimazole (1) in Boiling Dry 1,2-Dichlorethane
3.3.4. 7-Methyl-2H, 3H, 7H-imidazo[2,1-b]thiazol-4-ium chloride (4a)
3.3.5. 7-Methyl-2H, 3H, 7H -imidazo[2,1-b]thiazol-4-ium tetrafluoroborate (4b)
3.3.6. Bis-{2-[(chloroethyl)thio]-1-methyl-1H-imidazole}-silver(I) Tetrafluoroborate (6)
3.3.7. Reaction of 2-[(Chloroethyl)thio]-1-methyl-1H-imidazole (3) with Methimazole (1)
3.3.8. 1-Chloroethyl-2,3-dihydro-3-methyl-1H-imidazole-2-thione (7)
3.3.9. 2,3-Dihydro-3-methyl-1-[(1-methyl-1H-imidazole-2-yl)thioethyl]-1H-imidazole-2-thione (8)
3.3.10. 1,2-Bis(2,3-dihydro-3-methyl-1H-imidazole-2-thione-1-yl)ethane (9)
3.4. NMR Search for the Thiiranium Intermediate 5
3.4.1. Kinetics of 2-[(Chloroethyl)thio]-1-methyl-1H-imidazole (3) Isomerization in DMSO-d6
3.4.2. Search for Thiiranium Intermediate 5 in the Reaction of 2-[(Chloroethyl)thio]-1-methyl-1H-imidazole 3 with Silver Tetrafluoroborate in Toluene-d8
3.4.3. Search for the Thiiranium Intermediate 5 in the Reaction of 2-[(Chloroethyl)thio]-1-methyl-1H-imidazole 3 with Silver Tetrafluoroborate in DMSO-d6
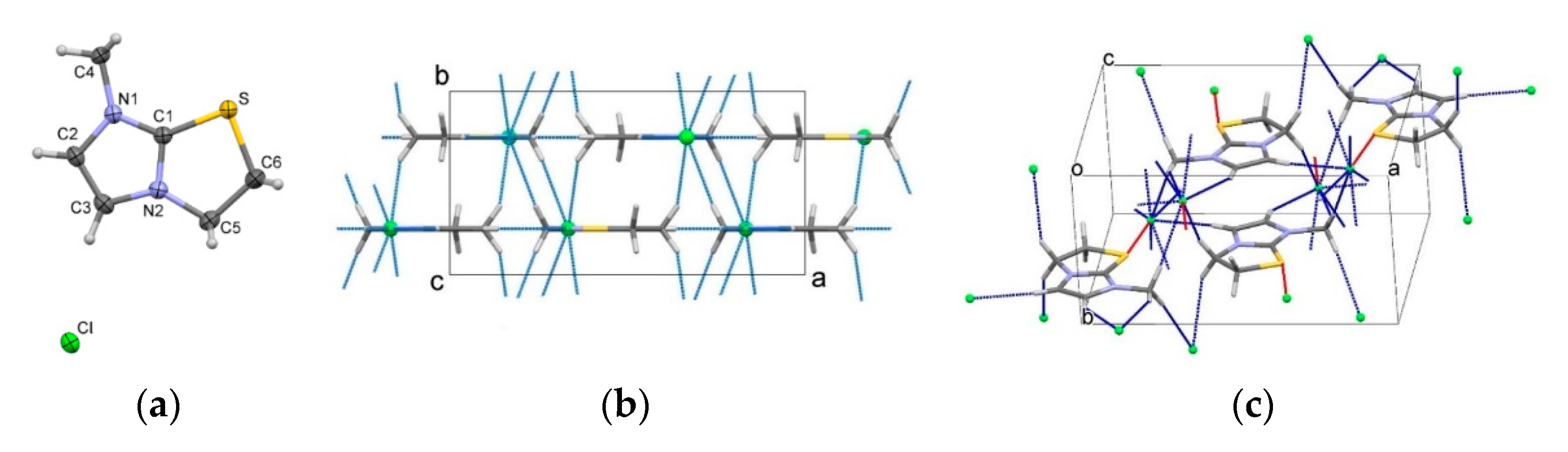
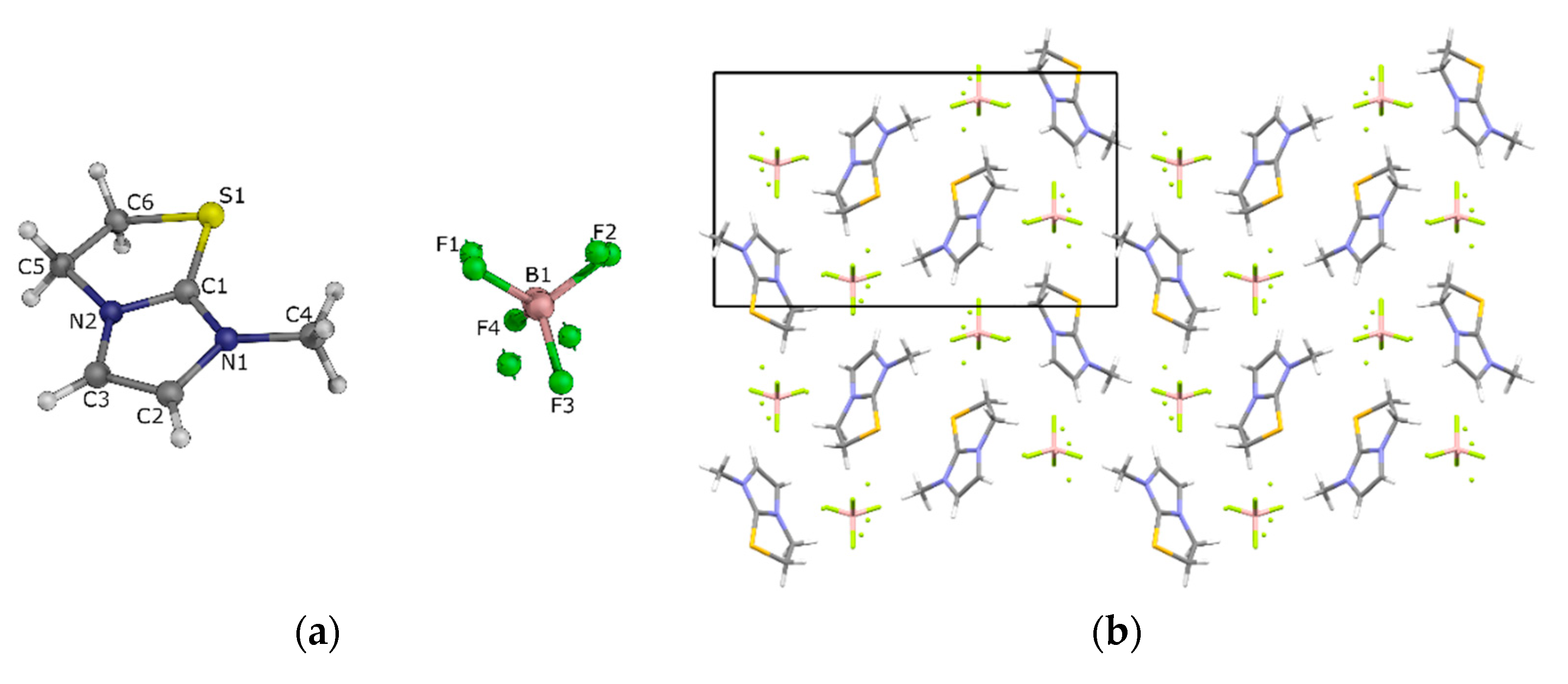
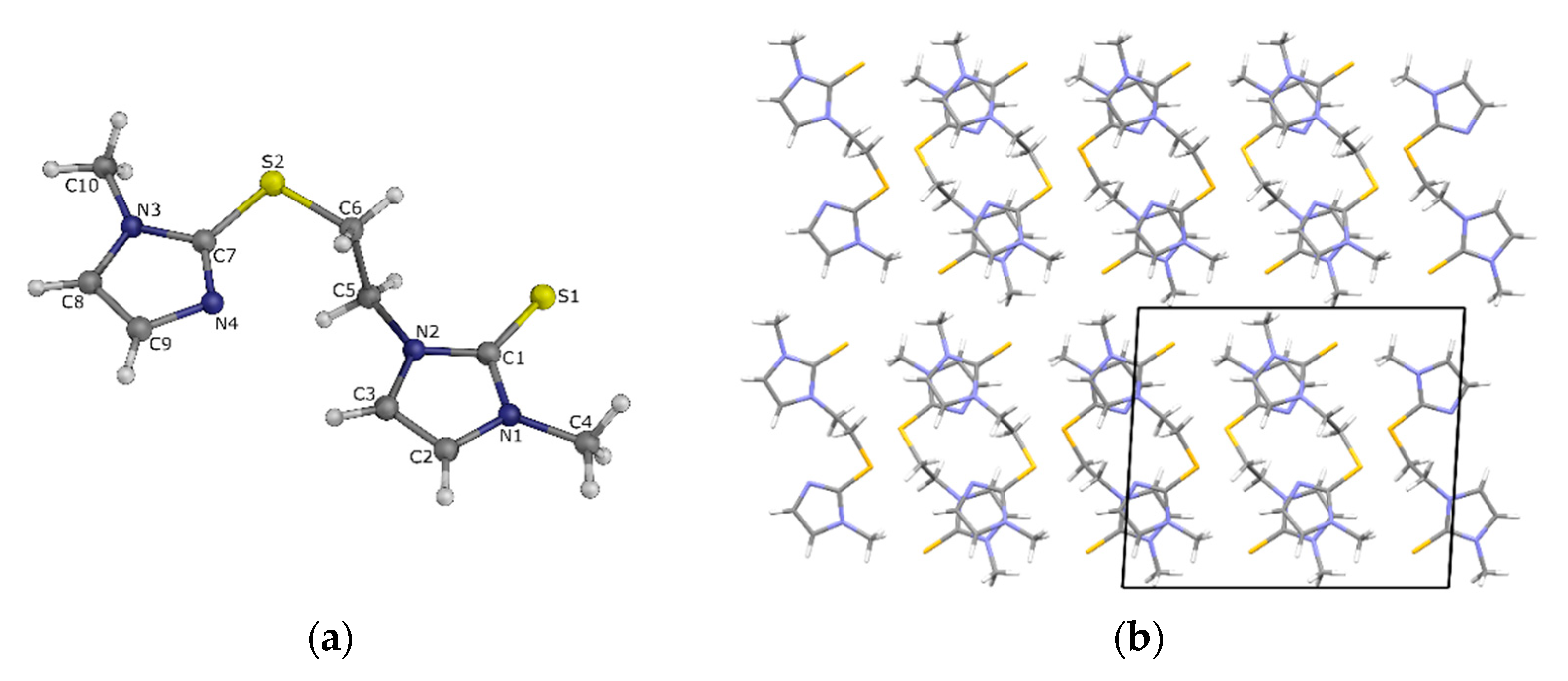
3.5. Single Crystal X-ray Diffraction
3.6. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aboul-Enein, H.Y.; Al-Badr, A.A. Analytical Profiles of Drug Substances; Florey, K., Ed.; Academic Press: New York, NY, USA, 1979; Volume 8, pp. 351–370. [Google Scholar]
- Qingjian, L.; Mingli, S. Synthesis of noncyclic crown ethers with methimazole heterocycle as a terminal group. Youji Huaxue 1992, 12, 509–513. [Google Scholar]
- Qingjian, L.; Mingli, S.; Chongqiu, J.; Fengling, L. Syntheses and coordination properties of bridged bis(methimazole) compounds. Gaodeng Xuexiao Huaxue Xuebao 1992, 13, 328–331. [Google Scholar]
- Štefan, L.; Matković-Čalogović, D.; Filić, D.; Dumić, M. Synthesis, Crystal Structure and Solid State Transformation of 1,2-Bis[(1-methyl-1H-imidazole-2-yl)thio]ethane. Crystals 2020, 10, 667. [Google Scholar] [CrossRef]
- Tafeenko, V.A.; Schenk, H.; Paseshnichenko, K.A.; Aslanov, L.A. 7-Methyl-6-phenylimidazo[2,1-b]thiazolium Iodide. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1996, 52, 729–731. [Google Scholar] [CrossRef]
- Modena, G.; Pasquato, L.; Lucchini, V. Different Approaching Directions of σ and π Nucleophiles to the Sulfur Atom of Thiiranium and Thiirenium Ions. Chem. A Eur. J. 2000, 6, 589–590. [Google Scholar] [CrossRef]
- Destro, R.; Lucchini, V.; Modena, G.; Pasquato, L. X-ray structures and anionotropic rearrangements of Di-tert-butyl-substituted thiiranium and thiirenium ions. A structure-reactivity relationship. J. Org. Chem. 2000, 65, 3367–3370. [Google Scholar] [CrossRef]
- Denmark, S.E.; Collins, W.R.; Cullen, M.D. Observation of Direct Sulfenium and Selenenium Group Transfer from Thiiranium and Seleniranium Ions to Alkenes. J. Am. Chem. Soc. 2009, 131, 3490–3492. [Google Scholar] [CrossRef]
- Sølling, T.I.; Wild, S.B.; Radom, L. Are Pi-Ligand Exchange Reactions of Thiirenium and Thiiranium Ions Feasible? An Ab Initio Investigation. Chem. Eur. J. 2000, 5, 509–514. [Google Scholar] [CrossRef]
- Alom, N.E.; Rina, Y.A.; Li, W. Intermolecular Regio- and Stereoselective Sulfenoamination of Alkenes with Thioimidazoles. Org. Lett. 2017, 19, 6204–6207. [Google Scholar] [CrossRef] [PubMed]
- Enoch, S.J.; Ellison, C.M.; Schultz, T.W.; Cronin, M. A review of the electrophilic reaction chemistry involved in covalent protein binding relevant to toxicity. Crit. Rev. Toxicol. 2011, 41, 783–802. [Google Scholar] [CrossRef] [PubMed]
- Dohn, D.R.; Casida, J.E. Thiiranium ion intermediates in the formation and reactions of S-(2-haloethyl)-l-cysteines. Bioorg. Chem. 1987, 15, 115–124. [Google Scholar] [CrossRef]
- Bamford, C.H.; Tipper, C.F.H. (Eds.) Comprehensive Chemical Kinetics. In The Theory of Kinetics, 1st ed.; Elsevier Scientific Publishing: Amsterdam, Netherlands, 1969; Volume 2. [Google Scholar]
- Denmark, S.E.; Vogler, T. Synthesis and Reactivity of Enantiomerically Enriched Thiiranium Ions. Chem. Eur. J. 2009, 15, 11737–11745. [Google Scholar] [CrossRef]
- Liu, Q.; Shi, D.; Yu, K.; Xu, J. 1,1’-(1,2-Ethanediyl) bis(2,3-di hydro-3-methyl-1H-imidazole-2-thione). Acta Crystallogr. Sect. E Struct. Rep. Online 2003, E59, 356–357. [Google Scholar] [CrossRef]
- Silva, R.M.; Smith, M.D.; Gardinier, J.R. Unexpected New Chemistry of the Bis(thioimidazolyl)methanes. J. Org. Chem. 2005, 70, 8755–8763. [Google Scholar] [CrossRef]
- Converso, A.; Saaidi, P.L.; Sharpless, K.B.; Finn, M.G. Nucleophilic Substitution by Grignard Reagents on Sulfur Mustards. J. Org. Chem. 2004, 69, 7336–7339. [Google Scholar] [CrossRef]
- Guthrie, J.P.; Pike, D.C. Hydration of acylimidazoles: Tetrahedral intermediates in acylimidazole hydrolysis and nucleophilic attack by imidazole on esters. The question of concerted mechanisms for acyl transfers. Can. J. Chem. 1987, 65, 1951–1969. [Google Scholar] [CrossRef]
- Lõkov, M.; Tshepelevitsh, S.; Heering, A.; Plieger, P.G.; Vianello, R.; Leito, I. On the Basicity of Conjugated Nitrogen Heterocycles in Different Media. Eur. J. Org. Chem. 2017, 30, 4475–4489. [Google Scholar] [CrossRef]
- Congdon, W.I.; Edward, J.T. Mono-and diprotonation of N-acylthioureas in aqueous sulfuric acid. J. Am. Chem. Soc. 1972, 94, 6096–6099. [Google Scholar] [CrossRef]
- Tshepelevitsh, S.; Kütt, A.; Lõkov, M.; Kaljurand, I.; Saame, J.; Heering, A.; Plieger, P.G.; Vianello, R.; Leito, I. On the Basicity of Organic Bases in Different Media. Eur. J. Org. Chem. 2019, 2019, 6735–6748. [Google Scholar] [CrossRef]
- CrysAlisPro Software System, version 1.171.39.46; Rigaku Oxford Diffraction: Oxford, UK, 2018.
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, 71, 3–8. [Google Scholar]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; Van De Streek, J.; Wood, P.A. Mercury CSD 2.0—new features for the visualization and investigation of crystal structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar]
- Hok, L.; Vianello, R. Direct metal-free transformation of alkynes to nitriles: Computational evidence for the precise reaction mechanism. Int. J. Mol. Sci. 2021, 22, 3193. [Google Scholar] [CrossRef] [PubMed]
- Ptiček, J.; Hok, L.; Grbčić, P.; Topić, F.; Cetina, M.; Rissanen, K.; Kraljević Pavelić, S.; Vianello, R.; Racané, L. Amidino substituted 2-aminophenols: Biologically important building blocks for the amidino-functionalization of 2-substituted benzoxazoles. Org. Biomol. Chem. 2021, 19, 2784–2793. [Google Scholar] [CrossRef] [PubMed]
- Roca, S.; Hok, L.; Vianello, R.; Borovina, M.; Đaković, M.; Karanović, L.; Vikić-Topić, D.; Popović, Z. The role of non-covalent interactions on dimensionality of the supramolecular structures of silver nitrate complexes with dihalopyridine derivatives and identification of the species in solution. Cryst. Eng. Comm. 2020, 22, 7962–7974. [Google Scholar] [CrossRef]
- Juraj, N.P.; Miletić, G.I.; Perić, B.; Popović, Z.; Smrečki, N.; Vianello, R.; Kirin, S.I. Stereochemistry of hexacoordinated Zn(II), Cu(II), Ni(II), and Co(II) complexes with iminodiacetamide ligands. Inorg. Chem. 2019, 58, 16445–16457. [Google Scholar] [CrossRef]
- Tandarić, T.; Vianello, R. Computational insight into the mechanism of the irreversible inhibition of monoamine oxidase enzymes by the anti-parkinsonian propargylamine inhibitors rasagiline and selegiline. ACS Chem. Neurosci. 2019, 10, 3532–3542. [Google Scholar]
- Maršavelski, A.; Vianello, R. What a difference a methyl group makes: The selectivity of monoamine oxidase B towards histamine and N-methylhistamine. Chem. Eur. J. 2017, 23, 2915–2925. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]



















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Štefan, L.; Čikoš, A.; Vianello, R.; Đilović, I.; Matković-Čalogović, D.; Dumić, M. Chemistry of Spontaneous Alkylation of Methimazole with 1,2-Dichloroethane. Molecules 2021, 26, 7032. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26227032
Štefan L, Čikoš A, Vianello R, Đilović I, Matković-Čalogović D, Dumić M. Chemistry of Spontaneous Alkylation of Methimazole with 1,2-Dichloroethane. Molecules. 2021; 26(22):7032. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26227032
Chicago/Turabian StyleŠtefan, Leo, Ana Čikoš, Robert Vianello, Ivica Đilović, Dubravka Matković-Čalogović, and Miljenko Dumić. 2021. "Chemistry of Spontaneous Alkylation of Methimazole with 1,2-Dichloroethane" Molecules 26, no. 22: 7032. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26227032