
Synthetic and Structural Study of peri-Substituted Phosphine-Arsines

 , , and
, , and
Abstract
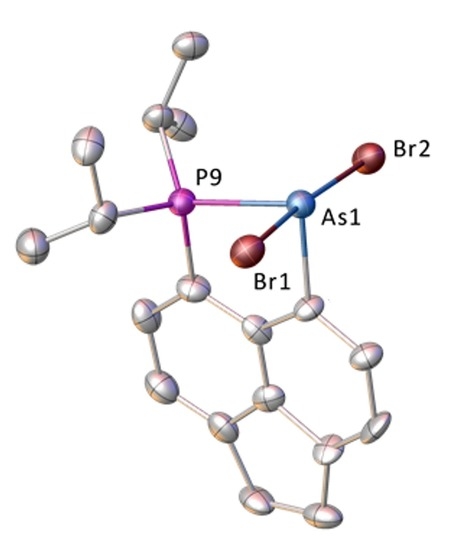
:
1. Dedication
2. Introduction
3. Results & Discussion
3.1. Reactions Involving PBr3 and AsBr3
3.2. Reactions Involving EtAsI2
4. Experimental
4.1. General Considerations
4.2. Synthetic Methods
4.2.1. iPr2PAcenapPBr2 (3)
4.2.2. iPr2PAcenapAsBr2 (4)
4.2.3. [iPr2PAcenapAsBr]+Br–∙AsBr3 (5) and [(iPr2PAcenap)2As]2[As6Br8] (6)
4.2.4. (BrAcenap)2AsEt (7)
4.2.5. Cyclo-PhP(Acenap)2AsEt (8)
4.2.6. (Ph2PAcenap)2AsEt (9)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Peruzzini, M.; Gonsalvi, L. Phosphorus Compounds; Springer: Berlin/Heidelberg, Germany, 2011; Volume 37. [Google Scholar]
- Greenwood, N.N.; Earnshaw, A. Chemistry of the Elements, 2nd ed.; Elsevier: Oxford, UK, 1997. [Google Scholar]
- Crabtree, R.H. The Organometallic Chemistry of the Transition Metals, 5th ed.; John Wiley and Sons Inc.: Hoboken NJ, USA, 2009. [Google Scholar]
- Holmes, R.R.; Bertaut, E.F. The Reduction of Phosphorus and Antimony Chlorides by Trimethylarsine and Trimethylstibine. J. Am. Chem. Soc. 1958, 80, 2983–2985. [Google Scholar] [CrossRef]
- Holmes, R.R.; Bertaut, E.F. The Reactions of Phosphorus and Antimony Chlorides with Trimethylamine, Triethylamine and Trimethylphosphine1. J. Am. Chem. Soc. 1958, 80, 2980–2983. [Google Scholar] [CrossRef]
- Spangenberg, S.F.; Sisler, H.H. Reactions of some tri-n-alkylphosphines with some chlorophosphines. Inorg. Chem. 1969, 8, 1006–1010. [Google Scholar] [CrossRef]
- Muller, G.; Matheus, H.J.; Winkler, M. Donor-Acceptor Complexes between Simple Phosphines. First Structural Data for an Almost Forgotten Class of Compounds. Z. Nat. B J. Chem. Sci. 2001, 56, 1155–1162. [Google Scholar] [CrossRef]
- Wawrzyniak, P.; Fuller, A.L.; Slawin, A.M.Z.; Kilian, P. Intramolecular Phosphine-Phosphine Donor-Acceptor Complexes. Inorg. Chem. 2009, 48, 2500–2506. [Google Scholar] [CrossRef] [PubMed]
- Summers, J.C.; Sisler, H.H. Reactions of some trialkyls of phosphorus, arsenic, or antimony with some organohalophosphines, -arsines, or -stibines. Inorg. Chem. 1970, 9, 862–869. [Google Scholar] [CrossRef]
- Chitnis, S.S.; Burford, N.; McDonald, R.; Ferguson, M.J. Prototypical Phosphine Complexes of Antimony(III). Inorg. Chem. 2014, 53, 5359–5372. [Google Scholar] [CrossRef] [PubMed]
- Hill, N.J.; Levason, W.; Reid, G. Arsenic(iii) halide complexes with phosphine and arsine co-ligands: Synthesis, spectroscopic and structural properties. J. Chem. Soc. Dalton Trans. 2002, 1188–1192. [Google Scholar] [CrossRef]
- Burt, J.; Levason, W.; Reid, G. Coordination chemistry of the main group elements with phosphine, arsine and stibine ligands. Coord. Chem. Rev. 2014, 260, 65–115. [Google Scholar] [CrossRef] [Green Version]
- Robertson, A.P.M.; Gray, P.A.; Burford, N. Interpnictogen Cations: Exploring New Vistas in Coordination Chemistry. Angew. Chem. Int. Ed. 2014, 53, 6050–6069. [Google Scholar] [CrossRef] [PubMed]
- Chitnis, S.S.; Burford, N. Phosphine complexes of lone pair bearing Lewis acceptors. Dalton Trans. 2015, 44, 17–29. [Google Scholar] [CrossRef]
- Chalmers, B.A.; Bühl, M.; Athukorala Arachchige, K.S.; Slawin, A.M.Z.; Kilian, P. Geometrically Enforced Donor-Facilitated Dehydrocoupling Leading to an Isolable Arsanylidine-Phosphorane. J. Am. Chem. Soc. 2014, 136, 6247–6250. [Google Scholar] [CrossRef] [Green Version]
- Chalmers, B.A.; Bühl, M.; Athukorala Arachchige, K.S.; Slawin, A.M.Z.; Kilian, P. Structural, Spectroscopic and Computational Examination of the Dative Interaction in Constrained Phosphine–Stibines and Phosphine–Stiboranes. Chem. Eur. J. 2015, 21, 7520–7531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hupf, E.; Lork, E.; Mebs, S.; Chęcińska, L.; Beckmann, J. Probing Donor–Acceptor Interactions in peri-Substituted Diphenylphosphinoacenaphthyl–Element Dichlorides of Group 13 and 15 Elements. Organometallics 2014, 33, 7247–7259. [Google Scholar] [CrossRef]
- Carmalt, C.J.; Cowley, A.H.; Culp, R.D.; Jones, R.A.; Kamepalli, S.; Norman, N.C. Synthesis and Structures of Intramolecularly Base-Coordinated Group 15 Aryl Halides. Inorg. Chem. 1997, 36, 2770–2776. [Google Scholar] [CrossRef] [PubMed]
- Taylor, L.J.; Bühl, M.; Wawrzyniak, P.; Chalmers, B.A.; Woollins, J.D.; Slawin, A.M.Z.; Fuller, A.L.; Kilian, P. Hydride Abstraction and Deprotonation—An Efficient Route to Low Coordinate Phosphorus and Arsenic Species. Eur. J. Inorg. Chem. 2016, 2016, 659–666. [Google Scholar] [CrossRef] [Green Version]
- Ray, M.J.; Slawin, A.M.Z.; Buehl, M.; Kilian, P. peri-Substituted Phosphino-Phosphonium Salts: Synthesis and Reactivity. Organometallics 2013, 32, 3481–3492. [Google Scholar] [CrossRef]
- Taylor, L.J.; Buhl, M.; Chalmers, B.A.; Ray, M.J.; Wawrzyniak, P.; Walton, J.C.; Cordes, D.B.; Slawin, A.M.Z.; Woollins, J.D.; Kilian, P. Dealkanative Main Group Couplings across the peri-Gap. J. Am. Chem. Soc. 2017, 139, 18545–18551. [Google Scholar] [CrossRef] [Green Version]
- Nejman, P.S.; Curzon, T.E.; Bühl, M.; McKay, D.; Woollins, J.D.; Ashbrook, S.E.; Cordes, D.B.; Slawin, A.M.Z.; Kilian, P. Phosphorus–Bismuth Peri-Substituted Acenaphthenes: A Synthetic, Structural, and Computational Study. Inorg. Chem. 2020, 59, 5616–5625. [Google Scholar] [CrossRef]
- Kilian, P.; Slawin, A.M.Z.; Woollins, J.D. Preparation and structures of 1,2-dihydro-1,2-diphosphaacenaphthylenes and rigid backbone stabilized triphosphenium cation. Dalton Trans. 2006, 2175–2183. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, B.A.; Meigh, C.B.E.; Nejman, P.S.; Bühl, M.; Lébl, T.; Woollins, J.D.; Slawin, A.M.Z.; Kilian, P. Geminally Substituted Tris(acenaphthyl) and Bis(acenaphthyl) Arsines, Stibines, and Bismuthine: A Structural and Nuclear Magnetic Resonance Investigation. Inorg. Chem. 2016, 55, 7117–7125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furan, S.; Hupf, E.; Boidol, J.; Brunig, J.; Lork, E.; Mebs, S.; Beckmann, J. Transition metal complexes of antimony centered ligands based upon acenaphthyl scaffolds. Coordination non-innocent or not? Dalton Trans. 2019, 48, 4504–4513. [Google Scholar] [CrossRef]
- Gabes, W.; Olie, K. Refinement of the crystal structure of phosphorus pentabromide, PBr5. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1970, 26, 443–444. [Google Scholar] [CrossRef]
- Hubert, S.; Wolfgang, B.; Brigitte, H.; Gerhard, M. Arene Adducts with Weak Interactions: Hexaethylbenzene–bis(tribromoarsane). Angew. Chem. Int. Ed. Engl. 1987, 26, 234–236. [Google Scholar] [CrossRef]
- The WBI is a measure for the covalent character of a bond and adopts values close to 1 and 2 for true single and double bonds, respectively: Wiberg, K.B. Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [CrossRef]
- Müller, U.; Sinning, H. Octabromocyclohexaarsenate, [As6Br8]2⊖. Angew. Chem. Int. Ed. Engl. 1989, 28, 185–186. [Google Scholar] [CrossRef]
- Ellis, B.D.; Macdonald, C.L.B. Stabilized Arsenic(I) Iodide: A Ready Source of Arsenic Iodide Fragments and a Useful Reagent for the Generation of Clusters. Inorg. Chem. 2004, 43, 5981–5986. [Google Scholar] [CrossRef] [PubMed]
- Batsanov, S.S. Van der Waals Radii of Elements. Inorg. Mater. 2001, 37, 871–885. [Google Scholar] [CrossRef]
- Tanaka, N.; Kasai, T. Reactions of 5,6-Dilithioacenaphthene-N,N,N′,N′-Tetramethyl-1,2-ethanediamine Complex with α-Diketones. I.cis-Directing 1:1 Cyclic Additions with Acyclic and Cyclic α-Diketones and Related Compounds. Bull. Chem. Soc. Jpn. 1981, 54, 3020–3025. [Google Scholar] [CrossRef] [Green Version]
- Armarego, W.L.F.; Chai, C.L.L. Purification of Laboratory Chemicals, 6th ed.; Elsevier: Burlington, VT, USA, 2009. [Google Scholar]
- Neudorff, W.D.; Lentz, D.; Anibarro, M.; Schlüter, A.D. The Carbon Skeleton of the Belt Region of Fullerene C84 (D2). Chem. Eur. J. 2003, 9, 2745–2757. [Google Scholar] [CrossRef]
- Arnaiz, F.J.; Miranda, M.J.; Rheingold, A.L. Arsenic (III) Bromide. Inorg. Synth. 2002, 33, 203. [Google Scholar] [CrossRef]
- Matuska, V.; Slawin, A.M.Z.; Woollins, J.D. Five-Membered Arsenic−Sulfur−Nitrogen Heterocycles, RAs(S2N2) (R = Me, Et, iPr, tBu, Ph, Mes). Inorg. Chem. 2010, 49, 3064–3069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalmers, B.A.; Somisara, D.M.U.K.; Surgenor, B.A.; Athukorala Arachchige, K.S.; Woollins, J.D.; Buehl, M.; Slawin, A.M.Z.; Kilian, P. Synthetic and Structural Study of peri-Substituted Phosphine-Arsines (Dataset); University of St Andrews Research Portal: St Andrews, UK, 2021. [Google Scholar] [CrossRef]
- CrystalClear 2.0; Rigaku Americas: The Woodlands, TX, USA; Rigaku Corporation: Tokyo, Japan, 2007.
- CrystalClear Software User’s Guide; Molecular Structure Corportation, 2017.
- Sheldrick, G. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CrystalStructure 4.3.0; Rigaku Americas: The Woodlands, TX, USA; Rigaku Corporation: Tokyo, Japan, 2018.
- Bruno, I.J.; Cole, J.C.; Edgington, P.R.; Kessler, M.; Macrae, C.F.; McCabe, P.; Pearson, J.; Taylor, R. New software for searching the Cambridge Structural Database and visualizing crystal structures. Acta Crystallogr. Sect. B 2002, 58, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Thomas, I.R.; Bruno, I.J.; Cole, J.C.; Macrae, C.F.; Pidcock, E.; Wood, P.A. WebCSD: The online portal to the Cambridge Structural Database. J. Appl. Crystallogr. 2010, 43, 362–366. [Google Scholar] [CrossRef] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Risthaus, T.; Grimme, S. Benchmarking of London Dispersion-Accounting Density Functional Theory Methods on Very Large Molecular Complexes. J. Chem. Theory Comput. 2013, 9, 1580–1591. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D.; Johnson, E.R. Exchange-hole dipole moment and the dispersion interaction. J. Chem. Phys. 2005, 122, 154104. [Google Scholar] [CrossRef]
- Johnson, E.R.; Becke, A.D. A post-Hartree-Fock model of intermolecular interactions: Inclusion of higher-order corrections. J. Chem. Phys. 2006, 124, 174104. [Google Scholar] [CrossRef] [PubMed]
- Binning, R.C.; Curtiss, L.A. Compact contracted basis sets for third-row atoms: Ga–Kr. J. Comput. Chem. 1990, 11, 1206–1216. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 3·THF (E = P) | 4·C6H6 [b] (E = As) | 5 (E = As) | 6 [c] (E = As) |
| peri-region bond distances | ||||
| P9–E1 | 2.2701(16) | 2.405(3) [2.407(3)] | 2.3978(15) | 2.365(4) [3.145(4)] |
| E1–Br1 | 2.6864(13) | 2.6337(19) [2.6639(19)] | 2.4375(9) | – |
| E1–Br2 | 2.4376(13) | 2.6400(18) [2.634(2)] | 3.000(9) | – |
| peri-region angles | ||||
| C9–P9–E1 | 97.92(14) | 97.9(3) [98.4(3)] | 97.78(18) | 98.7(4) |
| C1–E1–P9 | 88.82(13) | 84.9(3) [85.3(3)] | 86.25(17) | 85.7(3) |
| Br1–E1–Br2 | 172.22(5) | 173.95(6) [172.27(6)] | 175.3(2) | – |
| P9–E1–Br1 | 91.13(5) | 96.01(9) [91.02(9)] | 95.57(4) | – |
| P9–E1–Br2 | 96.38(5) | 90.00(9) [95.95(9)] | 88.77(4) | – |
| splay angle [a] | –7.8(5) | –5(1) [–5(1)] | –4(1) | –5(1) [15(1)] |
| out-of-plane displacements | ||||
| P9 | 0.23 | 0.07 [0.10] | 0.09 | 0.21 [0.12] |
| E1 | –0.03 | –0.36 [–0.26] | –0.01 | –0.11 [–0.05] |
| dihedral angles | ||||
| P9–C9···C1–E | 7.7(2) | 10.9(4) [9.2(4)] | 2.4(2) | 2.8(5) [1.2(5)] |
| Compound | 71/2CH2Cl2 (E = Br) | 8∙CH2Cl2 (E = P) | 9-O (E = P) [f] | |
| peri-region bond distances | ||||
| As1–E9 | 3.23(1) | 3.004(6) | 3.176(5) | |
| As1–E29 | 3.28(1) | – | 3.273(5) | |
| peri-region angles | ||||
| splay angle [a,d] | 17(1) | 11.7(4) | 16.1(8) | |
| splay angle [a,e] | 19(1) | 11.4(4) | 15.2(8) | |
| out-of-plane displacements | ||||
| As1 | 0.35, 0.02 | 0.19, 0.02 | 0.04, 0.71 | |
| E9 | –0.08 | −0.03 | −0.40 | |
| E29 | –0.01 | −0.23 | 0.41 | |
| dihedral angles | ||||
| As1–C1···C9–E9 | 12(1) | 3.3(6) | 8(1) | |
| As1–C21···C29–E29 | 1(1) | 5.0(6) | 27(1) | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chalmers, B.A.; Somisara, D.M.U.K.; Surgenor, B.A.; Athukorala Arachchige, K.S.; Woollins, J.D.; Bühl, M.; Slawin, A.M.Z.; Kilian, P. Synthetic and Structural Study of peri-Substituted Phosphine-Arsines. Molecules 2021, 26, 7222. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26237222
Chalmers BA, Somisara DMUK, Surgenor BA, Athukorala Arachchige KS, Woollins JD, Bühl M, Slawin AMZ, Kilian P. Synthetic and Structural Study of peri-Substituted Phosphine-Arsines. Molecules. 2021; 26(23):7222. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26237222
Chicago/Turabian StyleChalmers, Brian A., D. M. Upulani K. Somisara, Brian A. Surgenor, Kasun S. Athukorala Arachchige, J. Derek Woollins, Michael Bühl, Alexandra M. Z. Slawin, and Petr Kilian. 2021. "Synthetic and Structural Study of peri-Substituted Phosphine-Arsines" Molecules 26, no. 23: 7222. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26237222