
Visible-Light-Induced Catalytic Selective Halogenation with Photocatalyst


1
Department of Nuclear Medicine, Molecular Imaging & Therapeutic Medicine Research Center, Jeonbuk National University Medical School and Hospital, Jeonju 54907, Korea
2
Research Institute of Clinical Medicine of Jeonbuk National University, Biomedical Research Institute of Jeonbuk National University Hospital, Jeonju 54907, Korea
3
Department of Applied Chemical Engineering, Korea University of Technology and Education, Cheonan 31253, Korea
*
Author to whom correspondence should be addressed.
Molecules 2021, 26(23), 7380; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26237380
Submission received: 10 November 2021
/
Revised: 29 November 2021
/
Accepted: 2 December 2021
/
Published: 5 December 2021
(This article belongs to the Special Issue Photoredox Catalysis for Sustainable Chemical Synthesis/Medicinal Chemistry)
{kind=link}
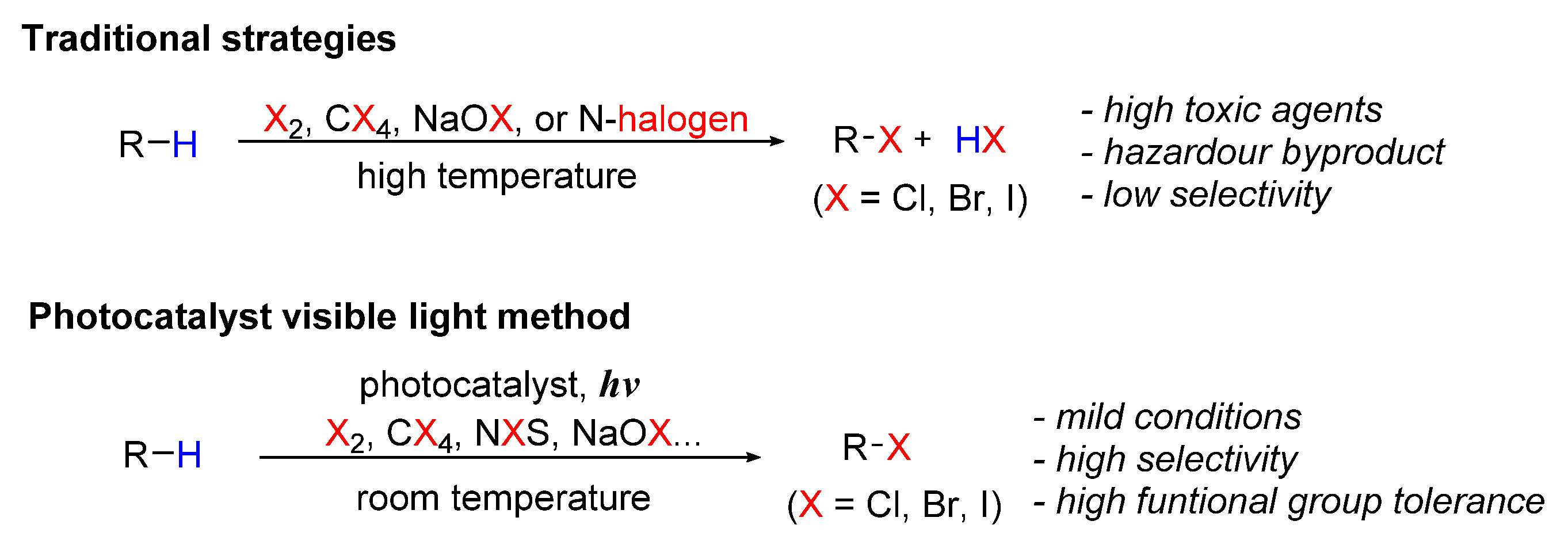{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Halide moieties are essential structures of compounds in organic chemistry due to their popularity and wide applications in many fields such as natural compounds, agrochemicals, and pharmaceuticals. Thus, many methods have been developed to introduce halides into various organic molecules. Recently, visible-light-driven reactions have emerged as useful methods of organic synthesis. Particularly, halogenation strategies using visible light have significantly improved the reaction efficiency and reduced toxicity, as well as promoted reactions under mild conditions. In this review, we have summarized recent studies in visible-light-mediated halogenation (chlorination, bromination, and iodination) with photocatalysts.
1. Introduction
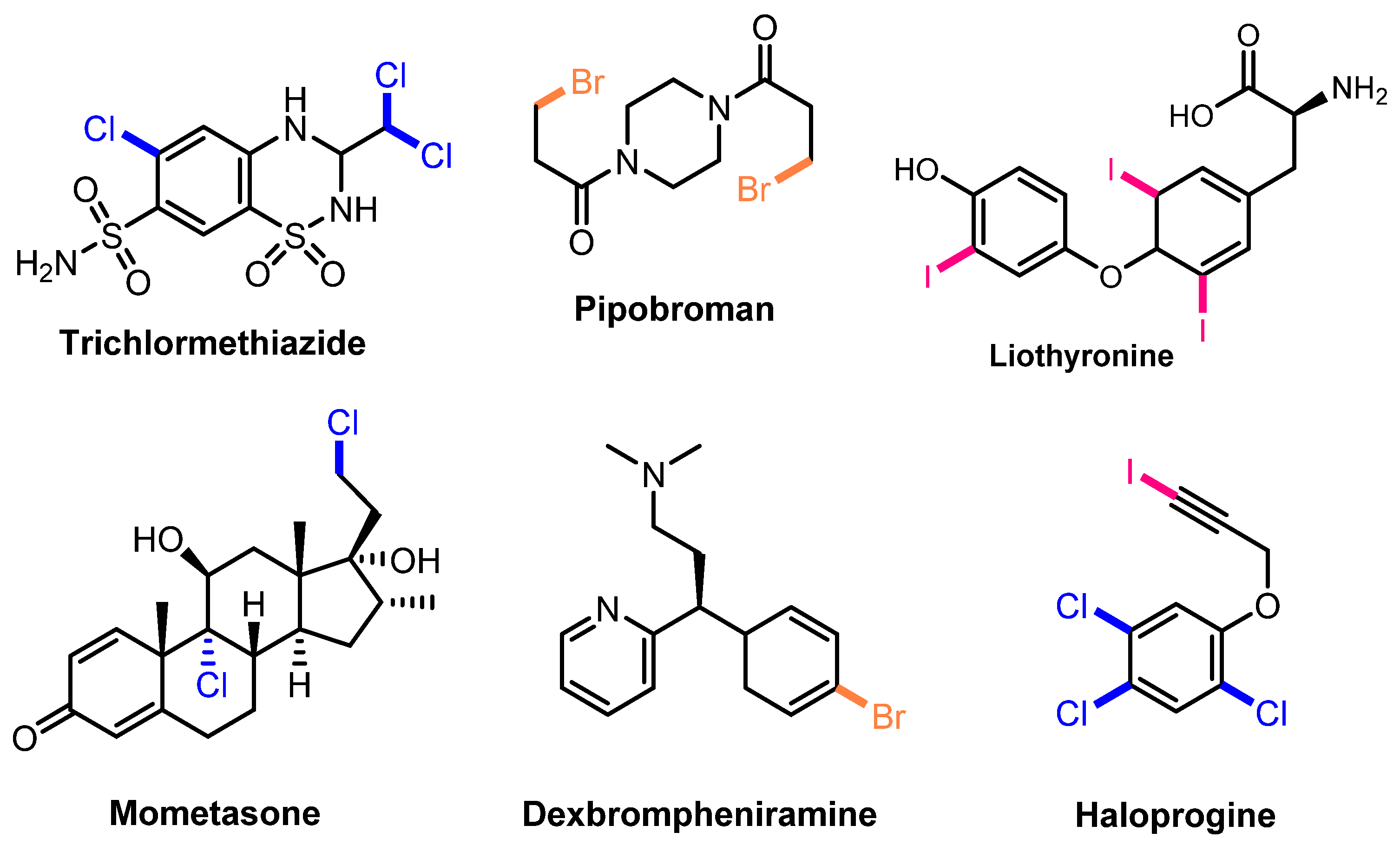
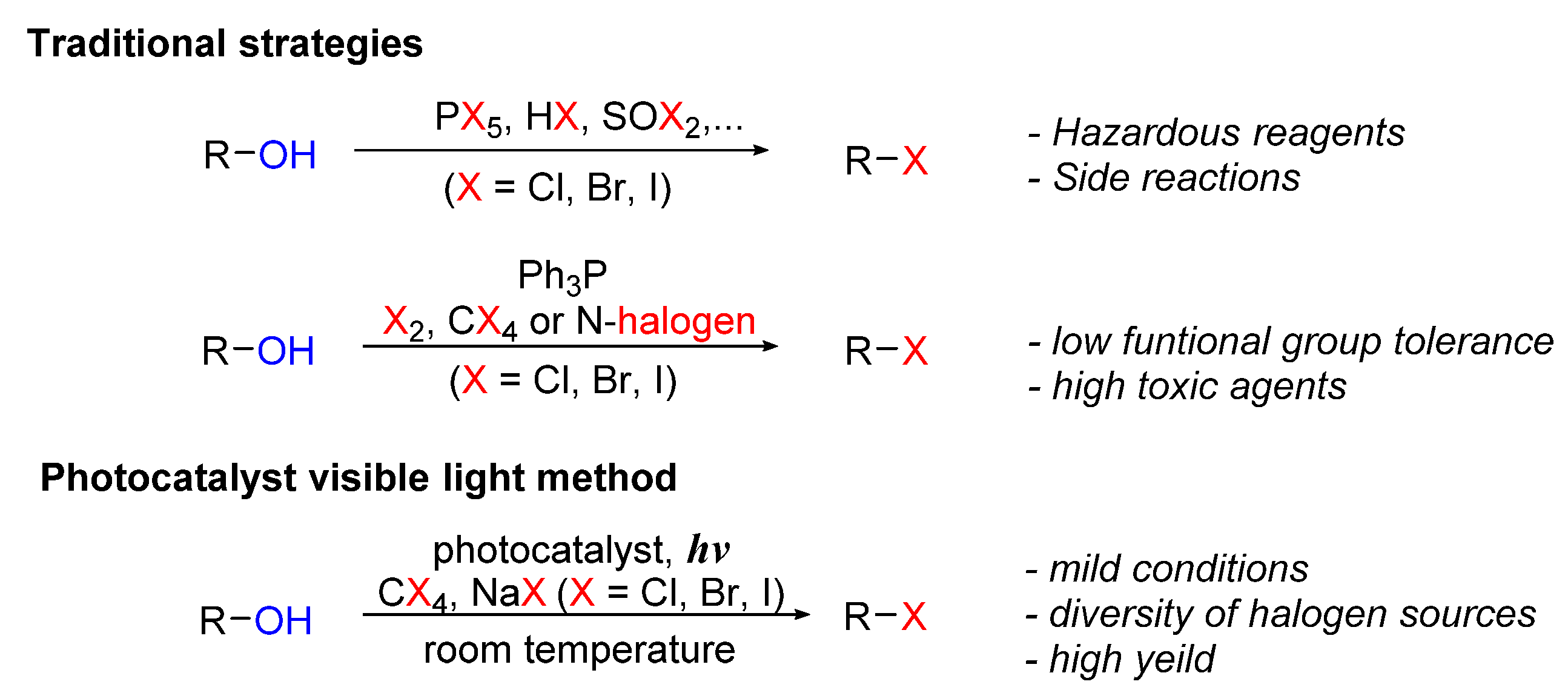
Halogenation is one of the most important modifications in organic synthesis because of its extremely wide applications. Halogen derivatives are useful building blocks in organic synthesis for the construction of complicated, high-activity molecules [1,2,3]. Moreover, as halogenation can be applied to a wide variety of organic compounds without altering their basic structures, halogen-substituted compounds have become popular intermediates for transformation to create different functional groups [4,5,6]. Various areas such as pharmaceuticals, material sciences, industrial chemicals, and bioactive compounds have all benefited from halogen-containing compounds [7,8,9,10]. So far, more than 5000 halogenated natural compounds have been identified, with several of them exhibiting intriguing pharmacological characteristics (Figure 1) [11]. Thus, developing halogenation methods is an interesting area of research, which has received the attention of scientists for decades.
Traditional halogenation methods include addition reactions to multiple bonds, nucleophilic substitution, or radical substitution reactions [12,13,14,15,16,17,18]. One of the most fundamental halogenation reactions in organic chemistry is the addition of halide reagents to C-C multiple bonds. Halogen electrophiles are the most common type of electrophile, and they are commonly employed to generate electrophilic addition reactions to unsaturated carbon [12]. Multiple-bond compounds (alkenes and alkynes) are easily transformed directly to halogenated products by reacting with halogen molecules or hydrohalic acids. However, this technique has significant weaknesses, such as low selectivity, extremely volatility, and the toxic nature of some halogens, and environmental risks [13].
On the other hand, electrophilic substitution and radical substitution reactions are the most feasible and well-recognized approaches for the production of aryl halides, alkyl halides, and many other halide compounds [14]. To produce a carbon-halogen bond, the C-H bond was broken, and then the hydrogen atom was replaced by a halide anion or radical. Halide substitution reactions often require harsh reaction conditions such as high temperature, inert pressure, or an excess of halogen agent and initiator compounds [13,14,15,16,17,18,19,20]. These requirements have increased the purification and treatment costs of the actual halogenation process.
Several classic or modified halogen sources including N-bromo- and N-chlorosuccinimide, plus Selectfluor for halogenation, have also been used. However, the existence of special reagents has limited the application scope and decreased functional group tolerance. Besides, these reactions have been generally carried out under difficult circumstances with poor atom economy [13,14].
Photocatalysis refers to chemical reactions that use light as an energy source. Under irradiation of light, the ground state photocatalyst receives or releases one electron to transfer to the excited state, which subsequently interacts with the substrates or reagents to cause chemical reactions. Generally, photocatalysts can be divided into three main types, including metal complexes, organic dyes, and heterogeneous catalysts [21,22].
In recent years, visible light photocatalysis has emerged as an effective alternative in organic synthesis. Many studies have demonstrated the effectiveness of the visible-light-mediated method in overcoming the inherent disadvantages of traditional organic synthesis methods, such as proceeding under milder reaction conditions, reducing the amount of initiator, introducing outstanding functional group tolerance, and maintaining good regioselectivity [23]. Using photocatalysts for halogenation improves selectivity, allows better reaction control, and lowers costs [24,25].
Many new discoveries in halogenation utilizing visible light via photoredox catalysis have been made in the last decade, and many positive results from reactions with a variety of substrates involving alkyl, aryl, alcohol, carboxyl, etc., have been achieved. In this review, recent advances in halogenation (chlorination, bromination, and iodination) of a variety of organic molecules via photocatalysis are presented.
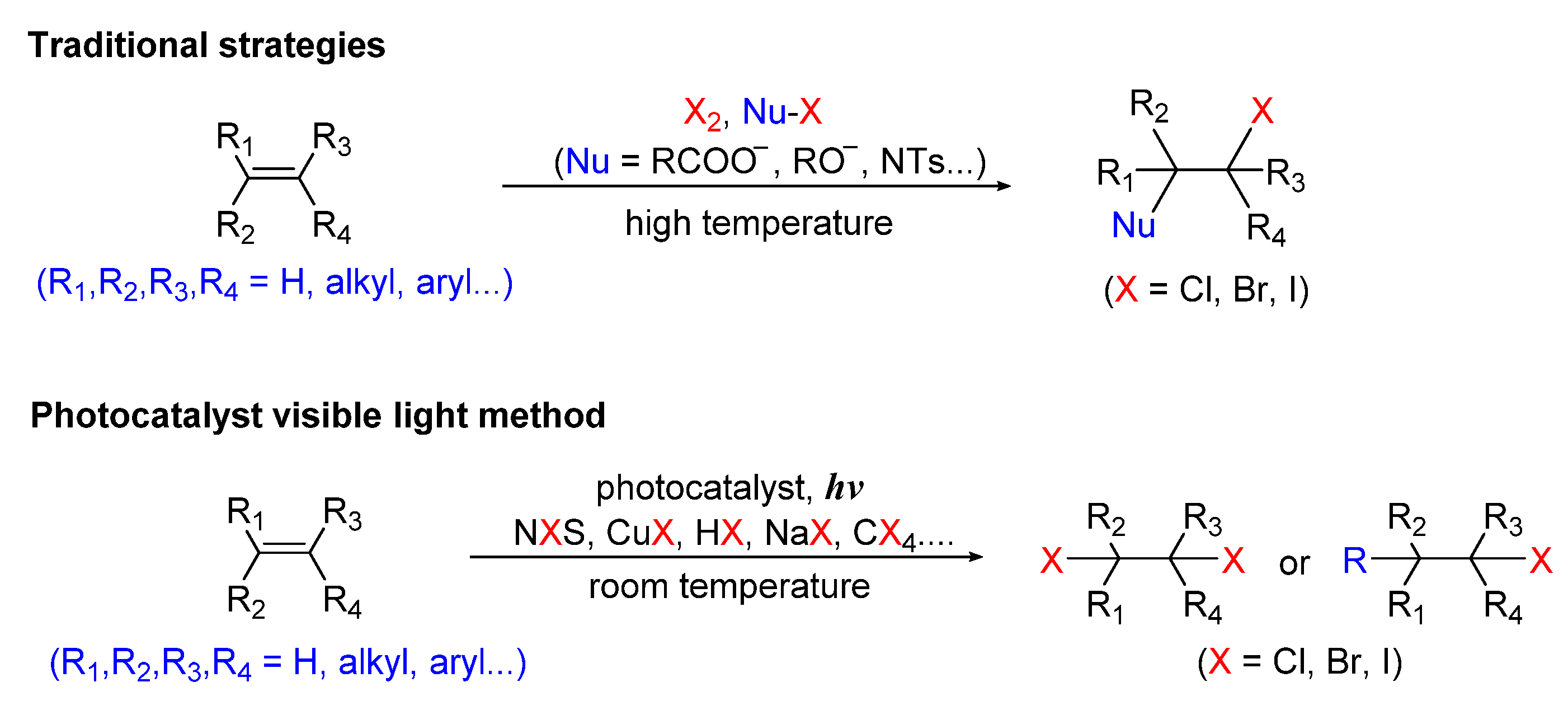
2. Photo-Catalyzed Halogenation of Aliphatic C-H Bonds
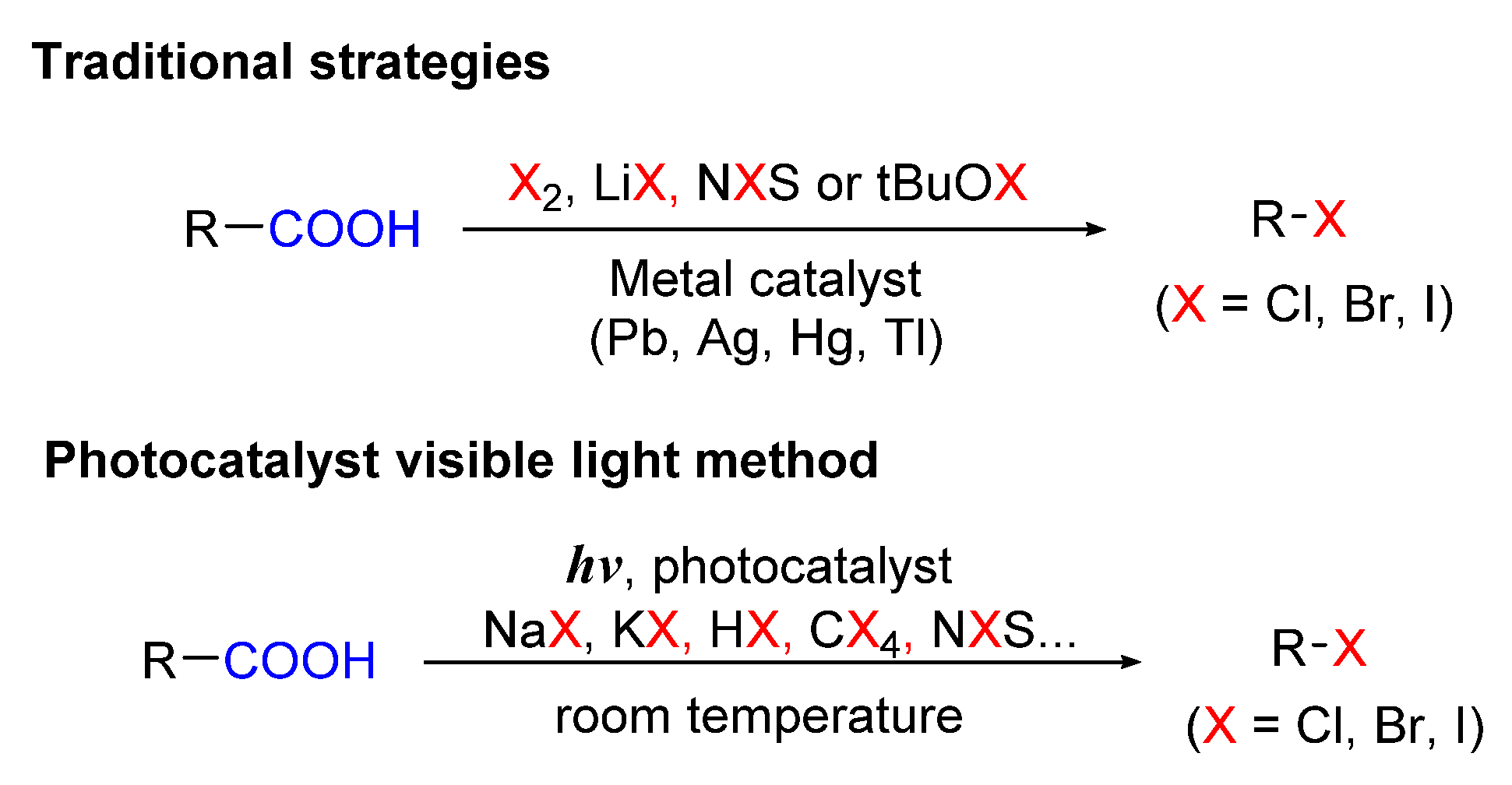
Scheme 1 shows schematic diagrams of the comparison of the traditional methods with visible-light-induced halogenation of aliphatic C-H bonds.
2.1. Chlorination of Aliphatic C-H bBonds
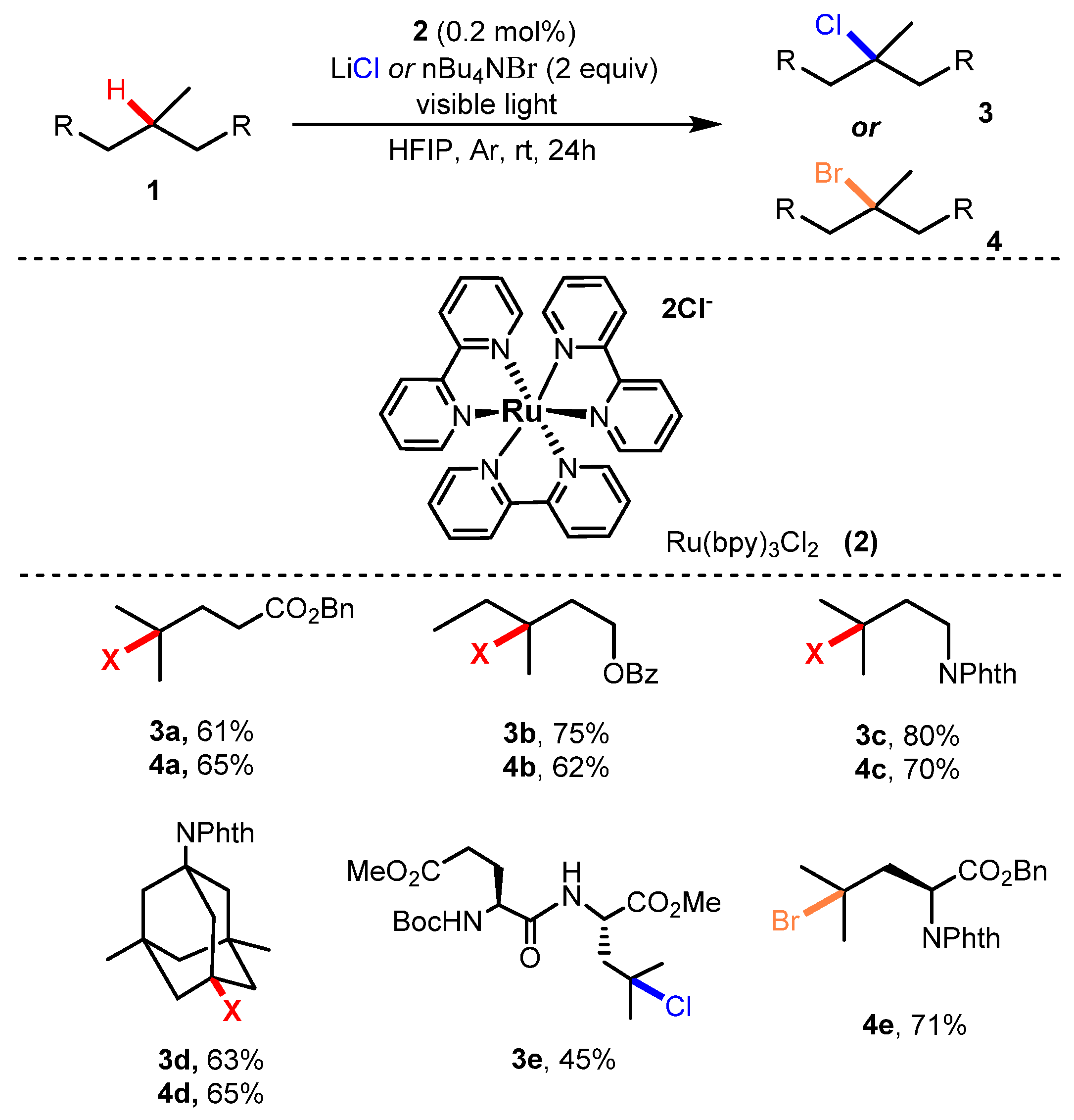
In 2016, Gong Chen and co-workers developed nucleophilic halogenation of tertiary aliphatic C-H bonds [26]. In the reaction, starting substance 1 reacted with LiCl as a chlorinating source in the presence of (Ru(bpy)3Cl2) 2 as a photocatalyst and azidoiodane in hexafluoroisopropanol (HFIP) under the irradiation of a fluorescent bulb at room temperature to give the corresponding products (Scheme 2). This protocol successfully demonstrated site-selectivity for specific tertiary C-H bonds and functional group tolerance. Substrates bearing functional groups such as ester (3a, 4a), ether (3b, 4b), and amide (3c–3e; 4c–4e) provided the corresponding products with moderate to excellent yields (45–80%). This method was also applied for bromination of tertiary C-H bonds. n-Bu4NBr was employed as a brominating source, and the bromination was proved to be more efficient than chlorination and showed better yields.
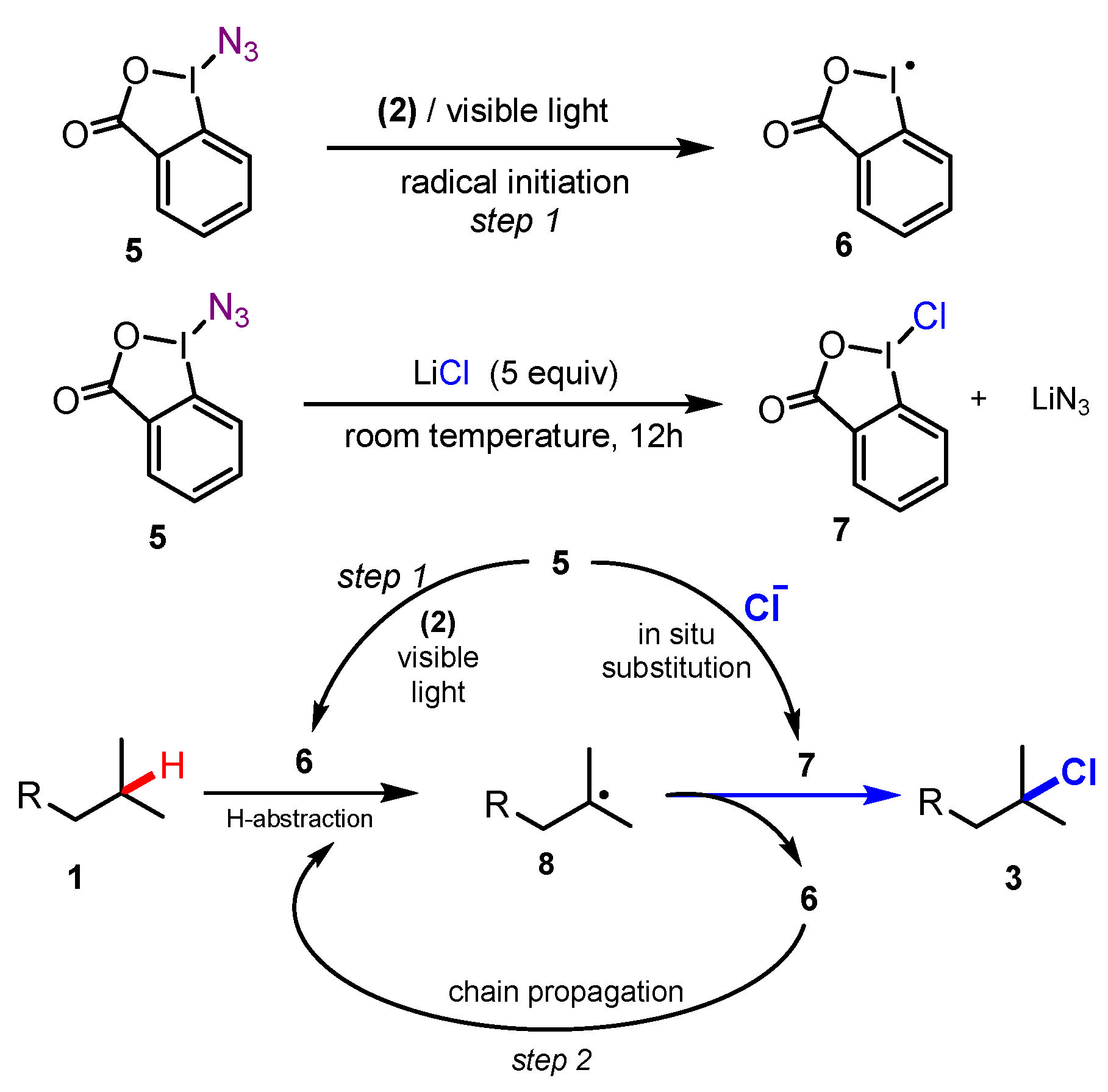
A plausible mechanism for chlorination is illustrated in Scheme 3, in which azidoiodane 5 participated in two processes simultaneously. Under irradiation of visible light and photocatalyst Ru(bpy)3Cl2 2, homolytic break of the I–N3 bond of azidoiodane 5 yielded an iodanyl radical 6 and an azido radical. Azidoiodane 5 also reacted with chlorinating source LiCl to generate chloroiodane 7. Capture of an H atom of substrate 1 by radical 6 gave the intermediate radical 8. In the meanwhile, chloroiodane 7 provided a Cl atom to radical 8 to form the desired product 3 and then recovered iodanyl radical 6.
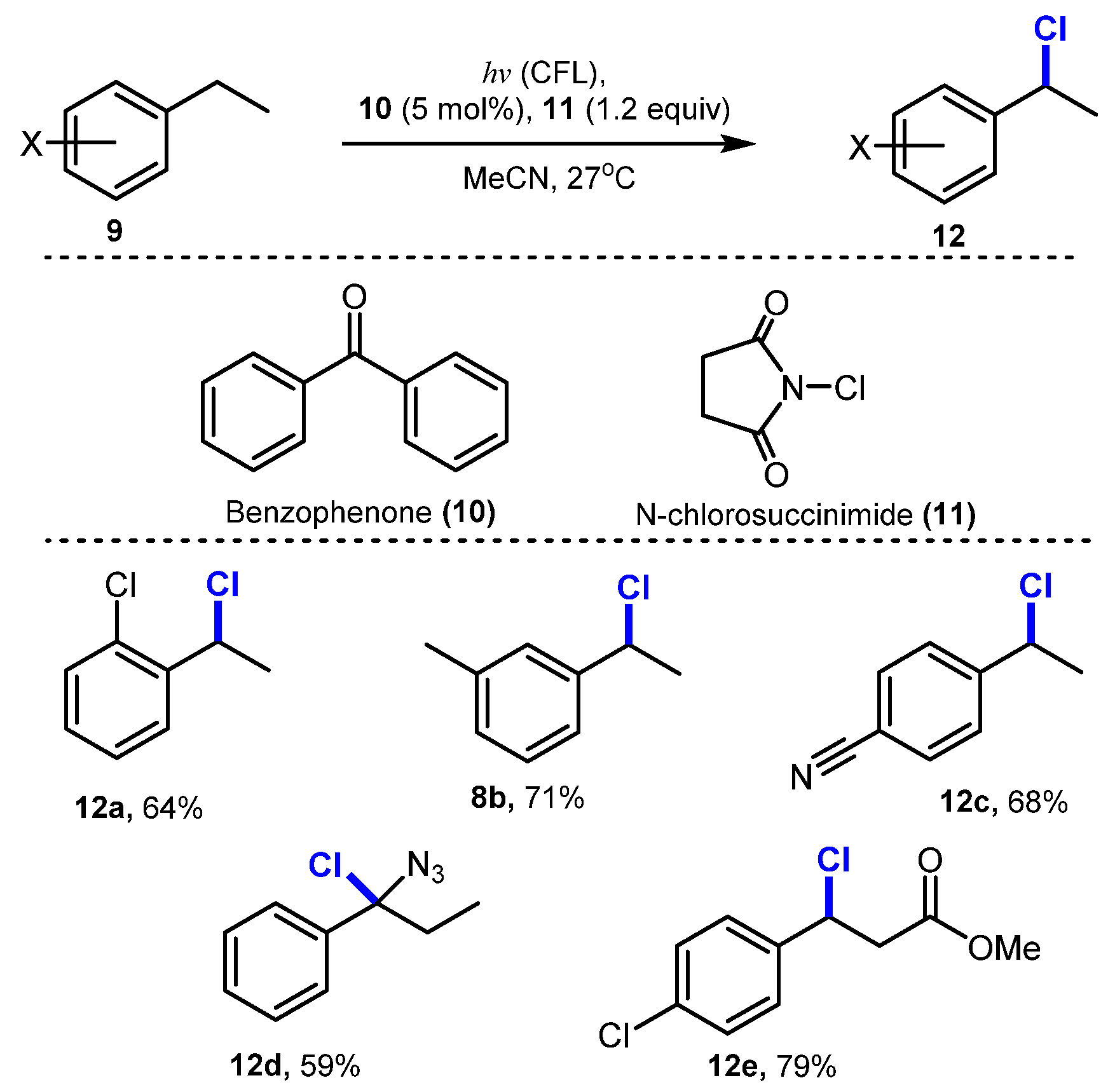
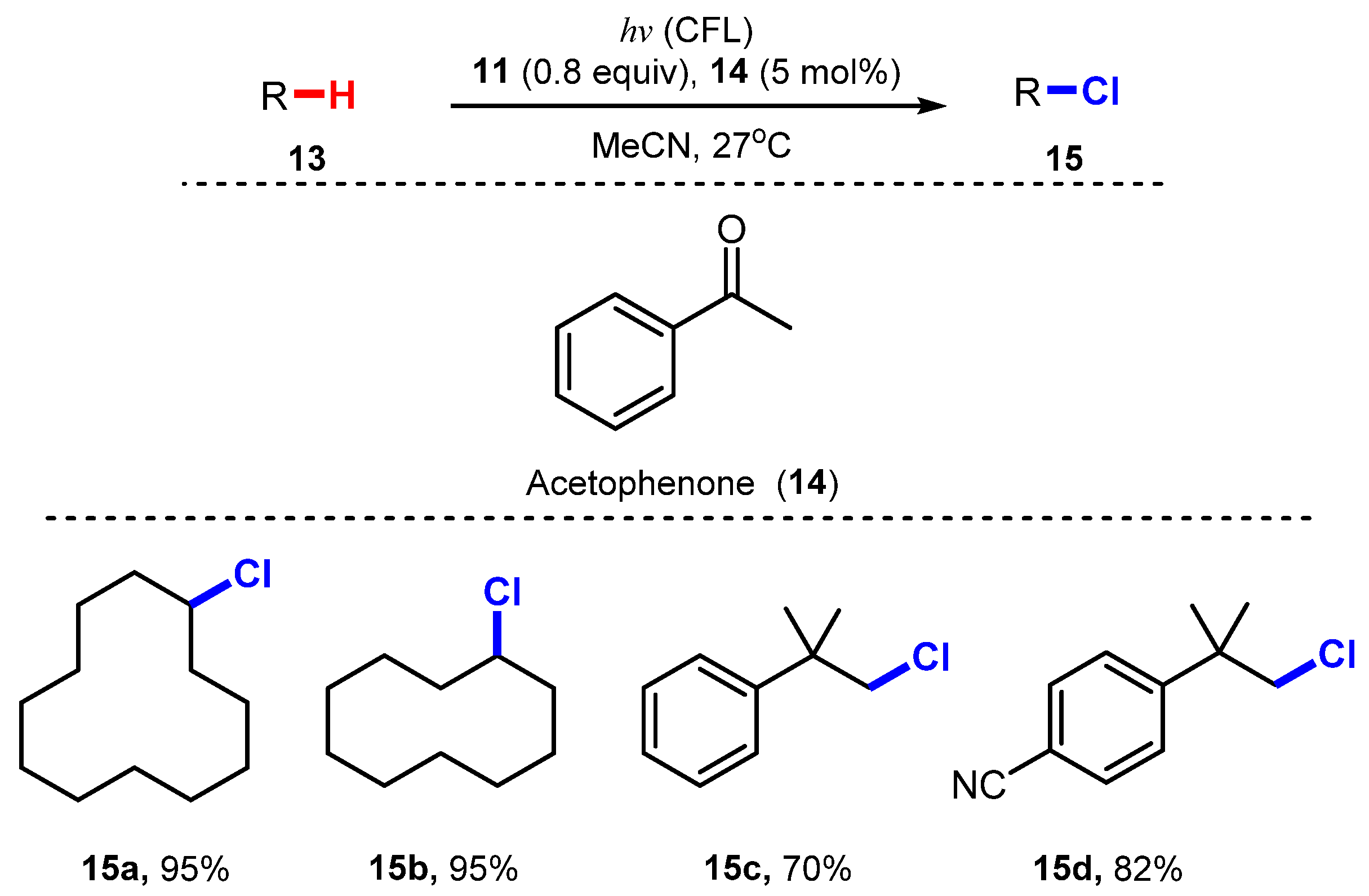
Another photo-mediated C(sp3)-H chlorination was reported by Chuo Chen and co-workers in 2017 [27]. In this reaction, aryl ketones such as benzophenone were employed as a photocatalyst to assist in the chlorination of C-H groups in the presence of N-chlorosuccinimide (NCS) as a chloride source under irradiation of a household compact fluorescence lamp (CFL) in acetonitrile at room temperature (Scheme 4). The benzylic C-H chlorination was readily performed regardless of the position (ortho, meta, or para) of an electron-withdrawing group on the benzene ring (12a–c). Chlorinations of the primary and tertiary benzylic C-H groups (12d) were successfully achieved, and the ester group at the β-position (12e) was tolerated for this protocol. This method was also highly effective for non-benzylic chlorination, when acetophenone was used as the photocatalyst instead of benzophenone (Scheme 5). Particularly, chlorination of cyclo-compounds was carried out smoothly at high yield (15a–b), whereas the dichlorination of the tert-butyl group (15c–d) was conducted with a lower yield.
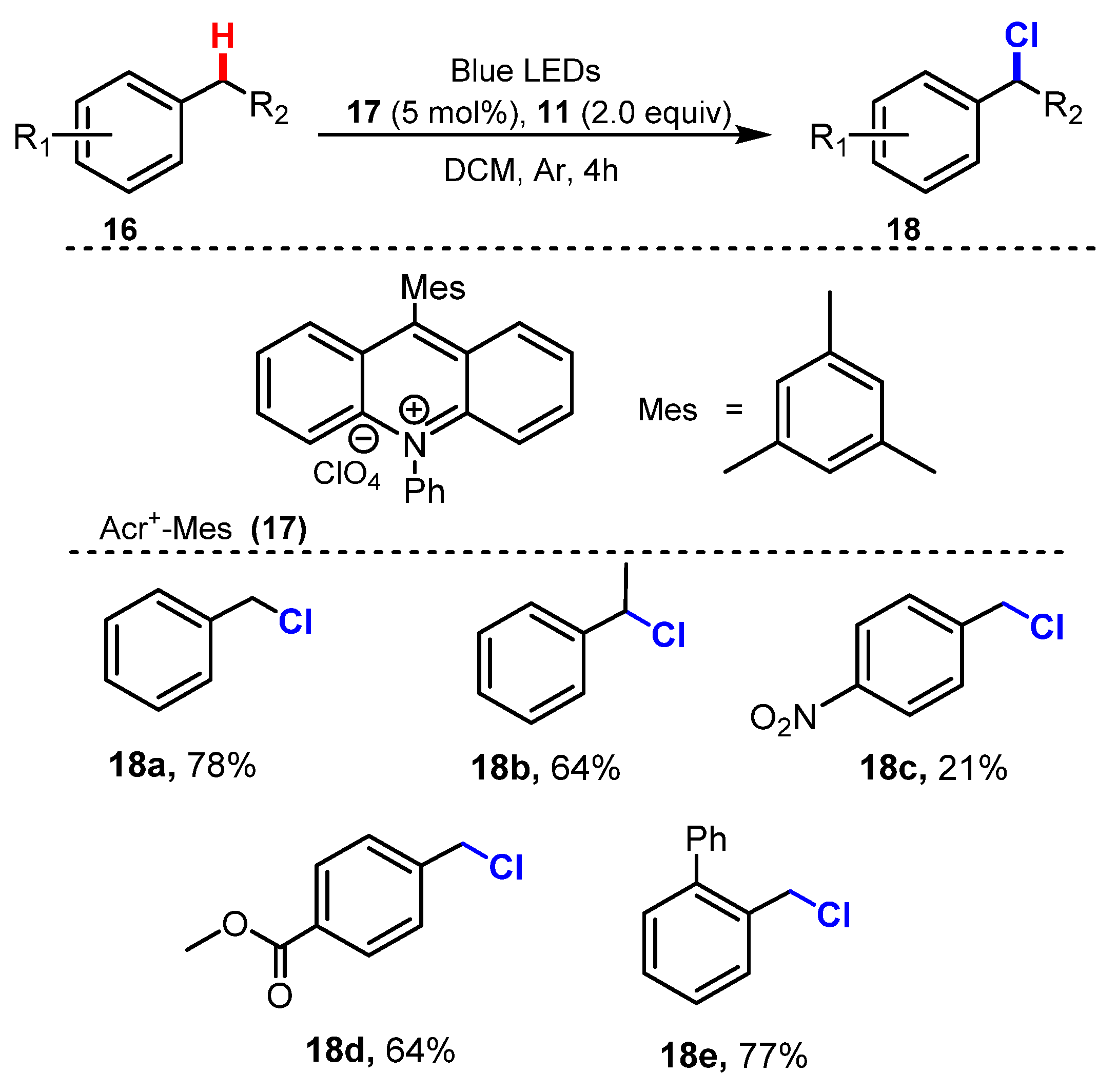
In 2020, Wu and co-workers reported a novel strategy for benzylic chlorination using N-chlorosuccinimide (NCS) as a chloride source and Acr+-Mes as a photocatalyst under radiation of blue LED light in dichloromethane (Scheme 6) [28]. Several typical alkylbenzene derivatives were tested to assess the scope of this chlorination method. Reaction of toluene 18a had a higher reaction yield than that of ethylbenzene 18b (78% for 18a and 64% for 18b). Substrates containing different groups such as phenyl 18e and carbonyl 18d on the aromatic ring were smoothly converted to target chlorides in moderate to good yields (64–77%), while reaction of nitro group 18c on the aromatic ring achieved a lower yield (21%).
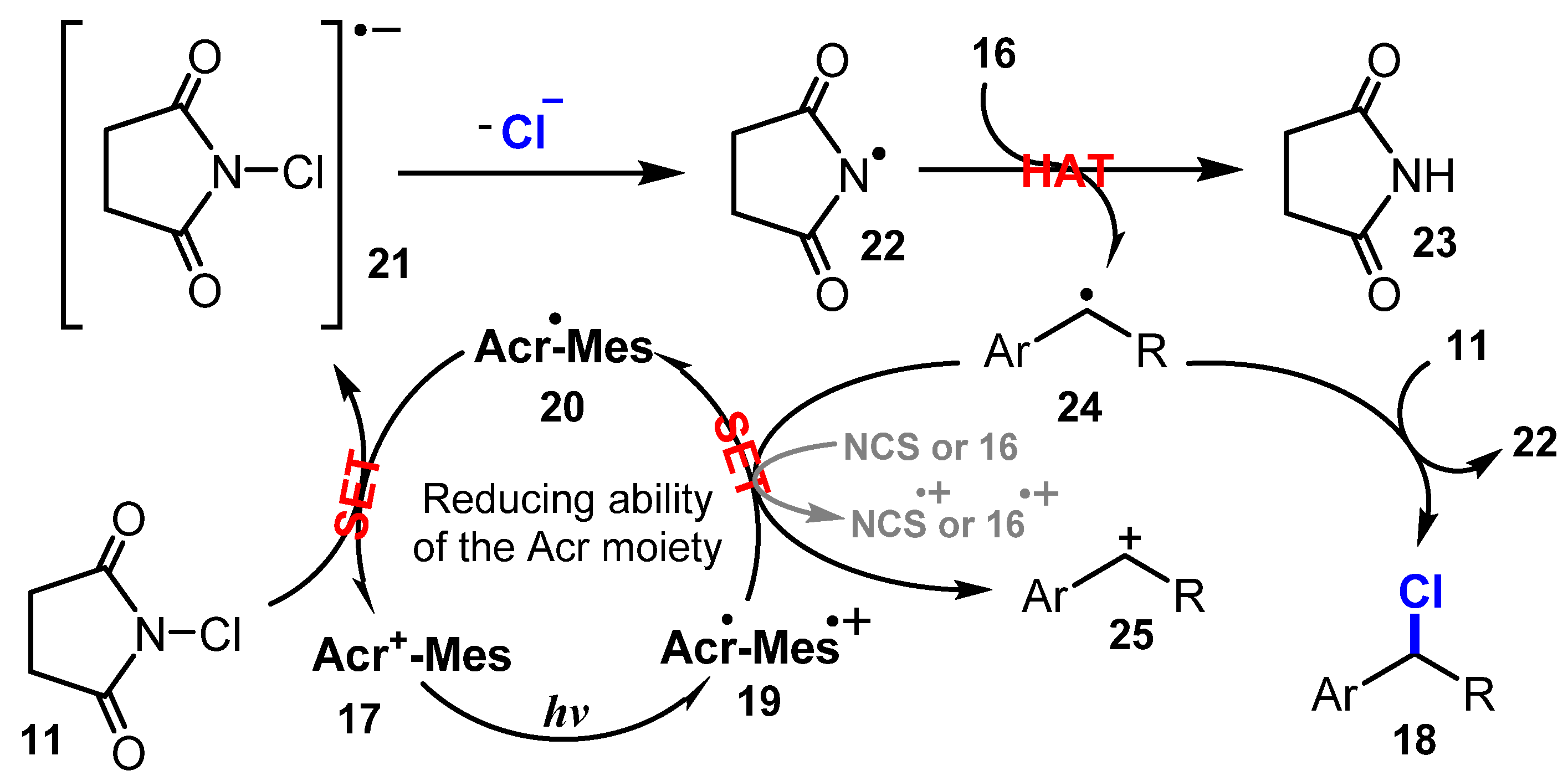
A plausible mechanism for chlorination proposed by Wu and co-workers is depicted in Scheme 7. Visible light excited Acr+-Mes 17 to give the charge state Acr•-Mes•+ 19, which caused the oxidization of N-chlorosuccinimide (NCS) or substrate 16 to provide Acr•-Mes radical 20. This radical 20 reacted with NCS 11 to yield NCS•− 21 via the SET process and to recover Acr+-Mes 17. Then, NCS•− 21 lost a chlorine anion to give N-centered radical 22, which underwent the hydro atom transfer (HAT) process with substrate 16 to afford benzylic radical 24. The radical 24 captured a chloride atom of NCS 11 to form benzylic chloride product 18.
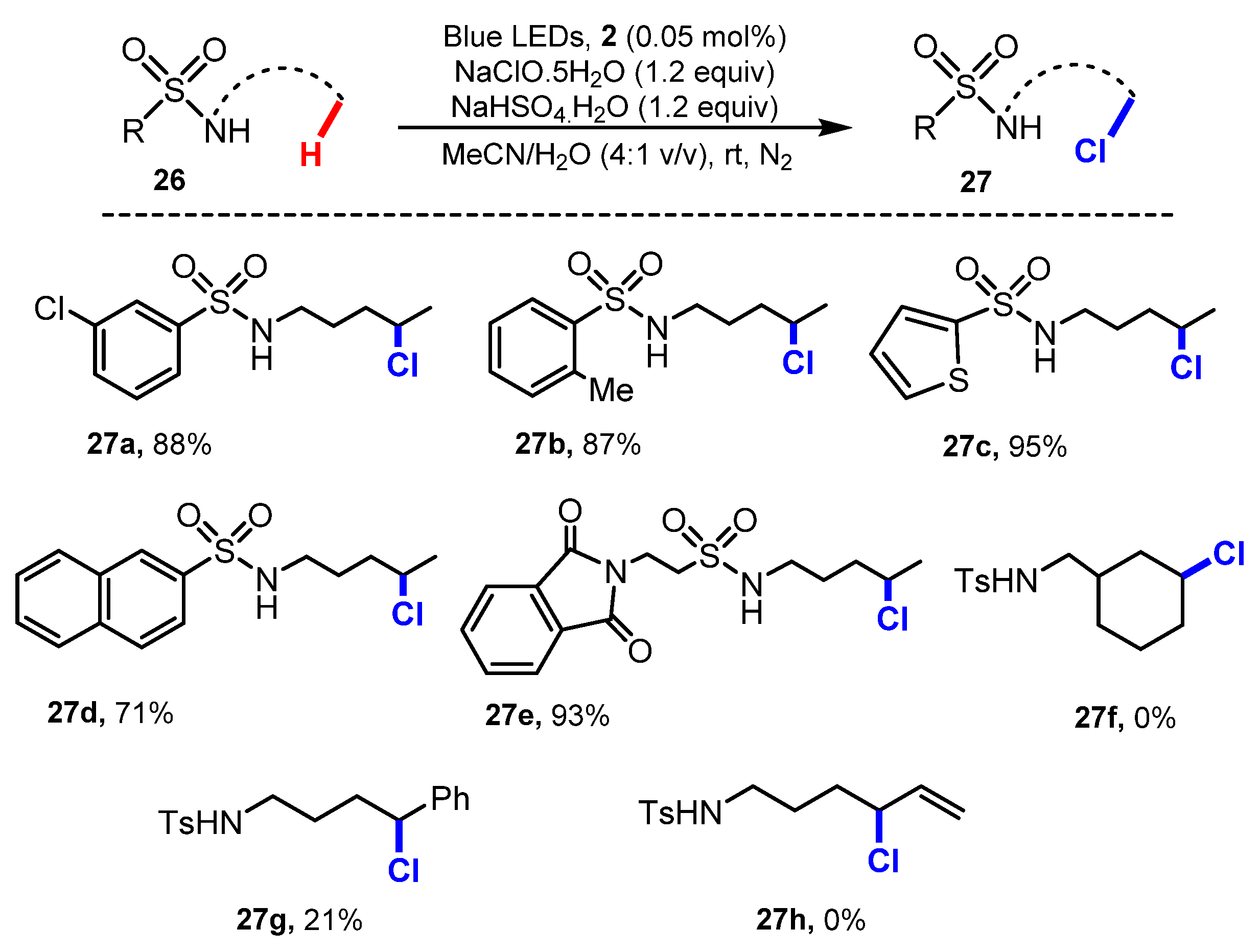
In the same year, Wei Yu and co-workers developed a method for the chlorination of aliphatic sulfonamides [29]. The chlorination was achieved via a reaction with NaOCl·5H2O crystals as a chlorinating agent, NaHSO4, and Ru(bpy)3Cl2 as a photocatalyst under blue LED irradiation at room temperature in a mixture of acetonitrile and water (4:1) (Scheme 8). A wide range of sulfonamide substitutes with variations at the sulfonyl moiety were chlorinated at the δ-position with 71% to 95% yields (27a–27e), while the substituents on the amide moiety led to a significant decrease in reaction yield (27f–27h).
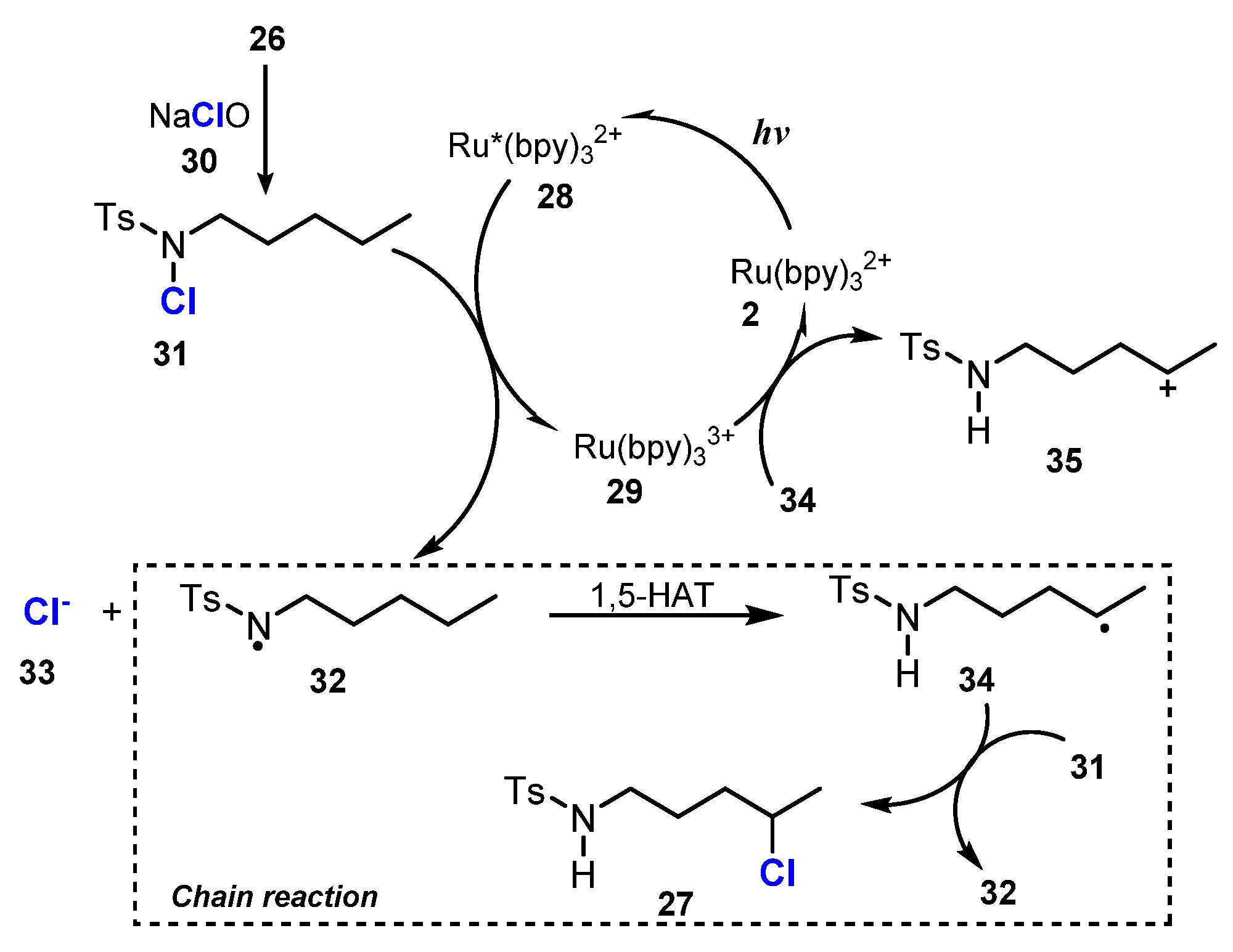
A probable mechanism of the reaction is presented in Scheme 9. Substrate 26 reacted with NaOCl 30 to form N-chlorosulfonamides 31, which was transformed to sulfonamide radical 32 under the effect of the photocatalyst Ru*(bpy)32+ 28, which was generated from Ru(bpy)32+ 2 by light. This radical 32 underwent the 1,5-hydrogen atom transfer (1,5-HAT) process to form carbon-centered radical 34 at the C5 position. The carbon-centered radical 34 then participated in two reactions. Firstly, radical 34 was oxidized by photocatalyst Ru(bpy)33+ 29 to give carbocation 35 and Ru(bpy)32+ 2, and then carbocation 35 obtained Cl anions to form the final product 27. On the other hand, radical 34 also picked the chloride atom of compound 31 to generate the final compound 27 and sulfonamide radical 32.
2.2. Bromination of Aliphatic C-H Bonds
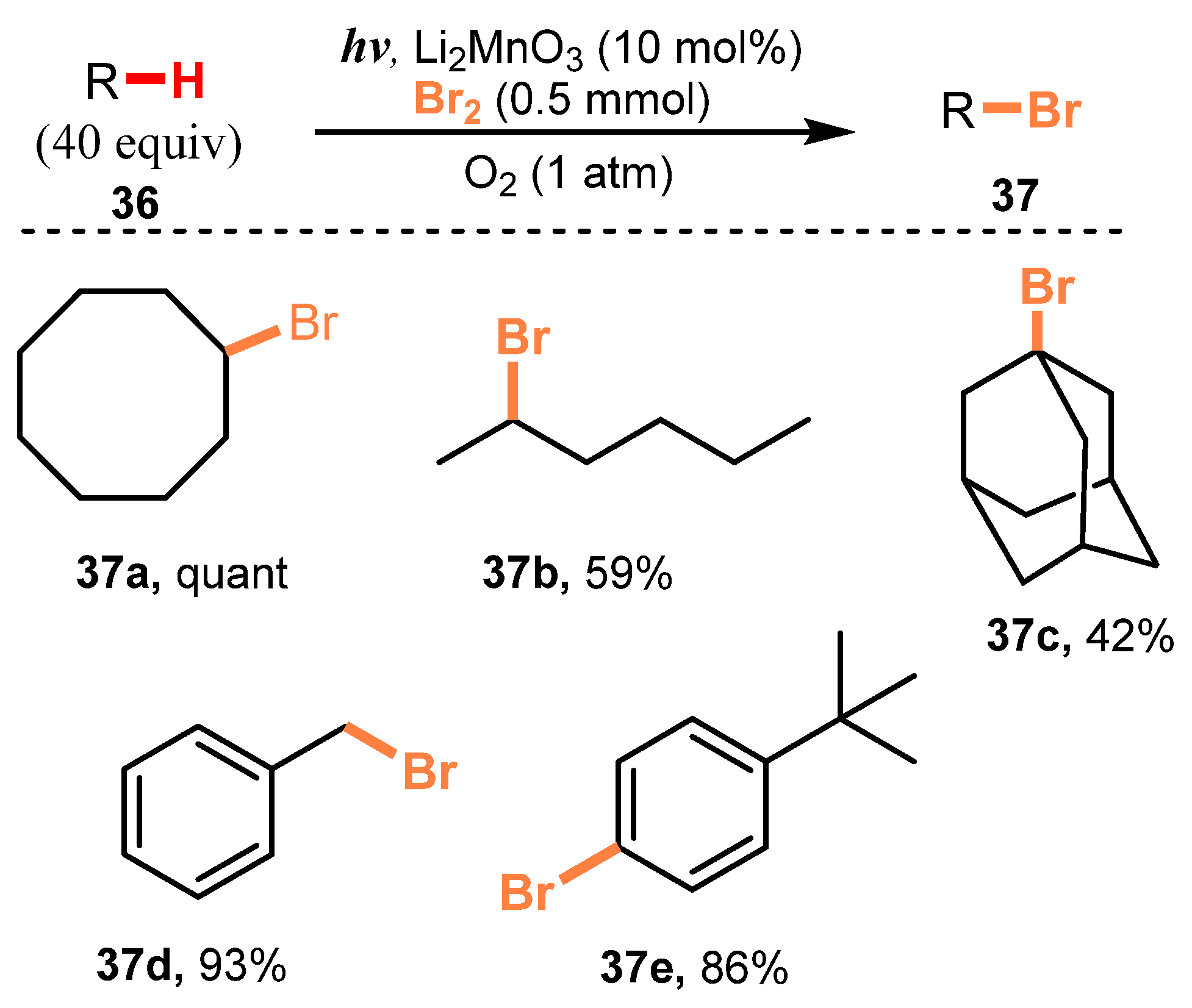
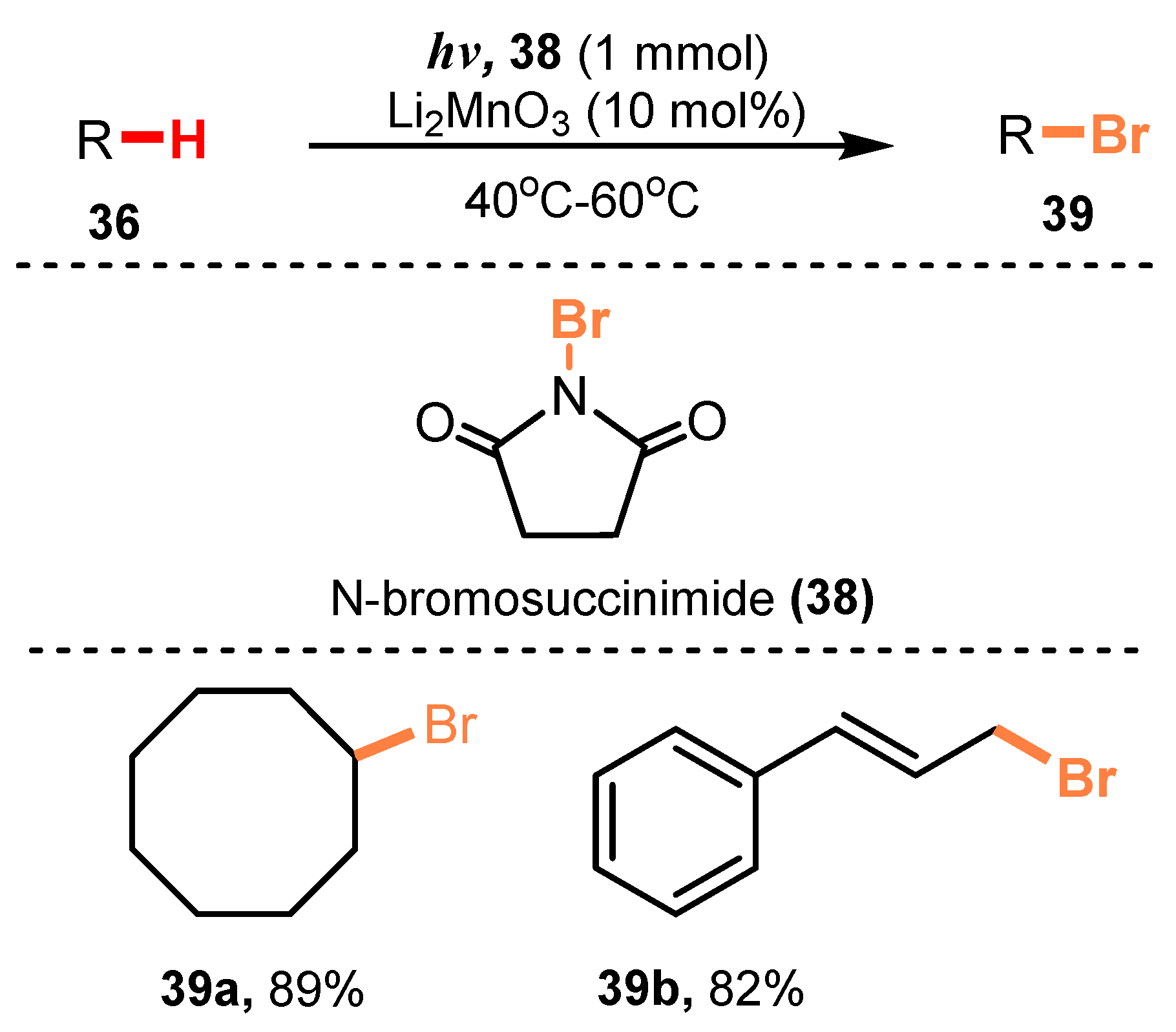
In 2013, Nishina and co-workers reported mono-bromination of hydrocarbons [30]. In the reaction, starting substances reacted with Br2 in the presence of Li2MnO3 as a photocatalyst under irradiation of fluorescent light under O2 pressure to give brominated products (Scheme 10). This reaction showed higher selectivity to the secondary C-H bonds of n-hexane than to the primary C-H bonds (37b), and bromination of the 2-position had priority over that of the 3-position with a ratio of 2:1. Some other compounds, such as adamantane (37c), benzine (37d), and tert-butylbenzene (37e), were all tolerated for this method with good to excellent yield (42–93%). Furthermore, N-bromosuccinimide, a bromine source, could be used rather than Br2 to brominate a wide range of substrates, which broadened the scope and applicability of this method without the need for harsh reaction conditions (39a–b) (Scheme 11).
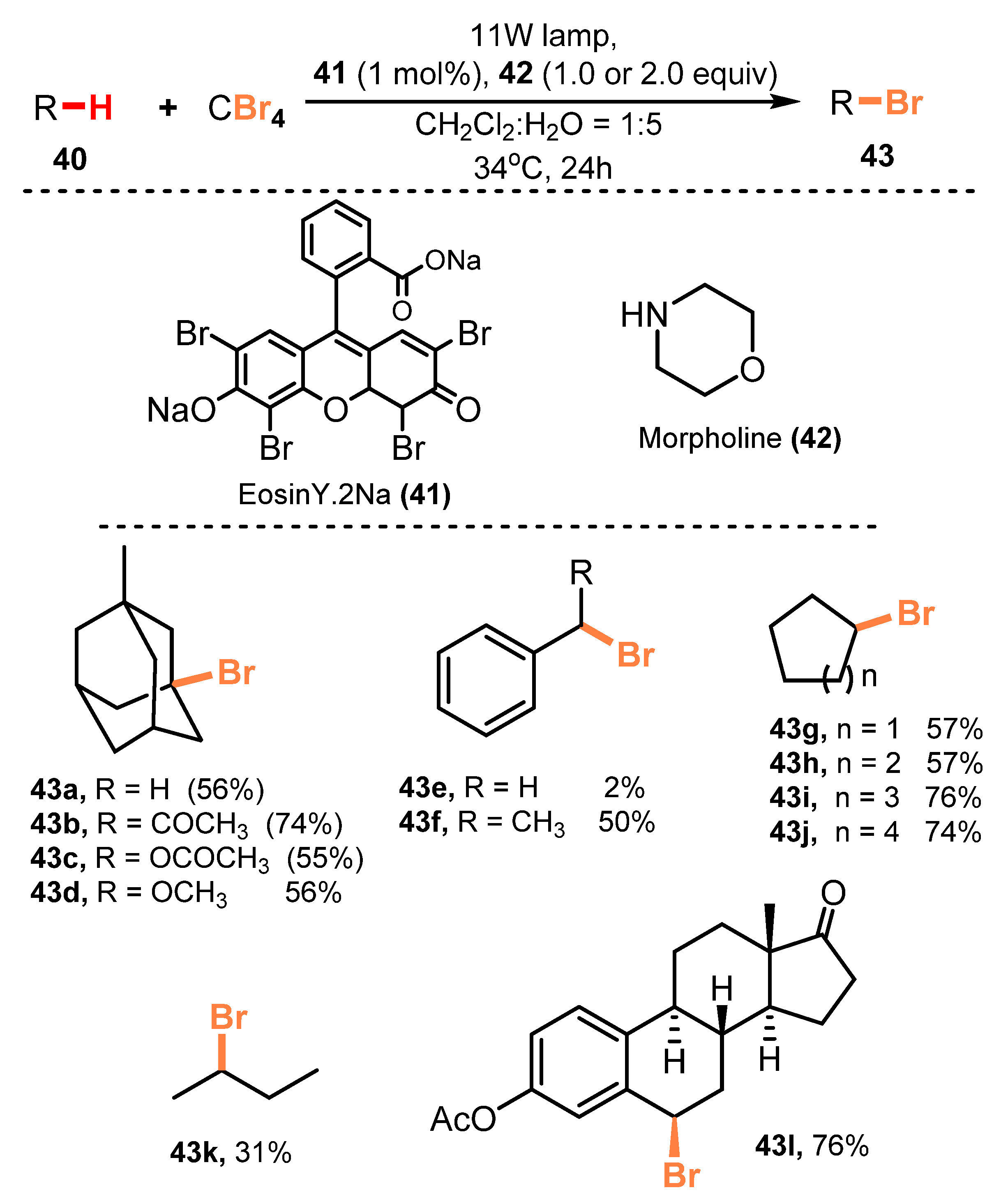
In 2014, an efficient C-H bond bromination process on aliphatic and benzylic compounds, without the use of an inert environment or anhydrous solvent, was reported by Tan and co-workers [31]. Eosin Y disodium salt, as a photoredox catalyst, and reductive compound morpholine were employed to perform bromination of aliphatic and benzylic compounds under irradiation of an 11 W lamp in a mixture of dichloromethane and water (1:1) at 34 °C for 24 h (Scheme 12). Bromination of adamantane derivatives containing ketones, esters, and ether functional groups were successfully achieved (43a–d) (55–74% yield). The reaction did not occur with unsaturated C-H, but C(sp3)-H on toluene derivatives (43e–f) and alkyls (43g–k) were brominated with 2–76% yields. Additionally, they applied this bromination method to some useful compounds that could be utilized in the pharmaceutical and medical fields, and a Terpenoid and an Estrone derivative (43l) were brominated with good efficiency.
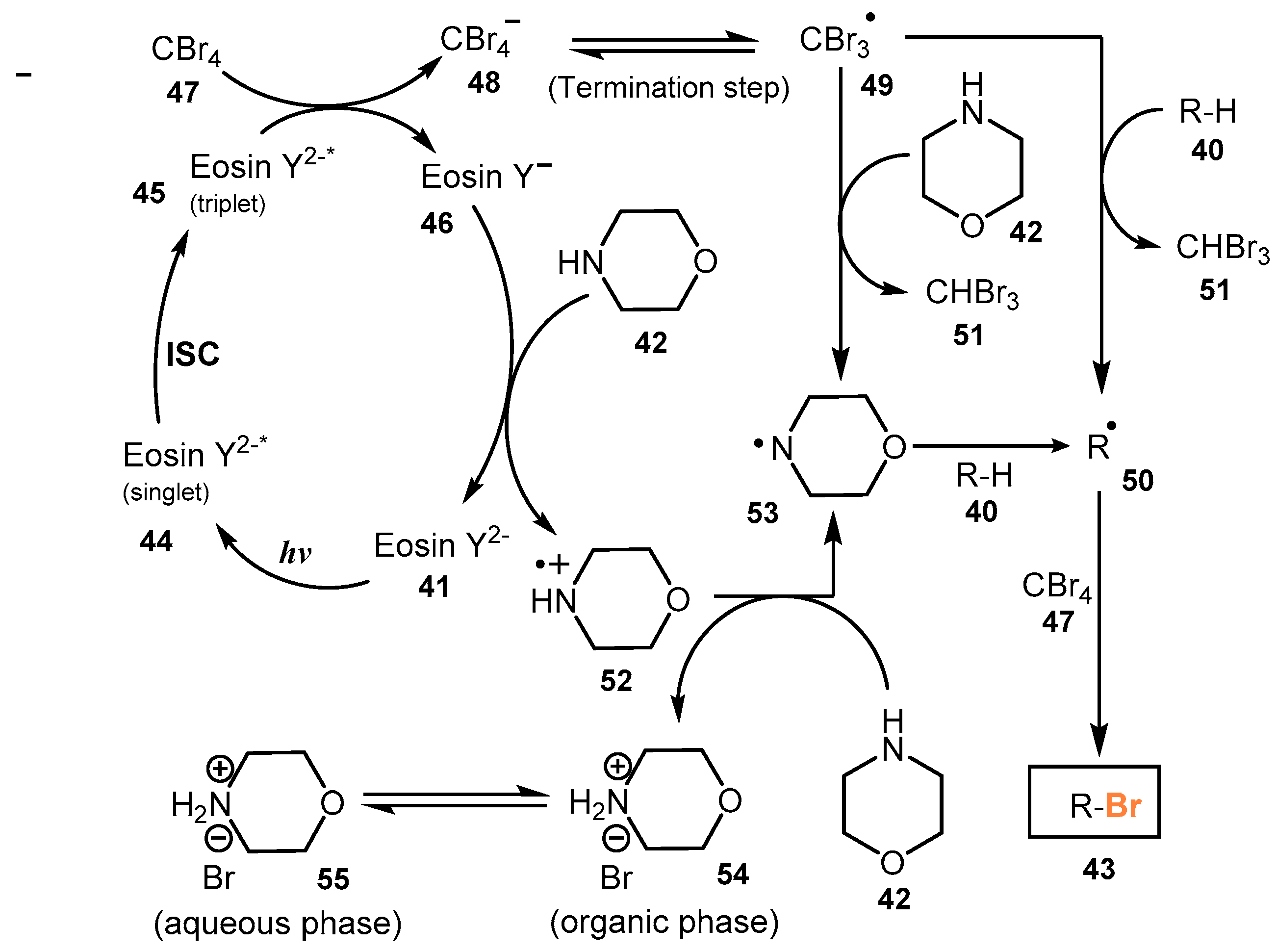
The proposed mechanism of this reaction is shown in Scheme 13. Absorbing light of photocatalyst Eosin Y2− 41 formed Eosin Y2−* (singlet) 44, which underwent an intersystem crossing process (ISC) to generate Eosin Y2−* (triplet) 45. When CBr4 47 was reduced to CBr4− 48 by Eosin Y2−* (triplet) 45, the C-Br bonds of CBr4− 48 became less stable, and a Br atom of CBr4− 48 was lost to form CBr3 radical 49. The CBr3 radical 49 then captured a proton from substrate 40 to create CHBr3 51 and carbon-radical R• 50, which was linked to the free Br atom or received Br from CBr4 to give the product RBr 43. Morpholine 42 reduced Eosin Y− 46 to Eosin Y2− 41 and afforded compound 52. Compound 52 reacted with morpholine 42 to give radical 53, which captured a proton from substrate 40 to afford carbon radical R• 50.
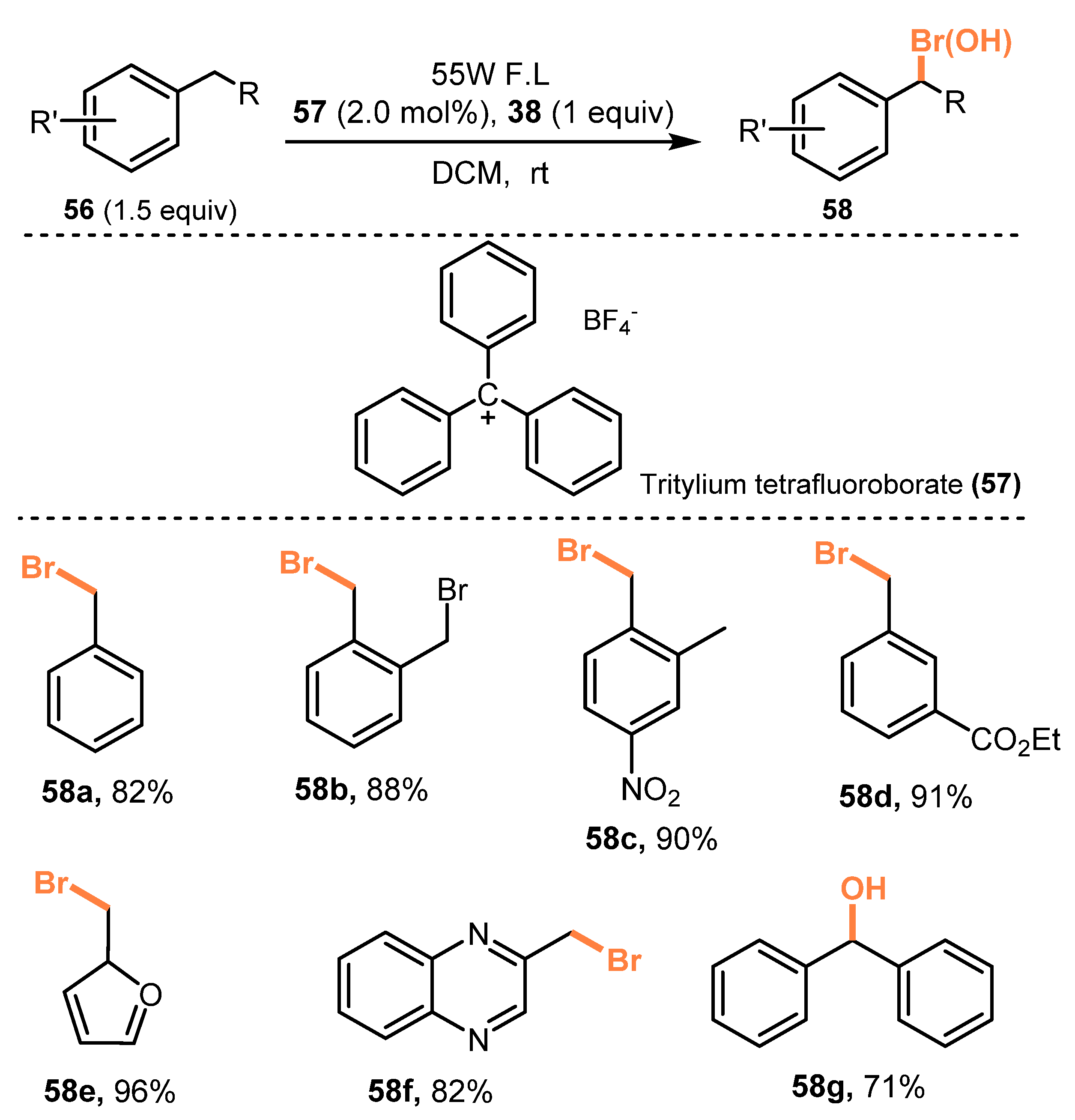
In 2018, Franzén and co-workers reported a chemoselective protocol for benzylic C(sp3)-H bromination without observation of competing arene C(sp2)-H bromination [32]. This process was carried out in the presence of NBS as a bromide source, trityl cation (TrBF4) as a Lewis acid organocatalyst, and in dichloromethane under irradiation of fluorescent light (55W F. L.) at room temperature (Scheme 14). In the reaction of toluene, benzyl bromide was generated in 82% yield (58a). The toluene derivatives with different substitutes (including halogen, nitrozo, cyanide, ester, and sulfochloride) were tolerated for this protocol with good to excellent yields (88–91%) (58b–d). Reaction of ethylbenzene also gave the corresponding product in 92% yield. Naphthalene and heterocycle derivatives were smoothly brominated via this process to produce target compounds in good yield (82–96%) (58e–f). For the reaction of diphenylmethane (58g), the desired benzyl bromide could be observed by 1H NMR. However, this bromide was spontaneously hydrolyzed during isolation and purification to produce the corresponding alcohols.
3. Photo-Catalyzed Halogenations of Aliphatic Multiple Bonds
Scheme 15 shows schematic diagrams of the comparison of the traditional methods with visible-light-induced halogenation of aliphatic multiple bonds.
3.1. Chlorination of Aliphatic Multiple Bonds
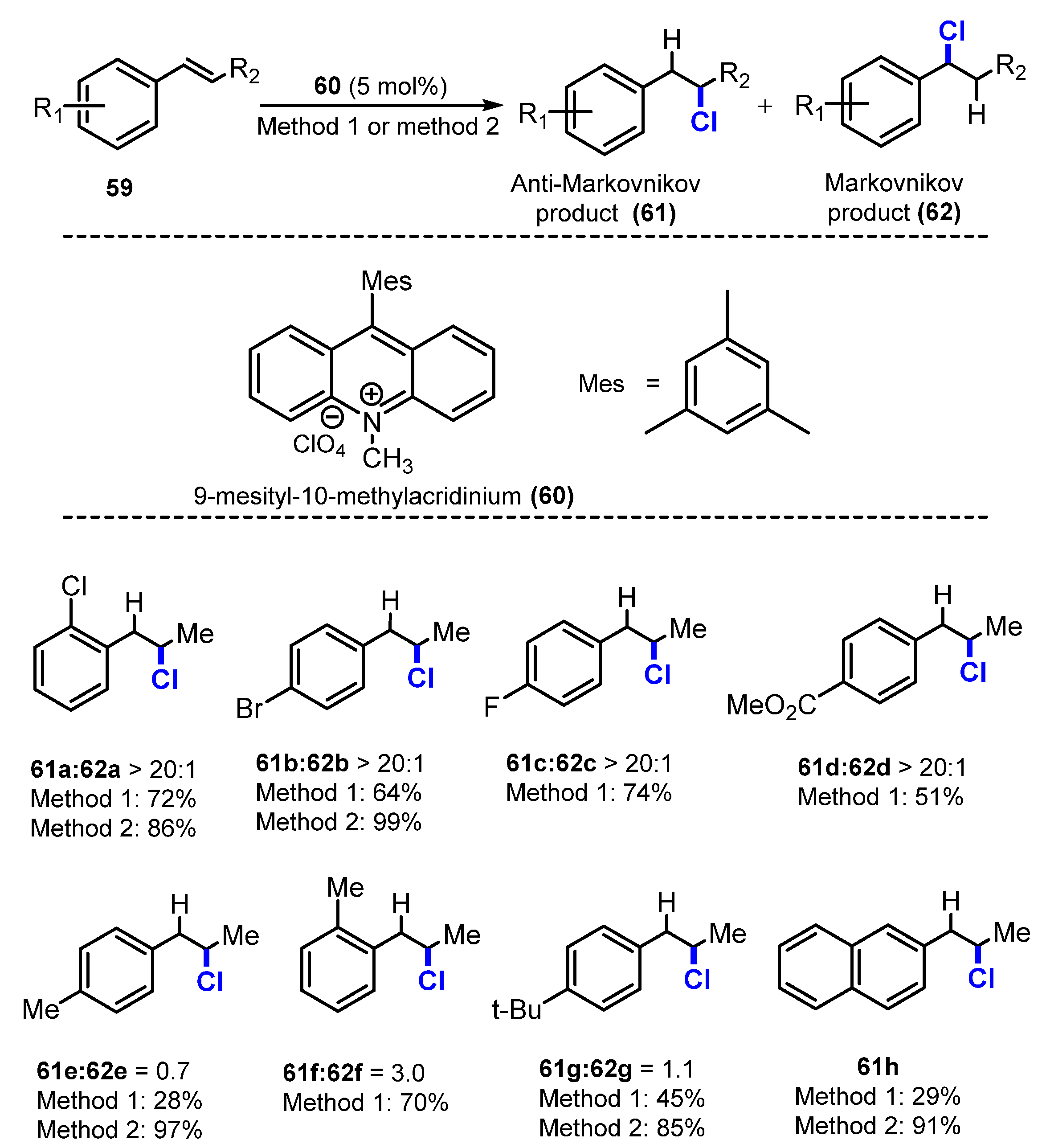
In 2020, Nicewicz and co-workers developed an organic photoredox catalyst system for the regioselective addition of strong Bronsted acidic nucleophiles such as HCl to alkenes (Scheme 16) [33]. Two different techniques were employed for chlorinating β-methylstyrene derivatives using 9-mesityl-10-methylacridinium as a catalyst under irradiation of 450 nm light. In the first method, a reaction with in situ anhydrous HCl (from pivaloyl chloride and 2,2,2-trifluoroethanol (TFE)) in thiophenol and 2,6-lutidine in chloroform was performed under irradiation of 450 nm light. In the second method, substrates reacted with 2,6-lutidine HCl and 4-methoxythiophenol in a mixture of CHCl3 and TFE under irradiation of 450 nm light. Reaction using in situ anhydrous hydrogen chloride (HCl) yielded an anti-Markovnikov hydrohalogenation product. For the reaction of styrene substrates with electron-withdrawing groups, the corresponding products were generated with moderate yields (51–99%) (61a–d), and few to no Markovnikov addition compounds were observed. Chlorination of substrates containing electron-releasing substituents showed lower yields and favored the undesirable Markovnikov reaction (61e–g). The anti-Markovnikov hydrohalogenation of α-methylstyrene was completed in less than 5 h with a 93% yield using 2,6-lutidine hydrochloride. Reaction of several α-methylstyrene compounds gave the products with more than 60% yields, whereas reaction of mono-substituted styrenyl alkenes provided slightly lower yields.
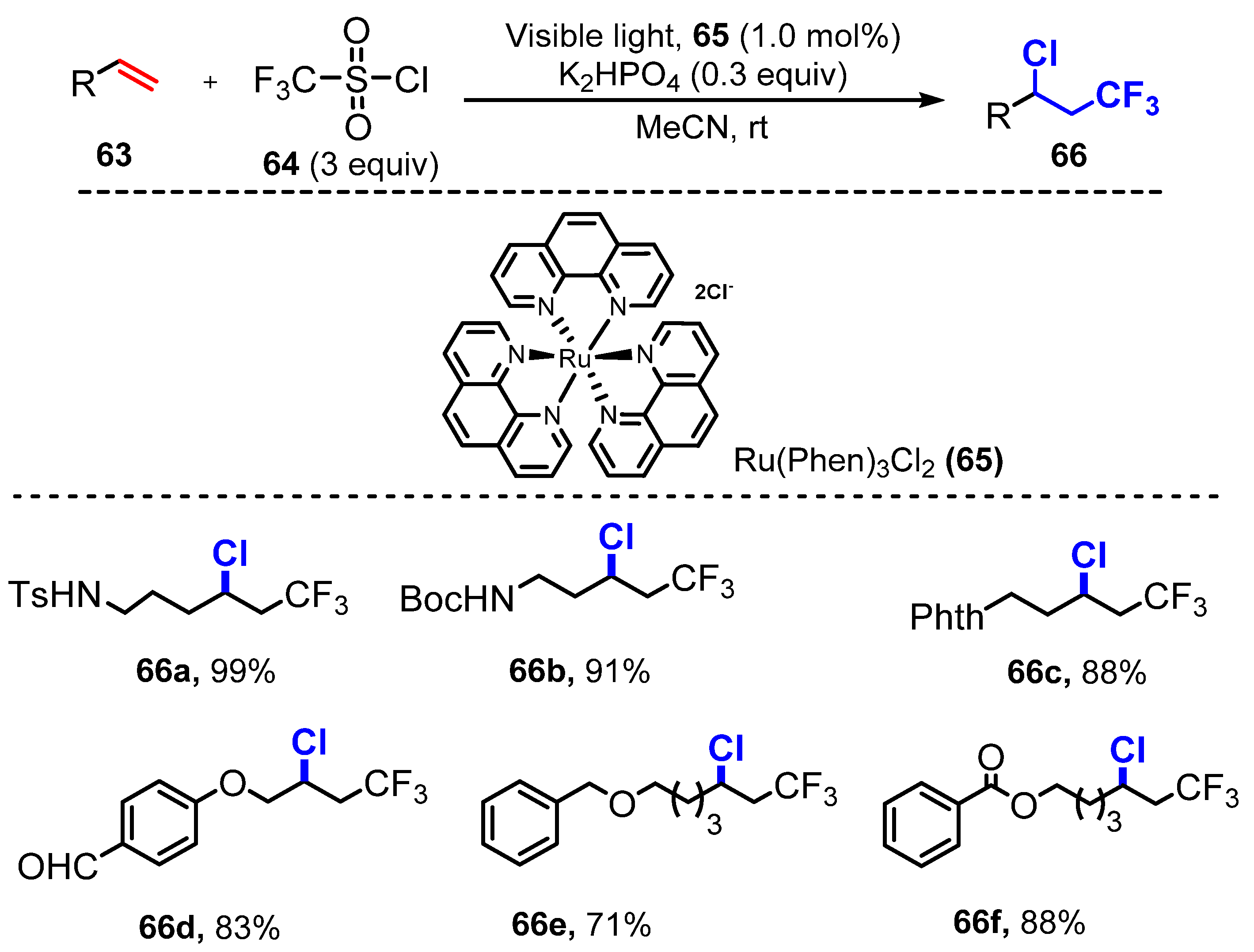
Vicinal chloro-trifluoromethylation of alkenes was reported by Han and co-workers in 2014 [34]. In the chloro-trifluoromethylation, alkenes reacted with CF3SO2Cl 64 in the presence of Ru(Phen)3Cl2 65 as a photocatalyst and K2HPO4 as an additive in acetonitrile under visible light at room temperature to give the corresponding products (Scheme 17). In general, terminal alkenes showed high reactivity. Alkenes containing N-tosyl- and N-Boc-protected amines were readily chloro-trifluoromethylated (66a–b) (99% and 91% yields, respectively), and the reaction of an alkene bearing a phthalimide group generated the corresponding product (66c) (88% yield). Notably, unprotected hydroxyl and formyl groups of alkenes (66d) were tolerated for the reaction procedure, giving 75% and 83% yields, respectively. Furthermore, reactions of alkenes containing ether (66e), ester (66f), amide, and halogen functional groups on the aromatic ring generated target compounds in high yields (71–88%).
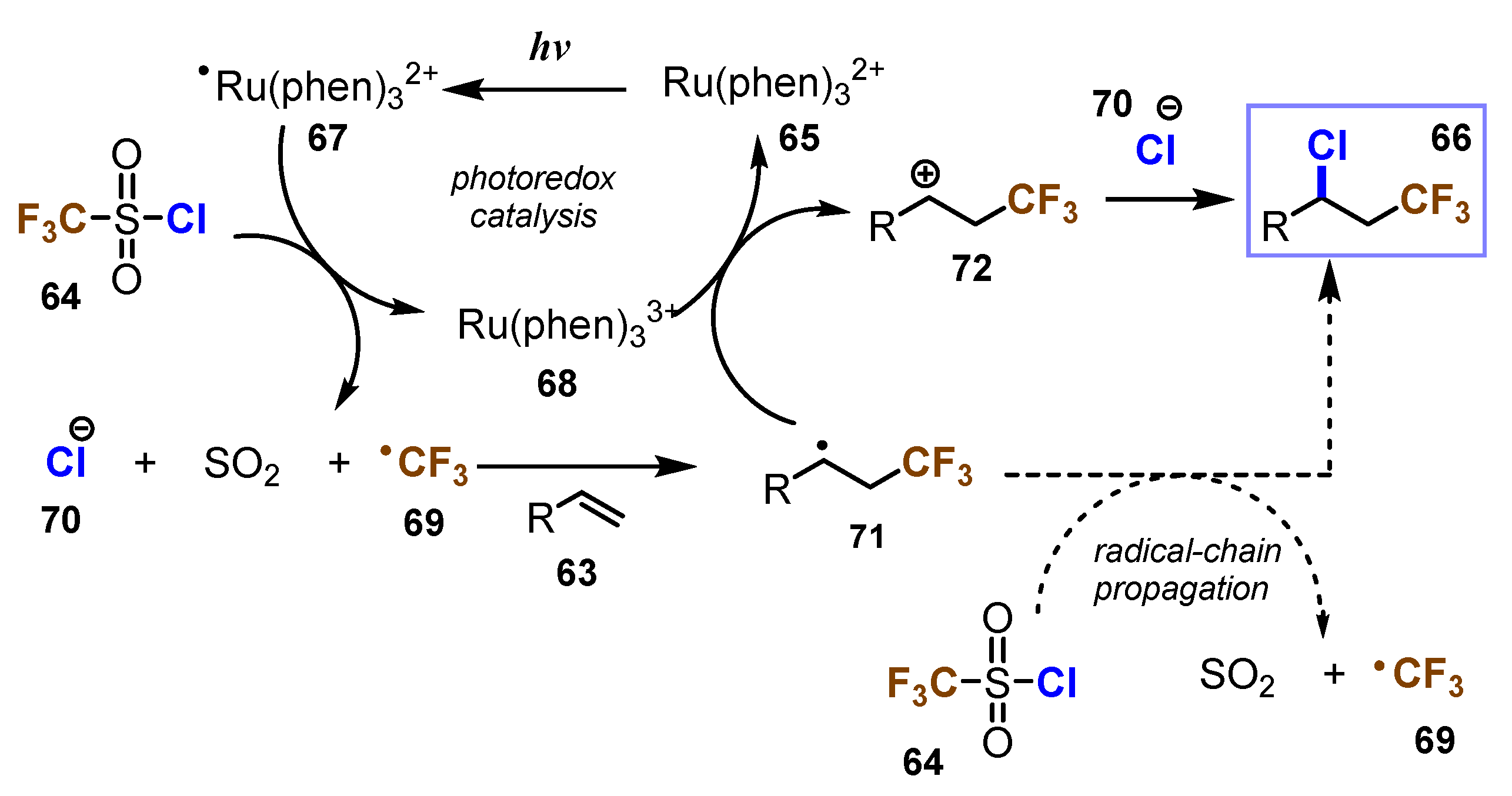
A proposed mechanism of this reaction is shown in Scheme 18. When being exposed to visible light, Ru(Phen)32+ 65 became the excited state *Ru(Phen)32+ 67. Reduction of triflyl chloride CF3SO2Cl 64 by *Ru(Phen)32+ 67 was then cleaved to CF3• 69, SO2, and Cl− 70. After that, *Ru(Phen)32+ 67 became the highest oxidation state Ru(Phen)33+ 68. CF3• radical 69 attacked alkene 63 to generate radical intermediate 71, which was later oxidized by Ru(Phen)33+ 68 to give the carbonation intermediate 72 and Ru(Phen)32+ 65. Finally, Cl− anion 70 was captured by carbonation intermediate 72 to produce product 66.
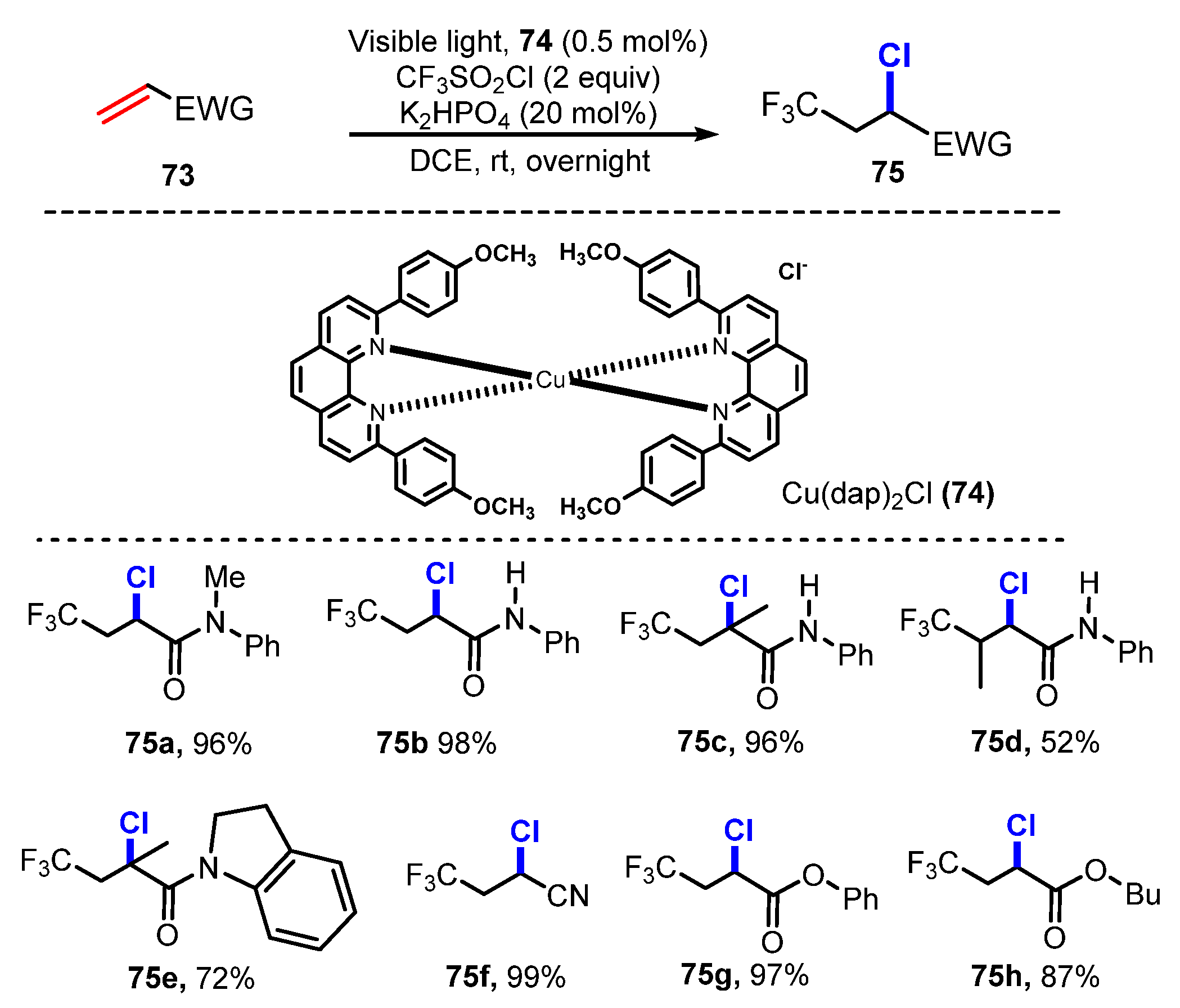
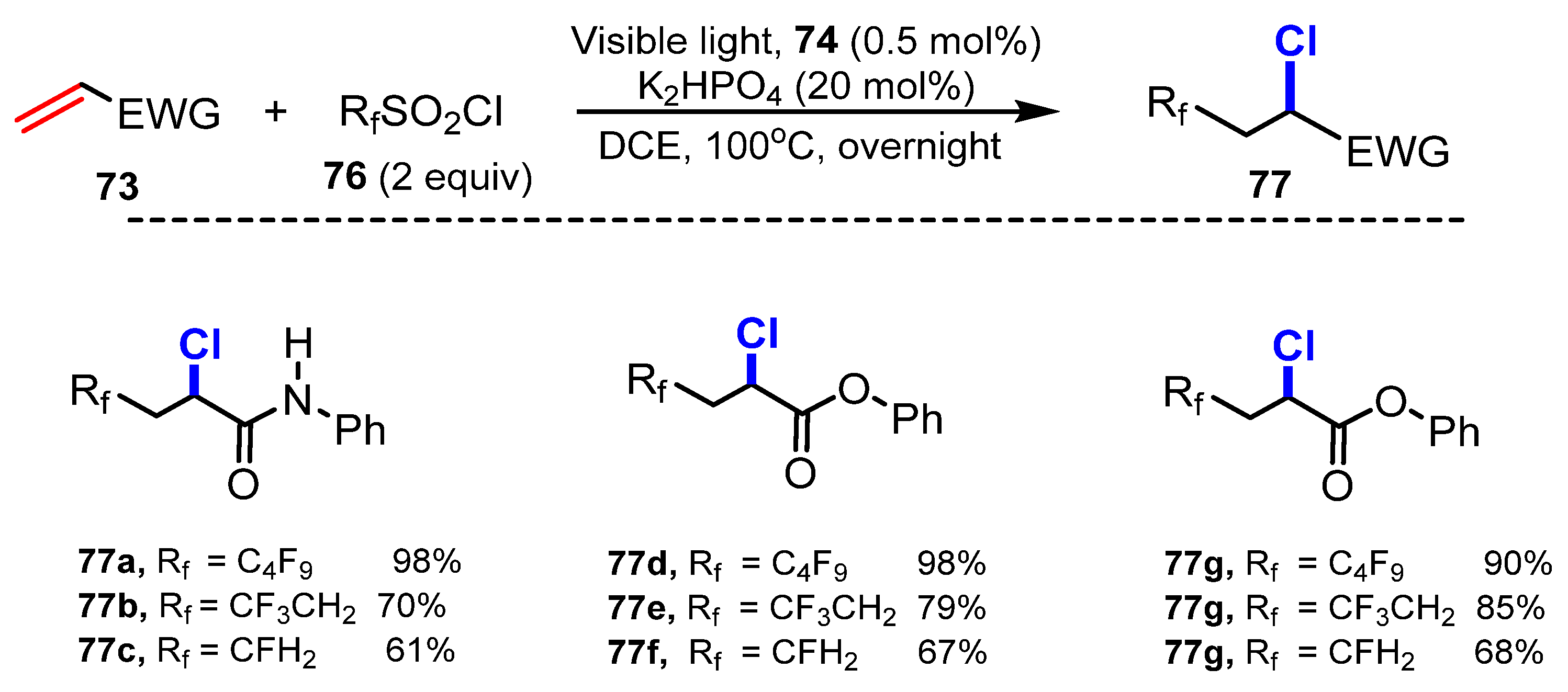
In 2015, Dolbier and co-workers reported photoinduced atom transfer radical addition (ATRA) reactions of alkenes using fluoroalkylsulfonyl chlorides (CF3SO2Cl) [35]. For chlorination, alkenes reacted with CF3SO2Cl in the presence of Cu(dap)2Cl 74 as an efficient photocatalyst and K2HPO4 as a promoter in dichloromethane under irradiation of visible light, which produced the corresponding products in high yields (Scheme 19). Various alkenes were successfully tested for the reaction with CF3SO2Cl to generate target products. Reactions of unsaturated carbonyl substrates such as amides (75a–e), esters (75g–75h), and cyanide (75f) led to the production of target products in moderate to excellent yields. Unsubstituted and α-substituted substrates smoothly underwent this process, while synthetic yields were considerably decreased to 52%, when the substrate was replaced at the β-position (75d). Other fluoroalkylsulfonyl chlorides, such as HCF2SO2Cl, H2CFSO2Cl, and CF3CH2SO2Cl, were tested in this reaction process. Even though, it was discovered that their reactions required higher temperatures (108 °C), this reaction procedure of alkenes provided desired products with good to excellent yields (61–98%) (Scheme 20). (77a–77i).
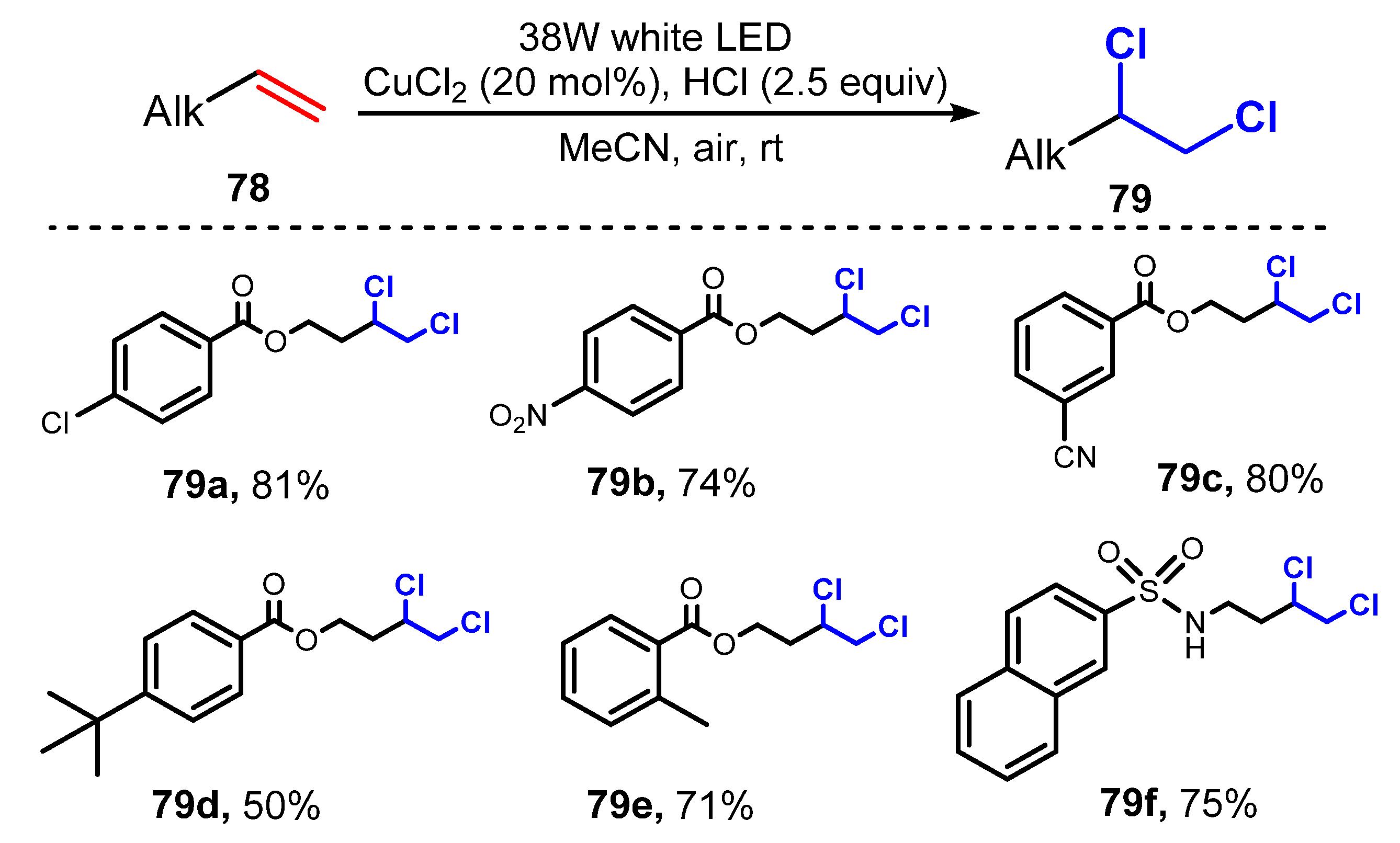
In 2020, Wan and co-workers demonstrated photoredox vicinal dichlorination of alkenes [36]. For this transformation, CuCl2 (20 mol%) as a catalyst and hydrochloric acid (2.5 equiv.) as a chlorine source were used for dichlorination in acetonitrile under irradiation of a 38W white LED (Scheme 21). A variety of phenolic esters with electron-withdrawing groups (NO2, SO2, carbonyl, CN, ester, CF3, and halides) (79a–c) and electron-donating groups (phthalimide, N-hydroxyphthalimide, acetal, Me, t-Bu, and ether) (79d–e) on benzene rings were well tolerated for this reaction with moderate to good yields (50–71%). Reaction of sulphonamides (79f) with free N-H groups was successfully conducted, providing dichlorinated compounds with acceptable yield (75%). The presence of heteroatoms such as oxygen and sulfur had no effect on the efficiency of this reaction, while alkenes with oxidatively labile amine groups were readily converted into dichloride products.
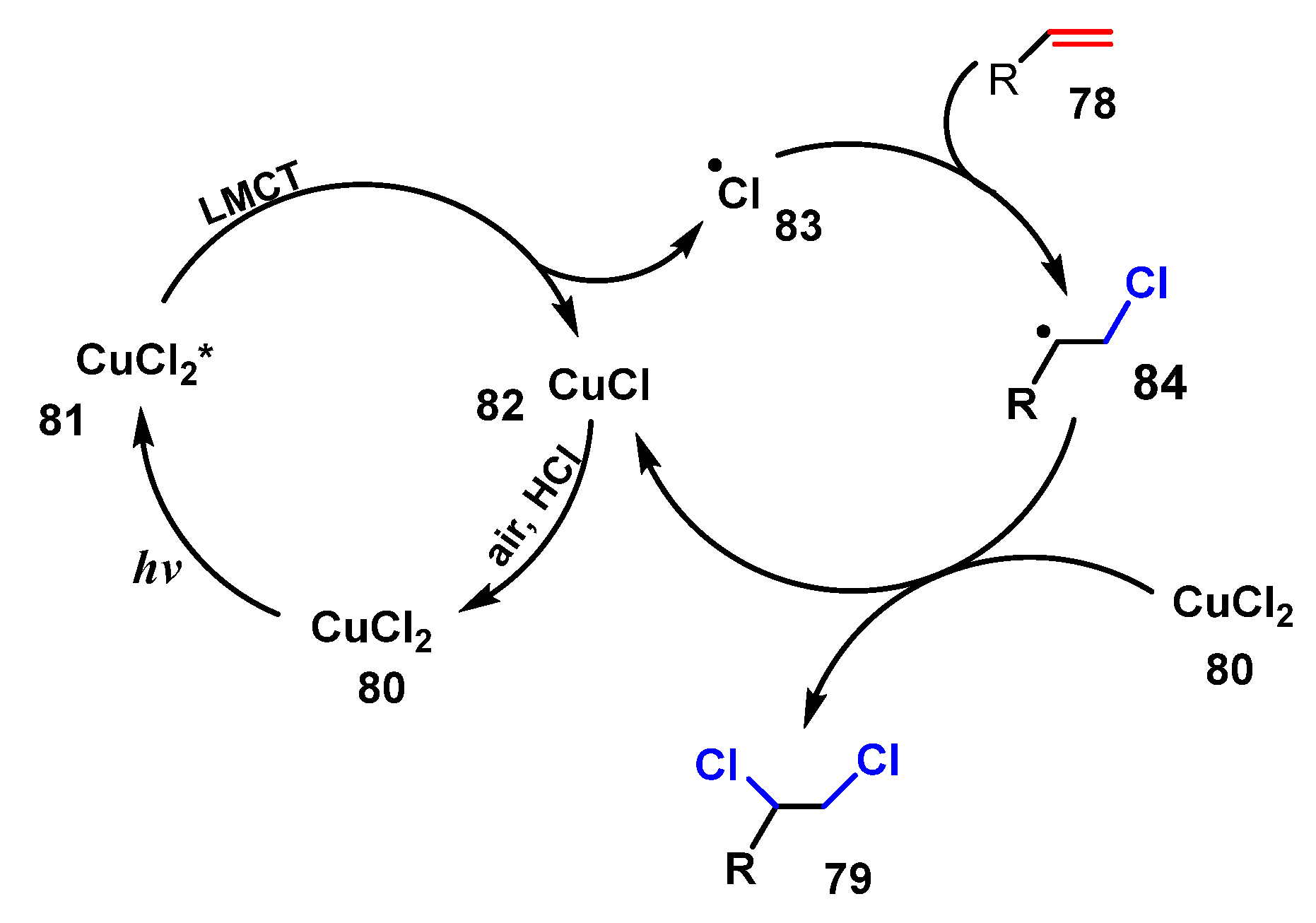
A proposed mechanism of this reaction is shown in Scheme 22. When CuCl2 80 was irradiated by visible light, it was excited to CuCl2* state 81. After that, ligand to metal charge transfer (LMCT) excitation occurred, forming chlorine radical 83, which quickly reacted with alkene 78 to give radical 84. Finally, radical 84 reacted with CuCl2 80 to afford the desired product 79 and CuCl. Then, oxidation of CuCl by HCl recovered CuCl2 80.
3.2. Bromination of Aliphatic Multiple Bonds
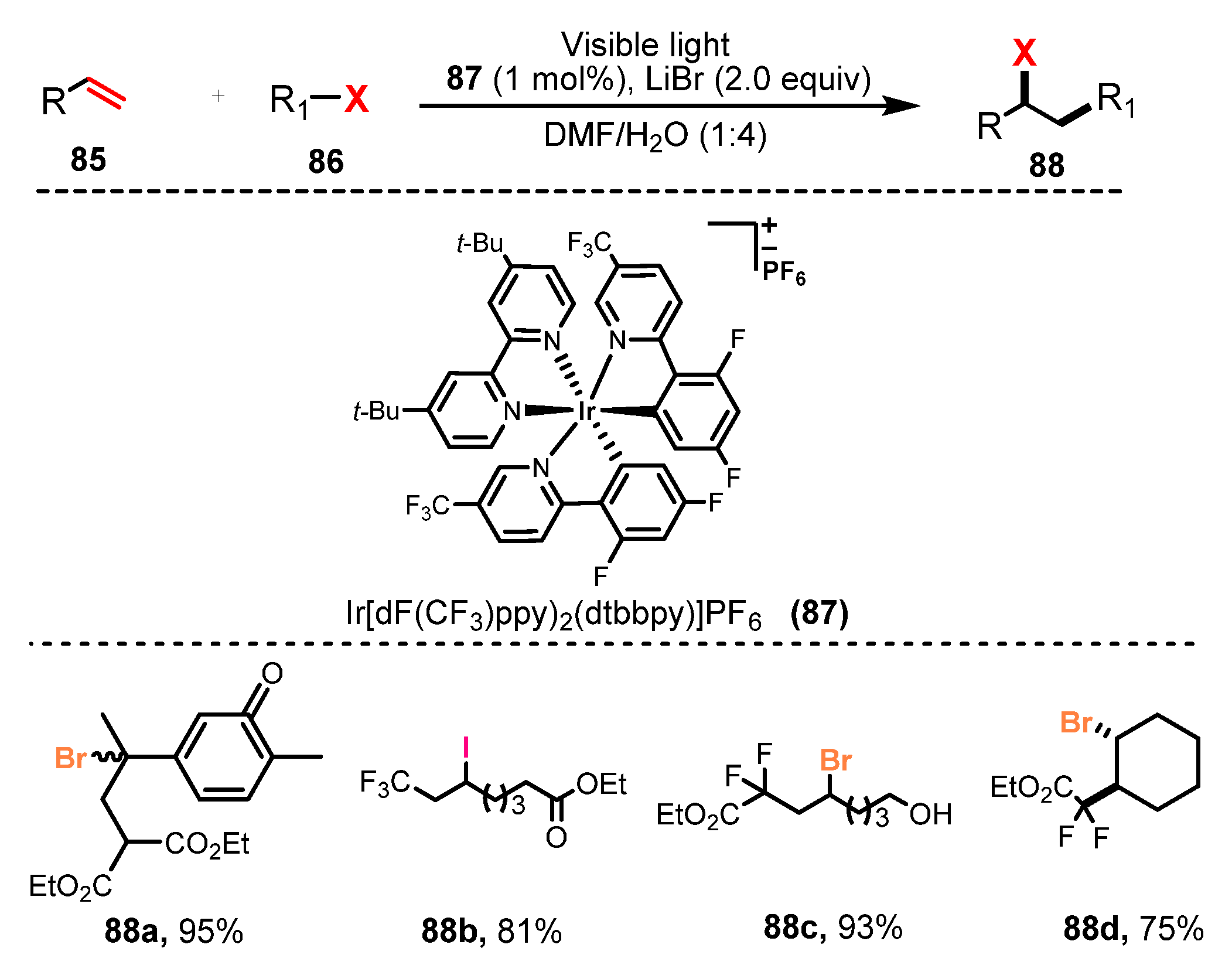
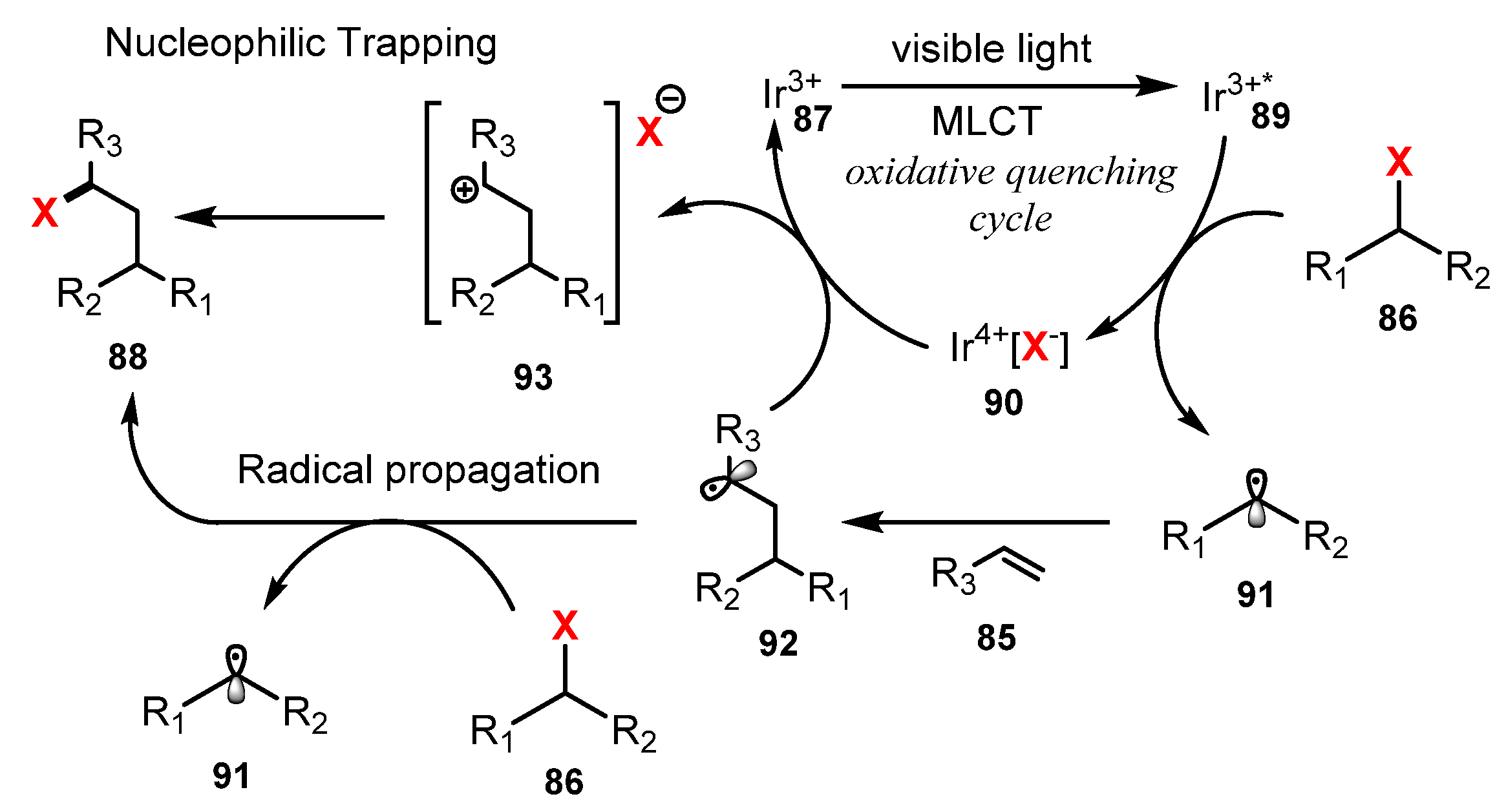
In 2011, Stephenson and co-workers developed a photoredox-catalyzed halogenation via atom transfer radical addition (ATRA) of haloalkanes and α-halocarbonyls to olefins [37]. By using Ir[(dF(CF3)ppy)2(dtbbpy)]PF6 as a photocatalyst and LiBF4 as a Lewis acid additive, they carried out the addition of various haloalkanes and α-halocarbonyls to different olefins under irradiation of visible light in a mixture of DMF and H2O (1:4) (Scheme 23). Using diethyl 2-bromomalonate as a halide source, the reaction of monosubstituted and 1,1-disubstituted olefins was carried out smoothly (67–99% yields). Olefin functional groups that were well tolerated included free alcohols, silyloxy ethers, benzyl ethers, alkyl bromides, esters, enones, carbamates, and aromatic rings (88a–b). A number of α-halocarbonyls and haloalkanes could be used as halogen sources. A variety of fluorinated compounds were successfully employed for this reaction, generating products with high yields (75–93%) (88c–d).
A mechanism of this reaction was proposed as shown in Scheme 24. Ir3+ 87 was changed to excited state Ir3+* 89 under irradiation of visible light, and Ir3+* 89 subsequently reacted with haloalkane 86 or α-halocarbonyl to give radical 91 and Ir4+[X−] complex 90. The electrophilic radical 91 then underwent an atom transfer radical addition (ATRA) process with olefin 85 to generate a new radical 92. This radical 92 was oxidized by Ir4+ and then captured X− to give the product 88. On other hand, the new radical 92 also received X− from haloalkane 86 or α-halocarbonyl to give the product 88.
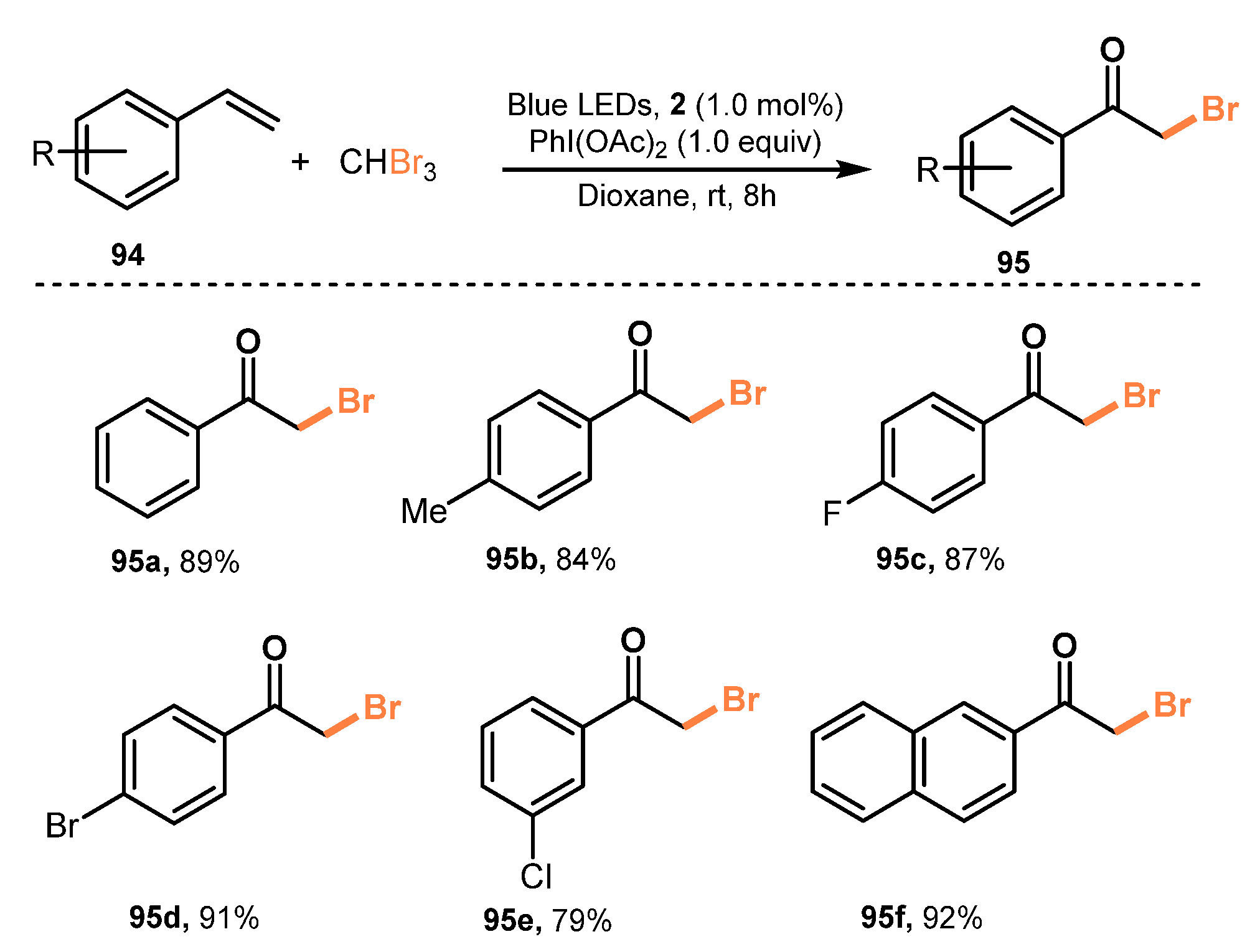
Synthesis of α-bromoketones from olefins was reported by Zhang and co-workers in 2021 [38]. In this method, the reactions of styrenes with CHBr3 in the presence of Ru(bpy)3Cl2 (1.0 mol%) as a photocatalyst and PhI(OAc)2 (1.0 equiv) as a promoter in dioxane under irradiation of a blue LED (450–455 nm) was carried out to produce α-bromoketone products in good yields (Scheme 25). Using this protocol, various olefin derivatives were transformed to α-bromoketones. Styrene with different substitutes such as methyl groups and halides were readily treated with tribromomethane to give the corresponding products in good to excellent yields (95a–e) (79–91%). In addition, 2-vinylnaphthalene was transformed to a desired product with high yield (92%) via this protocol (95f). This visible-light-irradiation protocol was also applied to the synthesis of α-iodo/chloroketones from olefins, and it successfully provided target products.
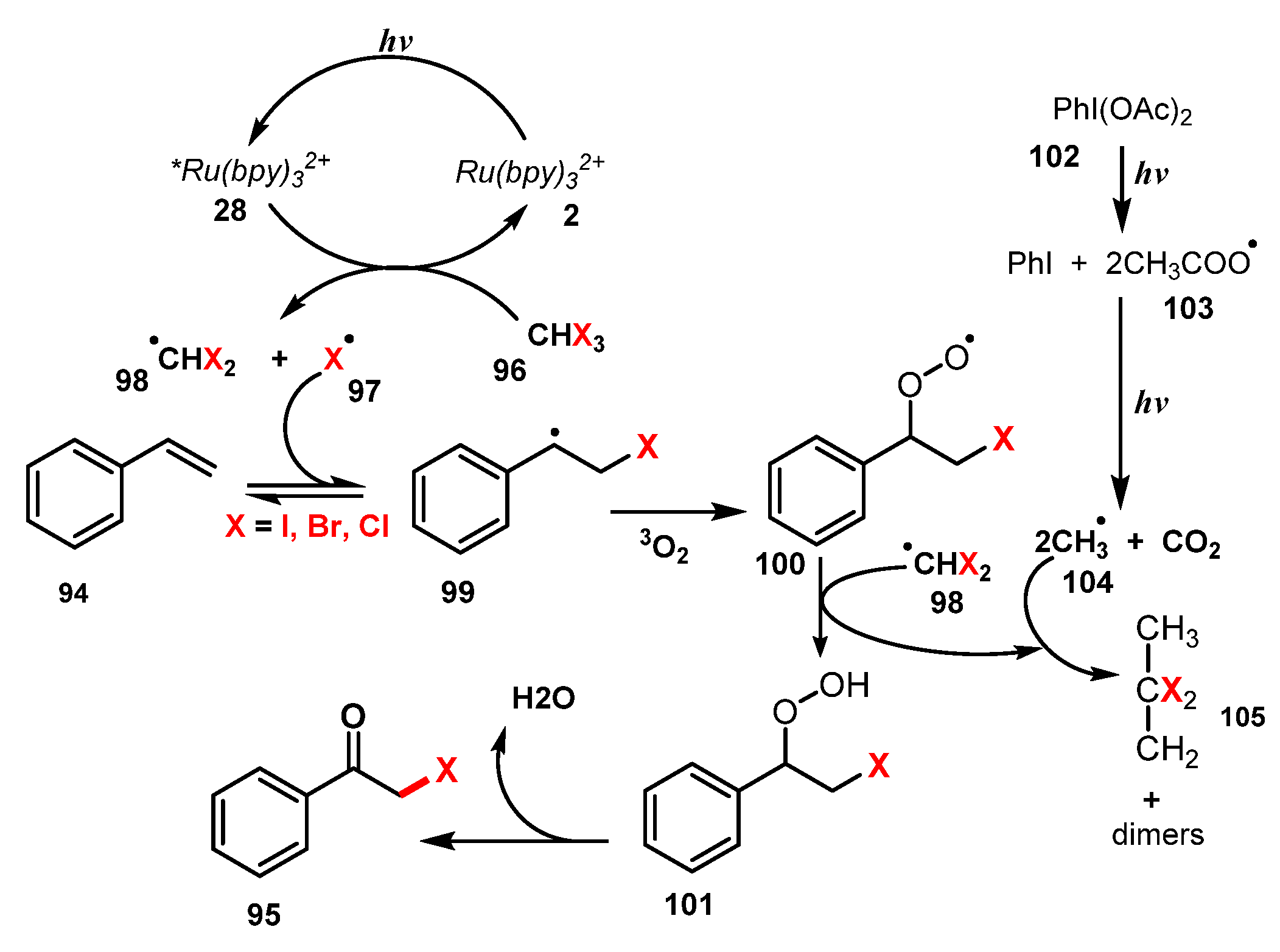
A proposed mechanism of this method is illustrated in Scheme 26. First, photocatalyst (PC) 2 was activated under irradiation of visible light to produce the excited state (PC)* 28. The generated (PC)* 28 then reacted with halide reagent 96, yielding halide radicals (X• 97 and (CHX2)• 98) through C-X bond cleavage. Addition of X• radical 97 to substrate 94 yielded radical intermediate 99, which was then incorporated with 3O2 to give intermediate radical 100. The radical 100 captured a hydrogen atom from (CHX2)• radical 98 to form compound 101, which subsequently underwent a dehydration process to provide the final product 95.
4. Photo-Catalyzed Halogenations of Alcohols
Scheme 27 shows schematic diagrams of the comparison of the traditional methods with visible light-induced halogenation of alcohols.
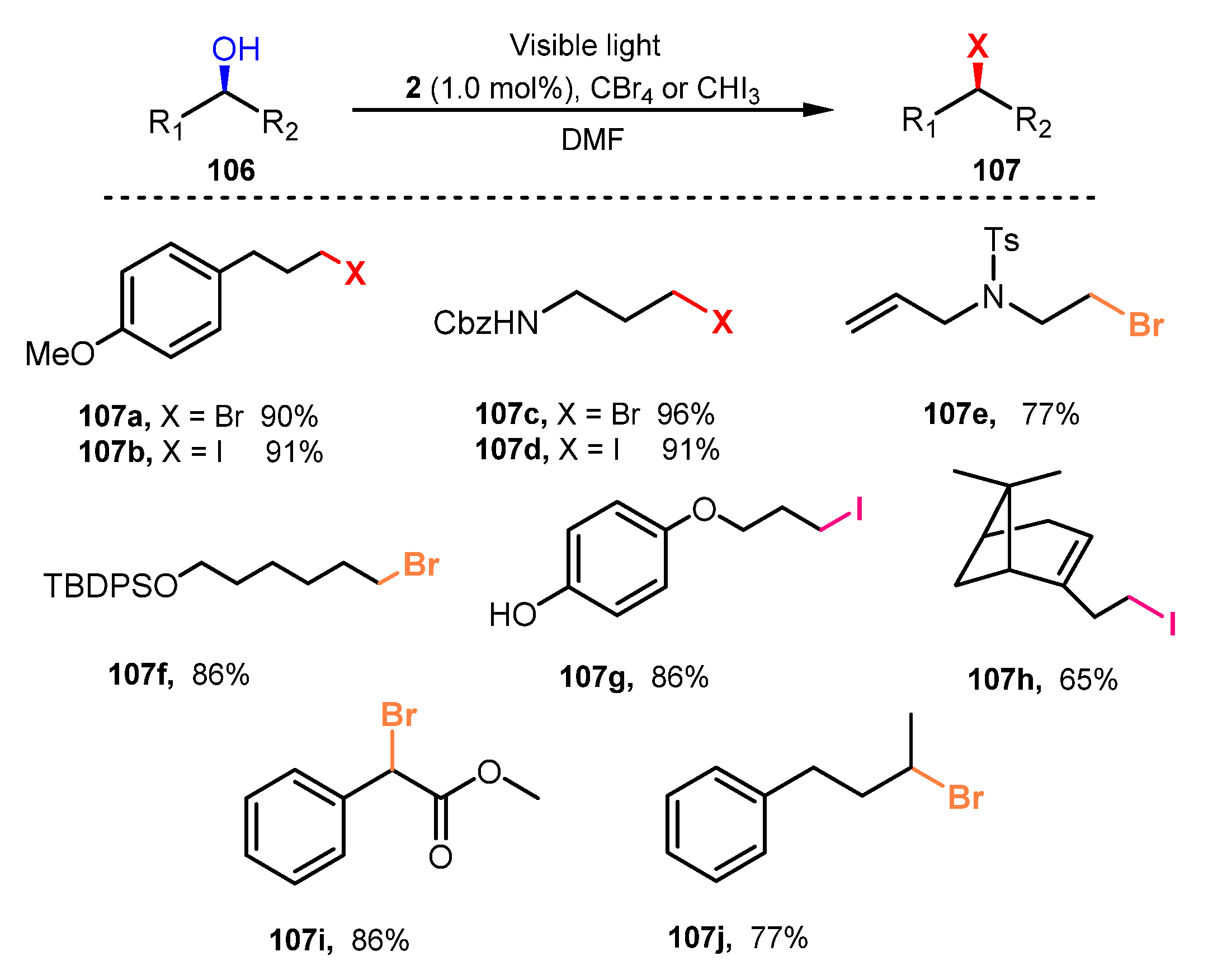
In 2011, Stephenson and co-workers performed halogenation of alcohols in the presence of CBr4 or CHI3 as halide sources and Ru(bpy)3Cl2 as a photocatalyst in DMF under radiation of blue LED irradiation at room temperature (Scheme 28) [39]. Substrates bearing various functional groups such as ethers, silyl ethers, alkene, alkynes, carbamates, and phenols were tolerated for this reaction procedure (107a–h). In this reaction, primary alcohols were successfully converted to the corresponding halides with yields ranging from 77 to 98%. Reactions of secondary alcohols were smoothly conducted for the bromination and iodination processes (107i–j), although the reaction rates were slower than those of primary alcohols.
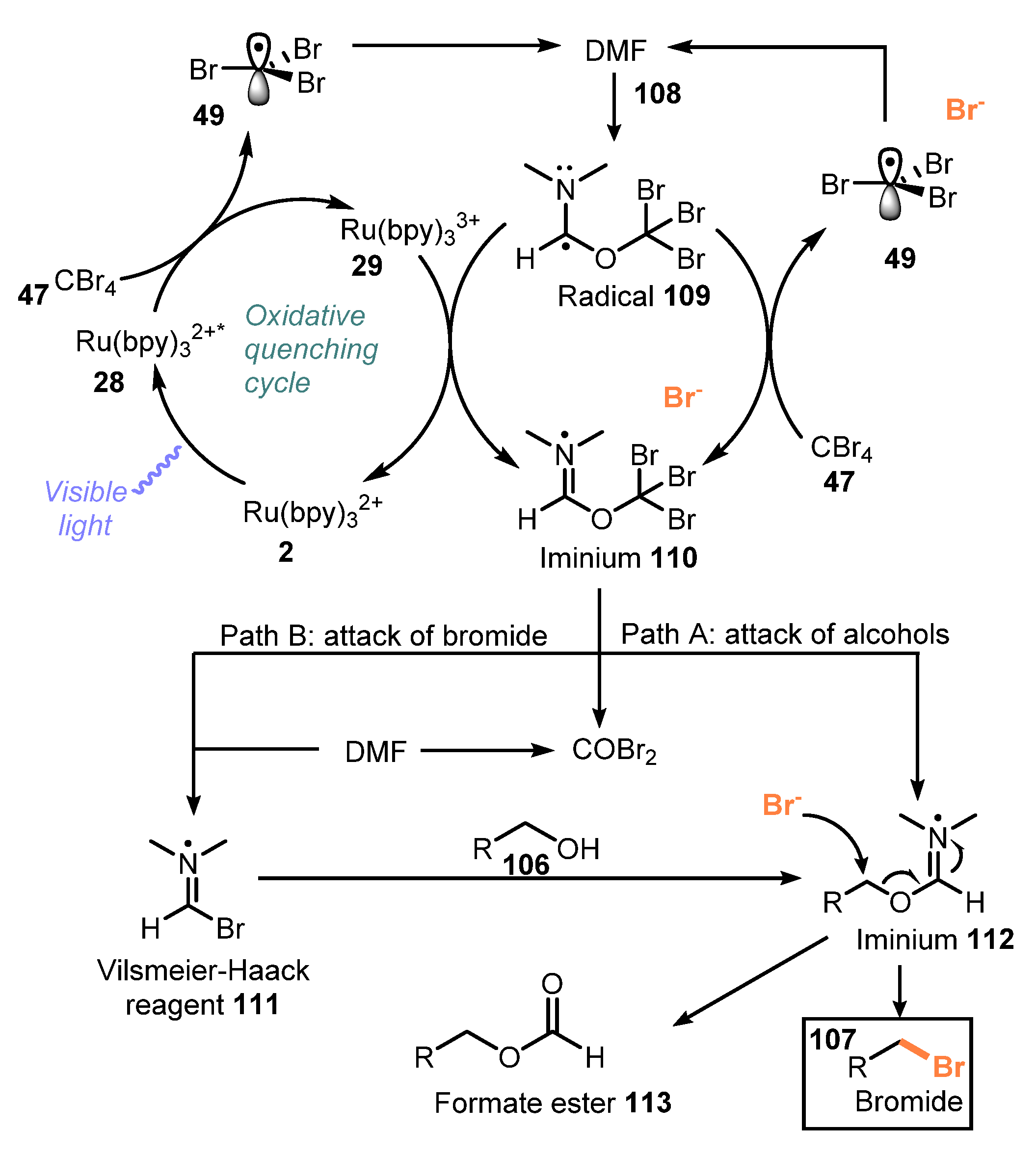
A possible mechanism was proposed as shown in Scheme 29. Under visible light irradiation, the photocatalyst Ru(bpy)32+ 2 was changed to excited state Ru(bpy)32+* 28, which underwent single-electron oxidation by CBr4 47 to generate Ru(bpy)33+ 29 and electron-deficient radical •CBr3 49. The •CBr3 radical 49 then reacted with DMF 108, resulting in stable radical 109. Ru(bpy)33+ 29 was reduced by radical 109 to return Ru(bpy)32+ 2 and produced intermediate 110. On the other hand, intermediate 110 was also generated through the reaction of radical 109 with CBr4 47. At this point, there are two possible ways to afford the target product. The first way involved the reaction of alcohol with compound 110 to create intermediate 112. In the second process, the bromide anion directly attacked intermediate 110 to generate Vilsmeier–Haack reagent 111, which then reacted with alcohol 106 to form intermediate 112. Finally, the SN2 substitution reaction of 112 with bromide anion provided the desired product 107.
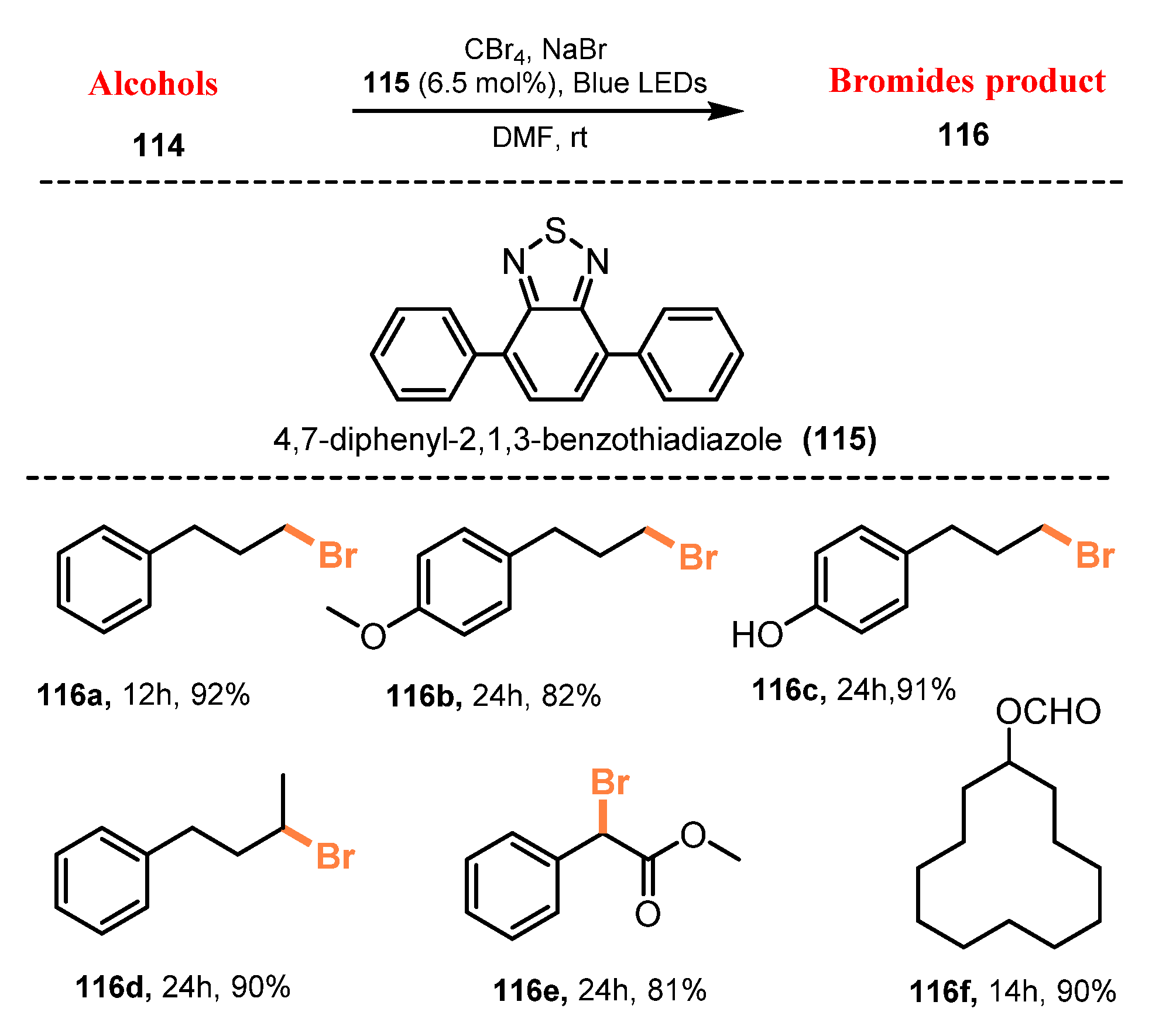
Bromination of alcohols using metal-free organic photocatalyst was demonstrated by Li and co-workers in 2019 [40]. The bromination reaction of alcohols was carried out in the presence of CBr4 as a bromide source and 4,7-diphenyl-2,1,3-benzothiadiazole (Ph-BT-Ph) as a photocatalyst under blue LEDs irradiation in DMF at room temperature to yield the corresponding products (Scheme 30). Both primary and secondary alcohols were readily converted into desired bromides in the use of Ph-BT-Ph. Reaction yields of primary alcohols (116a–c) were somewhat greater than those of secondary alcohols (116d–e). It was noted that formate ester was observed as a minor side product from the reaction of alcohols, and, in the reaction of cyclododecanol, cyclododecyl formate (116f) was generated as a main product. No photobleaching impact of photocatalyst was discovered.
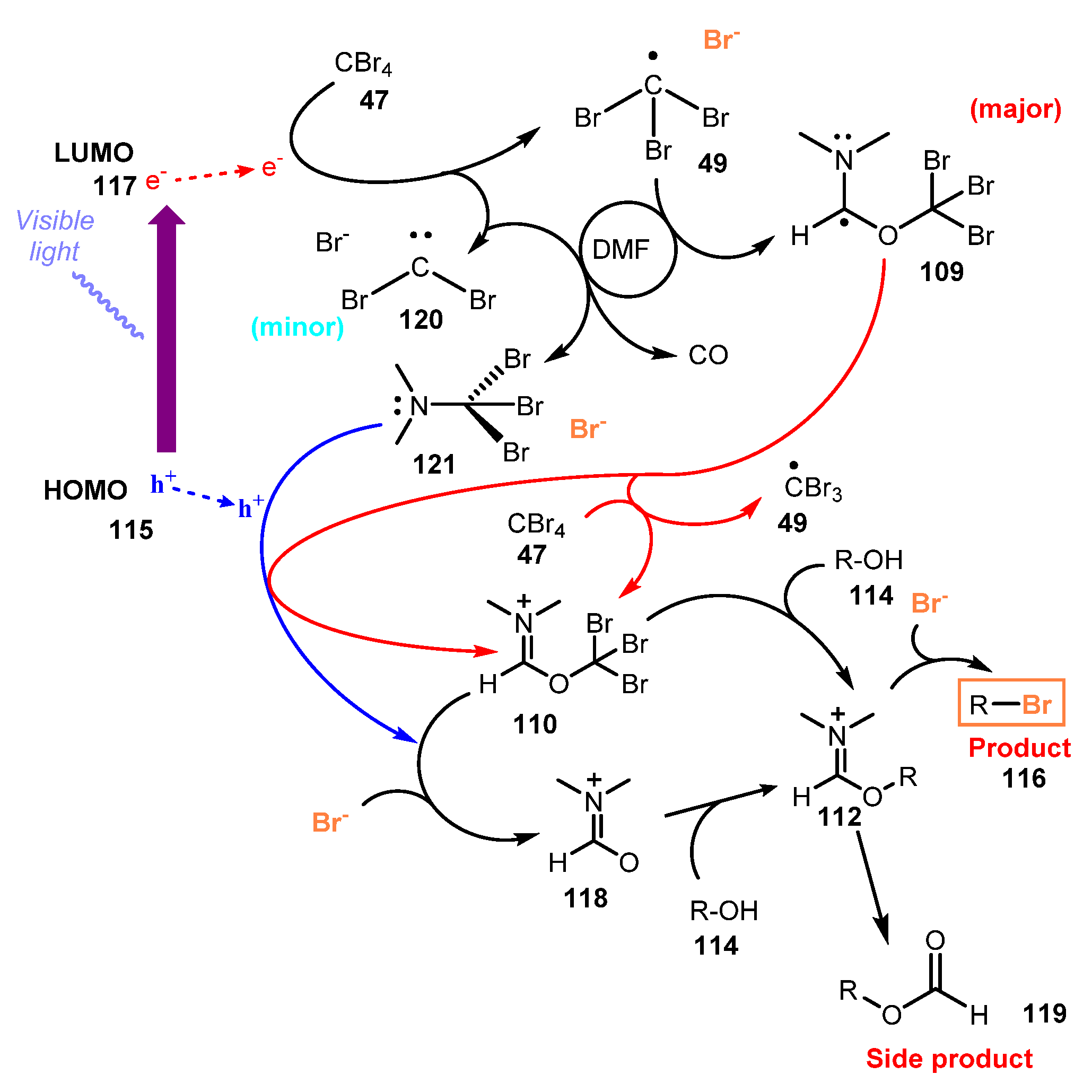
A mechanism was proposed as shown in Scheme 31. Under irradiation of visible light, one electron was transferred from the lowest unoccupied molecular orbital (LUMO) of the photocatalyst Ph-BT-Ph 115 to CBr4 47 to afford •CBr3 radical 49 and Br−. DMF captured radical 49 to give intermediate 109, which subsequently delivered an electron, resulting in iminium compound 110. The bromide ion reacted with intermediate 110 to generate Vilsmeier–Haack reagent 118, which then interacted with alcohol 114 to produce the desired compound 119. In another pathway, reduction of CBr4 47 by photocatalyst Ph-BT-Ph 115 gave carbene CBr2 120. Then, reaction of carbene CBr2 120 with DMF produced CO and (dibromomethyl) dimethylamine intermediate 121, which was also converted to Vilsmeier–Haack reagent 118 after losing one bromide atom.
5. Photo-Catalyzed Halogenations of Carboxylic Acids
Scheme 32 shows schematic diagrams of the comparison of the traditional methods with visible-light-induced halogenation of carboxylic acids.
5.1. Chlorination of Carboxylic Acids
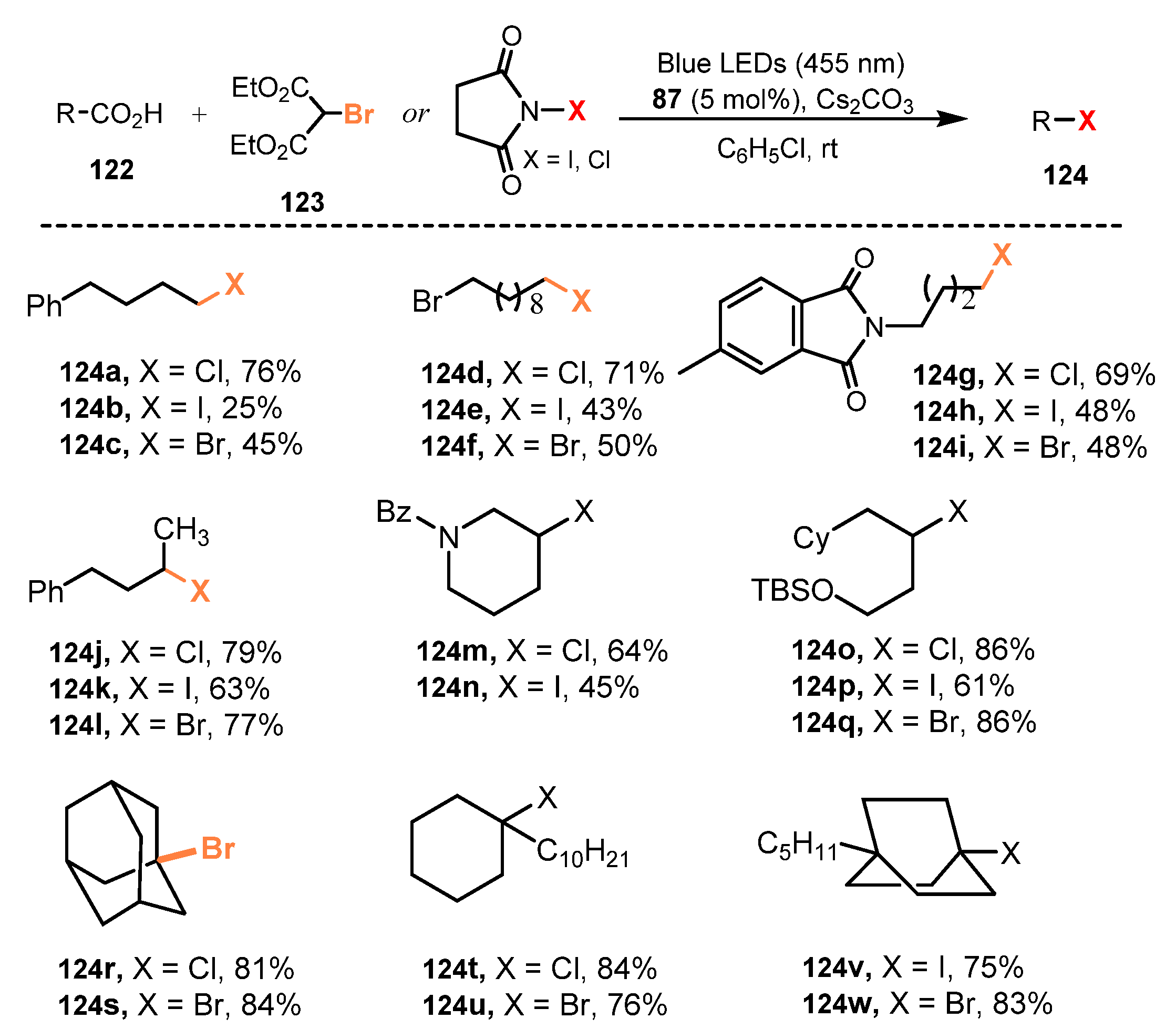
In 2016, Glorius and co-workers reported photocatalytic Hunsdiecker-type decarboxylative halogenation (bromination, chlorination and iodination) of alkyl carboxylic acids [41]. Diethyl bromomalonate, N-chlorosuccinimide (NCS), and N-iodosuccinimide (NIS) were used as halide sources, Cs2CO3 was a promoter, and [Ir(dF(CF3)ppy)2(dtbbpy)]PF6 was a photocatalyst in chlorobenzene to perform this decarboxylative halogenation of carboxylic acids under irradiation of blue LEDs (lmax = 455nm) (Scheme 33). Primary, secondary, and tertiary carboxylic acid substrates were all tolerated for this method. A broad range of functional groups such as esters, protected amines, aryl, and silyl ethers were successfully used in this protocol to produce target products with good to high yields (124a–w) (25–86%). It was discovered that the reaction could also be achieved with excellent product yields in ethyl acetate instead of chlorinated solvents. Besides, this reaction could be conducted on a gram scale (5 mmol) without reducing the yield, even though a longer reaction time was required.
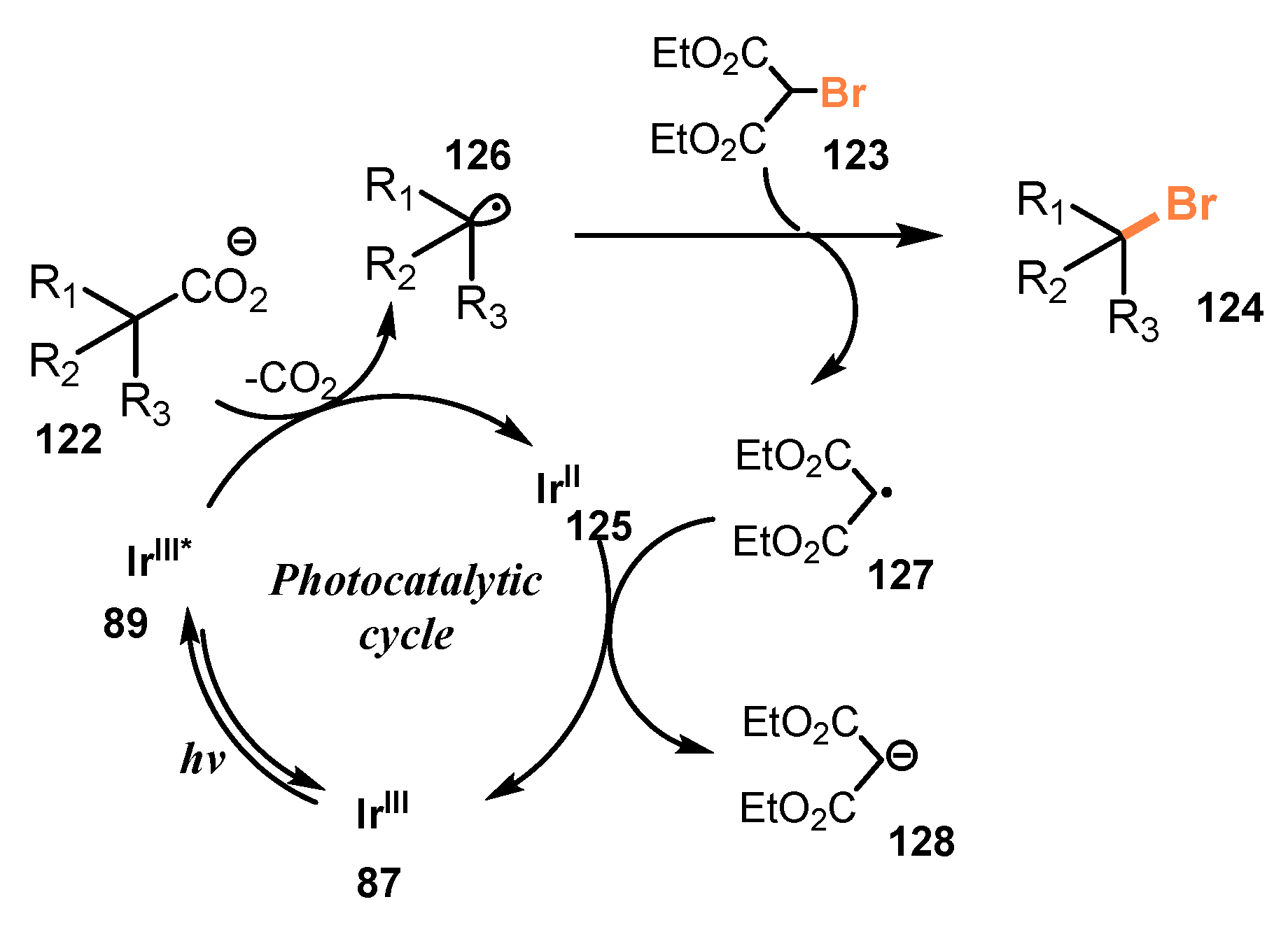
A proposed mechanism for this method is illustrated in Scheme 34. Photocatalyst IrIII 87 was transformed to IrIII* 89 under the irradiation of visible light. Photoexcitation of photocatalyst IrIII* 89 facilitated decarboxylation of substrate 122 to give IrII 125 and appropriate alkyl radical 126, which captured a halide atom from the halide source 123 to give the final product 124 and malonyl radical 127 as a byproduct. The malonyl radical 127 received one electron from IrII 125 to recover photocatalyst IrIII 87 and yielded the malonate anion 128.
5.2. Bromination of Carboxylic Acids
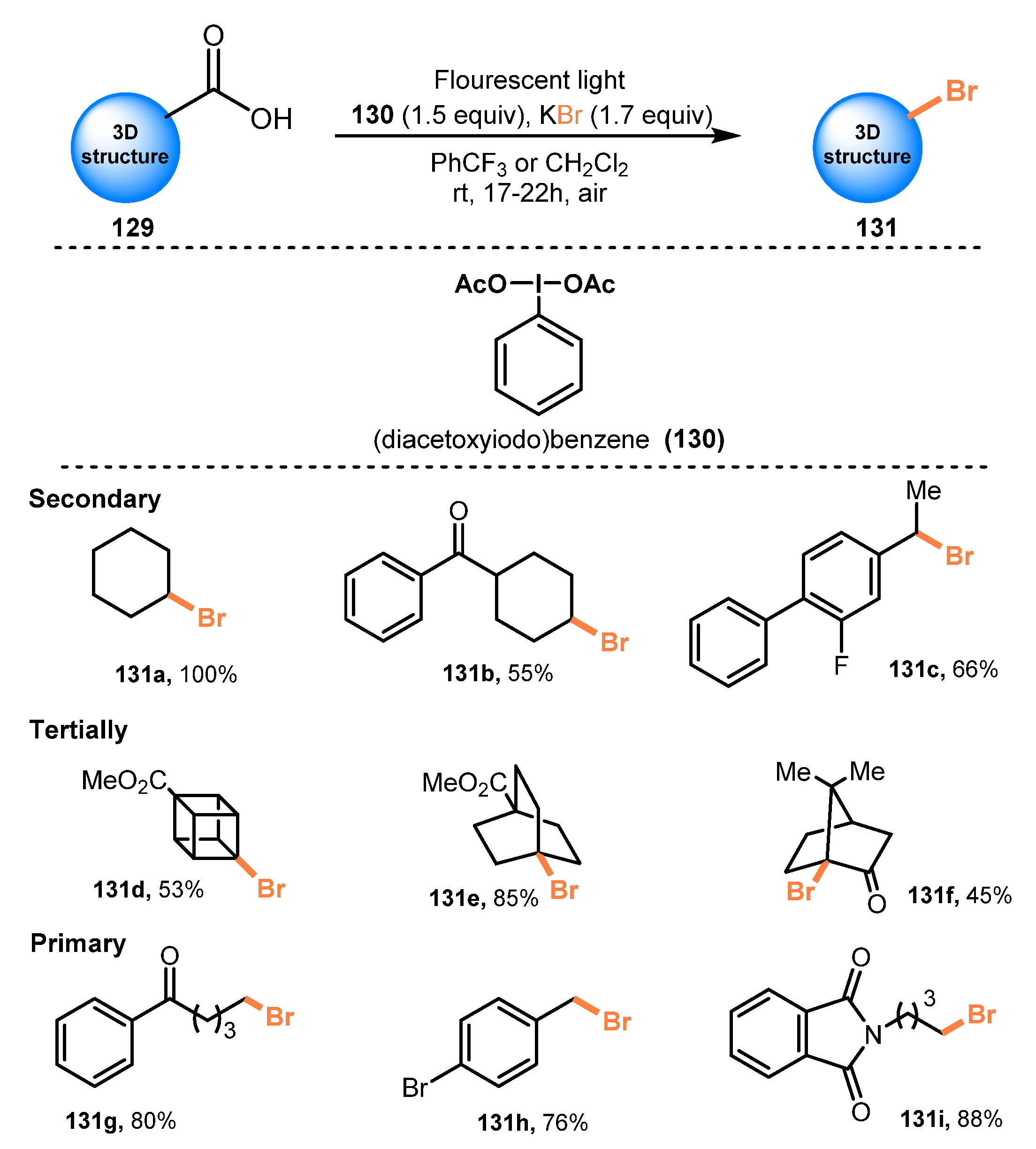
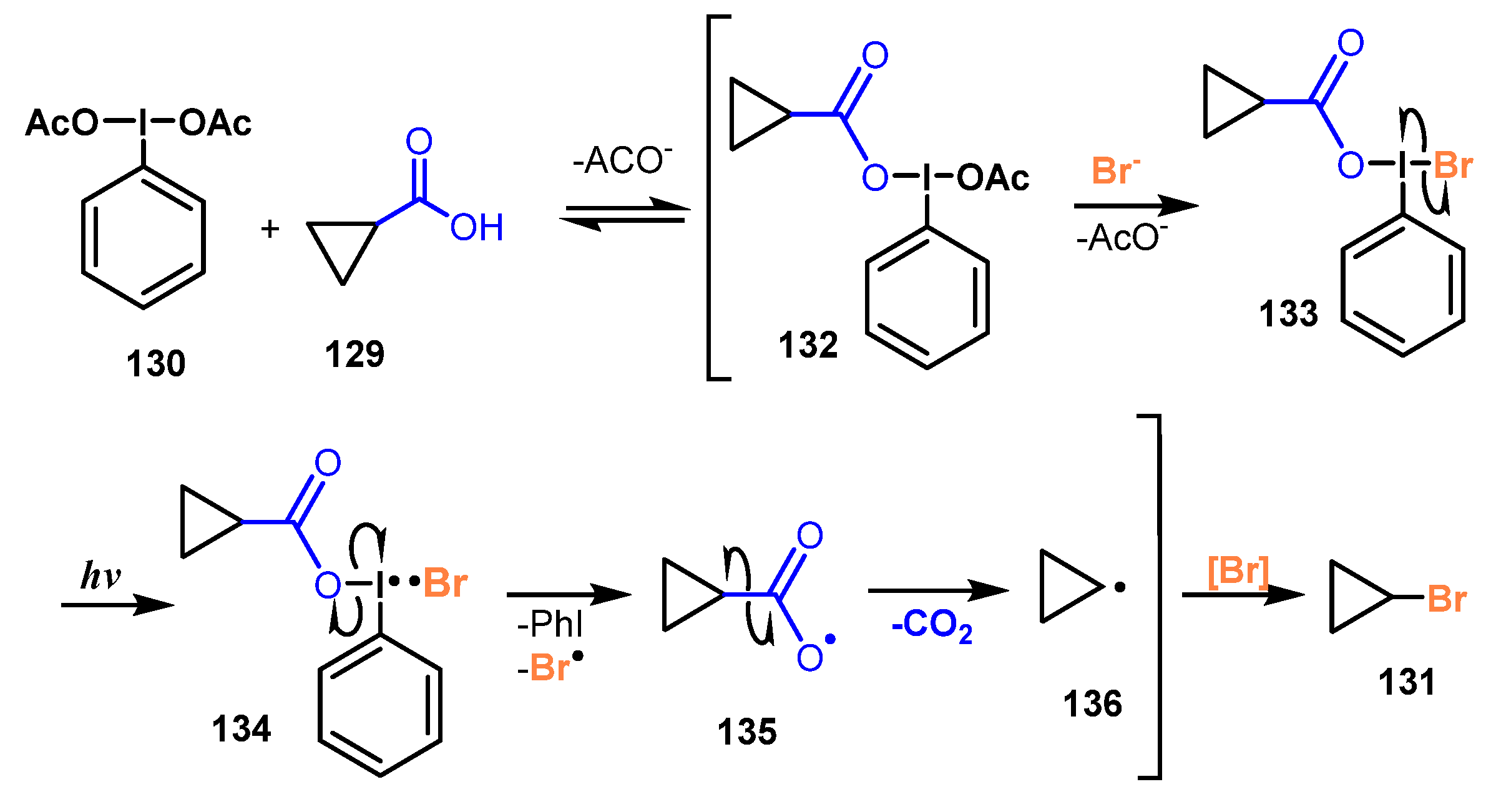
Another decarboxylative bromination of carboxylic acids using potassium bromide was reported by Uchiyama and co-workers in 2020 [42]. In this protocol, a series of sterically hindered primary, secondary, and tertiary carboxylic acids, bearing different structures such as acyclic, cyclic, caged, and bridgehead, were treated with potassium bromide in the presence of (diacetoxyiodo)benzene as a photocatalyst in CH2Cl2 or PhCF3 under irradiation of a ceiling fluorescent light at room temperature to generate the corresponding products without rearrangement or fragmentation (Scheme 35). Substrates bearing nitro, ester/lactone (131d–e), amide/sulfonamide/2-nitrophenylsulfonyl (nosyl)/imide (131i), ketone (131b, 131f–g), and bromide/fluoride (131h) functionalities were tolerated for this brominating procedure. Additionally, reaction of carboxylic acid with the extremely radical-sensitive ether group was also successfully achieved with 87% yield.
A possible mechanism was proposed as shown in Scheme 36. The hypervalent iodine reagent PhI(OAc)2 130 reacted with substrate 129, followed by treatment with KBr, to provide intermediate 133. Intermediate 133 was triggered by visible light to yield •Br radical and iodo-radical intermediate 134. The I-O bond in radical 134 was cleaved to afford acyloxy radical 135. Removal of CO2 from 135 gave cyclopropyl radical 136. At last, radical 136 captured the bromide source to generate the desired product 131.
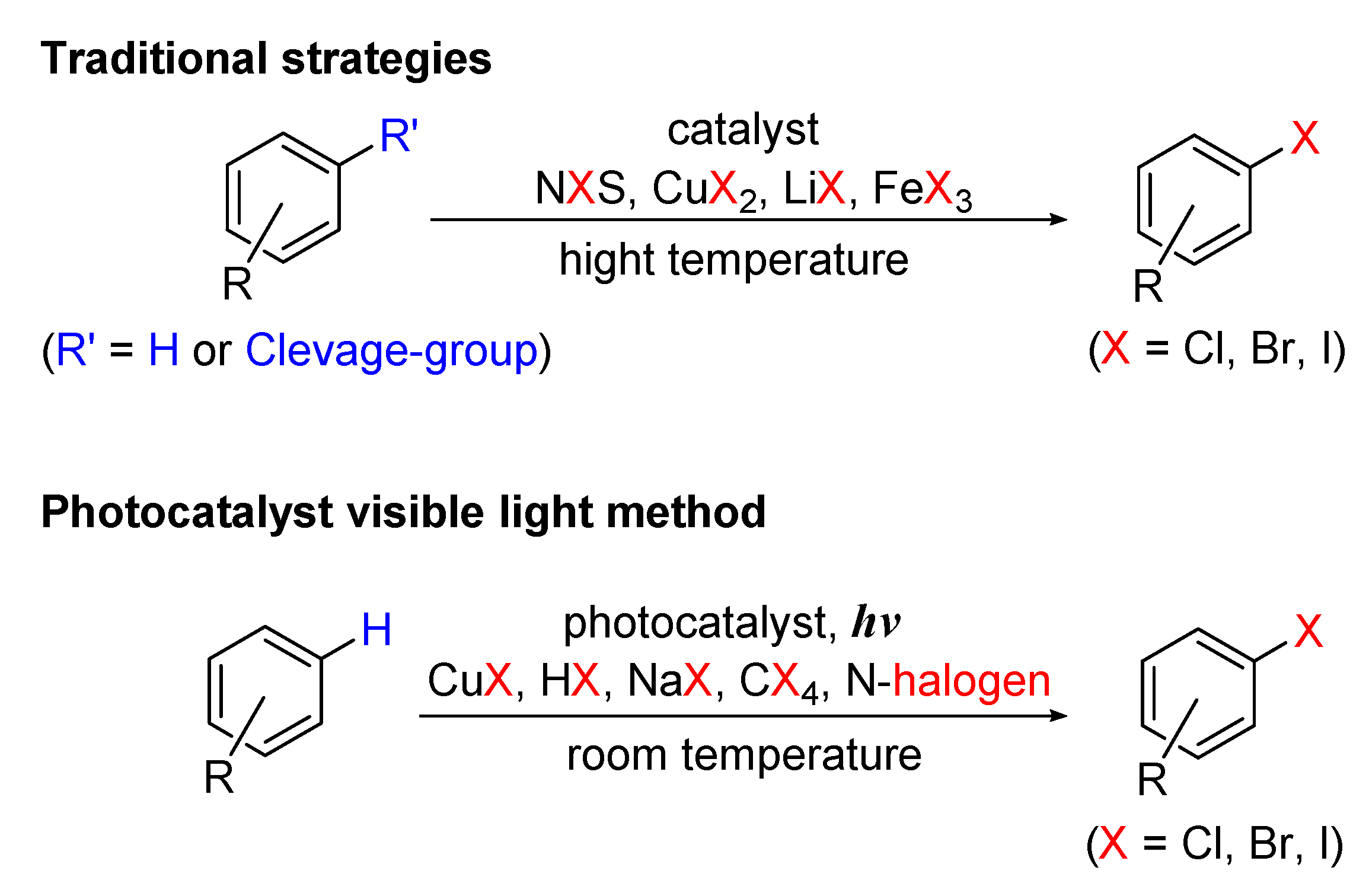
6. Photo-Catalyzed Halogenations of Aromatic C-H Bonds
Scheme 37 shows schematic diagrams of the comparison of the traditional methods with visible-light-induced halogenation of aromatic C-H bonds.
6.1. Halogenation of Aromatic C-H Bonds
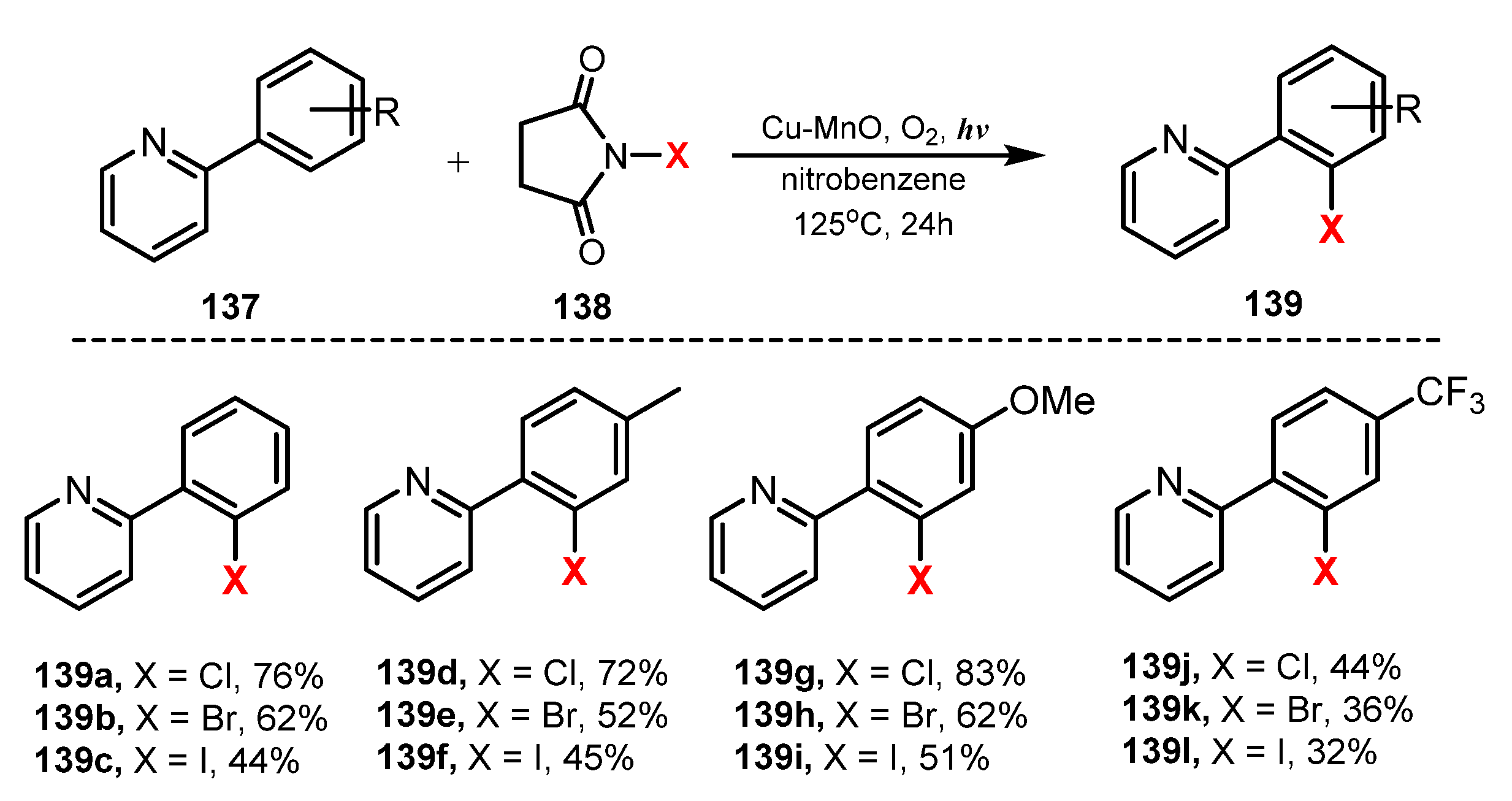
In 2015, Ghosh and co-workers reported halogenation of aromatic C-H bonds utilizing Cu-MnO as a heterogeneous catalyst [43]. In this methodology, Cu-MnO as a catalyst, N-halogen succinimide as a halide source, and O2 as an oxidant reagent were employed to perform the halogenation in nitrobenzene at 125 °C under irradiation of visible light to provide the corresponding products (Scheme 38). It was reported that good yield and high regioselective halogenation of aromatic C-H bonds can be well achieved with other transition metal catalysts (Pd, Au, Ru, Co) [44,45,46,47]. However, in this study, a heterogeneous Cu-MnO catalyst was employed due to the cost-efficiency, ubiquity, and versatility properties of Cu. This halogenation (chlorination, bromination, and iodination) produced monohalogenated products selectively in moderate to high yields (32–83%). In this method, chlorination performance was generally better than that of bromination and ionization. Using this protocol, the monohalogenated products from substrates bearing electron-donating groups such as methyl and methoxy groups in the para position of the aromatic ring were prepared with high yield (136d–i), whereas the monohalogenated products from substrates bearing an electron-withdrawing group, such as the trifluoro-methyl group (139j–l), were obtained with moderate to good yields (32–44%).
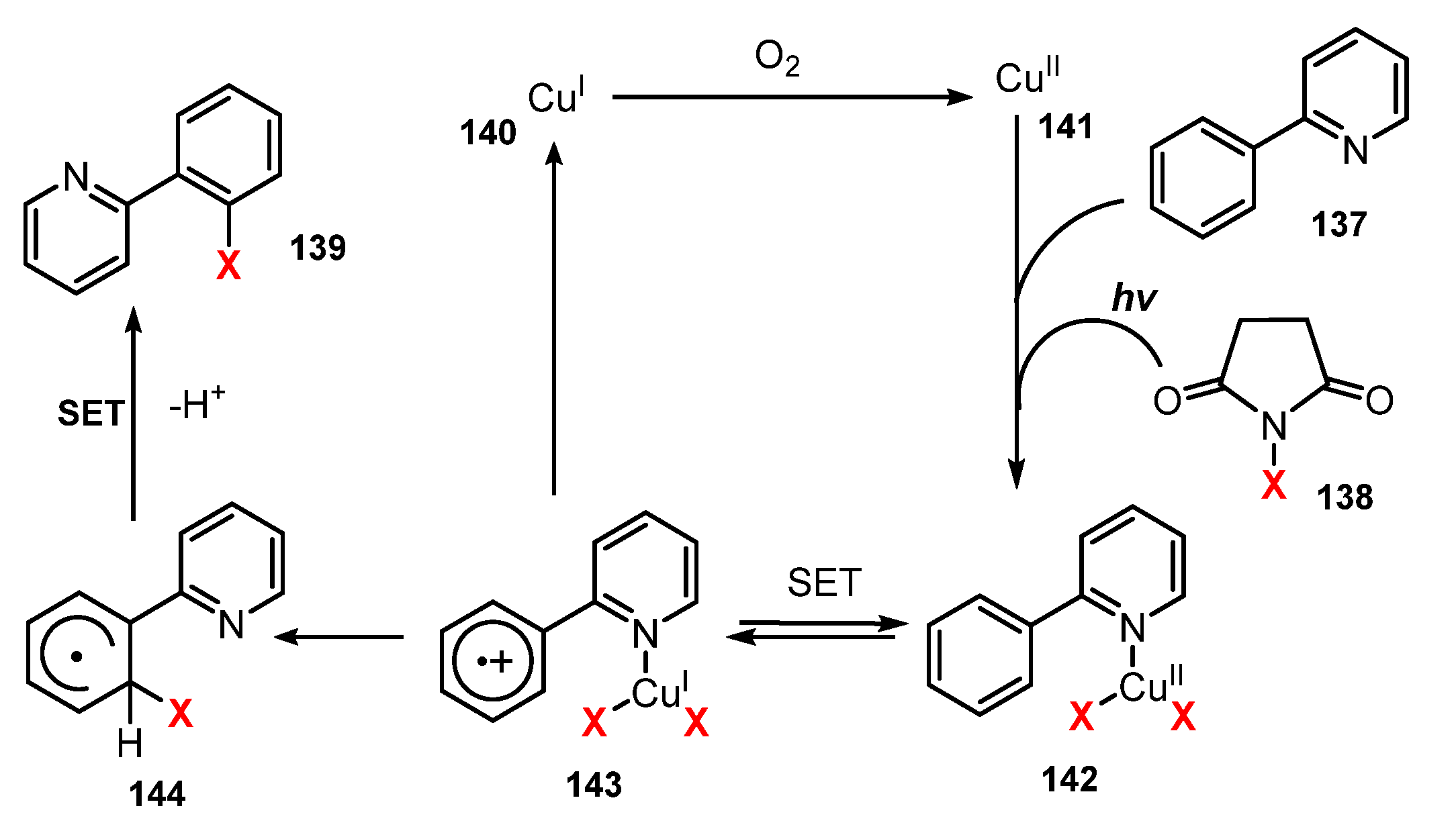
A plausible mechanism of this halogenation as proposed by Ghosh and co-workers is shown in Scheme 39. After oxidization of CuI 140 to CuII 141 by O2, CuII 141 reacted with 2-phenylpyridine 137 and halide ion, which was generated from NXS 138 under irradiation of visible light to produce complex 142. A single electron transfer (SET) process between the phenyl ring and CuII caused complex 142 to become cationic radical 143. Then, intramolecular transfer of the halide anion to the phenyl ring of 143 yielded compound 144 and recovered CuI 140. Finally, 144 underwent another single electron transfer (SET) process to lose a proton to give the desired product 139.
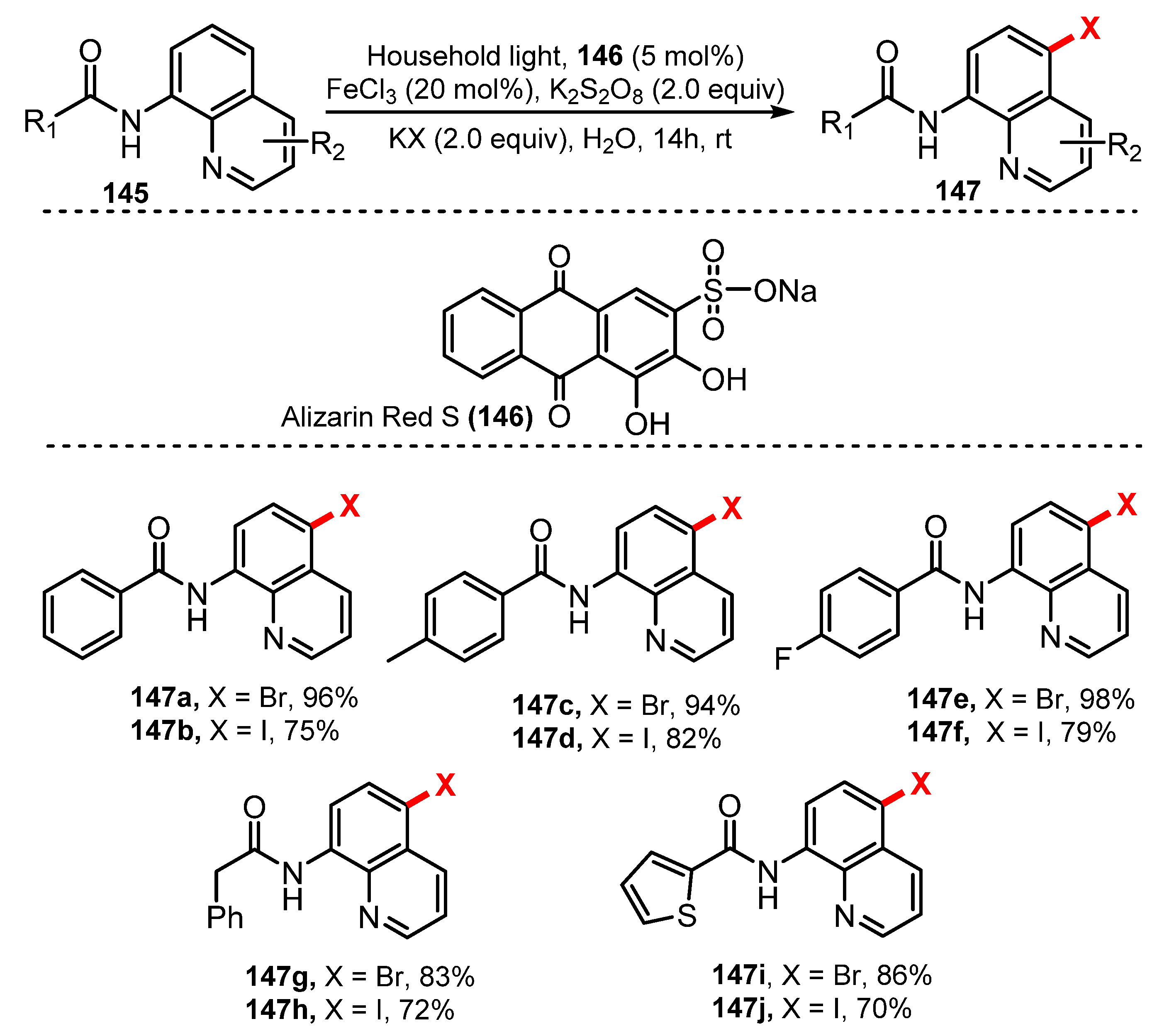
In 2017, Wu and co-workers demonstrated halogenation of quinoline using a photoredox process in mild conditions [48]. This halogenation was achieved in the presence of alizarin red S as a photocatalyst, FeCl3 as a catalyst, K2S2O8 as an oxidant, and potassium halides as halogen sources under irradiation of CFL in water at room temperature (Scheme 40). Being abundant, readily obtainable, inexpensive, and non-toxic, water is more environmentally friendly compared to other organic solvents. However, most organic substrates are poorly soluble in neat water, and, thus, the reactions that take place in neat water are often inefficient and generate the products with low yields. On the other hand, organic substrates are easily soluble in organic solvents such as DMF, and organic solvents help increase the reaction yields even though they are harmful and not environmentally friendly. Wu’s method used water as a solvent, but could overcome the disadvantage of aqueous solvents to still achieve high yields. Using this reaction procedure, all target compounds were readily prepared in good to outstanding yields, but the effect of substitute groups on the benzene ring of benzamides on the reaction efficiency was not clearly understood. Bromination was conducted more effectively than iodination. Substrates with methoxy, methyl, and halide groups on the quinoline ring yielded the desired products in good yields (75–98%) (147a–f). Additionally, the reaction of heterocyclic amide substrate produced the halogenated product in high yields (70–83%) (147g–j).
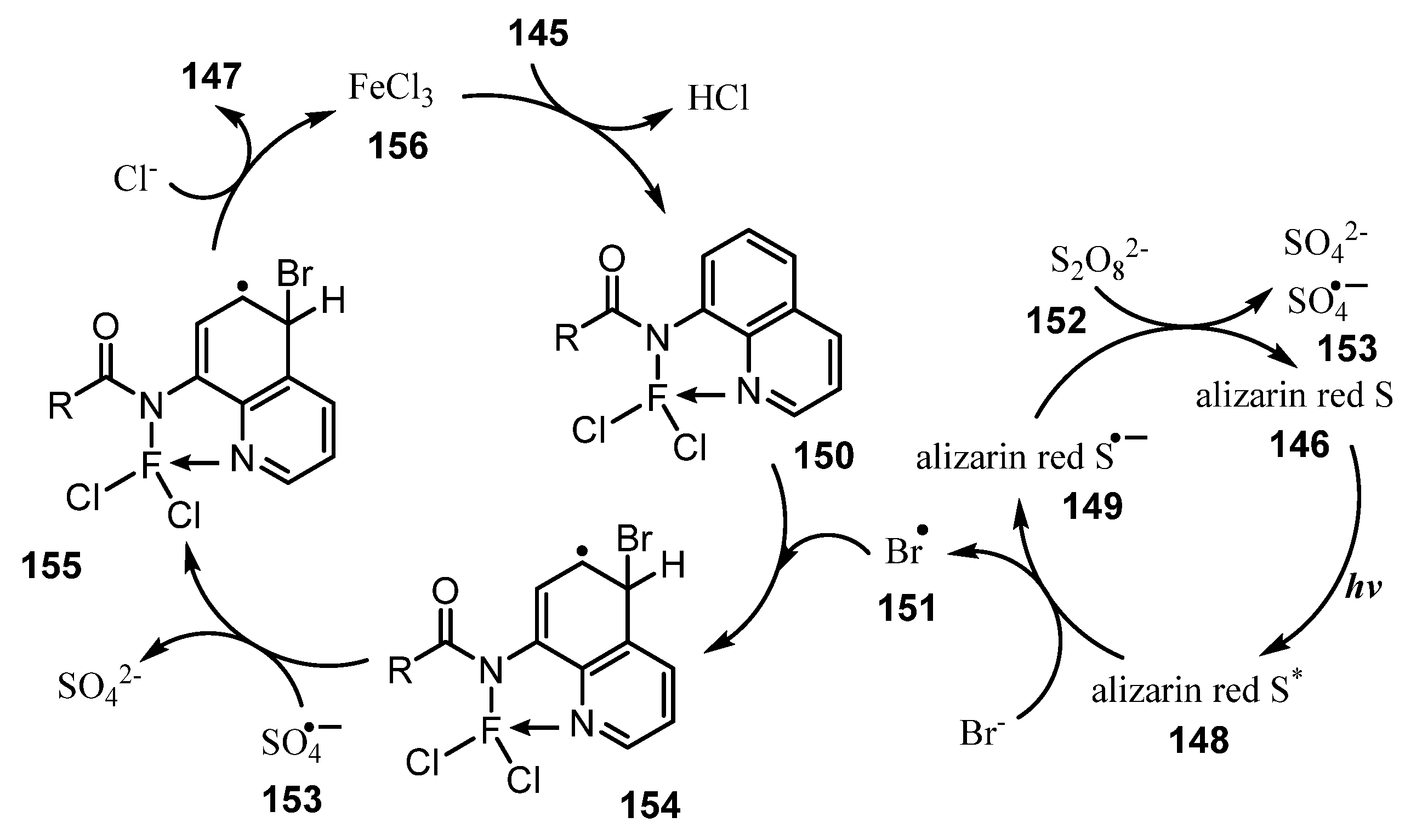
A plausible mechanism is illustrated in Scheme 41. Initially, the household light excited photocatalyst alizarin red S 146, resulting in the excited state alizarin red S* 148. Alizarin red S* 148 was reductively quenched by Br-, affording a •Br radical 151 and an alizarin red S*− 149. K2S2O8 152 then oxidized alizarin red S•− 149 to recover the ground state alizarin red S 146 and provided SO4•− 153. FeCl3 incorporated with substrate 145 to give chelated compound 150, which reacted with •Br radical to give intermediate radical 154. Then, intermediate radical 154 was oxidized by SO4•− 153 to generate cation intermediate 155. Finally, intermediate 155 interacted with a chloride anion to give the final product 147, while recovering FeCl3 156.
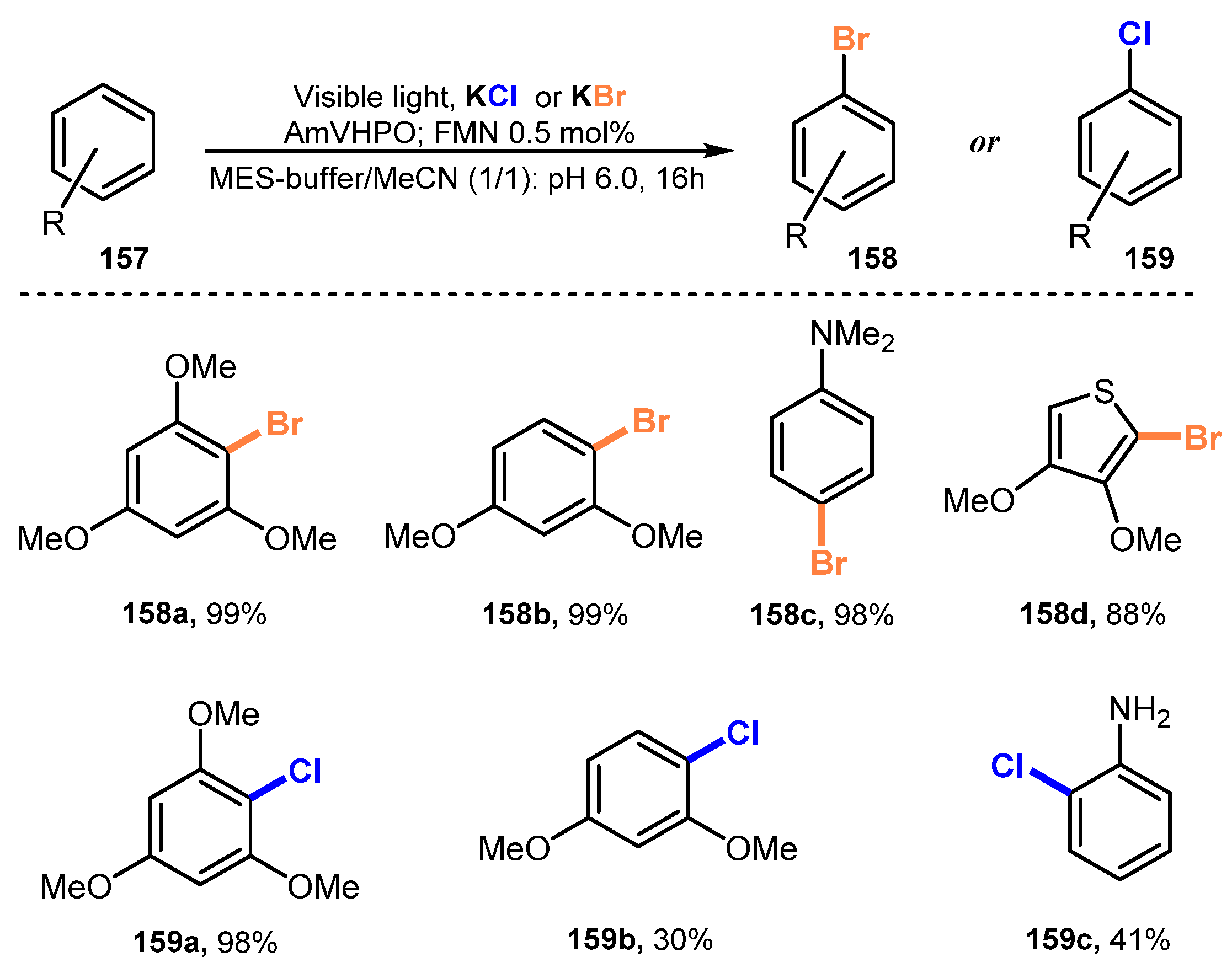
Combining photocatalysis and biocatalysis for halogenation of aromatic compounds was described by Gulder and co-workers in 2018 [49]. They employed vanadium-dependent haloperoxidase (VHPO) from Acaryochloris marina (AmVHPO) and Curvularia inaequalis (CiVHPO) as biocatalysts, flavin mononucleotide (FMN) as a photocatalyst, and KBr or KCl as halide providers for halogenation under irradiation of blue LEDs in a mixture of MES buffer (pH = 6.0) and MeCN to yield the corresponding products (Scheme 42). The method was highly effective for the bromination process. Reactions of substrates with benzene ring containing methoxy substituents provided target products in excellent yield (99%) (158a–b). Substrates with a heterocycle ring were halogenated with moderate performance (158d). Regarding chlorination, this method was less efficient for one- and two-substituent derivatives (159b–c).
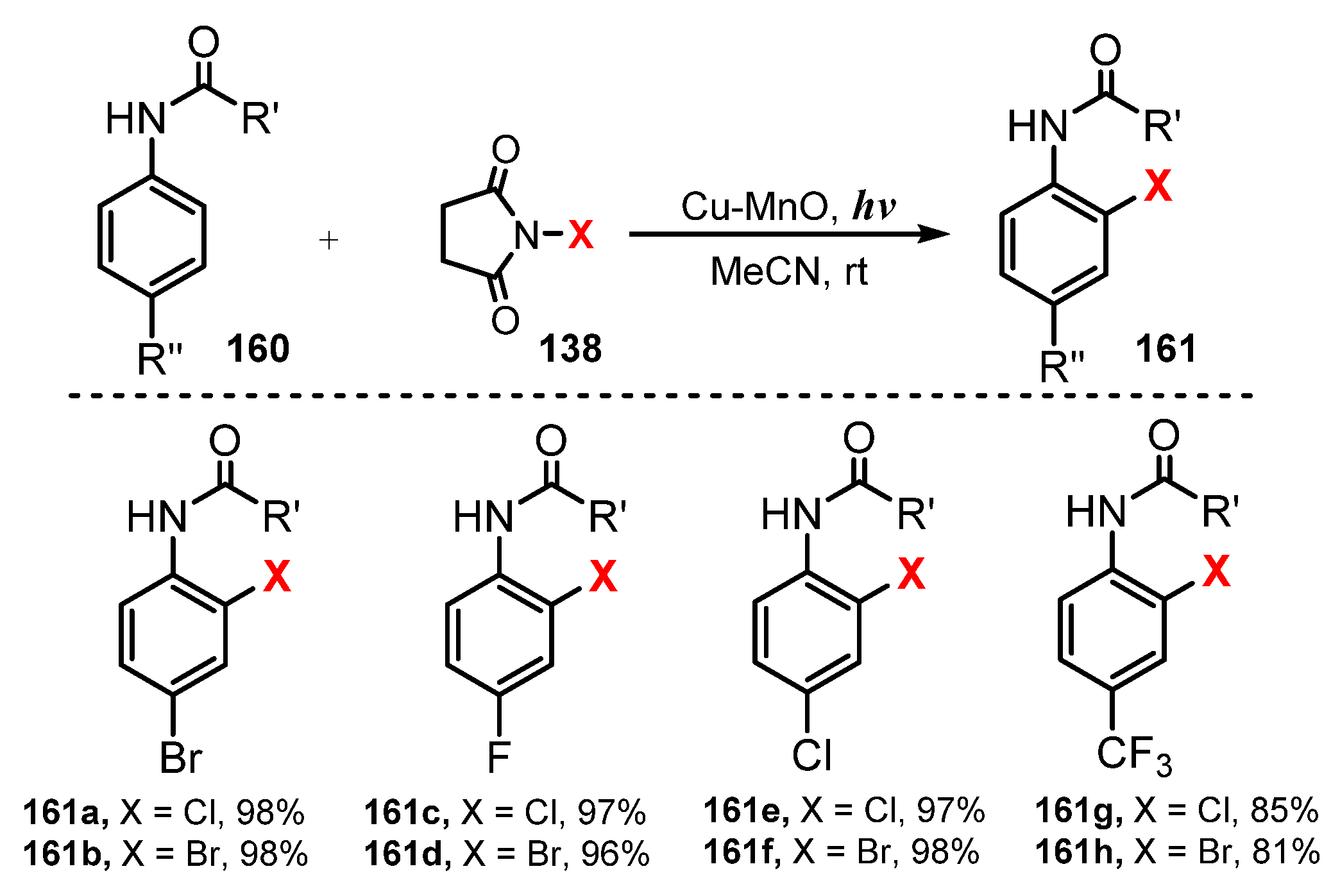
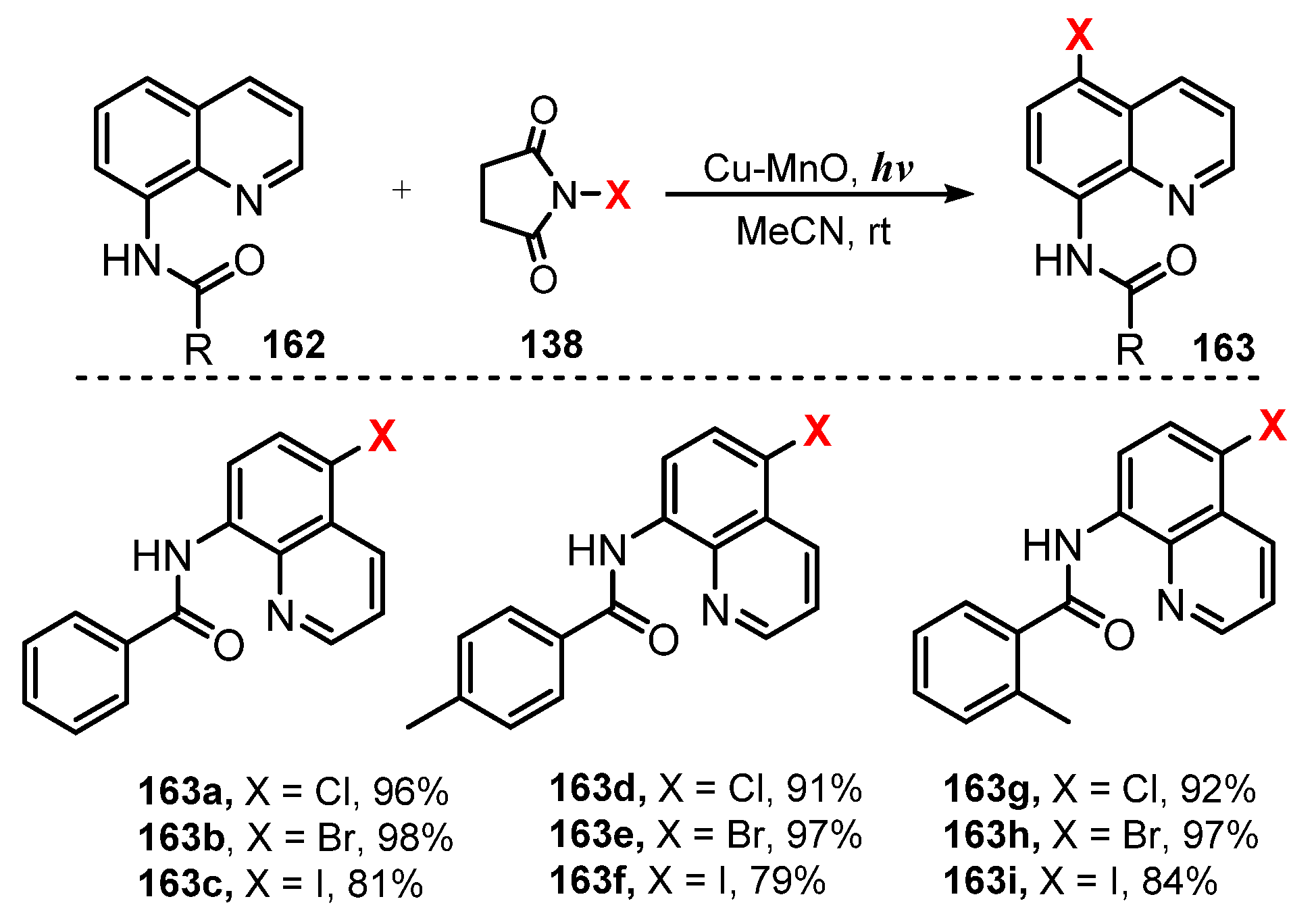
In 2018, Ghosh and co-workers reported extensive application of Cu-MnO catalyst for halogenations of anilides and quinolones [50]. They used Cu-MnO as a catalyst and N-halosuccinimide as a halogenating source in acetonitrile under visible light irradiation to achieve halogenations of anilides and quinolines with good regioselectivity (Scheme 43 and Scheme 44). For anilide derivatives, reaction of para-substituted substrates containing both electron-withdrawing and electron-donating groups, such as anilides bearing isopropyl, tert-butyl, hexyl, and cyclohexyl groups in the amide chain and chloro, bromo, fluoro, and trifluoromethyl groups in the phenyl ring in acetanilide, successfully produced mono ortho-halogenated products in high yields (81–98%) (161a–h). The protocol showed that halogenation of 8-aminoquinoline amides with a variety of functional groups was also successful and worked well with an aryl amide group and an alkyl amide group (163a–i). Benzamides containing both electron-donating and electron-withdrawing groups were readily halogenated. In addition, the reactions of alkyl amides such as acetamide, cyclopentanecarboxamide, and decanamide were successfully conducted to give desired products.
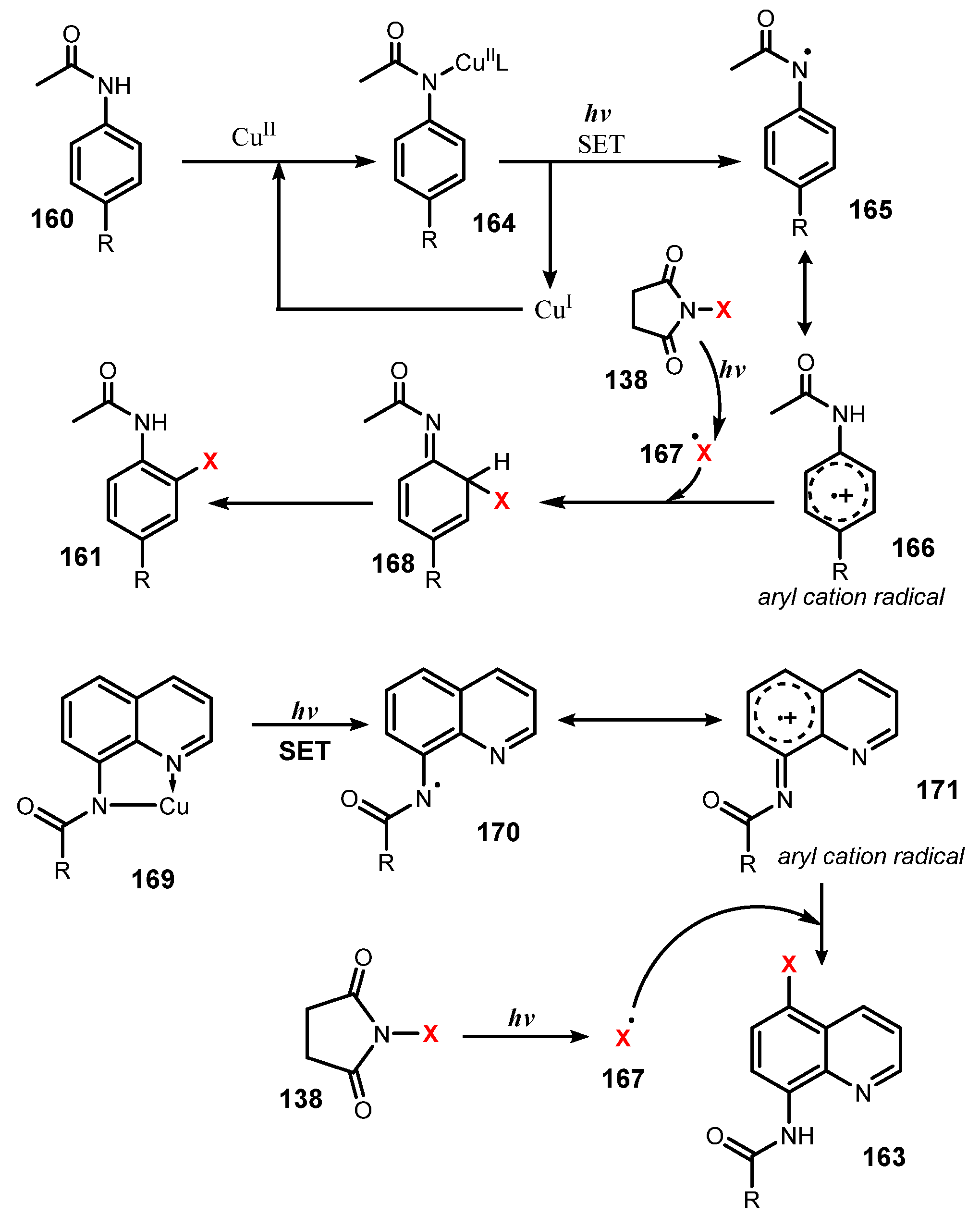
A possible mechanism for this halogenation was proposed as shown in Scheme 45. For anilides, nitrogen of substrate 160 was coordinated with CuII to generate complex 164, and then visible light caused the N–Cu bond of 164 to homolytically break to give the nitrogen radical 165 and CuI, which was oxidized by O2 to recover CuII. The radical 165 was subsequently converted to intermediate aryl cation radical 166. Reaction of 166 with the halide radical produced from N-halosuccinimide 138 under visible light occurred, followed by rearomatization to afford the final product 161. For the 8-amidoquinoline derivatives, a similar chemical mechanism was presented. This method showed regioselective addition of halide radical to the amide group at ortho- and para-positions, the most electrophilic locations.
6.2. Chlorination of Aromatic C-H Bonds
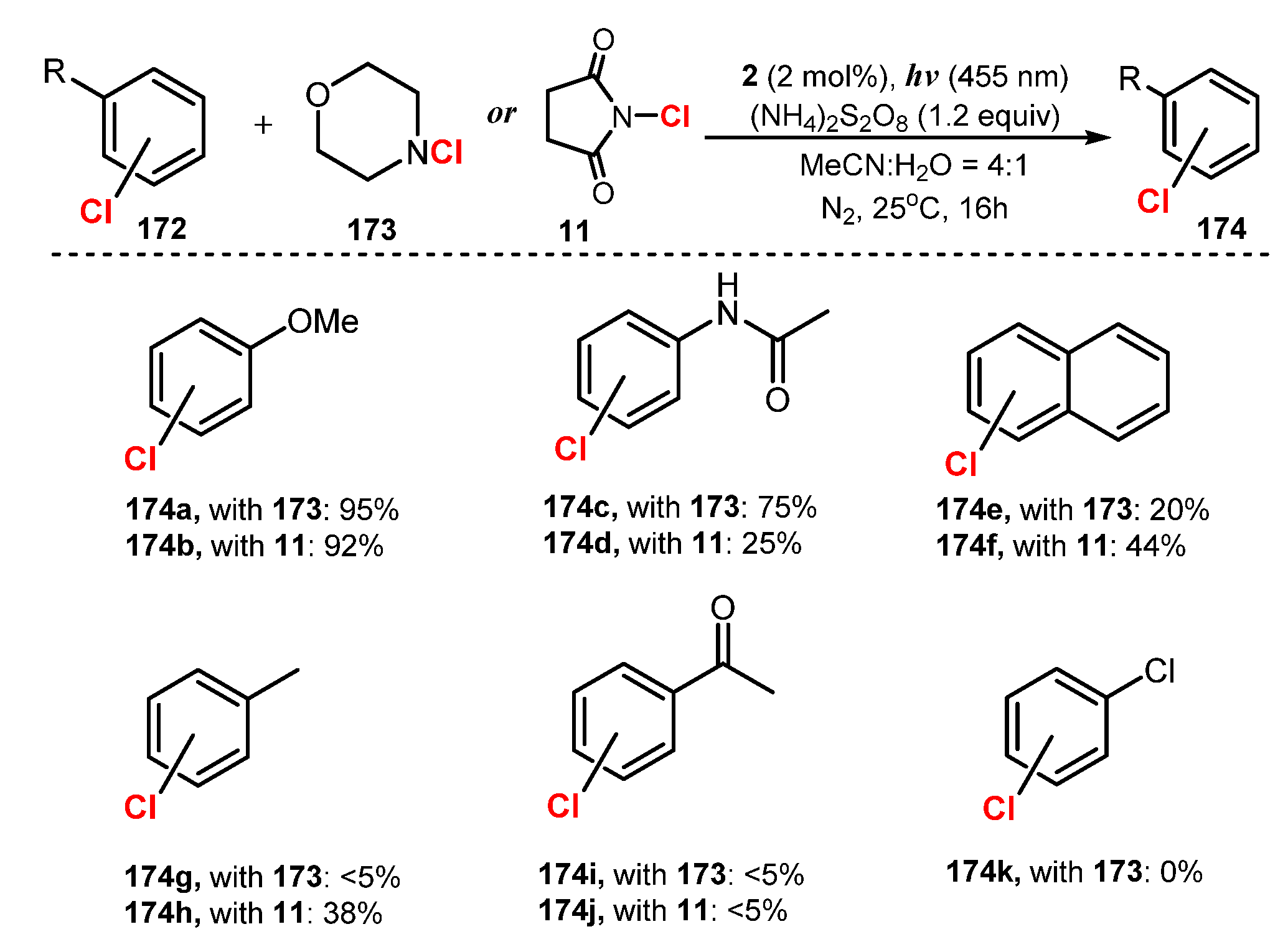
In 2016, Konig and co-workers proposed chlorination of arenes via reaction with N-chlorosuccinimide (NCS) or N-chloramines in the presence of [Ru(bpy)3]Cl2 as a photocatalyst and ammonium peroxodisulfate as an oxidant under irradiation of blue LEDs in a mixture of acetonitrile and water (4:1) (Scheme 46) [51]. In these reaction conditions, substrates with electron-donating groups such as anisole (174a–b), methoxybenzene, phenol, and acetanilide were chlorinated in good yields (92–95%) via treatment of both N-chlorosuccinimide 11 and N-chloramines 173. However, reaction of aromatic amines (174c), xylene, and toluene (174e) provided chlorinated products with low yields when N-chloramines were used. Besides, electron-poor substrates such as chlorobenzene (174k) did not provide chlorination. When compared to N-chloramines 173, the electron density on the nitrogen atom was significantly lowered by two electron withdrawing groups in N-chlorosuccinimide 11, and it was able to chlorinate fewer electron-dense substrates such as xylene and toluene by NCS.
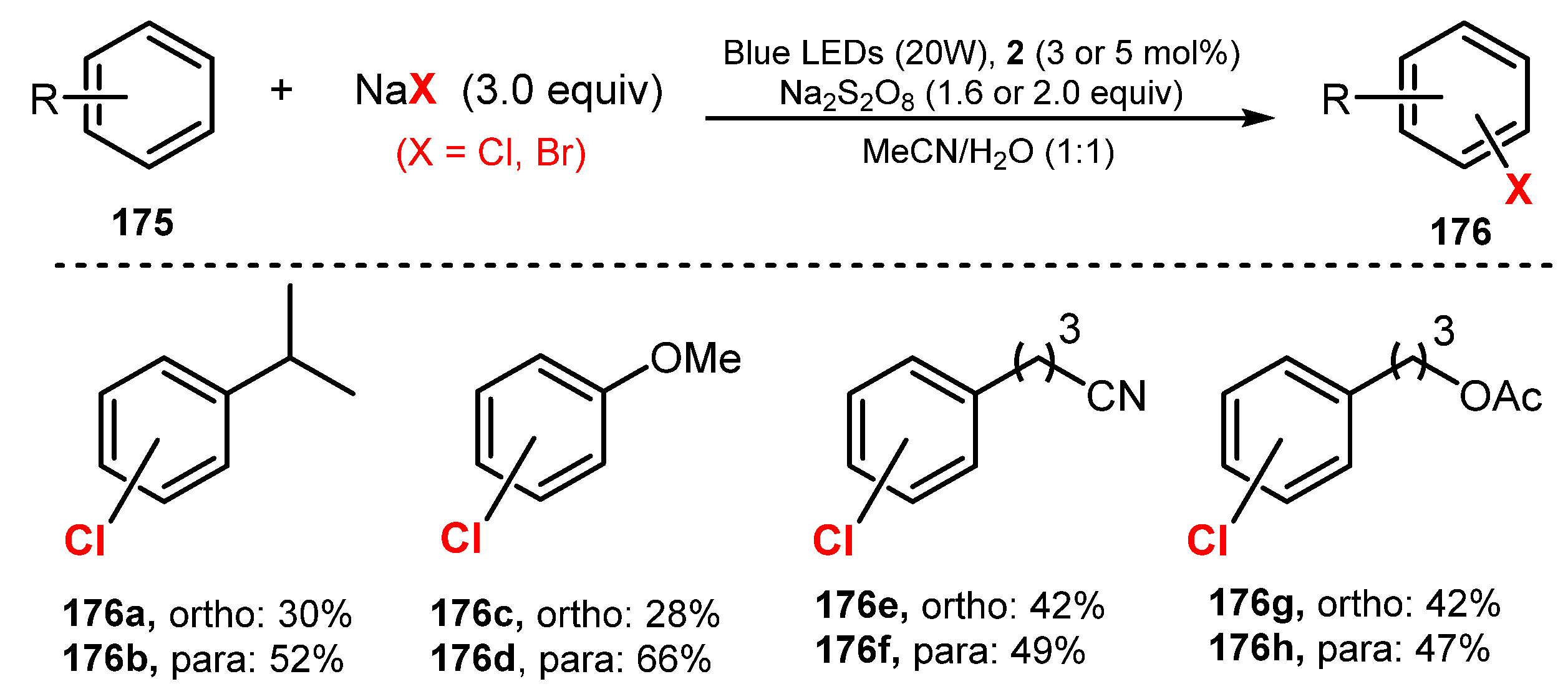
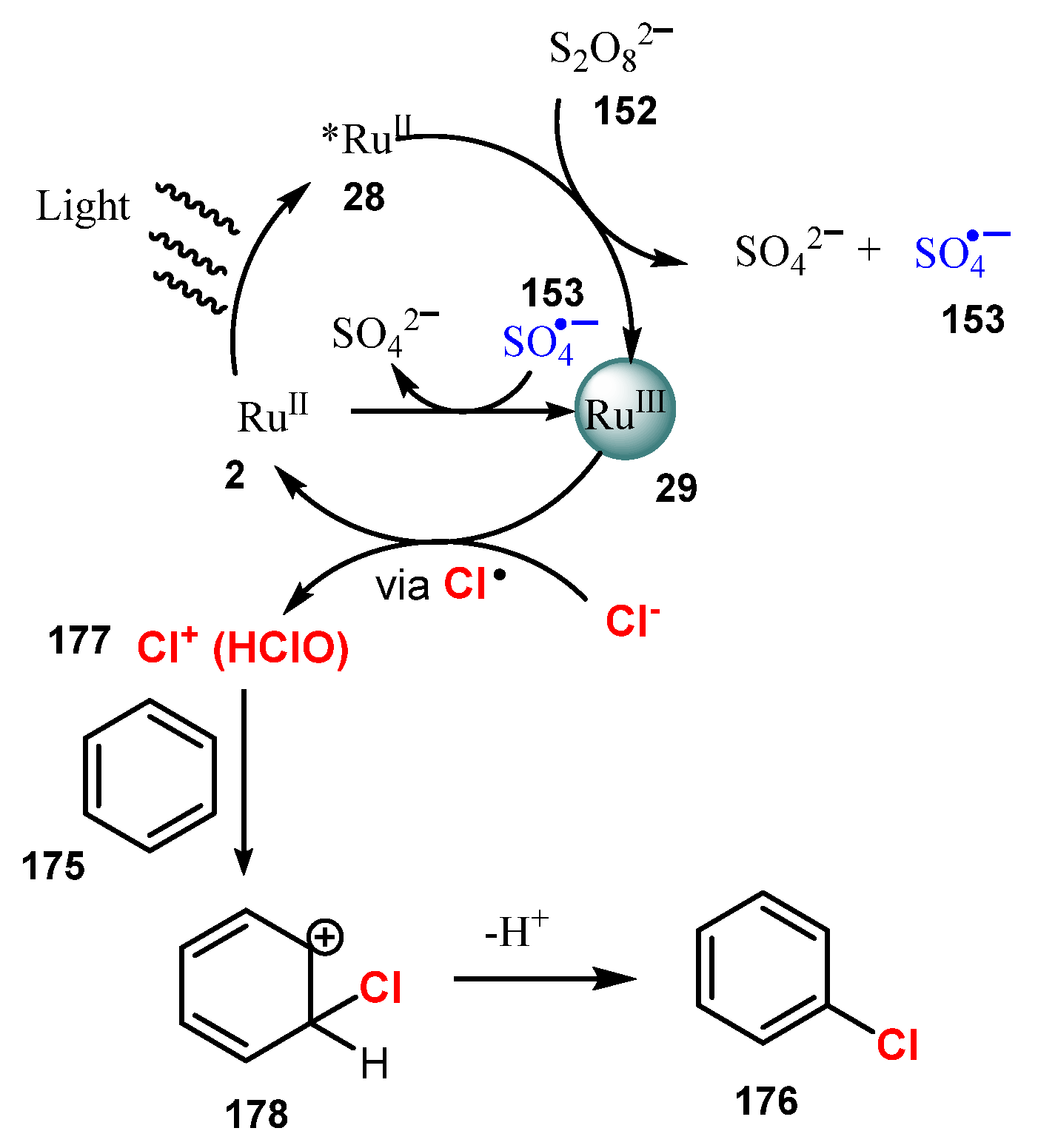
Selective chlorination of aryl C-H bonds using NaCl as a chlorine source, Ru(bpy)3Cl2∙6H2O as a photocatalyst, and Na2S2O8 as an oxidant under irradiation of a blue LED in a mixture of acetonitrile and water (1:1) at room temperature and air pressure was reported by Hu and co-workers in 2017 (Scheme 47) [52]. In this reaction, substrates bearing electron-donating groups such as isopropyl (176a–b) and methoxy groups (176c–d) on the phenyl ring were chlorinated in good yields (82–94%). Both para- and ortho-chlorinated products were also readily formed. Chlorination of substrates bearing -CN, an ester, or a halogen connected to the benzene ring via an alkyl chain worked well in good yields (89–91%) (176e–h). On the other hand, the substrates containing only an electron-withdrawing group such as nitrobenzene and (trifluoromethoxy)benzene were not tolerated for this method. However, the yields from these substrates were improved when combining an electron-withdrawing group and an election-donating group on the aryl ring.
A proposed mechanism of this reaction was presented in Scheme 48. Under light, the photocatalyst RuII 2 was excited to *RuII 28, which reacted with Na2S2O8 152 to generate RuIII 29 and SO4•−. SO4•− directly oxidized RuII 29 to give RuIII 2. Then, RuIII 29 reacted with Cl− to regenerate RuII 2 and form Cl+ 177. Cl+ 177 then reacted with aromatic compounds 175 to give the chlorination products 176 through the electrophilic addition of 178.
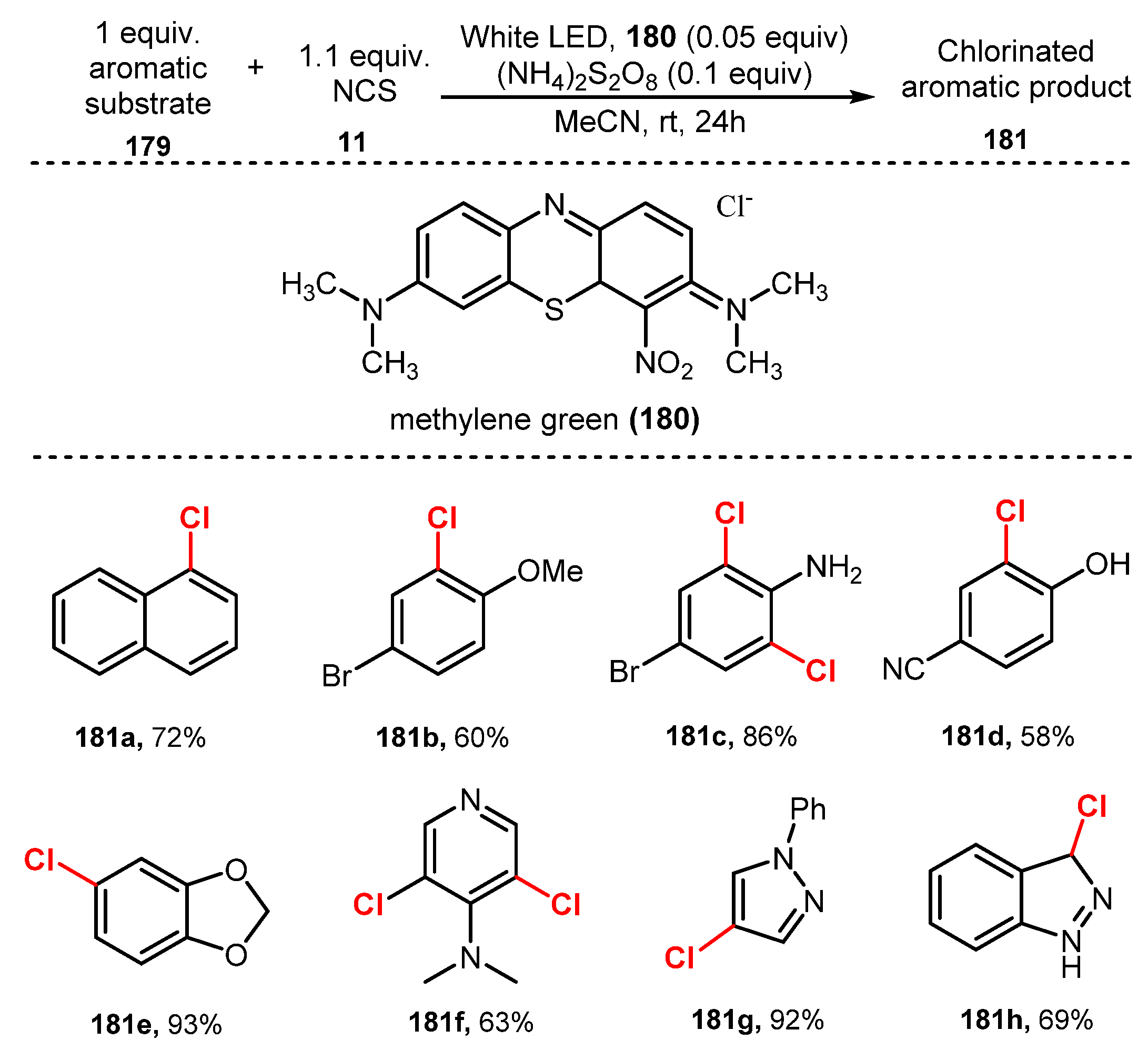
Lamar and his co-workers developed chlorination of arenes and heteroarenes using organic dyes as visible light photoredox catalysts in 2019 [53]. The reaction was carried out via treatment with NCS as a chlorine source in the presence of methylene green as an organic dye photocatalyst under irradiation of a white LED in acetonitrile (Scheme 49). Under the optimized conditions, reactions of disubstituted benzene derivatives bearing activating (electron-donating) and deactivating (electron-withdrawing) groups readily provided the corresponding products with good to high yields (58–86%) (181b–d). Various heteroarenes such as pyridine, pyrrole, indazole, and indole were tolerated for the reaction (181e–h), giving chlorinated products with moderate to excellent yields (63–93%).
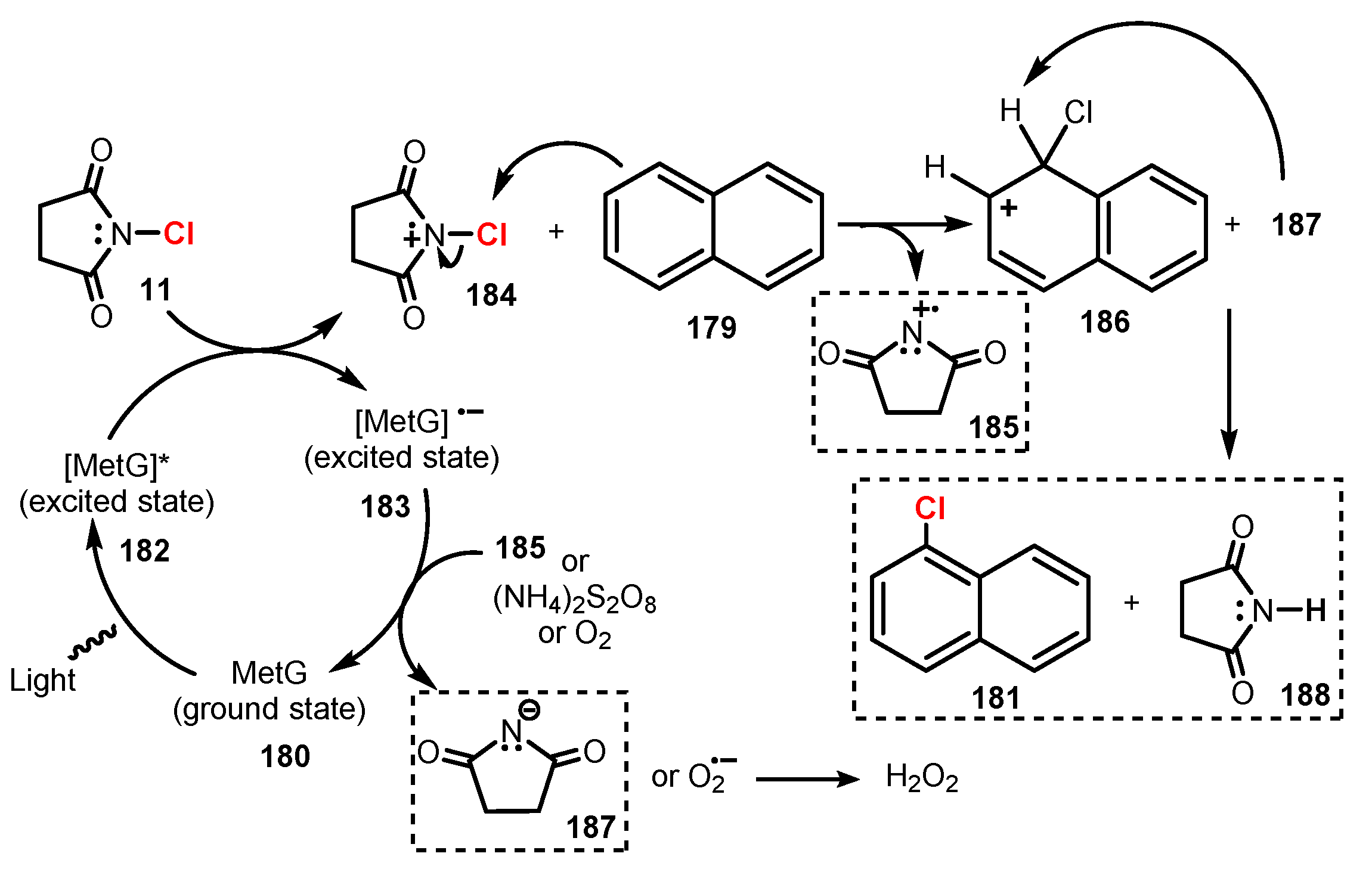
A plausible reaction mechanism was proposed as shown in Scheme 50. Irradiation of light allowed methylene green to transfer from ground state methylene green 180 to excited state methylene green 182. Then, 182 led to the single-electron oxidation of NCS 11, giving cationic radical 184 and providing reduced state methylene green 183. The radical 184 reacted with substrate 179 to generate arene chloride cation intermediate 186 and charged succinimide 185. Capture of a proton from intermediate 186 by anion 187 yielded final product 181. In addition, the reduced state methylene green 183 was oxidized to give back ground state methylene green 180 and to provide succinimide anion 187 by other oxidants (for example ammonium peroxodisulfate or oxygen gas in air) or by charged succinimide 185.
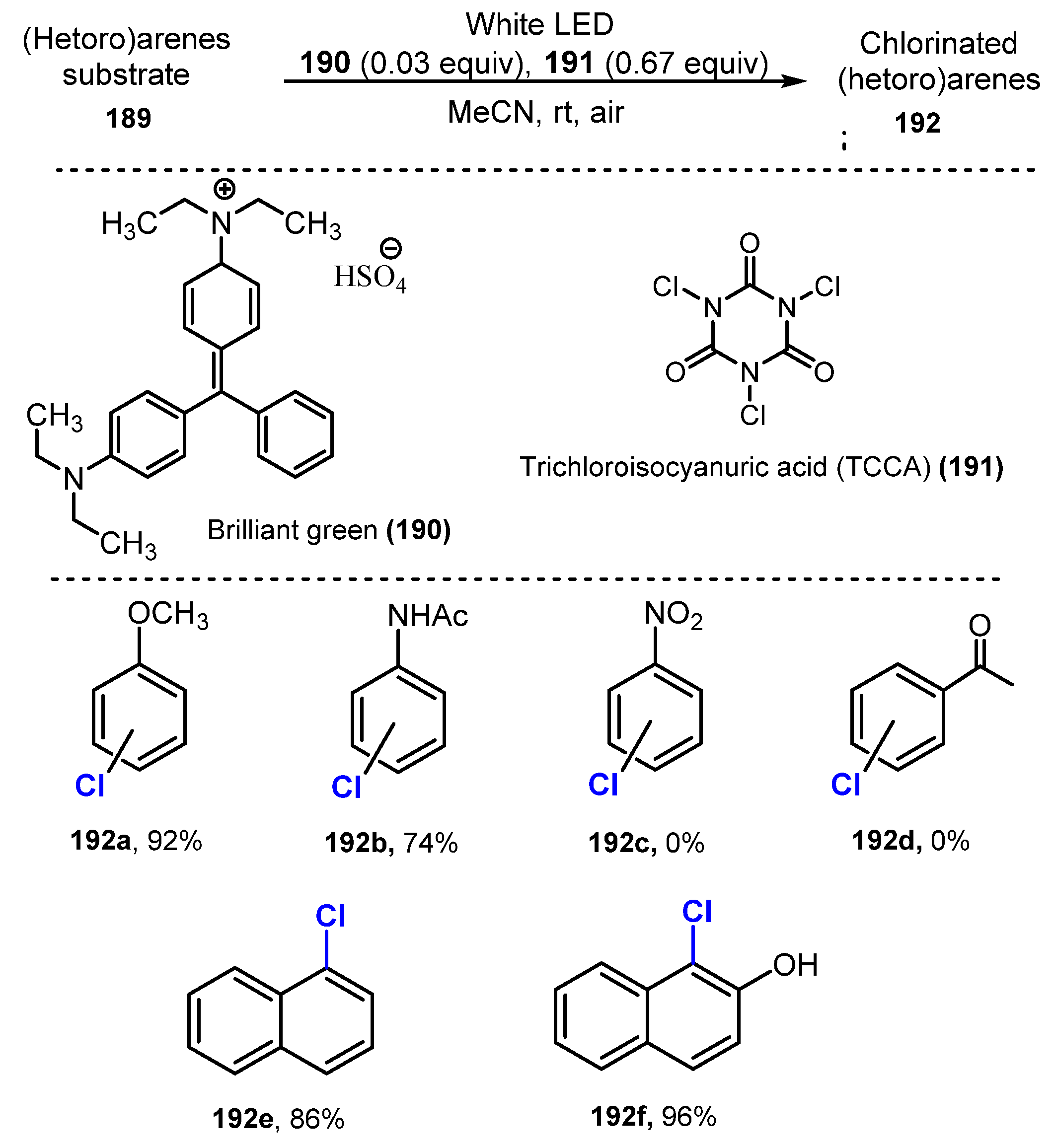
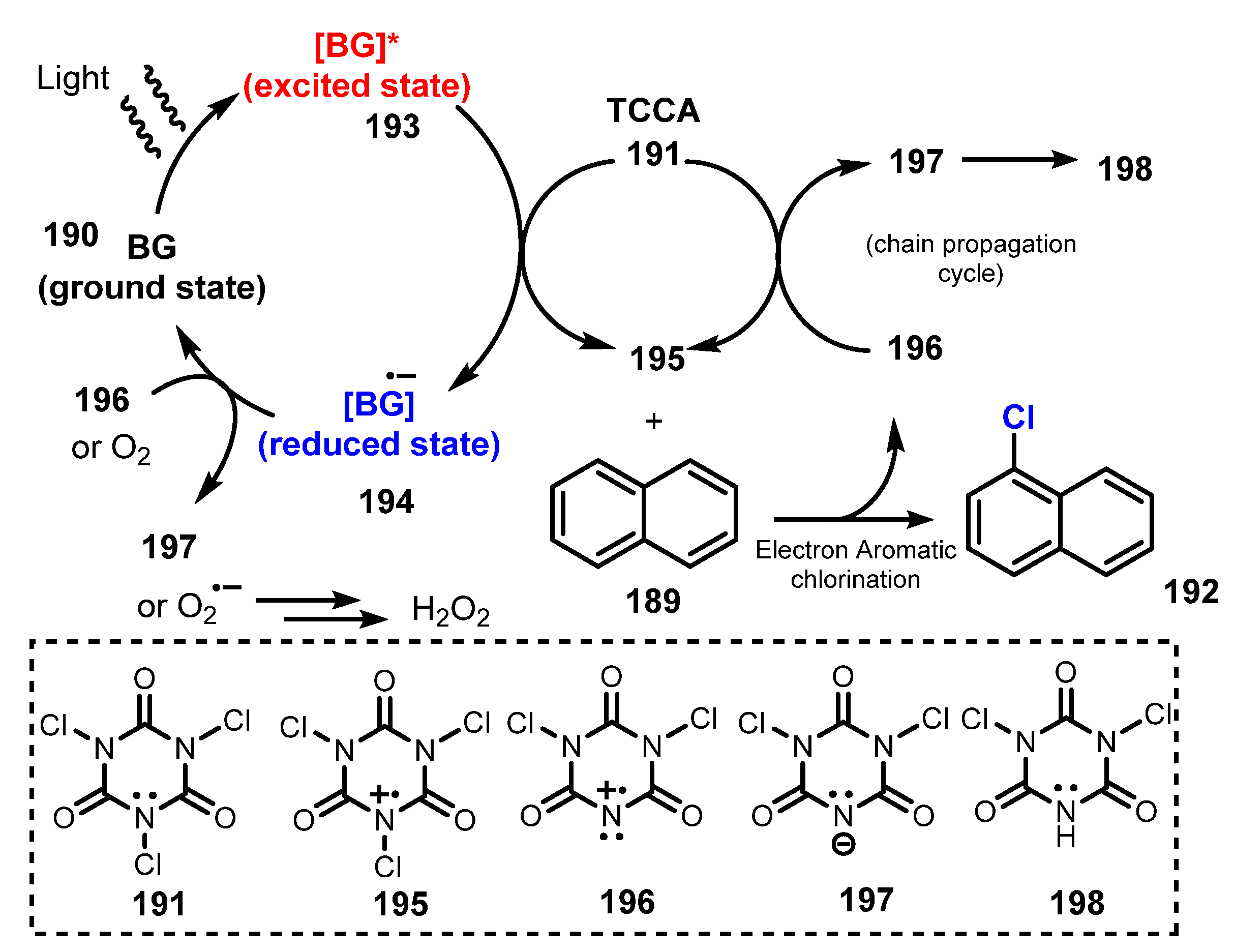
Hammond and co-workers proposed a novel strategy for chlorination of arenes and heteroarenes using brilliant green (BG) in 2019 [54]. The chlorination was conducted via treatment of trichloroisocyanuric acid (TCCA) as a chlorine source in the presence of brilliant green (BG) under irradiation of white LEDs in acetonitrile (Scheme 51). For monosubstituted benzene, reactions of substrates with an electron-donating group smoothly afforded the corresponding products with high yields (74–92%) (192a–b), while the chlorination of substrates with an electron-withdrawing group such as nitrobenzene (192c) and acetophenone (192d) was unsuccessful. On the other hand, reactions of naphthalene derivatives bearing electron-withdrawing groups or electron-donating groups successfully yielded monochlorinated products in good to high yields (63–96%) (192e–f). Various functional groups including halogens, carbonyls (ketone, aldehyde and amide), phenol, ethers, amines, nitro, nitrile, and benzylic C-Hs were tolerated for this method. Additionally, heterocyclic compounds were successfully chlorinated using this reaction as compared with the failure of the general reaction of TCCA using acidic conditions.
A possible mechanism was proposed as shown in Scheme 52. Photocatalyst brilliant green BG 190 was transferred to excited state BG* 193 by visible light, and reaction of excited state BG* 193 with TCCA 191 caused the single-electron oxidation to generate electrophilic chlorine species 195 and reduced state photocatalyst BG− 194. 195 reacted with substrate 189 to give chlorination product 192 via electrophilic aromatic chlorination, and to provide 196, which reacted with TCCA 191 to give 197. The reduced state photocatalyst BG− 194 was oxidized by 196 or O2 to return to its ground state BG 190.
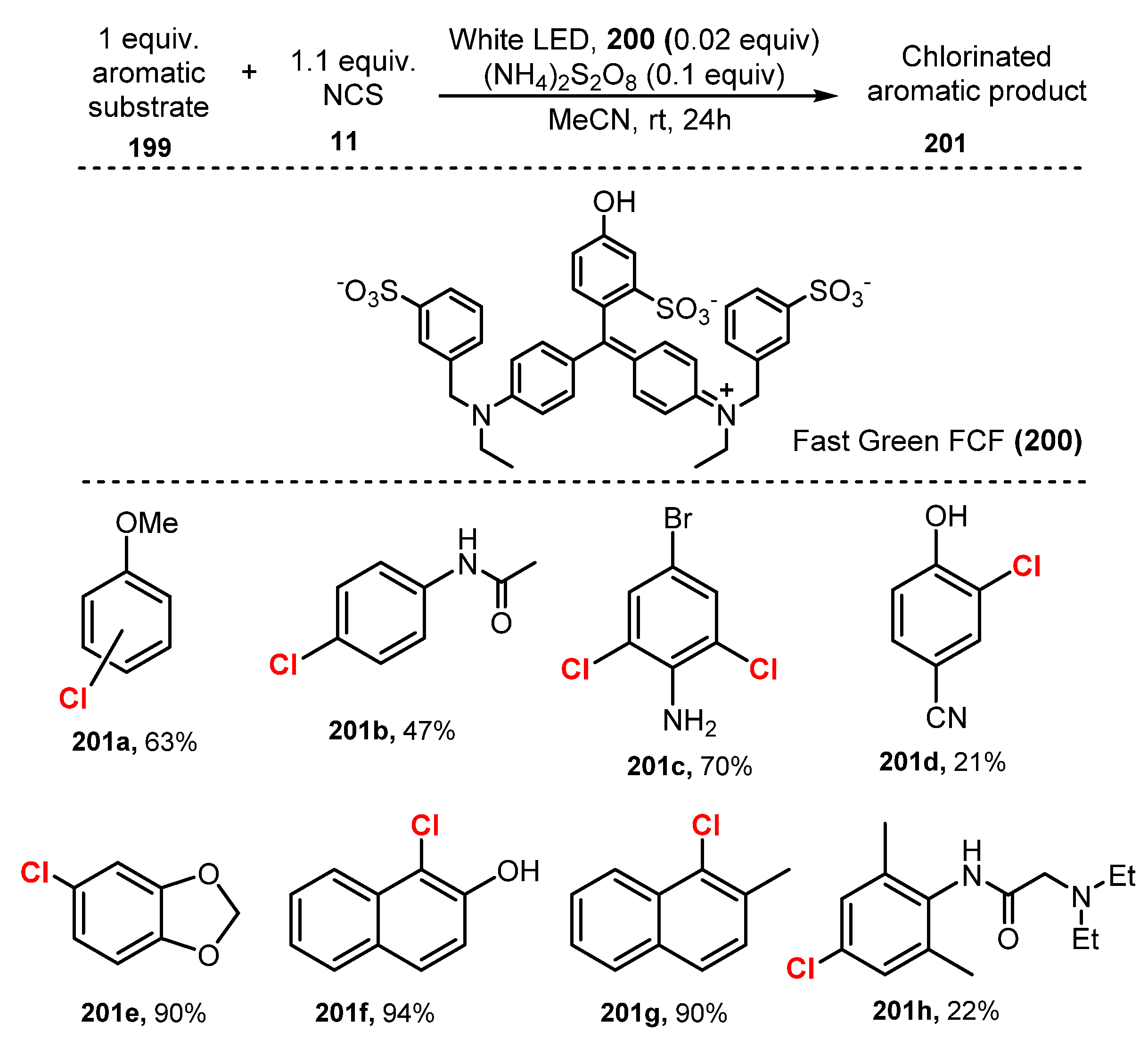
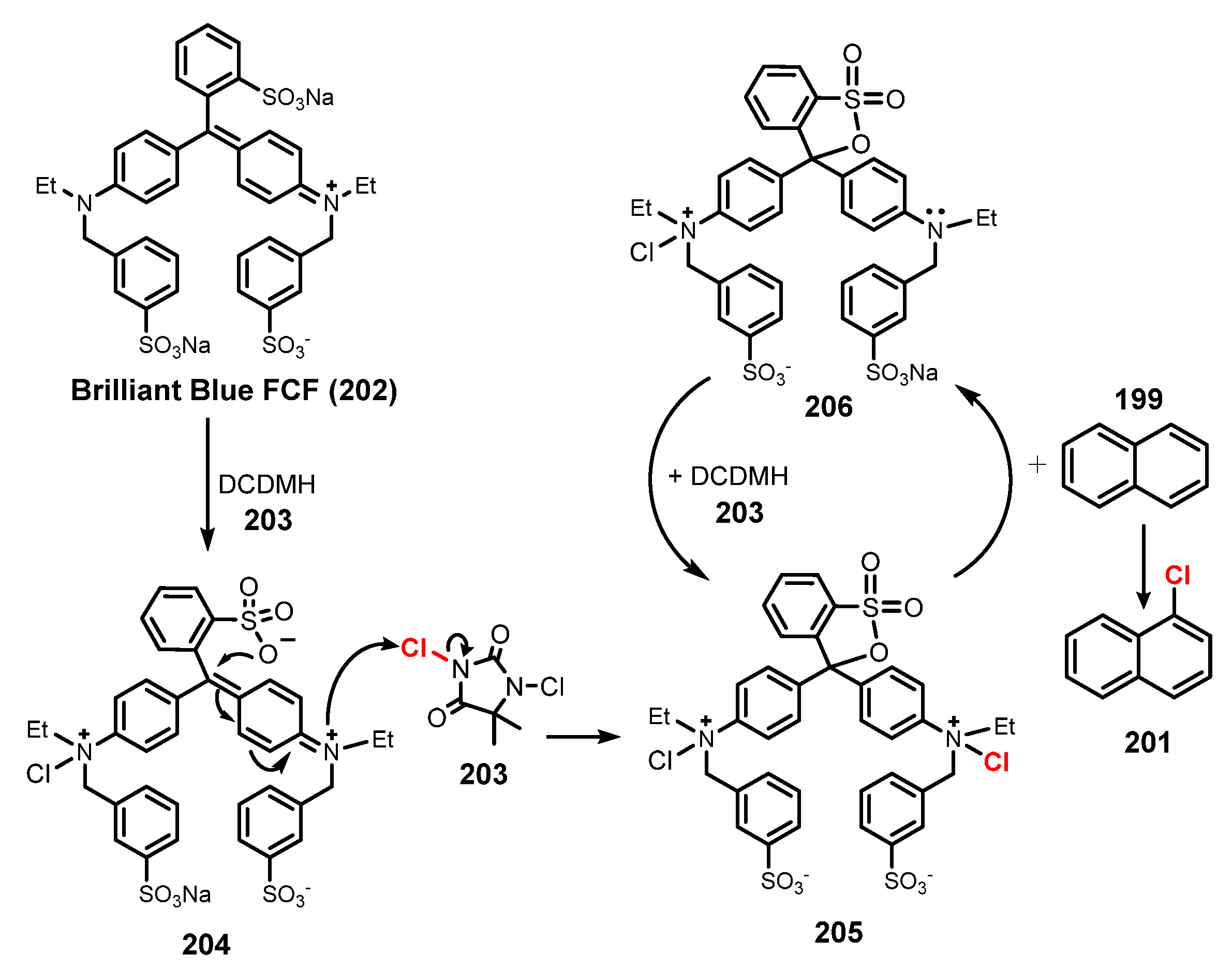
In 2020, Lamar and co-workers reported FDA-certified food dye mediated-chlorination of aromatic and heteroaromatic substrates [55]. The chlorination of aromatic compound was achieved via reaction with N-chlorosaccharin (NCS) as a chloride source in the presence of Fast Green FCF as photoredox catalyst under irradiation of white LED in acetonitrile (Scheme 53). Reaction of substrate with at least one electron-withdrawing group produced chlorinated product in moderate to good yields (47–70%). Electron-rich aromatics (201a) and naphthalene derivatives (201f–g) were readily chlorinated to give monochlorinated products in high to exceptional yields (90–94%). Pyrrole (201e), indole, indazole, and pyridine heteroaromatic substrates were also well tolerated for this reaction procedure, which showed considerably higher efficiency than uncatalyzed reaction processes. A reaction using 1,3-dichloro-5,5-dimethylhydantoin (DCDMH)/Brilliant Blue FCF system showed similar results to those of the NCS/Fast Green FCF system. However, a study of dichlorination reactions indicated that the chlorination using a DCDMH/Brilliant Blue FCF system was more efficient than chlorination using an NCS/Fast Green FCF system.
A plausible mechanism is shown in Scheme 54. Chlorination of brilliant blue 202 by DCDMH 203 was carried out to generate intermediate 204, which was subsequently converted to dichlorinated sulfonphthalein 205. Compound 205 then provided the electrophilic chlorine to aromatic compound 199 or heterocycle arenes to give chlorinated product 201, and, after that, 205 became a monochlorinated sulfonphthalein species 206, which obtained a chlorine atom from DCDMH 203 to recover dichlorinated compound 205.
6.3. Bromination of Aromatic C-H Bonds
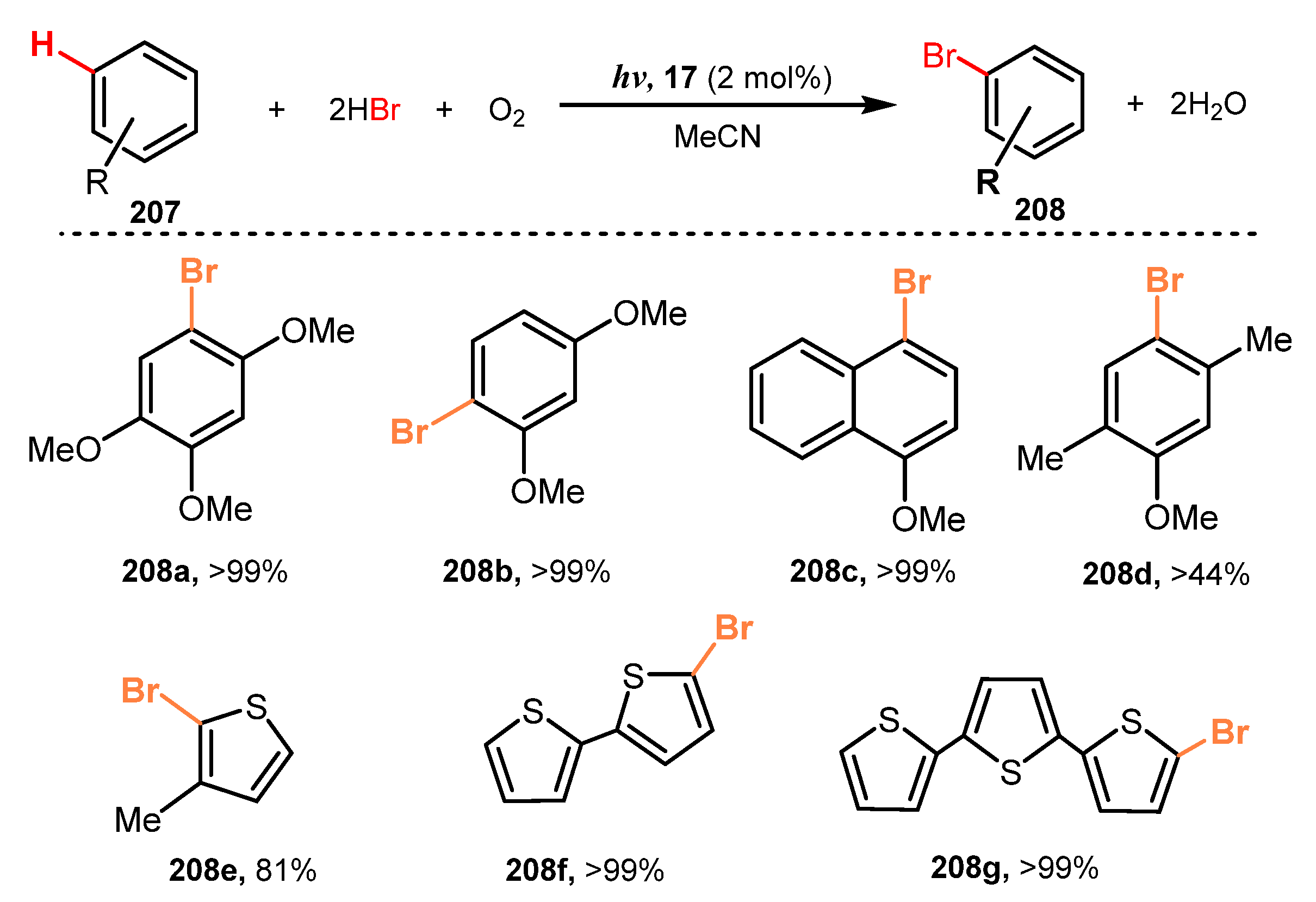
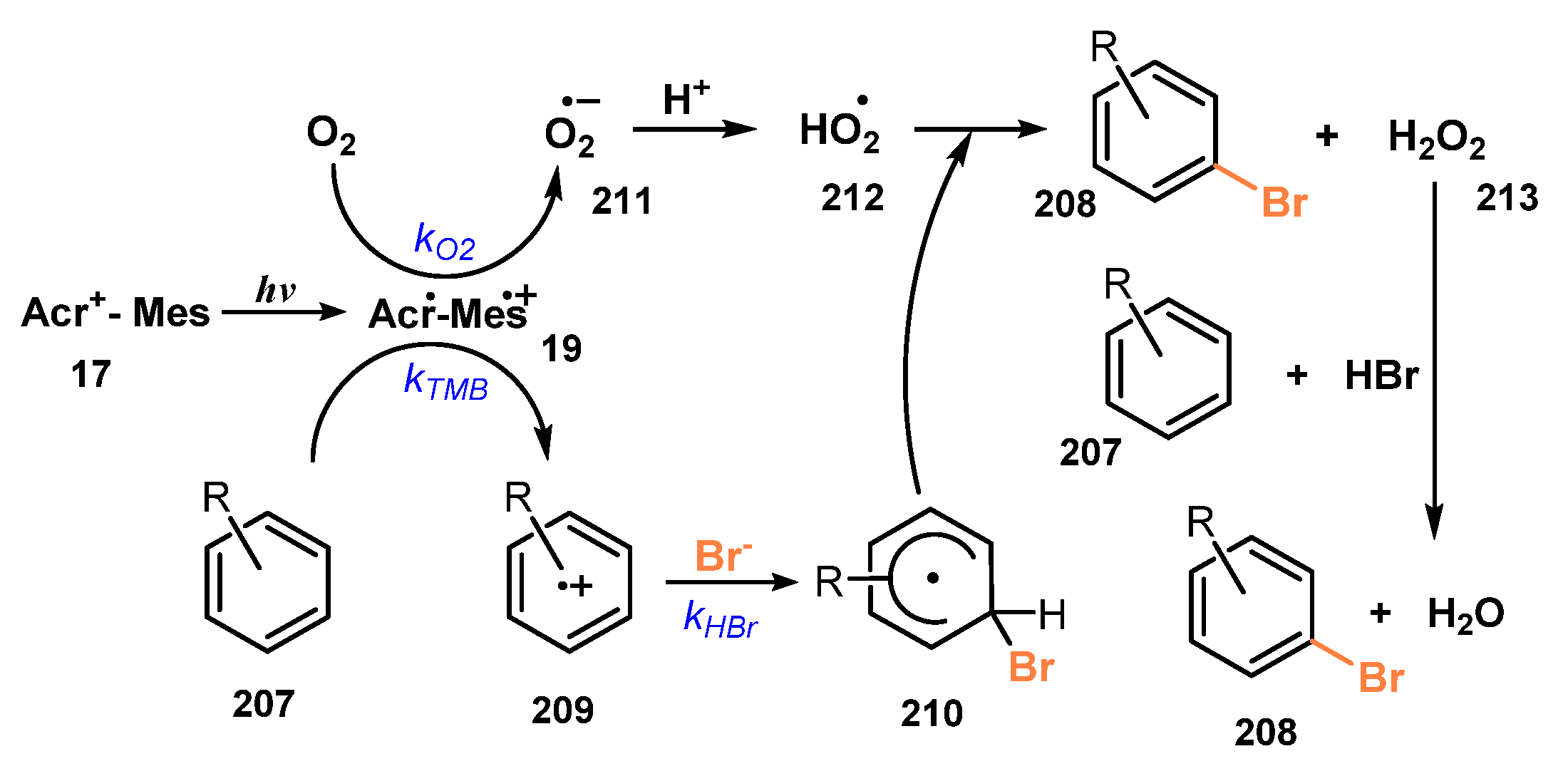
In 2011, Fukuzumi and co-workers developed selective bromination of aromatic hydrocarbons and thiophenes via reaction with a solution of 50% of HBr in O2-saturated acetonitrile in the presence of [Acr+-Mes][ClO4−] as a photocatalyst under irradiation of a xenon light (Scheme 55) [56]. For methoxy-substituted benzenes, bromination reactions were successfully achieved regardless of position or number of substituents. The reaction was highly selective, and the bromination yield was more than 99% (208a–b) without observation of dibromo- or tribromo-derivatives during the process. Toluene derivatives were also brominated (208c), even though the yield of bromination in the presence of methyl-substituted groups was lower than that of methoxy-substituted benzenes (208d). Besides, brominations of thiophenes were readily achieved with high yield (81–99%).
A plausible mechanism of this bromination of aromatic hydrocarbons is presented in Scheme 56. Under irradiation of light, intramolecular electron transfer caused Acr+-Mes 17 to become the excited state Acr•-Mes•+ 19, which oxidized substrate TMB 207 to give radical cation 209. At the same time, Acr•-Mes•+ 19 reduced O2 to provide radical HO2• 212. Reaction of 209 with Br− generated the aromatic ring radical 210, and then radical 210 reacted with HO2• 212 via dehydrogenated process to give brominated product 208 and H2O2. Besides, when H2O2 interacted with HBr and substrate 207, another brominated compound 208 and H2O were obtained.
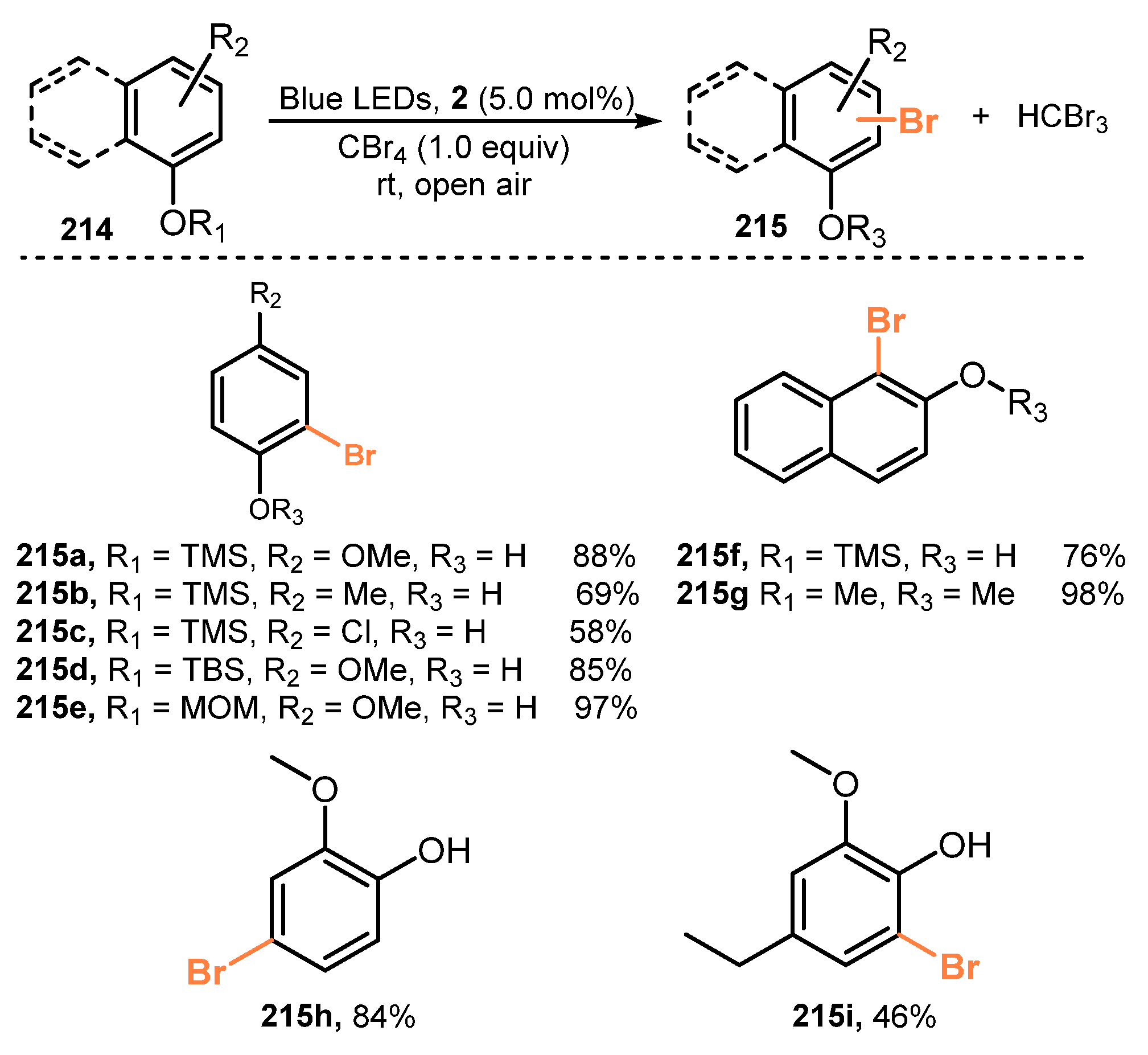
An efficient strategy for bromination of phenols was presented by Xia and co-workers in 2014 [57]. The reaction was carried out with CBr4 in the presence of Ru(bpy)3Cl2 (5.0 mol%) under visible light irradiation (blue LEDs, λmax = 435 nm) in acetonitrile (Scheme 57). Both electron-withdrawing and electron-donating groups as substituents in the benzene ring were tested. Reactions of aromatic substrates bearing TMS (trimethylsilyl), TBS (tert-butyldimethylsilyl), MOM (methoxymethyl), and THP (tetrahydropyranyl) groups (215a–e) at the para- and ortho-positions yielded 2- and 4-bromophenol in good to outstanding yields (58–97%), respectively. TMS and methyl-protected naphthalen-2-ol were readily employed to produce 1-bromonaphthalen-2-ol and 1-bromo-2-methoxynaphthalene in high yields (76–98%), with great selectivity (215f–g). Additionally, reactions of Bn or Ms-protected phenols gave target 2- and 4-bromophenol derivatives with no loss of Bn or Ms groups in good yields (215h–i).
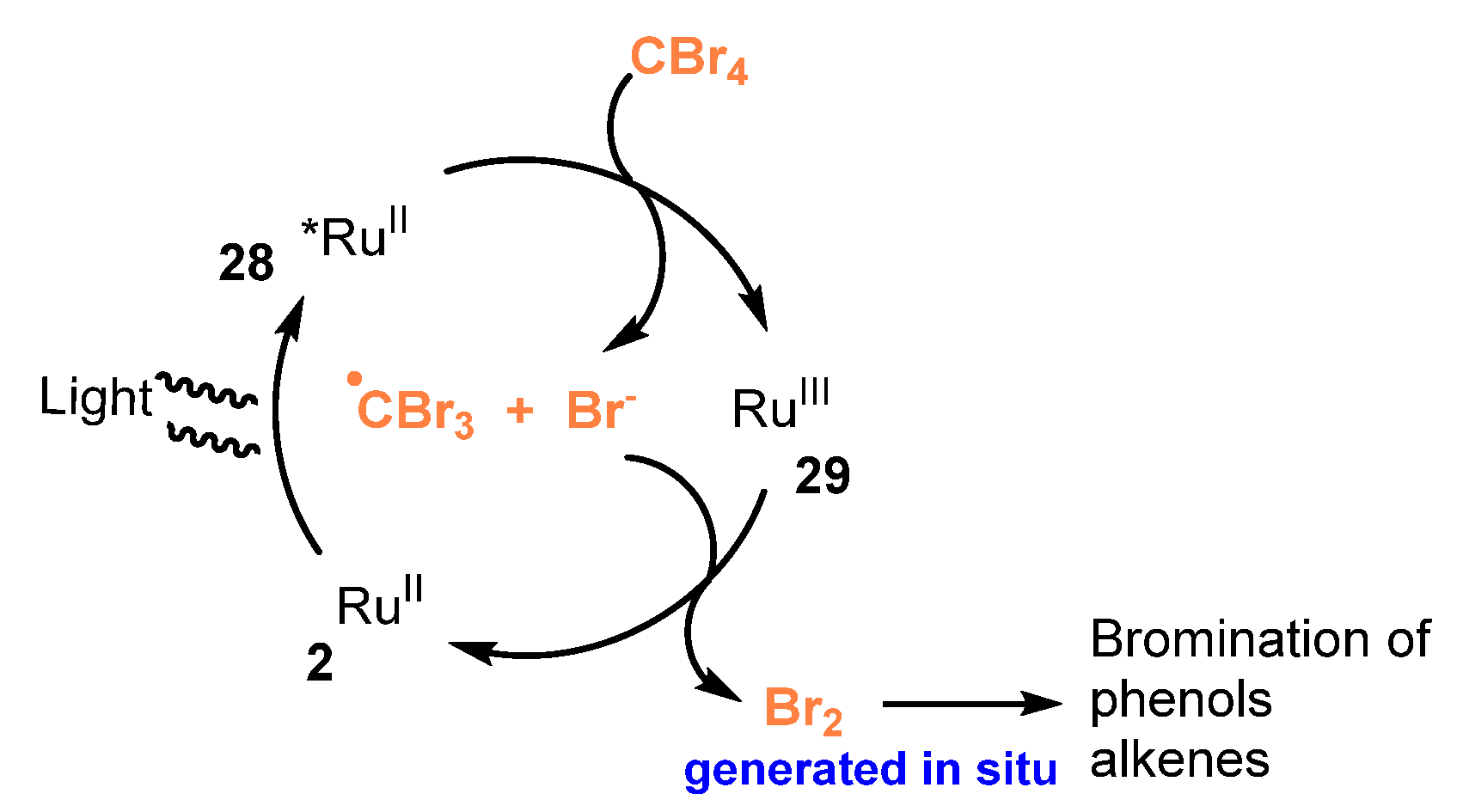
A mechanism of this conversion was shown in Scheme 58. First, visible light excited Ru(bpy)32+ 2 to give Ru(bpy)32+* 28. Oxidation reaction of Ru(bpy)32+* 28 with CBr4 occurred as oxidative quenchers generated Br−, •CBr3 and Ru(bpy)33+ 29. Then, anion Br− was oxidized by Ru(bpy)33+ 29 to produce Br2 in situ for bromination of phenols and alkenes, and to regenerate Ru(bpy)32+ 2.
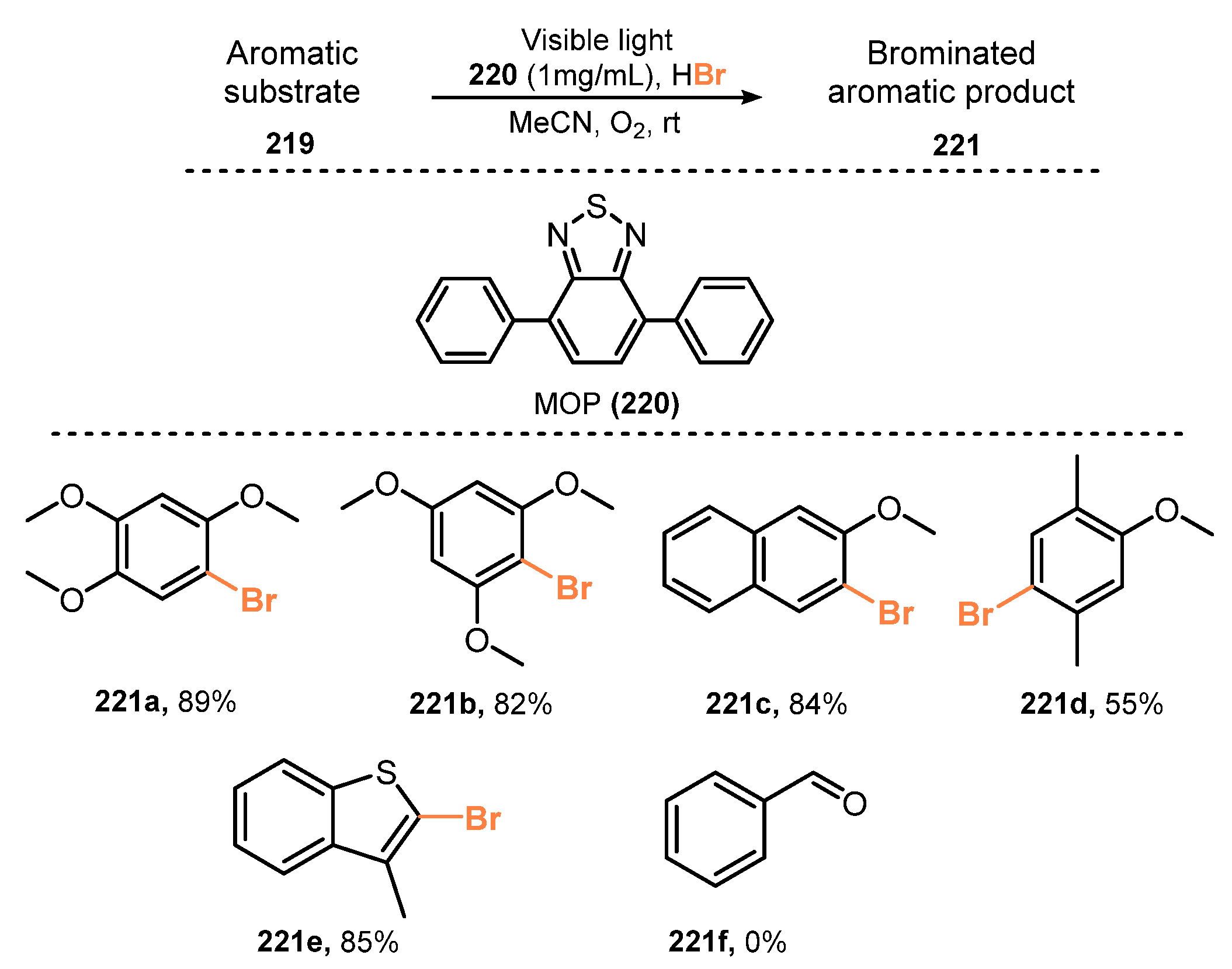
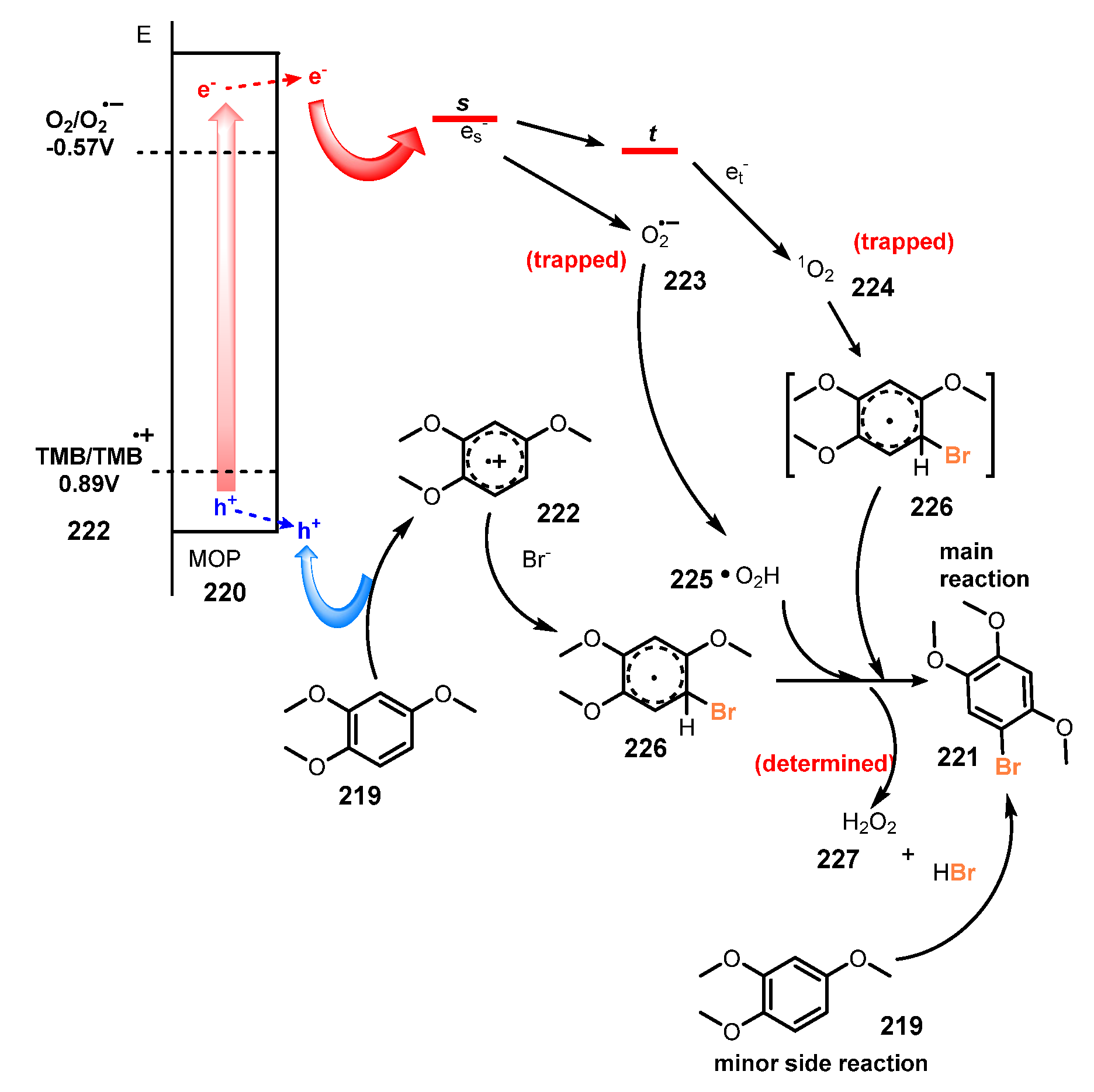
Employment of microporous organic polymers (MOPs) for selective bromination of aromatic compounds was reported by Zhang and co-workers in 2016 [58]. The bromination was achieved via reaction with HBr as a bromine source in the presence of MOPs as heterogeneous photocatalysts and molecular oxygen as a clean oxidant under irradiation of visible light in acetonitrile (Scheme 59). Electron-rich aromatic compounds were readily brominated in good to excellent yields (55–89%). In addition, benzene, naphthalene, thiophene, and 3-methylbenzo[b]thiophene derivatives were well tolerated for this protocol (221a–e). In this study, it was discovered that the methyl group on the aromatic ring led to lower bromination efficiency than that of methoxy groups. Toluene was not brominated under the same reaction conditions (221f).
A proposed mechanism of this reaction is shown in Scheme 60. Under irradiation of light, MOPs material 220 oxidized substrate (TMB) 219 to generate cationic radical TMB•+ 222. Reaction of TMB•+ 222 with Br− anion from HBr formed TMB•-Br radical 226. Besides, the activation of oxygen by MOPs material 220 gave its active forms of O2•− and 1O2. These activated oxygen species oxidized TMB•-Br radical 226 to create the desired product 221, along with H2O2 227 as a byproduct. However, H2O2 227 also reacted with substrate TMB 219 and HBr in a minor side reaction to form final product 221.
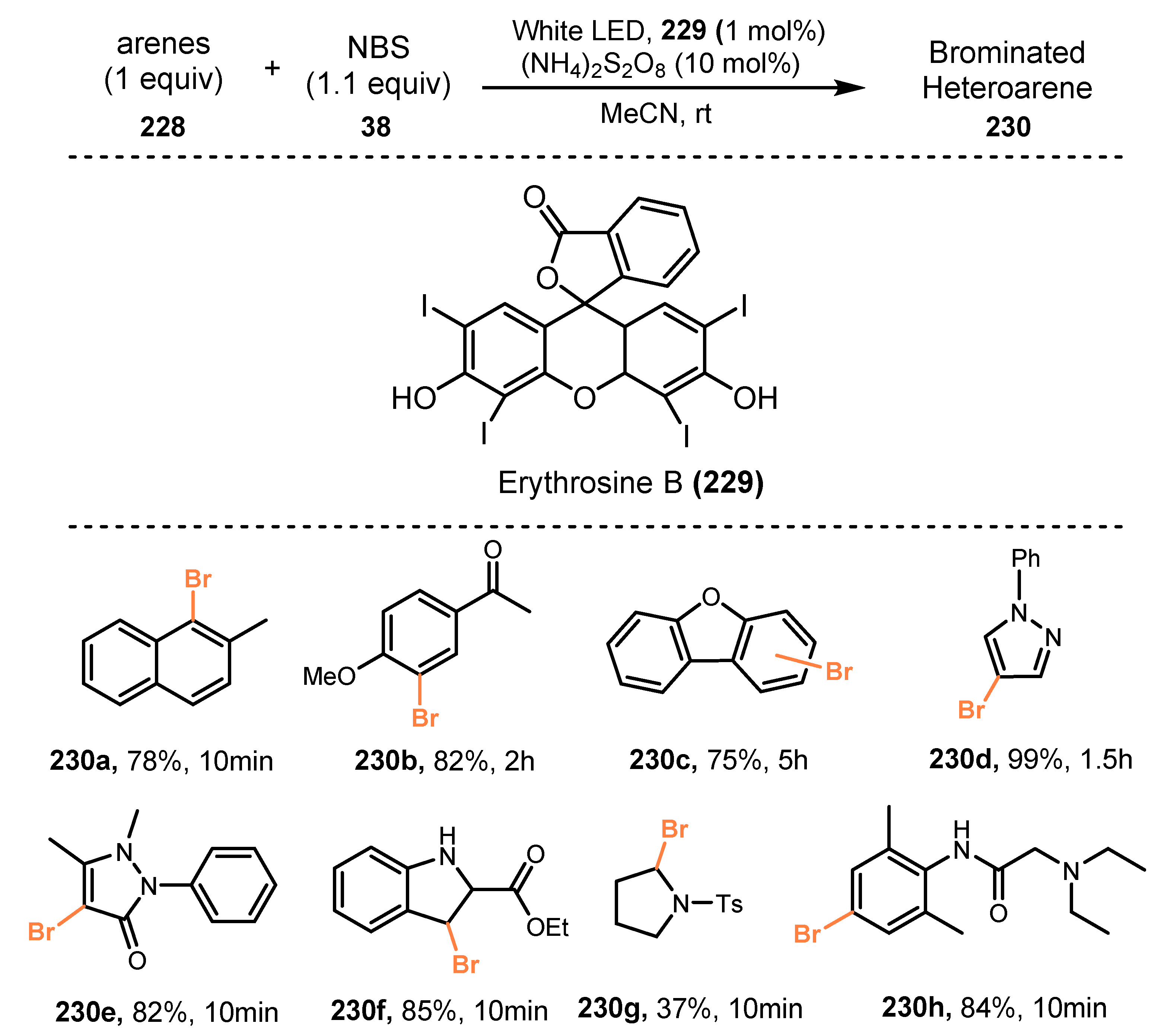
Organic dye-catalyzed bromination of arenes and heteroarenes was demonstrated by Lamar and co-workers in 2018 [59]. In this method, arenes reacted with N-bromosuccinimide (NBS) in the presence of erythrosine B as a photocatalyst and ammonium peroxodisulfate as an oxidant reagent under irradiation of white LED light in acetonitrile to give the corresponding products (Scheme 61). Erythrosine B is a xanthene dye, commonly used in daily life as a food colorant and a painting ink, and it is difficult to degrade under visible light without other supporting agents. In this study, the optimization reactions using Erythrosine B were performed in 2, 6, and 24 h, which gave the target products in high yields. Based on these results, it can be believed that it is stable under irradiation of light around 24 h. Under the optimized reaction conditions, naphthalene and anisole derivatives were efficiently brominated on the aromatic ring without byproducts. Brominations of aryl ether-containing substrates, phenol derivatives, aniline derivatives, acetanilide, and the anesthetic lidocaine were also successfully conducted. However, arenes with electron-withdrawing groups such as chlorobenzene and nitrobenzene did not work well. On the other hand, reactions of N-containing heteroarenes (pyrrole, pyrazole, indole, and indazole) successfully afforded brominated products in acceptable to good yields (37–99%).
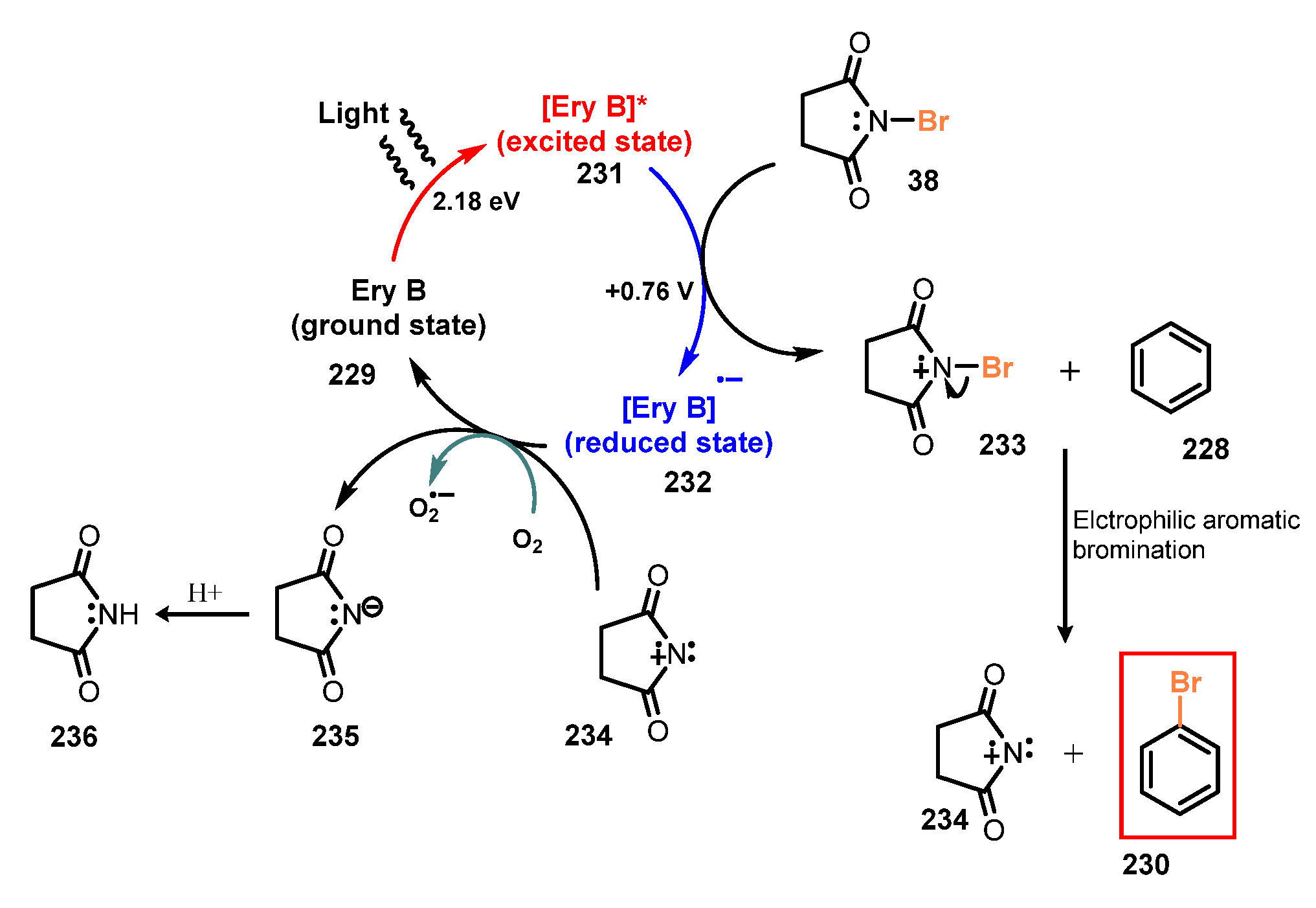
They proposed a possible reaction mechanism as shown in Scheme 62. Under irradiation of light, ground state erythrosine B 229 was converted to excited state erythrosine B 231, which oxidized the nitrogen of N-bromosuccinimide 38 to provide cationic radical 233 and reduced state erythrosine B 232. The cationic radical 233 reacted directly with arene substrate 228 to generate the desired product 230 and charged succinimide species 234 as a byproduct via electrophilic aromatic bromination. The succinimide 234 or external oxidant such as O2, (NH4)2S2O8, or H2O2 oxidized reduced state erythrosine B 232 to return to its ground state erythrosine B 229 and generate succinimide anion 236.
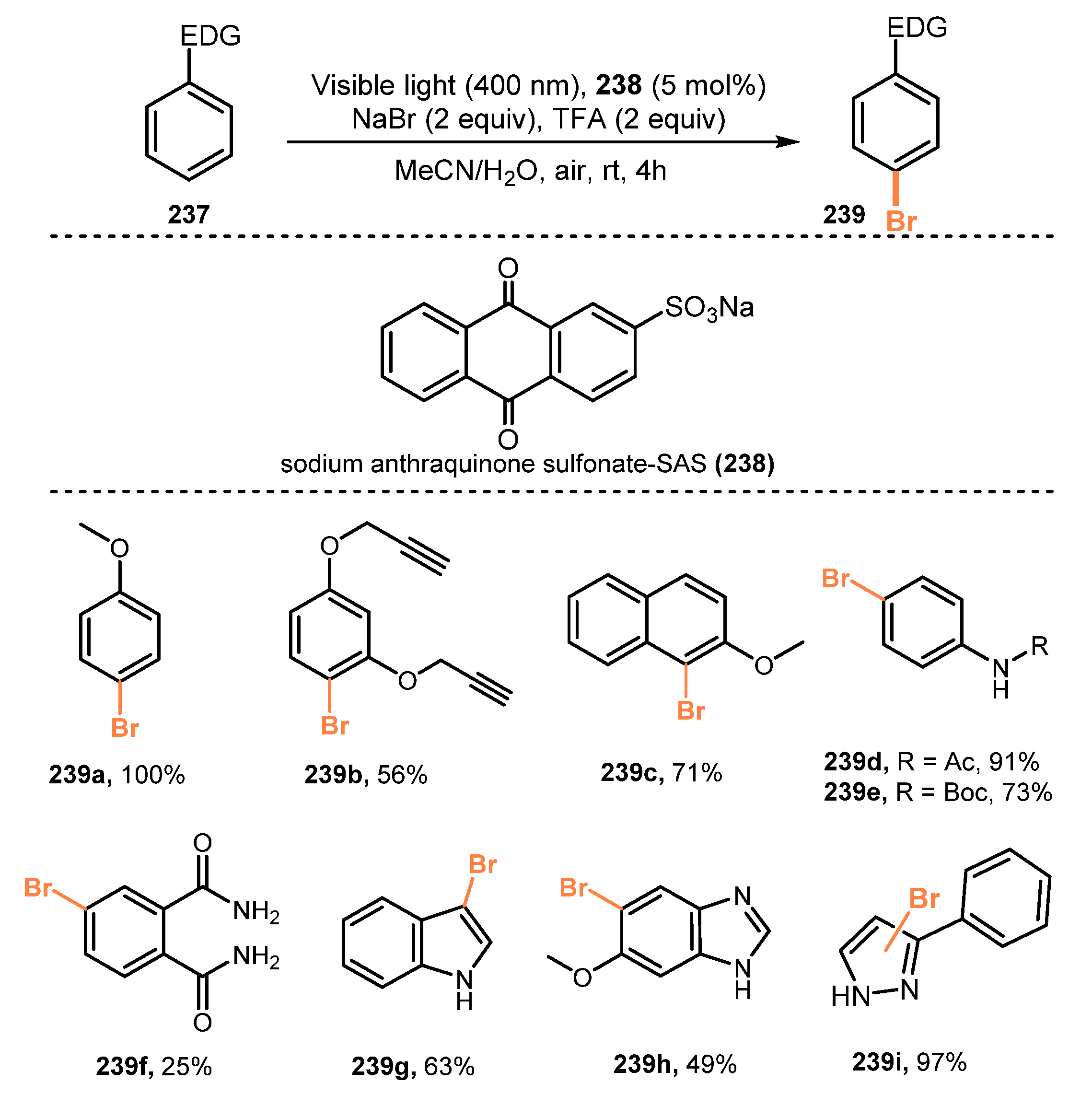
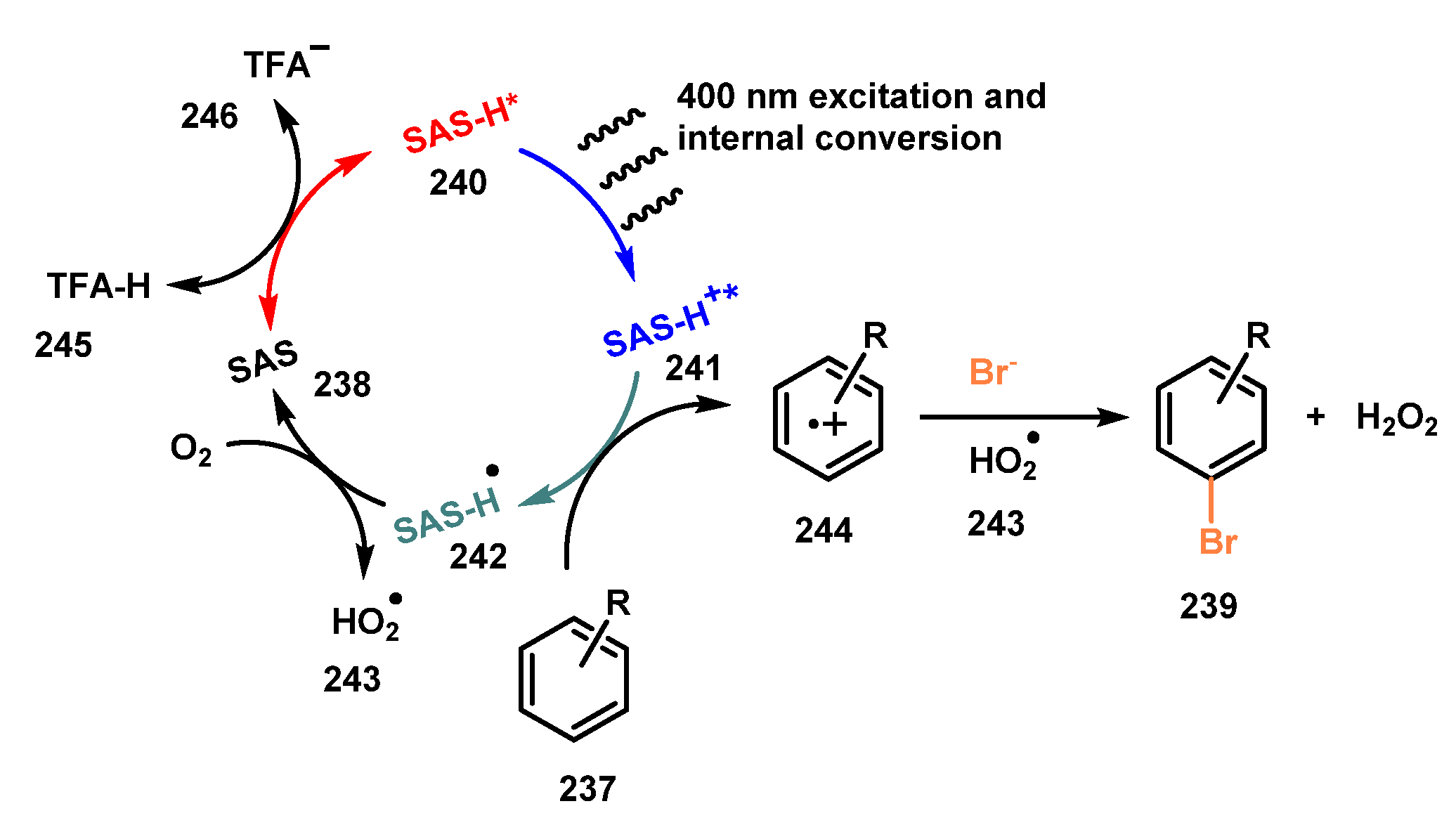
Anthraquinones were employed as photocatalysts in bromination of arenes and heteroarenes by König and co-workers in 2018 [60]. The bromination reaction was carried out by using sodium bromide as a bromide source, sodium anthraquinone sulfonate (SAS) as a photocatalyst, and TFA acid as an activator for anthraquinone under irradiation of LEDs in a mixture of acetonitrile and water (1:1) (Scheme 63). All the methoxy arenes were successfully brominated in high isolated yields (56–100%) (239a–c). Bromination of substrates bearing Boc-protected amine (239e) was readily achieved in good yield (73% yield), even though there was an acidic environment. In contrast, reactions of substrates with an electron-withdrawing substituent provided desired products with lower yields. In addition, heteroarenes such as indole derivative 239g and benzimidazole 239h derivatives were also brominated in modest yields (49–63%). This procedure using SAS was also successful to oxidize pyrazole derivatives to generate the corresponding products in high yields. Besides, several bioactive compounds such as phenazone, tramadol, and alkaloid strychnine were tested in this procedure and yielded the brominated products in good to excellent yields (25–97%).
A possible mechanism of this bromination reaction was proposed as shown in Scheme 64. TFA 245 reacted with SAS 238 to give the active state SAS-H+ 240, which was stimulated by visible light to yield the triplet state SAS-H+* 241. SAS-H+* 241 caused oxidation of arenes 237 to produce radical cation 244, which in turn reacted with bromide anions and HO2• 243 to afford the brominated product 239 and hydrogen peroxide. Besides, SAS-H• 242 radical was oxidized by O2 to regenerate SAS 238.
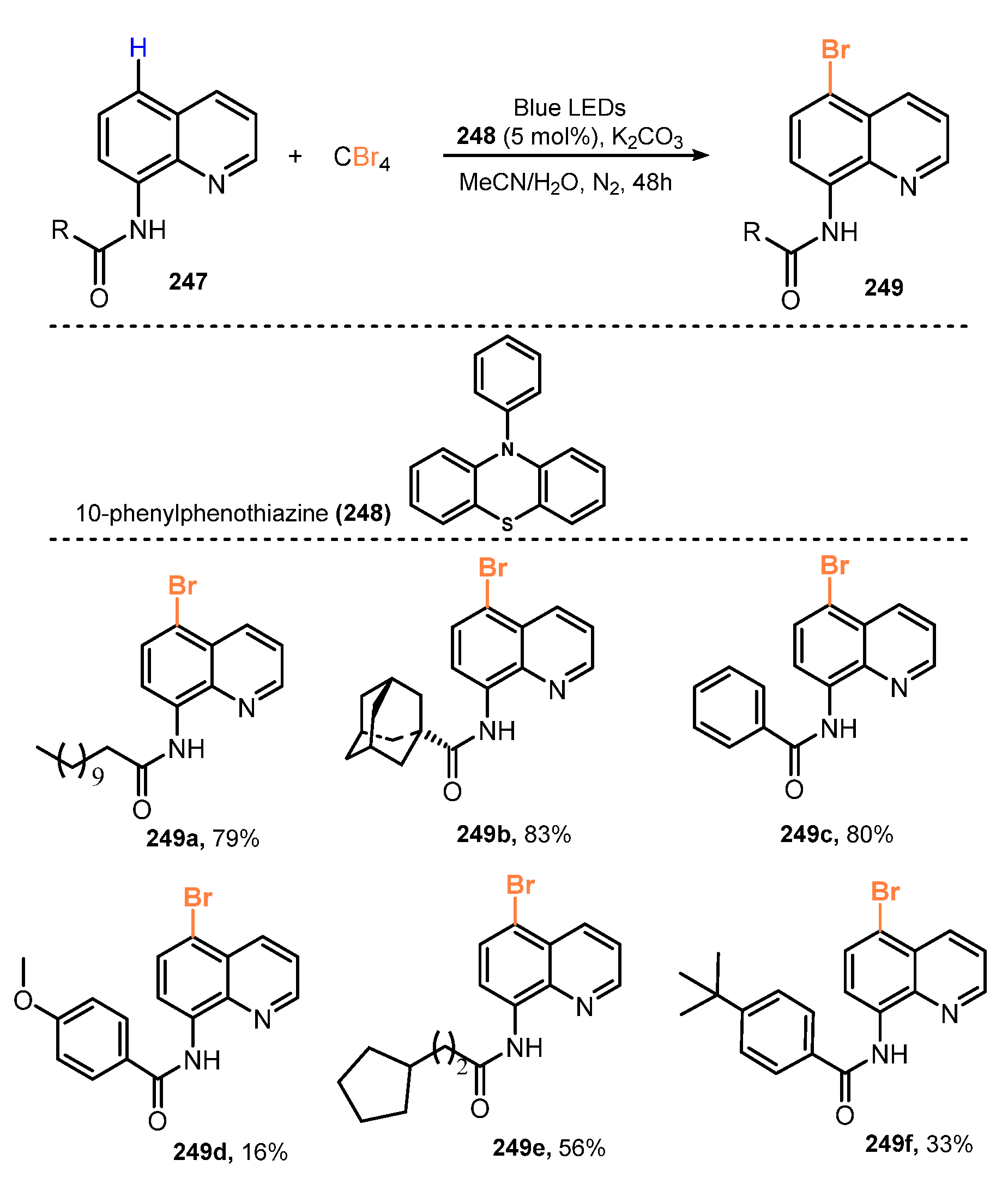
Lei and co-workers described selective bromination at the C5 position of 8-aminoquinoline amides in 2019 [61]. In the reaction, aminoquinoline amides reacted with CBr4 as a bromine source in the presence of 10-phenylphenothiazine (PTH) as an organophotoredox catalysis and K2CO3 as the base under the irradiation of blue light in acetonitrile to give the corresponding products (Scheme 65). Both linear and cyclo alkyl-substituted carboxamides were successfully brominated with good or moderate yields (56–83%). Reaction of dodecyl and bulky group-substituted carboxamides produced desired products in 88% and 83% yields, respectively (249a–b). However, this process was less effective for aryl-substituted carboxamide substrates. Target product was obtained in 80% yield by reaction of substrate with phenyl (249c), while brominated product was prepared in only 16% yield when the phenyl group was replaced by para methoxy groups (249d).
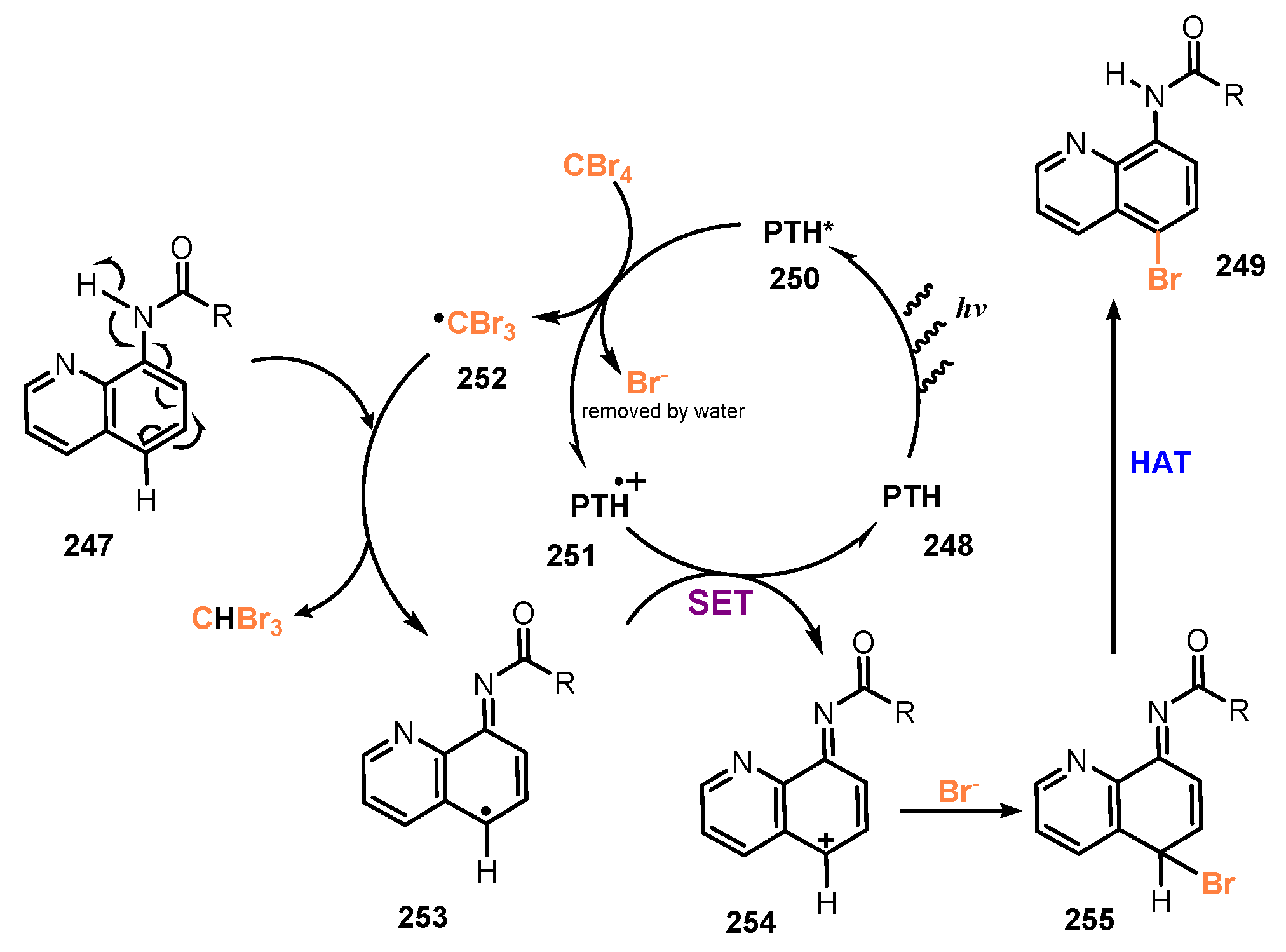
A plausible mechanism of this bromination reaction proposed by Lei and co-workers is shown in Scheme 66. Visible light excited photocatalyst PTH 248 to PTH* 250, which was then oxidatively quenched by CBr4 to provide •CBr3 radical 252 and bromide anion. •CBr3 radical 252 reacted with substrate 247 to give radical intermediate 253, which underwent single electron transfer with PTH*+ 251 to form radical intermediate 254 and recover ground state photocatalyst PTH 248. Reaction of radical 254 with Br− anion produced compound 255, which was then used in the hydro atom transfer (HAT) process to prepare the final product 249.
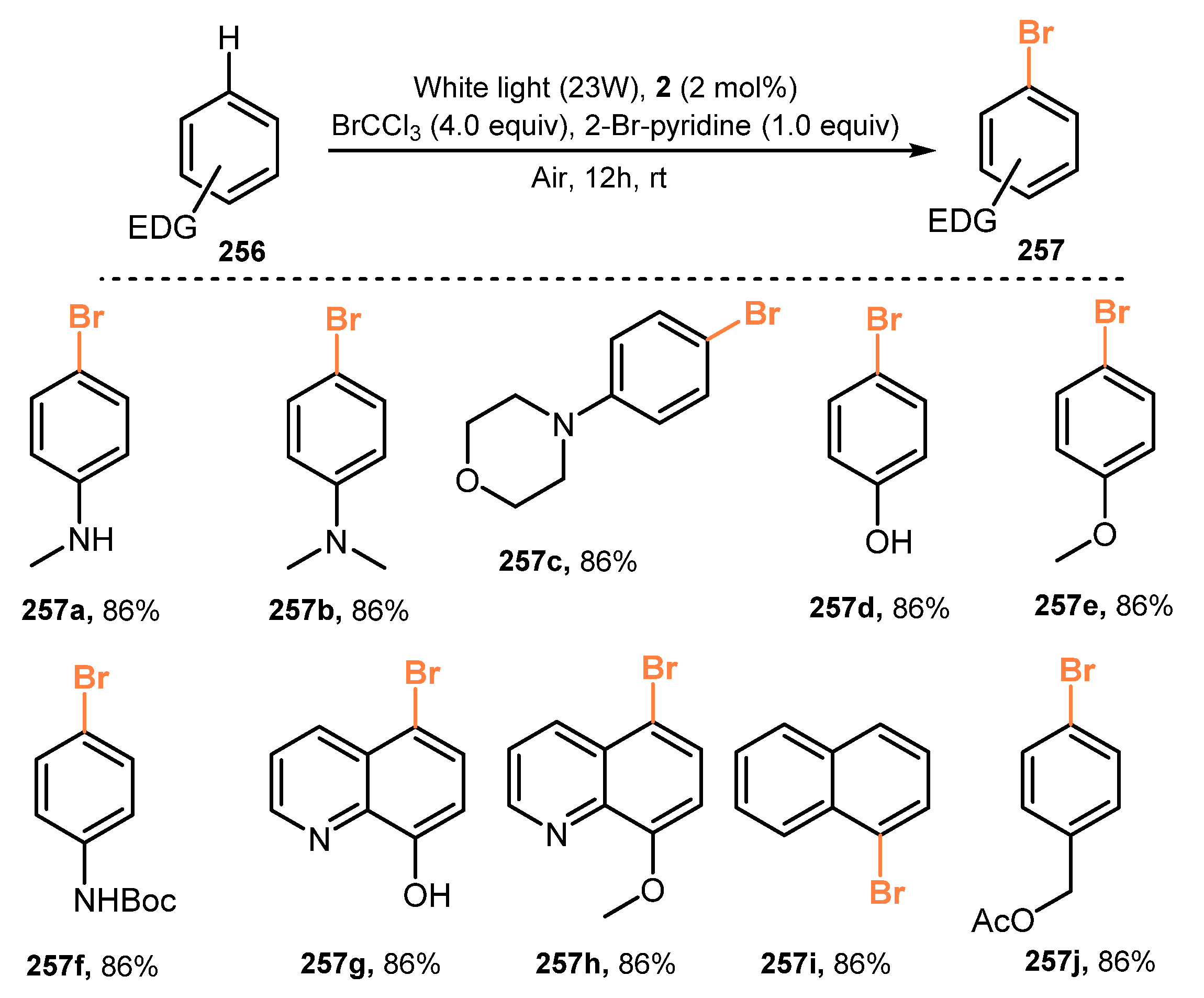
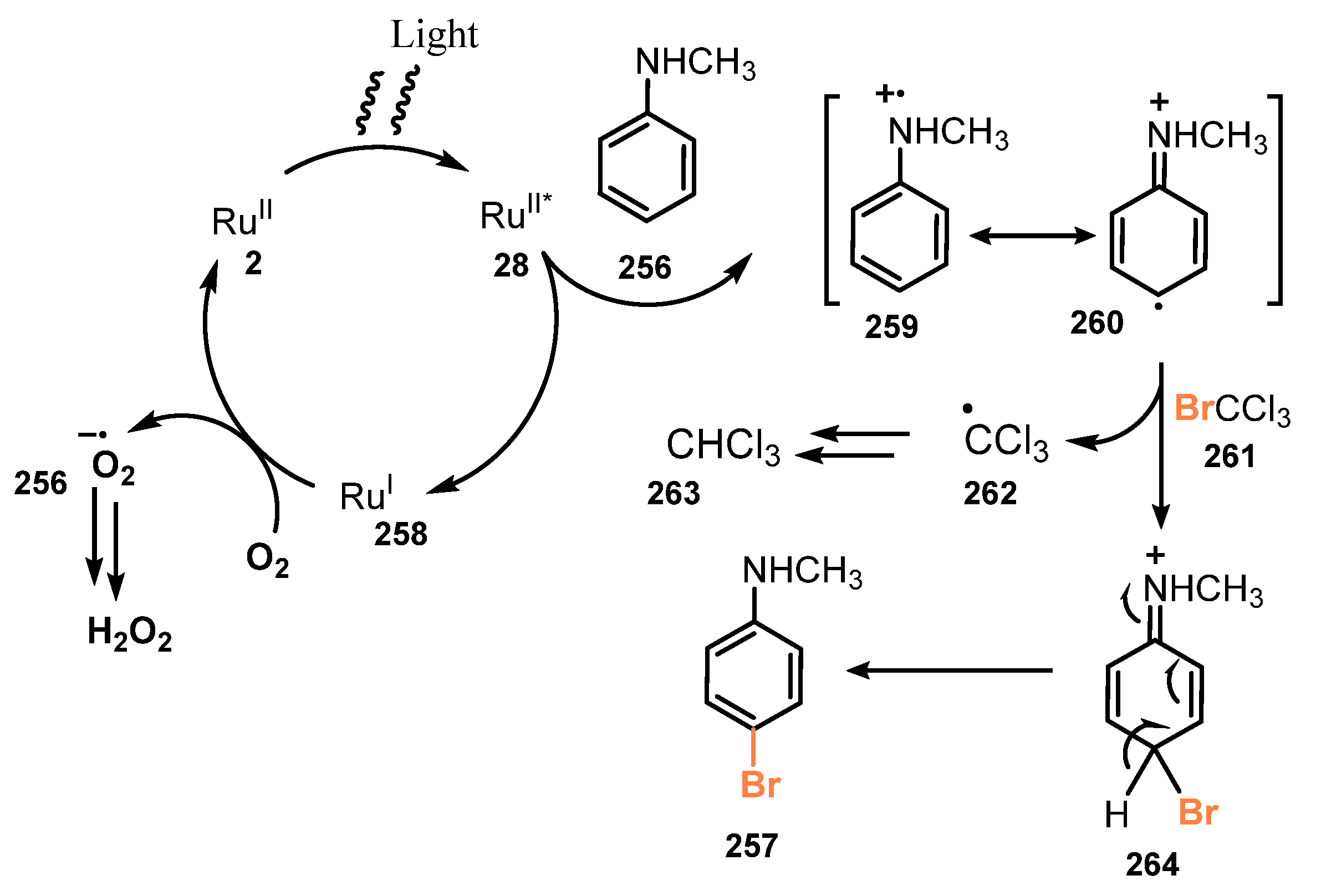
Bromotrichloromethane (BrCCl3)-mediated efficient and regioselective mono-bromination of electron-rich arenes was developed by Loh and co-workers in 2021 [62]. Reaction of arenes and heteroarene with BrCCl3 as a bromine source in the presence of RuII bipyridyl complex photocatalyst and 2-bromopyridine under irradiation of white light (23W) and air atmosphere gave the corresponding products (Scheme 67). Various electron-rich arenes and heteroarenes bearing different substitute groups, such as amino, N,N′-dimethyl, hydroxyl, methoxy, and other heterocyclic amino groups, were successfully brominated (257a–j). Brominations of protected anilines (257f) with acid-sensitive protective groups (BOC) and oxidation-sensitive groups (hydroxyl) of the phenol ring (257j) were also readily modified to provide target products. However, reactions of substrates baring various functional groups, such as naphthalene, weak electron-withdrawing groups (EDGs), or the combination of EDG and electron-withdrawing groups, did not work well to give desired products.
They proposed a probable mechanism for this protocol as described in Scheme 68. Light irradiation made photocatalyst RuII 2 excited to RuII* 28. The substrate 256 was oxidized by RuII* 28 to give radical cation 259 and RuI 258, which was then oxidized by O2 to RuII 2. The radical cation 259 was converted to cation radical 260, which in turn reacted with BrCCl3 261 to yield intermediate 264. Intermediate 264 was deprotonated by a base to afford the brominated product 257.
6.4. Iodination of Aromatic C-H Bonds
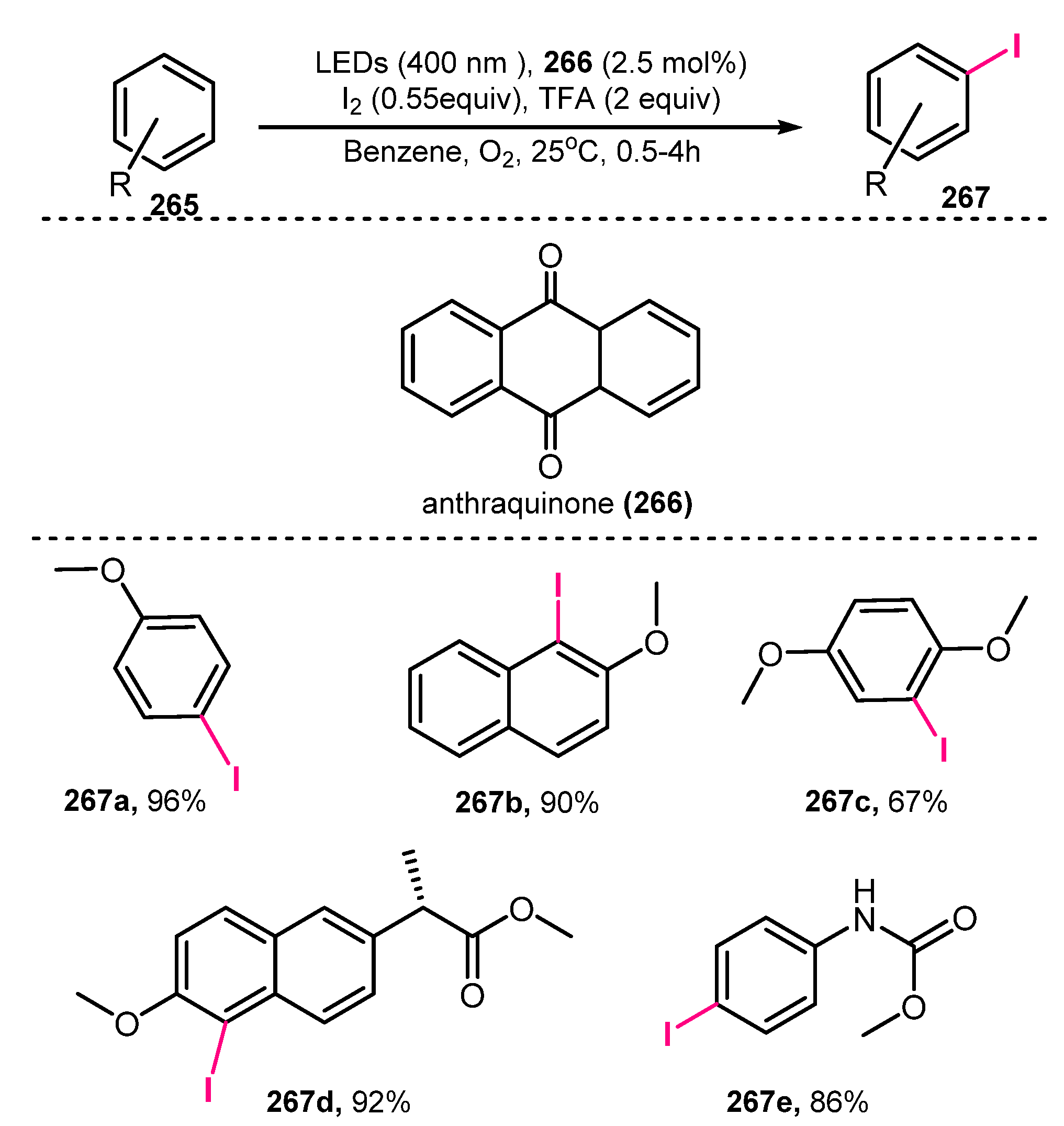
In 2019, König and co-workers demonstrated oxidative iodination of arenes [63]. The iodination was achieved via treatment of arenes with I2 as an iodine source, TFA, O2 as an oxidizing agent, and anthraquinone (AQ) as a photocatalyst under irradiation of LED (400 nm) in benzene (Scheme 69). In the reaction, synthetic protocol allowed arenes-bearing electron-donating groups to readily produce the desired iodinated products in good to high yields (56–96%). When arenes9bearing methyl groups were used, they were successfully iodinated without generation of any side reactions on the benzylic position. Besides, reaction of both trimethylbenzene and tert-butyl substituted arenes also worked well, although they have potential steric effects. It was found out that the acid-sensitive ester functionality (267d) was unchangeable when conducting this reaction method. In addition, nitrogen-containing compounds in the substrate (267e) were also stable during the reaction, which successfully afforded target products in high yields (86–92%).
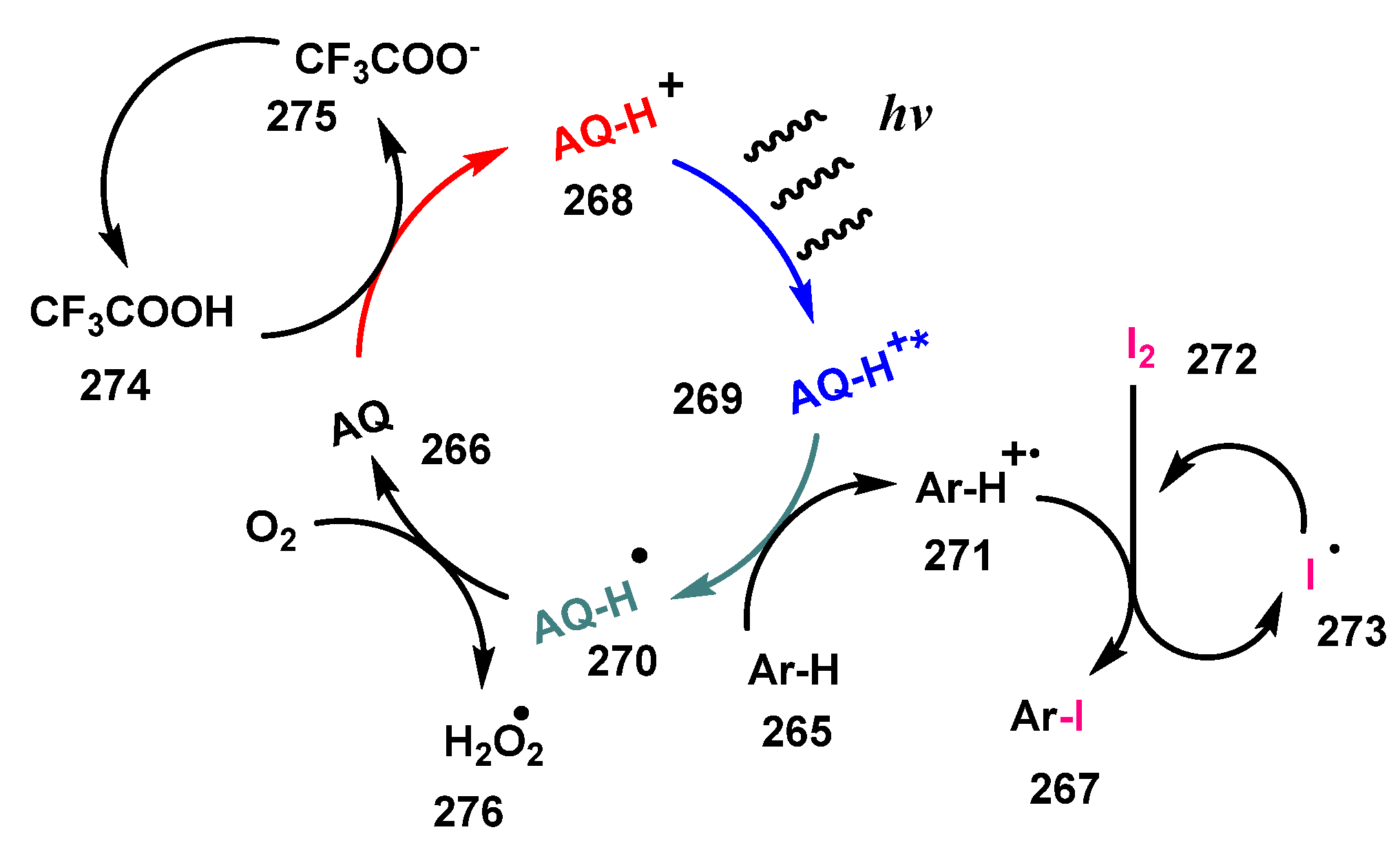
A plausible mechanism of this iodination was presented in Scheme 70. Reaction of photocatalyst AQ 266 with TFA 274 to receive a proton provided the protonated AQ-H+ 268. Light excited AQ-H+ 268 to the excited state AQ-H+* 269, which then oxidized arenes to give arene radical cation 271 and AQ-H• radical 270. The generated arene radical cation 271 then captured the iodine atom from iodine molecule 272 to produce iodinated product 267 and iodine radical I• 273, which recombined with another I• to give I2 272. AQ-H• 270 radical was oxidized by oxygen to regenerate AQ 266.
7. Conclusions
A lot of breakthroughs using photocatalytic reactions have been achieved in this decade. Current developments in the application of visible light photocatalysts for halogenation, including chlorination, bromination, and iodination, are described in this study.
Visible light and photocatalyst-mediated halogenation technologies have many attractive advantages that make them good candidates to replace the old methods. Mild reaction conditions are useful in multi-step synthetic chemistry and selective halogenation at specific positions. Being able to replace hazardous or expensive chemicals is another big advantage. In addition, the excellent functional group tolerance provides the possibility of applying this protocol to various organic compounds including aliphatic C-H bonds, multiple bonds, carboxylic acids, and aromatics. In particular, it can also be applied to halogenation of natural compounds involved in various pharmaceutical applications.
The visible-light-photocatalytic halogenation technique allows the use of several halogen sources (Br2, Cl2, CBr4, HCl, etc.) without the need for initiators or harsh reaction conditions, thereby reducing costs and making the reactions “greener”.
On the other hand, using green solvents is also a big challenge. Organic solvents were utilized in the majority of the reactions described in this investigation. In spite of improving the efficiency of halogenation processes, most of them have negative impacts on the environment and require higher solvent removal and recovery costs. Replacing organic solvents with water would be a challenging research field in the future.
Despite considerable advancements in this procedure, there is still a lot of room for development at this point. Nearly all photocatalysts contain iridium, ruthenium, lithium, and transition metals (Cu, etc), or have a very complicated structure (Eoxin Y), resulting in high prices that obstruct their commercial implementation. Besides, the removal of catalysts from the products should be taken into account. The heterogeneous catalysts can be easily separated through filtration. Generally, homogeneous metal catalysts are typically used in the form of soluble salts or chelating complexes and can be easily eluted via extraction with water. However, the criteria for product purity are becoming increasingly stringent, particularly for medicinal items. Therefore, it would be great to come up with approaches to employ other photocatalysts, which are more popular and cost effective.
Out of the studies on visible-light-mediated photocatalytic halogenation, those on halogenation of C-H aliphatic compounds and halogenation of arenes are dominant because aliphatic and aromatic halide derivatives have a wide range of applications in medicine and industrial chemistry. In the future, there will be a need to discover additional methods for direct halogenation from other functional groups.
In conclusion, the application of visible-light-mediated photocatalysis represents a major advance in the field of halogenation of organic compounds, offering a significant difference from traditional methods. Further research in the future may help overcome the limitations of photocatalytic halogenation and expand the applications to other functional groups.
Author Contributions
Conceptualization, H.-K.K.; writing–original draft preparation, T.G.L., Y.J. and H.-K.K.; writing–review and editing, T.G.L., Y.J. and H.-K.K.; funding acquisition, H.-K.K. and Y.J. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (NRF-2021R1A2C1011204), and by the Education and Research Promotion Program (2020) of KOREATECH.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rozhkov, A.V.; Eliseeva, A.A.; Baykov, S.V.; Galmes, B.; Frontera, A.; Kukushkin, V.Y. One-Pot Route to X-Perfluoroarenes (X = Br, I) Based on FeIII Assisted C−F Functionalization and Utilization of These Arenes as Building Blocks for Crystal Engineering Involving Halogen Bonding. Cryst. Growth Des. 2020, 20, 5908–5921. [Google Scholar] [CrossRef]
- Nemec, V.; Fotovic, L.; Tomislav, F.; Dominik, C. Large Family of Halogen-Bonded Cocrystals Involving Metal-Organic Building Blocks with Open Coordination Sites. Cryst. Growth Des. 2017, 17, 6169–6173. [Google Scholar] [CrossRef]
- Cotman, A.E.; Guérin, T.; Kovacevic, I.; Tiz, D.B.; Durcik, M.; Fulgheri, F.; Mozina, S.; Secci, D.; Sterle, M.; Ilas, J.; et al. Practical synthesis and application of halogen-doped pyrrole building blocks. ACS Omega 2021, 6, 9723–9730. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.L.; Wei, D.; Zhang, J.W.; Li, C.L.; Yu, W.; Han, B. Synthesis of halomethyl isoxazoles/cyclic nitrones via cascade sequence: 1,2-Halogen radical shift as a key link. Org. Lett. 2018, 20, 2906–2910. [Google Scholar] [CrossRef] [PubMed]
- Constantin, T.; Zanini, M.; Regni, A.; Sheikh, N.S.; Julia, F.; Leonori, D. Aminoalkyl Radicals as Halogen-Atom Transfer Agents for Activation of Alkyl and Aryl Halides. Science 2020, 367, 1021–1026. [Google Scholar] [CrossRef]
- Kazi, I.; Guha, S.; Sekar, G. Halogen Bond-Assisted Electron-Catalyzed Atom Economic Iodination of Heteroarenes at Room Temperature. J. Org. Chem. 2019, 84, 6642–6654. [Google Scholar] [CrossRef]
- Mendez, L.; Henriquez, G.; Sirimulla, S.; Narayan, M. Looking back, Looking forward at Halogen Bonding in Drug Discovery. Molecules 2017, 22, 1397. [Google Scholar] [CrossRef]
- Latham, J.; Brandenburger, E.; Shepherd, S.A.; Menon, B.R.K.; Micklefield, J. Development of Halogenase Enzymes for Use in Synthesis. Chem. Rev. 2018, 118, 232–269. [Google Scholar] [CrossRef] [Green Version]
- Saccone, M.; Catalano, L. Halogen Bonding beyond Crystals in Materials Science. J. Phys. Chem. B 2019, 123, 9281–9290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilcken, R.; Zimmermann, M.O.; Lange, A.; Joerger, A.C.; Boeckler, F.M. Principles and Applications of Halogen Bonding in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2013, 56, 1363–1388. [Google Scholar] [CrossRef] [PubMed]
- Gribble, G.W. Newly Discovered Naturally Occurring Organohalogens. Arkivoc Part I 2018, 372–410. Available online: http://www.arkat-usa.org/get-file/65071/ (accessed on 1 December 2021). [CrossRef]
- Hennecke, U. New Catalytic Approaches towards the Enantioselective Halogenation of Alkenes. Chem. Asian J. 2012, 7, 456–465. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, M. Visible Light-Mediated Installation of Halogen Functionalities into Multiple Bond Systems. Chem. Sel. 2017, 2, 9136–9146. [Google Scholar] [CrossRef]
- Das, R.; Kapur, M. Transition-Metal-Catalyzed Site-Selective C-H Halogenation Reactions. Asian J. Org. Chem. 2018, 7, 1524–1541. [Google Scholar] [CrossRef]
- Chung, W.J.; Vanderwal, C.D. Stereoselective Halogenation in Natural Product Synthesis. Angew. Chem. Int. Ed. 2016, 55, 4396–4434. [Google Scholar] [CrossRef]
- Tu, H.; Zhu, S.; Qing, F.L.; Chu, L. Visible-Light-Induced Halogenation of Aliphatic C-H Bonds. Tetrahedron Lett. 2018, 59, 173–179. [Google Scholar] [CrossRef]
- Neumann, C.S.; Fujimori, D.G.; Walsh, C.T. Halogenation Strategies in Natural Product Biosynthesis. Chem. Biol. 2008, 15, 99–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podgorsek, A.; Zupan, M.; Iskra, J. Oxidative Halogenation with “Green” Oxidants: Oxygen and Hydrogen Peroxide. Angew. Chem. Int. Ed. 2009, 48, 8424–8450. [Google Scholar] [CrossRef] [PubMed]
- Evano, G.; Nitelet, A.; Thilmany, P.; Dewez, D.F. Metal-Mediated Halogen Exchange in Aryl and Vinyl Halides: A review. Front. Chem. 2018, 6, 114. [Google Scholar] [CrossRef]
- Fredricks, P.S.; Tedde, J.M. Free-Radical Xubstitution in Aliphatic Compounds. Part II. Halogenation of the N-Butyl Halides. J. Chem. Soc. 1960, 144–150. [Google Scholar] [CrossRef]
- Marzo, L.; Pagire, S.K.; Reiser, O.; Burkhard, K. Visible-Light Photocatalysis: Does It Make a Difference in Organic Synthesis? Angew. Chem. Int. Ed. 2018, 57, 10034–10072. [Google Scholar] [CrossRef]
- Cavedon, C.; Seeberger, P.H.; Pieber, B. Photochemical Strategies for Carbon–Heteroatom Bond Formation. Eur. J. Org. Chem. 2019, 10, 1379–1392. [Google Scholar] [CrossRef] [Green Version]
- Fukuzumi, S.; Ohkubo, K. Organic Synthetic Transformations Using Organic Dyes as Photoredox Catalysts. Org. Biomol. Chem. 2014, 12, 6059. [Google Scholar] [CrossRef] [Green Version]
- Koike, T. Frontiers in Radical Fluoromethylation by Visible-Light Organic Photocatalysis. Asian J. Org. Chem. 2020, 9, 529–537. [Google Scholar] [CrossRef]
- Bui, T.T.; Hong, W.P.; Kim, H.K. Recent Advances in Visible Light-Mediated Fluorination. J. Fluor. Chem. 2021, 247, 109794. [Google Scholar] [CrossRef]
- Wang, Y.; Li, G.X.; Yang, G.; He, G.; Chen, G. A Visible-Light-Promoted Radical Reaction System for Azidation and Halogenation of Tertiary Aliphatic C-H Bonds. Chem. Sci. 2016, 7, 2679–2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, L.; Xia, J.B.; You, L.; Chen, C. Ketone-Catalyzed Photochemical C(sp3)-H Chlorination. Tetrahedron 2017, 73, 3696–3701. [Google Scholar] [CrossRef] [PubMed]
- Xiang, M.; Zhou, C.; Yang, X.L.; Chen, B.; Tung, C.H.; Wu, L.Z. Visible Light-Catalyzed Benzylic C-H Bond Chlorination by a Combination of Organic Dye (Acr+-Mes) and N-Chlorosuccinimide. J. Org. Chem. 2020, 85, 9080–9087. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, J.J.; Wu, D.; Yu, W. Visible-Light-Driven Remote C-H Chlorination of Aliphatic Sulfonamides with Sodium Hypochlorite. Asian J. Org. Chem. 2020, 9, 1650–1654. [Google Scholar] [CrossRef]
- Nishina, Y.; Morita, J.; Ohtani, B. Direct Bromination of Hydrocarbons Catalyzed by Li2MnO3 under Oxygen and Photo-Irradiation Conditions. RSC Adv. 2013, 3, 2158–2162. [Google Scholar] [CrossRef] [Green Version]
- Kee, C.W.; Chan, K.M.; Wong, M.W.; Tan, C.H. Selective Bromination of sp3 C-H Bonds by Organophotoredox Catalysis. Asian J. Org. Chem. 2014, 3, 536–544. [Google Scholar] [CrossRef]
- Ni, S.; El Remaily, M.A.E.A.A.A.; Franzén, J. Carbocation Catalyzed Bromination of Alkyl Arenes, a Chemoselective sp3 vs. sp2 C-H Functionalization. Adv. Synth. Catal. 2018, 360, 4197–4204. [Google Scholar] [CrossRef]
- Wilger, D.J.; Grandjean, J.M.M.; Lammert, T.R.; Nicewicz, D.A. The Direct Anti-Markovnikov Addition of Mineral Acids to Styrenes. Nat. Chem. 2014, 6, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.H.; Malpani, Y.R.; Ha, N.; Jung, Y.S.; Han, S.B. Vicinal Difunctionalization of Alkenes: Chlorotrifluoromethylation with CF3SO2Cl by Photoredox Catalysis. Org. Lett. 2014, 16, 1310–1313. [Google Scholar] [CrossRef]
- Tang, X.J.; Dolbier, W.R. Efficient Cu-catalyzed Atom Transfer Radical Addition Reactions of Fluoroalkylsulfonyl Chlorides with Electron-Deficient Alkenes Inducedby Visible Light. Angew. Chem. Int. Ed. 2015, 54, 4246–4249. [Google Scholar] [CrossRef]
- Lian, P.; Long, W.; Li, J.; Zheng, Y.; Wan, X. Visible-Light-Induced Vicinal Dichlorination of Alkenes through LMCT Excitation of CuCl2. Angew. Chem. Int. Ed. 2020, 59, 23603–23608. [Google Scholar] [CrossRef]
- Nguyen, J.D.; Tucker, J.W.; Konieczynska, M.D.; Stephenson, C.R.J. Intermolecular Atom Transfer Radical Addition to Olefins Mediated by Oxidative Quenching of Photoredox Catalysts. J. Am. Chem. Soc. 2011, 133, 4160–4163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wang, L.; Wang, Z.; Li, P.; Zhang, Y. A Practical Synthesis of α-Bromo/Iodo/Chloroketones from Olefins under Visible-Light Irradiation Conditions. Chin. Chem. Lett. 2021, 32, 429–432. [Google Scholar] [CrossRef]
- Dai, C.; Narayanam, J.M.R.; Stephenson, C.R.J. Visible-Light-Mediated Conversion of Alcohols to Halides. Nat. Chem. 2011, 3, 140–145. [Google Scholar] [CrossRef]
- Li, R.; Gehrig, D.W.; Ramanan, C.; Blom, P.W.M.; Kohl, F.F.; Wagner, M.; Landfester, K.; Zhang, K.A.I. Visible-Light-Mediated Conversion of Alcohols to Bromides by a Benzothiadiazole-Containing Organic Photocatalyst. Adv. Synth. Catal. 2019, 361, 3852–3859. [Google Scholar] [CrossRef]
- Candish, L.; Standley, E.A.; Suárez, A.G.; Mukherjee, S.; Glorius, F. Catalytic Access to Alkyl Bromides, Chlorides and Iodides via Visible Light-Promoted Decarboxylative Halogenation. Chem. Eur. J. 2016, 22, 9971–9974. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, A.; Koyamada, K.; Miyamoto, K.; Kanazawa, J.; Uchiyama, M. Decarboxylative Bromination of Sterically Hindered Carboxylic Acids with Hypervalent Iodine(III) Reagents. Org. Process. Res. Dev. 2020, 24, 1328–1334. [Google Scholar] [CrossRef]
- Pal, P.; Singh, H.; Panda, A.B.; Ghosh, S.C. Heterogeneous Cu-MnO Catalyzed Monoselective Ortho-Halogenation of Aromatic C-H Bonds under Visible Light. Asian J. Org. Chem. 2015, 4, 879–883. [Google Scholar] [CrossRef]
- Wang, X.C.; Hu, Y.; Bonacorsi, S.; Hong, Y.; Burrell, R.; Yu, J.Q. Pd (II)-Catalyzed C-H Iodination Using Molecular I2 as the Sole Oxidant. J. Am. Chem. Soc. 2013, 135, 10326–10329. [Google Scholar] [CrossRef] [Green Version]
- Mo, F.; Yan, J.M.; Qiu, D.; Li, F.; Zhang, Y.; Wang, J. Gold-Catalyzed Halogenation of Aromatics by N-Halosuccinimides. Angew. Chem. Int. Ed. 2010, 49, 2028–2032. [Google Scholar] [CrossRef]
- Wang, L.; Ackermann, L. Ruthenium-Catalyzed Ortho-C-H Halogenations of Benzamides. Chem. Commun. 2014, 50, 1083–1085. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.G.; Gensch, T.; de Azambuja, F.; Vasquez-Cespedes, S.; Glorius, F. Co (III)-Catalyzed C-H Activation/Formal SN-type Reactions: Selective and Efficient Cyanation, Halogenation, and Allylation. J. Am. Chem. Soc. 2014, 136, 17722–17725. [Google Scholar] [CrossRef]
- Qiao, H.; Sun, S.; Yang, F.; Zhu, Y.; Kang, J.; Wu, Y.; Wu, Y. Merging Photoredox Catalysis with Iron (III) Catalysis: C5-H Bromination and Iodination of 8-Aminoquinoline Amides in Water. Adv. Synth. Catal. 2017, 359, 1976–1980. [Google Scholar] [CrossRef]
- Seel, C.J.; Králík, A.; Hacker, M.; Frank, A.; König, B.; Gulder, T. Atom-Economic Electron Donors for Photobiocatalytic Halogenations. ChemCatChem 2018, 10, 3960–3963. [Google Scholar] [CrossRef]
- Singh, H.; Sen, C.; Sahoo, T.; Ghosh, S.C. A Visible Light-Mediated Regioselective Halogenation of Anilides and Quinolines by Using a Heterogeneous Cu-MnO Catalyst. Eur. J. Org. Chem. 2018, 34, 4748–4753. [Google Scholar] [CrossRef]
- Hering, T.; König, B. Photocatalytic Activation of N-Chloro Compounds for the Chlorination of Arenes. Tetrahedron 2016, 72, 7821–7825. [Google Scholar] [CrossRef]
- Zhanga, L.; Hu, X. Room Temperature C(sp2)-H Oxidative Chlorination via Photoredox Catalysis. Chem. Sci. 2017, 8, 7009–7013. [Google Scholar] [CrossRef]
- Rogers, D.A.; Gallegos, J.M.; Hopkins, M.D.; Lignieres, A.A.; Lamar, A.A. Visible-Light Photocatalytic Activation of N-Chlorosuccinimide by Organic Dyes for the Chlorination of Arenes and Heteroarenes. Tetrahedron 2019, 75, 130498. [Google Scholar] [CrossRef]
- Rogers, D.A.; Bensalah, A.T.; Espinosa, A.T.; Hoerr, J.L.; Refai, F.H.; Pitzel, A.K.; Alvarado, J.J.; Lamar, A.A. Amplification of Trichloroisocyanuric Acid (TCCA) Reactivity for Chlorination of Arenes and Heteroarenes via Catalytic Organic Dye Activation. Org. Lett. 2019, 21, 4229–4233. [Google Scholar] [CrossRef]
- Rogers, D.A.; Hopkins, M.D.; Rajagopal, N.; Varshney, D.; Howard, H.A.; LeBlanc, G.; Lamar, A.A. U.S. Food and Drug Administration-Certified Food Dyes as Organocatalysts in the Visible Light-Promoted Chlorination of Aromatics and Heteroaromatics. ACS Omega 2020, 5, 7693–7704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohkubo, K.; Mizushima, K.; Iwata, R.; Fukuzumi, S. Selective Photocatalytic Aerobic Bromination with Hydrogen Bromide via an Electron-Transfer State of 9-mesityl-10-methylacridinium ion. Chem. Sci. 2011, 2, 715–722. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, Z.; Yang, C.; Lin, R.; Xia, W. Visible-Light Photoredox Catalysis Enabled Bromination of Phenols and Alkenes. Beilstein J. Org. Chem. 2014, 10, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Wang, Z.J.; Wang, L.; Ma, B.C.; Ghasimi, S.; Lu, H.; Landfester, K.; Zhang, K.A.I. Photocatalytic Selective Bromination of Electron-Rich Aromatic Compounds Using Microporous Organic Polymers with Visible Light. ACS Catal. 2016, 6, 1113–1121. [Google Scholar] [CrossRef]
- Rogers, D.A.; Brown, R.G.; Brandeburg, Z.C.; Ko, E.Y.; Hopkins, M.D.; LeBlanc, G.; Lamar, A.A. Organic Dye-Catalyzed, Visible-Light Photoredox Bromination of Arenes and Heteroarenes Using N-Bromosuccinimide. ACS Omega 2018, 3, 12868–12877. [Google Scholar] [CrossRef] [Green Version]
- Petzold, D.; König, B. Photocatalytic Oxidative Bromination of Electron-Rich Arenes and Heteroarenes by Anthraquinone. Adv. Synth. Catal. 2018, 360, 626–630. [Google Scholar] [CrossRef]
- Ma, B.; Lu, F.; Yang, H.; Gu, X.; Li, Z.; Li, R.; Pei, H.; Luo, D.; Zhang, H.; Lei, A. Visible Light Mediated External Oxidant Free Selective C5 Bromination of 8-Aminoquinoline Amides under Ambient Conditions. Asian J. Org. Chem. 2019, 8, 1136–1140. [Google Scholar] [CrossRef]
- Fan, J.; Wei, Q.; Zhu, E.; Gao, J.; Cheng, X.; Lu, Y.; Loh, T.P. Visible Light-Induced Mono-Bromination of Arenes with BrCCl3. Chem. Commun. 2021, 57, 5977–5980. [Google Scholar] [CrossRef] [PubMed]
- Narobe, R.; Düsel, S.J.S.; Iskra, J.; König, B. Photocatalytic Oxidative Iodination of Electron-Rich Arenes. Adv. Synth. Catal. 2019, 361, 3998–4004. [Google Scholar] [CrossRef]
Figure 1.
Several halogenated compounds in pharmaceuticals.

Scheme 1.
Synthetic methods for halogenation of aliphatic C-H bonds.

Scheme 2.
Photocatalysis halogenation of tertiary aliphatic C-H bonds.

Scheme 3.
Proposed mechanism of photocatalyzed tertiary aliphatic C-H bond chlorination.

Scheme 4.
Benzophenone-catalyzed photochemical C(sp3)-H chlorination.

Scheme 5.
Acetophenone-catalyzed photochemical C(sp3)-H chlorination.

Scheme 6.
Visible-light-catalyzed benzylic C-H bond chlorination.

Scheme 7.
Proposed mechanism of benzylic C-H bond chlorination method.

Scheme 8.
Chlorination of aliphatic sulfonamides.

Scheme 9.
Mechanism of the aliphatic sulfonamide chlorination.

Scheme 10.
Li2MnO3-induced photocatalytic bromination of hydrocarbons.

Scheme 11.
Bromination of hydrocarbons by N-bromosuccinimide.

Scheme 12.
Selective bromination of sp3 C-H bonds by organophotoredox catalysis.

Scheme 13.
Mechanism for bromination of sp3 C-H bond promoted by Eosin Y and morpholine.

Scheme 14.
Conversion of alkyl arenes C(sp3)-H bond to brominated products.

Scheme 15.
Synthetic methods for halogenation of aliphatic multiple bonds.

Scheme 16.
Photoredox anti-Markovnikov hydrochlorination of styrenes.

Scheme 17.
Chlorotrifluoromethylation of alkenes with CF3SO2Cl by photoredox catalysis.

Scheme 18.
Proposed mechanism of chlorotrifluoromethylation of alkenes.

Scheme 19.
Conversion of electron-deficient alkenes to chlorinated products.

Scheme 20.
Another preparation of β-chlorinated phenyl amine and phenyl ester.

Scheme 21.
Visible-light-induced dichlorination of alkenes.

Scheme 22.
Proposed mechanism for conversion of alkenes to alkyldichlorides.

Scheme 23.
Photocatalysis halogenation of several olefins.

Scheme 24.
Proposed mechanism for conversion of olefins to products.

Scheme 25.
Several α-bromoketones from olefin sunder visible light irradiation.

Scheme 26.
The synthesis mechanism of α-bromoketones from styrenes.

Scheme 27.
Synthetic methods for halogenation of alcohols.

Scheme 28.
Conversion of alcohols to bromides or iodides using photoredox catalysis.

Scheme 29.
Mechanism of alcohols halogenation reaction.

Scheme 30.
Conversion of alcohols to bromides by metal-free organic photocatalyst.

Scheme 31.
Proposed mechanism of photocatalytic direct conversion of alcohols to bromides.

Scheme 32.
Synthetic methods for halogenation of carboxylic acids.

Scheme 33.
Decarboxylative halogenation reaction via visible light.

Scheme 34.
Proposed mechanism for the visible-light-promoted bromination.

Scheme 35.
Decarboxylative bromination of sterically hindered carboxylic acids.

Scheme 36.
Mechanism of decarboxylative bromination using hypervalent iodine(III) reagents.

Scheme 37.
Synthetic methods for halogenation of aromatic C-H bonds.

Scheme 38.
Photocatalysis halogenation of 2-arylpyridines.

Scheme 39.
Proposed mechanism for 2-arylpyridines C-H halogenation.

Scheme 40.
Halogenation of 8-aminoquinoline amide compounds.

Scheme 41.
General mechanism for halogenation of quinolone.

Scheme 42.
Photobiocatalytic aromatic bromination using AmVHPO.

Scheme 43.
Chlorination and bromination of anilide compounds.

Scheme 44.
Chlorination, bromination, and iodination of quinoline derivatives.

Scheme 45.
Proposed reaction mechanism for regioselective halogenation of anilides and quinolines.

Scheme 46.
The visible-light-mediated chlorination of arenes with N-chloramines.

Scheme 47.
Arene oxidative chlorination via photoredox catalysis.

Scheme 48.
Proposed mechanism of the photocatalytic oxidative chlorination reaction.

Scheme 49.
Chlorination of arenes and heteroarenes via organic dyes.

Scheme 50.
Mechanism for chlorination of arenes and heteroarenes by N-chlorosuccinimide.

Scheme 51.
Chlorination of arenes and heteroarenes via catalytic organic dye activation.

Scheme 52.
Mechanism for conversion of arenes and heteroarenes to chlorinated products using organic dye.
Scheme 52.
Mechanism for conversion of arenes and heteroarenes to chlorinated products using organic dye.

Scheme 53.
Visible-light-promoted chlorination of aromatics and heteroaromatics.

Scheme 54.
Mechanism of brilliant blue/DCDMH chlorination of arenes.

Scheme 55.
Selective photocatalytic bromination with hydrogen bromide.

Scheme 56.
Proposed photocatalytic reaction mechanism of bromination with hydrogen bromide.

Scheme 57.
Visible-light-photoredox catalysis enabled bromination of phenols.

Scheme 58.
General mechanism for in situ generation of bromine.

Scheme 59.
Photocatalytic-selective bromination of aromatic compounds using MOPs.

Scheme 60.
Mechanism of photocatalytic bromination of TMB using the MOPs as photocatalyst.

Scheme 61.
Visible-light-photoredox bromination of arenes and heteroarenes.

Scheme 62.
General mechanism for bromination reaction using N-bromosuccinimide and organic dye catalysis.
Scheme 62.
General mechanism for bromination reaction using N-bromosuccinimide and organic dye catalysis.

Scheme 63.
Bromination of electron-rich arenes and heteroarenes by anthraquinone.

Scheme 64.
Mechanism for the photocatalytic bromination of arenes and heteroarenes.

Scheme 65.
Selective C5 bromination of 8-aminoquinoline amides.

Scheme 66.
Proposed mechanism for transformation of 8-aminoquinoline amides to brominated product.

Scheme 67.
Visible-light-induced mono-bromination of arenes with BrCCl3.

Scheme 68.
Mechanism for para-bromination of active aniline.

Scheme 69.
Photocatalytic oxidative iodination of electron-rich arenes.

Scheme 70.
Proposed mechanism of the photocatalytic oxidative iodination reaction.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Luu, T.G.; Jung, Y.; Kim, H.-K. Visible-Light-Induced Catalytic Selective Halogenation with Photocatalyst. Molecules 2021, 26, 7380. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26237380
AMA Style
Luu TG, Jung Y, Kim H-K. Visible-Light-Induced Catalytic Selective Halogenation with Photocatalyst. Molecules. 2021; 26(23):7380. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26237380
Chicago/Turabian StyleLuu, Truong Giang, Yongju Jung, and Hee-Kwon Kim. 2021. "Visible-Light-Induced Catalytic Selective Halogenation with Photocatalyst" Molecules 26, no. 23: 7380. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26237380