
Synthesis, Structural Characterization, and In Vitro and In Silico Antifungal Evaluation of Azo-Azomethine Pyrazoles (PhN2(PhOH)CHN(C3N2(CH3)3)PhR, R = H or NO2)

 , , , and
, , , and 
Abstract
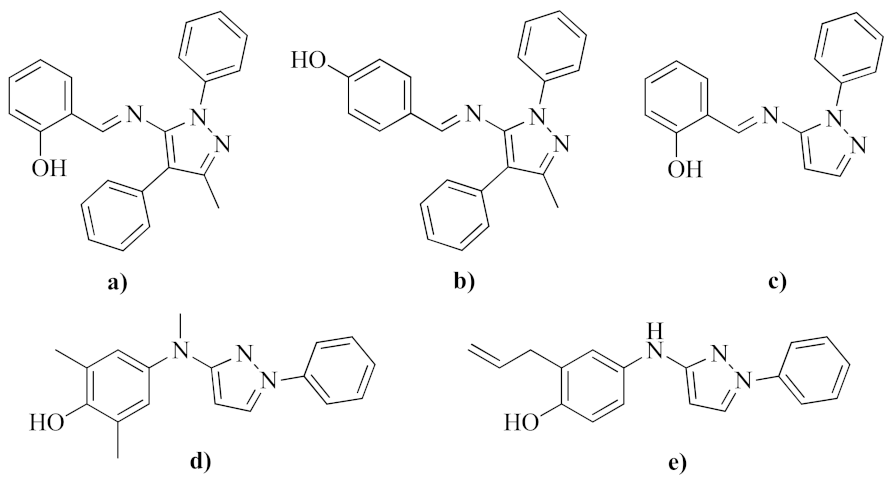
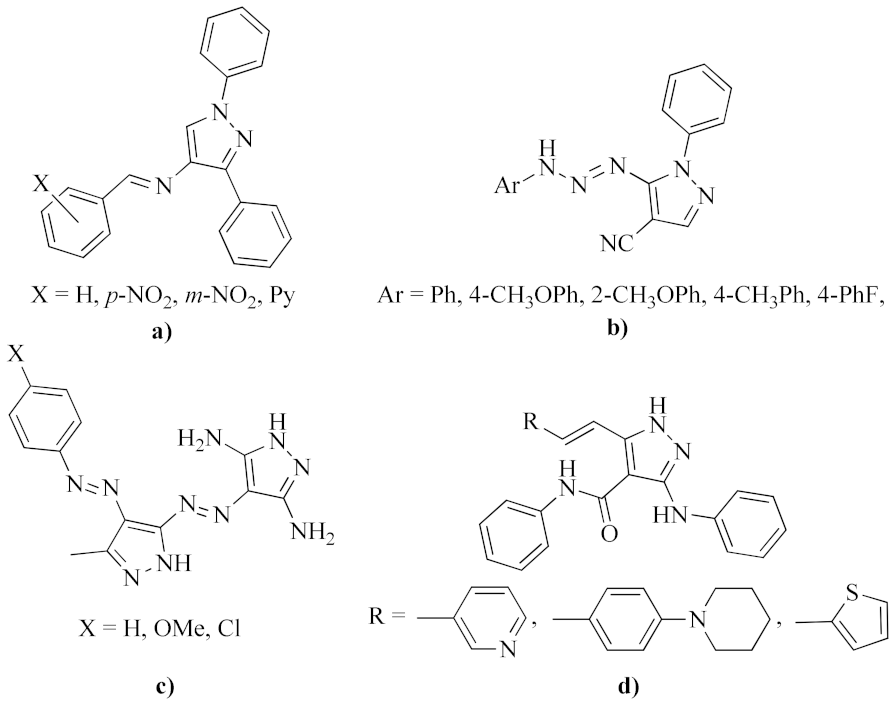
:1. Introduction
2. Results and Discussion
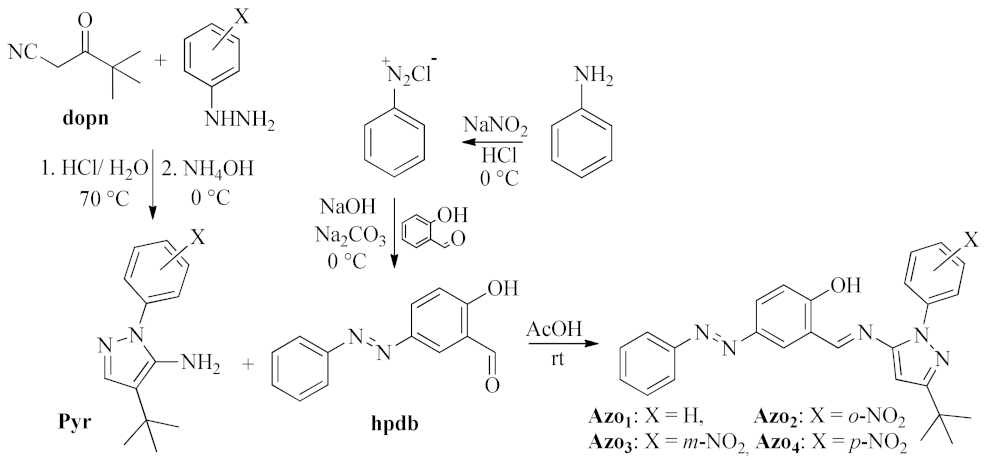
2.1. Synthesis and Molecular Identification
2.2. Spectroscopy Characterization
2.2.1. Fourier Transform Infrared (FTIR) Spectroscopy
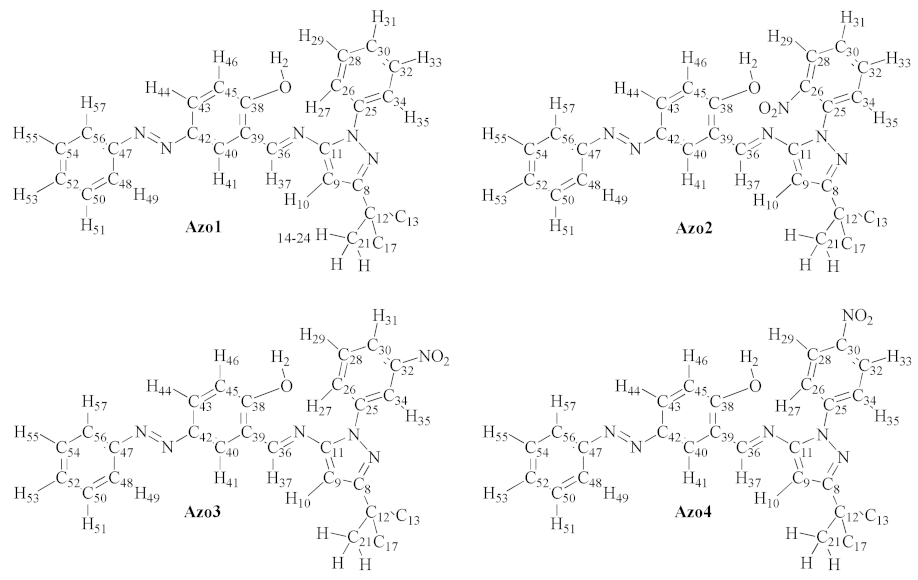
2.2.2. Nuclear Magnetic Resonance (NMR) Spectroscopy
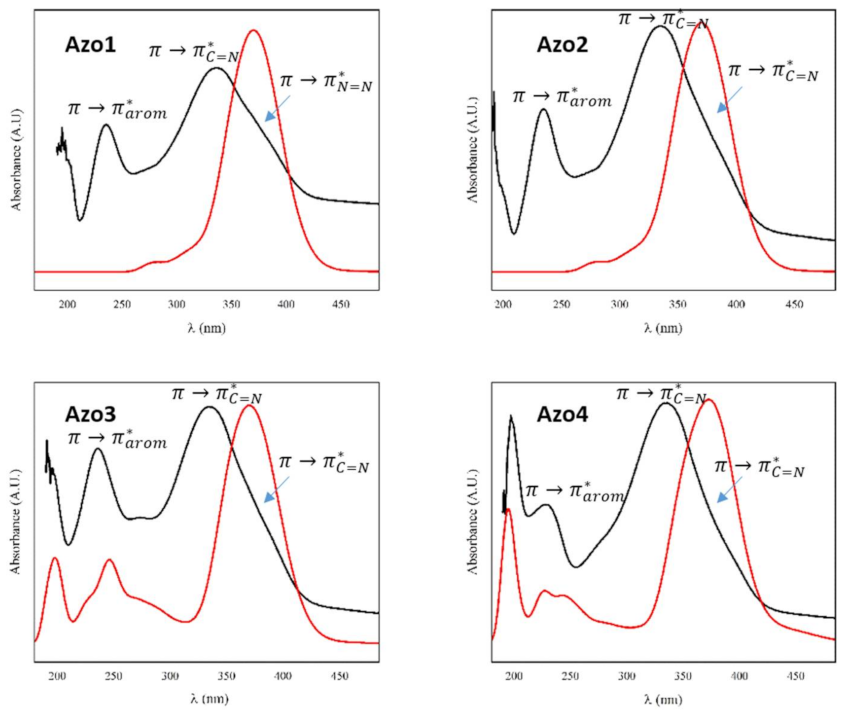
2.2.3. Absorption Electronic UV–Visible Spectroscopy
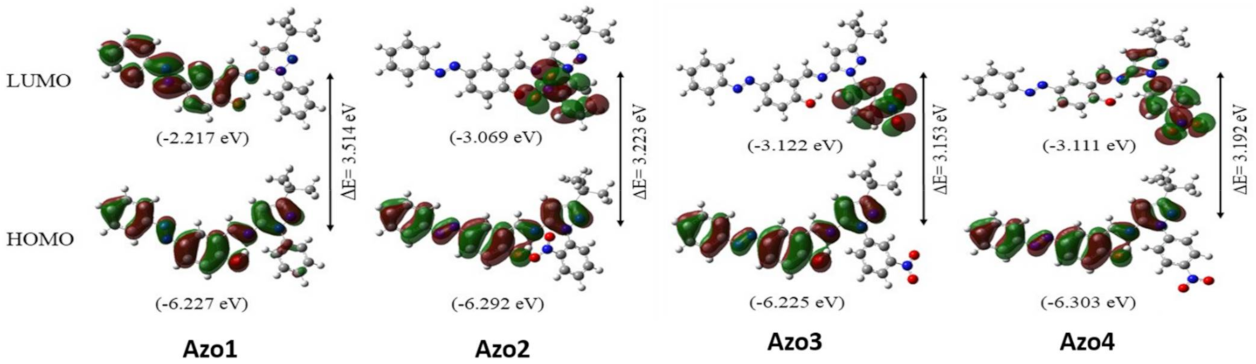
2.3. Frontier Molecular Orbitals
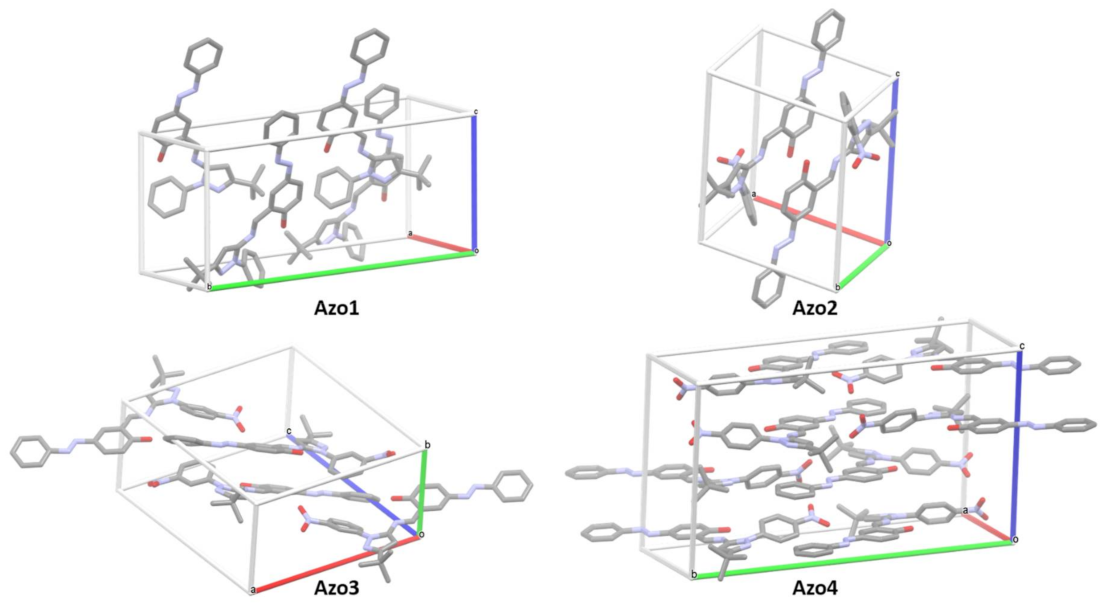
2.4. Crystalline Structural Analysis
2.4.1. Single Crystal X-ray Diffraction
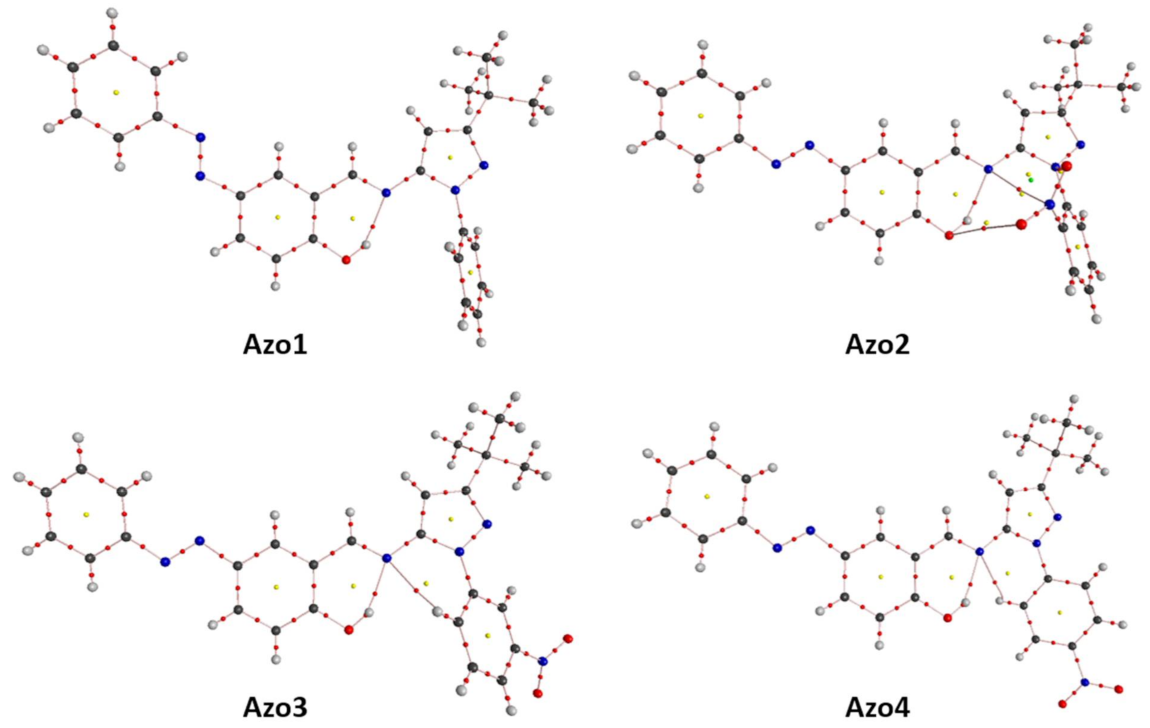
2.4.2. Topological Analysis on Electron Density (ρ)
2.5. In Vitro and In Silico Studies on the Antifungal Activity
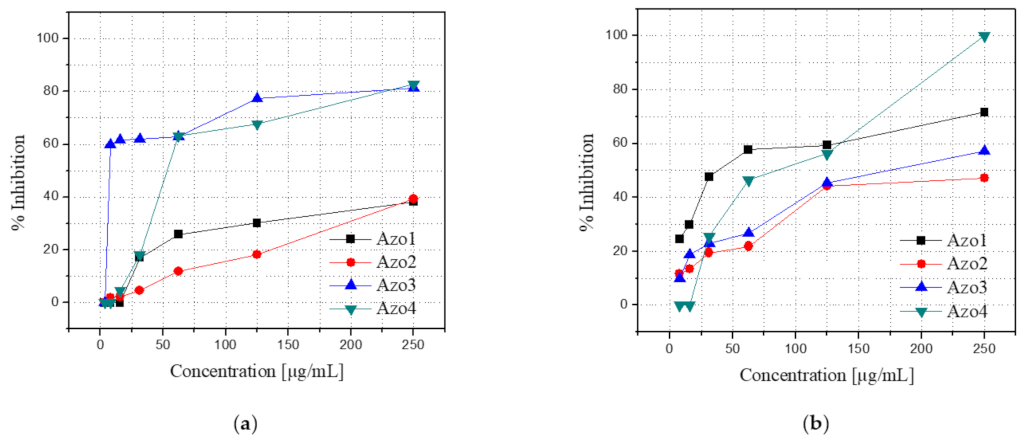
2.5.1. Antifungal Activity Evaluation
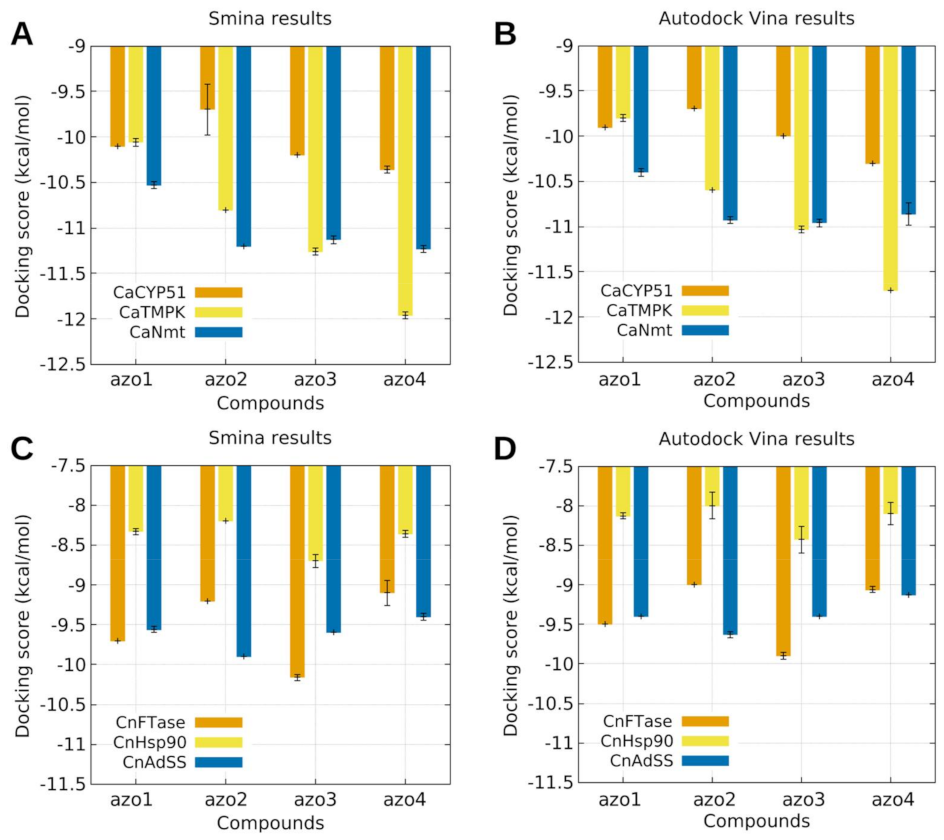
2.5.2. Molecular Docking Modelling
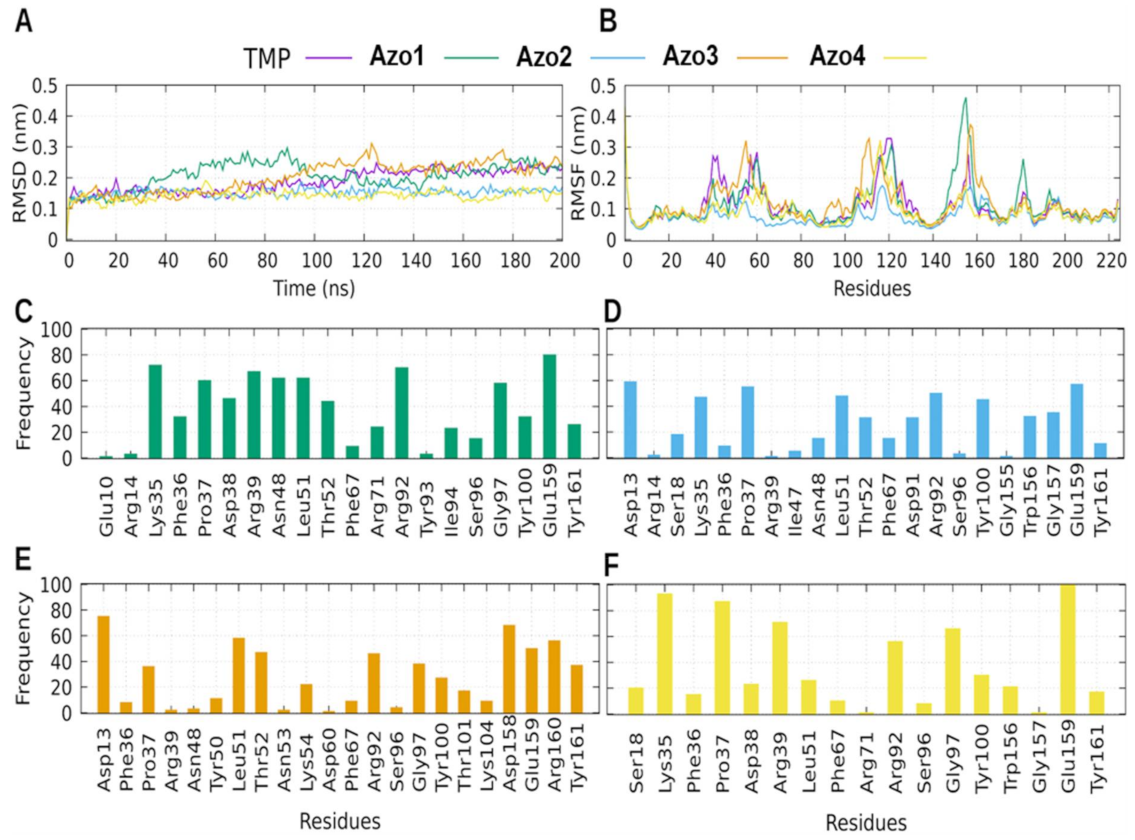
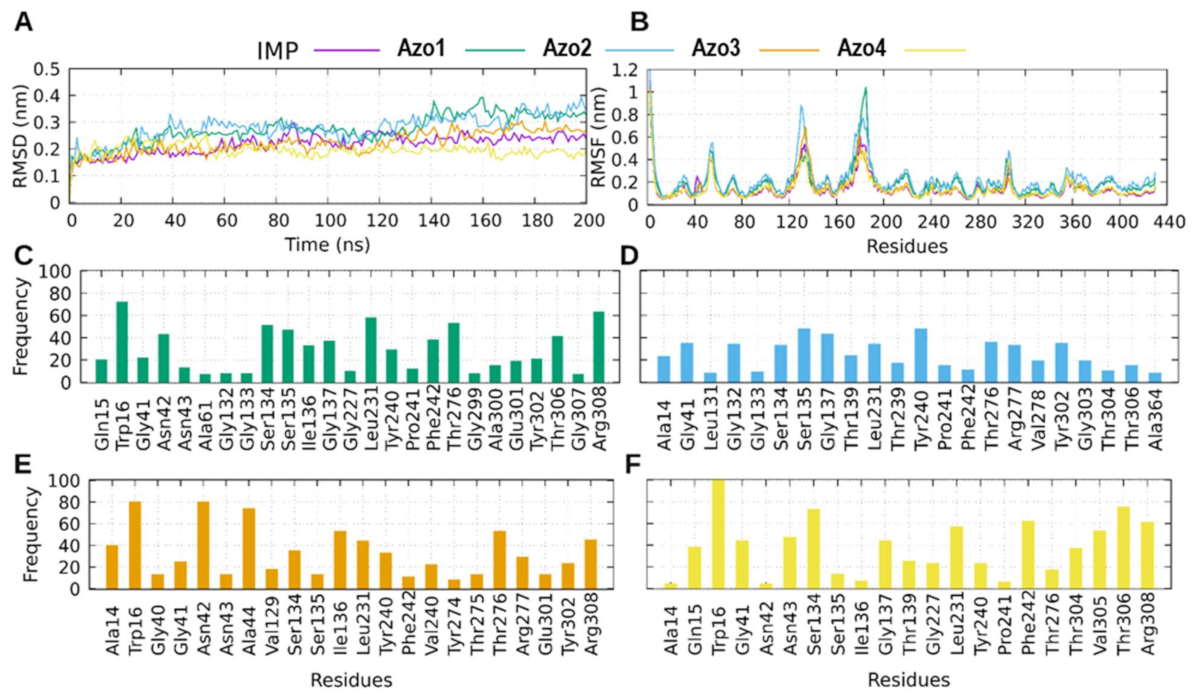
2.5.3. Molecular Dynamics (MD) Simulation
3. Materials and Methods
3.1. Synthesis
3.1.1. Synthesis of Aminepyrazole Derivatives
3.1.2. Synthesis of (E)-2-hydroxy-4-(phenyldiazenyl)benzaldehyde
3.1.3. Synthesis and Physicochemical Properties of Azo-imine-pyrazoles
3.2. Analytical and Physicochemical Measurements
3.3. X-ray Diffraction Analysis
3.4. Computational Studies
3.4.1. DFT Calculations
3.4.2. Topological Analysis
3.4.3. Molecular Docking Calculations
3.4.4. Molecular Dynamics Simulations
3.5. Antifungal Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Serbest, K.; Dural, T.; Emirik, M.; Zengin, A.; Faiz, Ö. Heteroligand bivalent transition metal complexes with an azo-oxime ligand and 1,10-phenanthroline: Synthesis, spectroscopy, thermal analysis, DFT calculations and SOD-mimetic activities. J. Mol. Struct. 2021, 1229, 129579–129585. [Google Scholar] [CrossRef]
- Khojasteh, V.; Kakanejadifard, A.; Zabardasti, A.; Azarbani, F. Spectral, structural, solvatochromism, biological and computational investigation of some new azo-azomethines containing N-alkylpyridinium salts. J. Mol. Struct. 2019, 1175, 261–268. [Google Scholar] [CrossRef]
- Karthikeyan, M.S.; Prasad, D.J.; Poojary, B.; Bhat, K.S.; Holla, B.S.; Kumari, N.S. Synthesis and biological activity of Schiff and Mannich bases bearing 2,4-dichloro-5-fluorophenyl moiety. Bioorg. Med. Chem. 2006, 14, 7482–7489. [Google Scholar] [CrossRef]
- Mladenova, R.; Ignatova, M.; Manolova, N.; Petrova, T.; Rashkov, I. Synthesis, structural features and evaluation of antibacterial activities of two Schiff bases derived from 3, 4-Dihydroxybenzhydrazide. Eur. Polym. J. 2002, 38, 989–999. [Google Scholar] [CrossRef]
- Zhang, Z.; Cao, P.; Fang, M.; Zou, T.; Han, J.; Duan, Y.; Xu, H.; Yang, X.; Li, Q.-S. Design, synthesis, and SAR study of novel 4,5-dihydropyrazole-Thiazole derivatives with antiinflammatory activities for the treatment of sepsis. Eur. J. Med. Chem. 2021, in press. [Google Scholar]
- Zhang, D.; Xu, G.; Zhao, J.; Wang, Y.; Wu, X.; He, X.; Li, W.; Zhang, S.; Yang, S.; Ma, C. Structure-activity relationship investigation for imidazopyrazole-3-carboxamide derivatives as novel selective inhibitors of Bruton’s tyrosine kinase. Eur. J. Med. Chem. 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Nayak, S.; Gaonkar, S.L.; Musad, E.A.; AL Dawsar, A.M. 1,3,4-Oxadiazole-containing hybrids as potential anticancer agents: Recent developments, mechanism of action and structure-activity relationships. J. Saudi. Chem. Soc. 2021, 25, 101284. [Google Scholar] [CrossRef]
- Viji, A.; Revathi, B.; Balachandrana, V.; Babiyana, S.; Narayana, B.; Salian, V.V. Analysis of spectroscopic, quantum chemical calculations, molecular docking, RDG, ELF, anticancer and antimicrobial activity studies on bioactive molecule 2-[3-(4-Chlorophenyl)-5-(4-(propane-2-yl) phenyl-4,5-dihydro-1H-pyrazol-1-yl]-4-(4-methoxyphenyl)-1,3-thiazol. Chem. Data Collect. 2021, 30, 100585. [Google Scholar]
- Zhu, Y.; Zhao, J.; Luo, L.; Gao, Y.; Bao, H.; Li, P.; Zhang, H. Research progress of indole compounds with potential antidiabetic activity. Eur. J. Med. Chem. 2021, 223, 113665. [Google Scholar] [CrossRef]
- da Silva, M.J.V.; Jacomini, A.P.; Gonçalves, D.S.; Pianoski, K.E.; Poletto, J.; Lazarin-Bidoia, D.; Volpato, H.; Nakamura, C.V.; Rosa, F.A. Discovery of 1,3,4,5-tetrasubstituted pyrazoles as anti-trypanosomatid agents: Identification of alterations in flagellar structure of L. amazonensis. Bioorg. Chem. 2021, 114, 105082. [Google Scholar] [CrossRef] [PubMed]
- Erkina, A.V.; Yurieva, A.V.; Yuzikhinb, O.S.; Gurzhiy, V.V.; Krutikov, V.I. 2-(2-Amino-6-methylpyrimidin-4-yl)-4-arylmethylidene5-methyl-2,4-dihydro-3H-pyrazol-3-ones: Design, synthesis, structure, in vitro anti-tubercular activity, and molecular docking study. J. Mol. Struct. 2021, 1243, 130863. [Google Scholar] [CrossRef]
- Gumus, A.; Bozdag, M.; Angeli, A.; Peat, T.S.; Carta, F.; Supuran, C.T.; Selleri, S. Privileged Scaffolds in Medicinal Chemistry: Studies on Pyrazolo[1,5-a]pyrimidines on Sulfonamide Containing Carbonic Anhydrase Inhibitor. Bioorg. Med. Chem. Lett. 2021, in press. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, Z.; Shi, J.; Liu, X.; Tang, W. Recent developments of synthesis and biological activity of sultone scaffolds in medicinal chemistry. Arab. J. Chem. 2021, 14, 103037. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 135464676. PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/135464676 (accessed on 4 September 2021).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 135464675. PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/135464675 (accessed on 4 September 2021).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 135473183, 2-[(E)-(2-Phenylpyrazol-3-yl)iminomethyl]phenol. PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/135473183 (accessed on 4 September 2021).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 54123178. PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/54123178 (accessed on 4 September 2021).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 54032820. PubChem. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/54032820 (accessed on 4 September 2021).
- Shetty, P.R.; Shivaraja, G.; Krishnaswamy, G.; Pruthviraj, K.; Mohan, V.C.; Sreenivasa, S. Pyrazole Schiff Bases: Synthesis, Characterization, Biological Screening, In Silico ADME and Molecular Docking Studies. Indian J. Het. Chem. 2020, 30, 123–130. [Google Scholar]
- Al-Azmi, A.; Mahmoud, H. Facile Synthesis and Antimicrobial Activities of Novel 1,4-Bis(3,5-dialkyl-4H-1,2,4-triazol-4-yl)benzene and 5-Aryltriaz-1-en-1-yl-1-phenyl-1H-pyrazole-4-carbonitrile Derivatives. ACS Omega 2020, 5, 10160–10166. [Google Scholar] [CrossRef] [Green Version]
- Karci, F.; Şener, N.; Yamaç, M.; Şener, I.; Demirçal, A. The synthesis, antimicrobial activity and absorption characteristics of some novel heterocyclic disazo dyes. Dyes Pigm. 2009, 80, 47–52. [Google Scholar] [CrossRef]
- Mukhtar, S.S.; Hassan, A.S.; Morsy, N.M.; Hafez, T.S.; Saleh, F.M.; Hassaneen, H.M. Design, synthesis, molecular prediction and biological evaluation of pyrazole-azomethine conjugates as antimicrobial agents. Synth. Commun. 2021, 51, 1564–1580. [Google Scholar] [CrossRef]
- Cuenú, F.; Londoño-Salazar, J.; Torres, J.E.; Abonia, R.; D’Vries, R.F. Synthesis, structural characterization and theoretical studies of a new Schiff base 4-(((3-(tert-Butyl)-(1-phenyl)pyrazol-5-yl) imino)methyl)phenol. J. Mol. Struct. 2018, 1152, 163–176. [Google Scholar] [CrossRef]
- Moreno-Fuquen, R.; Cuenú, F.; Torres, J.E.; De la Vega, G.; Galarza, E.; Abonia, R.; Kennedy, A.R. Presence of π…π and C H…π interactions in the new Schiff base 2-{(E)-[(3-tert-butyl-1-phenyl-1H-pyrazol-5-yl)imino]methyl}phenol: Experimental and DFT computational studies. J. Mol. Struct. 2017, 1150, 366–373. [Google Scholar] [CrossRef] [Green Version]
- Cuenú, F.; Restrepo-Acevedo, A.; Murillo, M.I.; Torres, J.E.; Moreno-Fuquen, R.; Abonia, R.; Kennedy, A.R.; Tenorio, J.C.; Lehmann, C.W. Synthesis, structural characterization, and theoretical studies of new pyrazole (E)-2-{[(5-(tert-butyl)-1H-pyrazol-3-yl)imino]methyl}phenol and (E)-2-{[(1-(4-bromophenyl)-3-(tert-butyl)-1H-pyrazol-5-yl] imino]methyl}phenol. J. Mol. Struct. 2019, 1184, 59–71. [Google Scholar] [CrossRef]
- Ozkınali, S.; Gür, M.; Şener, N.; Alkın, S.; Çavus, M.S. Synthesis of new azo schiff bases of pyrazole derivatives and their spectroscopic and theoretical investigations. J. Mol. Struct. 2018, 1174, 74–83. [Google Scholar] [CrossRef]
- Jamroz, M. Vibrational energy distribution analysis VEDA 4: Scopes and limitations. Spectrochim. Acta 2013, 114, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Cuenú, F.; Patiño, N.; Torres, J.; Abonia, R.; Toscano, R.; Cobo, J. The new 3-(tert- butyl)-1-(2-nitrophenyl)-1H-pyrazol-5-amine: Experimental and computational studies. J. Mol. Struct. 2017, 1148, 557–567. [Google Scholar] [CrossRef]
- Bader, R.F. Atom in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- CLSI, Clinical and Laboratory Standards Institute. Method M27-A3, 3rd ed.; CLSI: Wayne, PA, USA, 2008; Volume 28, pp. 1–25. [Google Scholar]
- Hargrove, T.Y.; Friggeri, L.; Wawrzak, Z.; Qi, A.; Hoekstra, W.J.; Schotzinger, R.J.; Lepesheva, G.I. Structural analyses of Candida albicans sterol 14α-demethylase complexed with azole drugs address the molecular basis of azole-mediated inhibition of fungal sterol biosynthesis. J. Biol. Chem. 2017, 292, 6728–6743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandeel, M.; Kitade, Y.; Al-Taher, A.; Al-Nazawi, M. The structural basis of unique substrate recognition by Plasmodium thymidylate kinase: Molecular dynamics simulation and inhibitory studies. PLoS ONE 2019, 14, e0212065. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.; Wang, W.; Fromm, H.; Honzatko, R.B. IMP Alone Organizes the Active Site of Adenylosuccinate Synthetase from Escherichia coli. J. Biol. Chem. 2001, 277, 5970–5976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, S.; Cuenú, F.; Abonia, R.; Cabrera, A. 3-tert-Butyl-1-(3-nitro phen yl)-1H-pyrazol-5-amine. Acta Cryst. E 2012, 68, o3171. [Google Scholar] [CrossRef] [PubMed]
- Quiroga, J.; Portilla, J.; Abonía, R.; Insuasty, B.; Nogueras, M.; Cobo, J. Synthesis of novel 5-amino-1-aroylpyrazoles. Tetrahedron Lett. 2008, 49, 5943–5945. [Google Scholar] [CrossRef]
- Choudhury, T.D.; Shen, Y.; Rao, N.V.; Clark, N.A. Dinuclear ortho-metallated palladium (II) azobenzene complexes with acetato and chloro bridges: Influence of polar substituents on the mesomorphic properties. J. Organomet. Chem. 2012, 712, 20–28. [Google Scholar] [CrossRef]
- Mahmoodi, N.; Ghavidast, A.; Mirkhaef, S.; Ali, M. Phorochromism of azobenzene-thiol-1,3-diazabicyclo-[3.1.0]hez-3-ene on silver nanoparticles. Dyes Pigm. 2015, 118, 110–117. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandenburg, K.; Putz, H. In diamond, impact, C. In Crystal Impact; Kreuzherrenstr. Bonn, Germany, 2006; Volume 102, p. 53227. [Google Scholar]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; Van De Streek, J.; Wood, P.A. Mercury CSD 2.0 e new features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision, D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Parr, R.G.; Weitao, Y. Density-Functional Theory of Atoms and Molecules; Oxford University Press: Oxford, UK, 1994; pp. 47–66. [Google Scholar]
- Dennington, R.; Keith, T.; Millam, J. GaussView 6.0.16; Semichem, Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Koopmans, T. About the assignment of wave functions and equities to the individual electronics of an atomic. Physica 1934, 1, 104–113. [Google Scholar] [CrossRef]
- Mulliken, R.S. A new electroaffinity scale; together with data on valence states and on valence ionization potentials and electron affinities. J. Chem. Phys. 1934, 2, 782–793. [Google Scholar] [CrossRef]
- Pearson, R.G. Absolute electronegativity and absolute hardness of Lewis acids and bases. J. Am. Chem. Soc. 1985, 107, 6801–6806. [Google Scholar] [CrossRef]
- Biegler-König, F.; Schönbohm, J. Update of the AIM2000-Program for atoms in molecules. J. Comput. Chem. 2002, 23, 1489–1494. [Google Scholar] [CrossRef] [PubMed]
- Sinha, K.; Rule, G.S. The structure of thymidylate kinase from Candida albicans reveals a unique structural element. Biochemistry 2017, 56, 4360–4370. [Google Scholar] [CrossRef] [PubMed]
- Sogabe, S.; Masubuchi, M.; Sakata, K.; Fukami, T.A.; Morikami, K.; Shiratori, Y.; Ohtsuka, Y.T. Crystal structures of Candida albicans N-Myristoyltransferase with two distinct inhibitors. Chem. Biol. 2002, 9, 1119–1128. [Google Scholar] [CrossRef] [Green Version]
- Hast, M.A.; Nichols, C.B.; Armstrong, S.M.; Kelly, S.M.; Hellinga, H.W.; Alspaugh, J.A.; Beese, L.S. Structures of Cryptococcus neoformans protein farnesyltransferase reveal strategies for developing inhibitors that target fungal pathogens. J. Biol. Chem. 2011, 286, 35149–35162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcyk, P.T.; LeBlanc, E.V.; Kuntz, D.A.; Xue, A.; Ortiz, F.; Trilles, R.; Bengtson, S.; Kenney, T.M.G.; Huang, D.S.; Robbins, N.; et al. Fungal-selective resorcylate aminopyrazole Hsp90 inhibitors: Optimization of whole-cell anticryptococcal activity and insights into the structural origins of cryptococcal selectivity. J. Med. Chem. 2021, 64, 1139–1169. [Google Scholar] [CrossRef] [PubMed]
- Blundell, R.D.; Williams, S.J.; Arras, S.D.M.; Chitty, J.L.; Blake, K.L.; Ericsson, D.J.; Fraser, J.A. Disruption of de novo adenosine triphosphate (ATP) biosynthesis abolishes virulence in Cryptococcus neoformans. ACS Infectious Diseases. 2016, 2, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koes, D.R.; Baumgartner, M.P.; Camacho, C.J. Lessons Learned in Empirical Scoring with Smina from the CSAR 2011 Benchmarking Exercise. J. Chem. Inf. Model. 2013, 53, 1893–1904. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins: Structure, Function, and Bioinformatics. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE - AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, P.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Alba-Ruiz, R. COVID-19, CORONAVIRUS PANDEMIC: Aproximación Bibliométrica y Revisión de los Resultados; Zenodo: Genève, Switzerland, 2020. [Google Scholar]
- Miller, B.R.; McGee, T.D.; Swails, J.M., Jr.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities, Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vibrational Mode | Azo1 | Azo2 | Azo3 | Azo4 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Exp | Theo | %PED 1 | Exp | Theo | %PED 1 | Exp | Theo | %PED 1 | Exp | Theo | %PED 1 | |
| ν(OH) | 3440 | 3152 | 99 | – | 3159 | 99 | – | 3157 | 99 | – | 3169 | 99 |
| ν(CH)pyrazole | 3127 | 3124 | 99 | 3122 | 3124 | 99 | 3123 | 3125 | 99 | 3120 | 3126 | 99 |
| ν(CH)aromatic | 3063 | 3068 | 98 | 3064 | 3067 | 98 | 3065 | 3068 | 98 | 3063 | 3068 | 98 |
| ν asym(CH3) | 2960 | 2968 | 98 | 2964 | 2974 | 97 | 2965 | 2965 | 99 | 2959 | 2966 | 93 |
| νsym(CH3) | 2862 | 2903 | 97 | 2861 | 2914 | 96 | 2861 | 2903 | 98 | 2863 | 2900 | 99 |
| ν(C=N)imine | 1598 | 1583 | 59 | 1604 | 1583 | 45 | 1604 | 1580 | 57 | 1605 | 1580 | 41 |
| νasym(NO2) | – | 1528 | 1523 | 57 | 1529 | 1525 | 75 | 1514 | 1515 | 55 | ||
| ν(N=N)azo | 1478 | 1477 | 52 | 1478 | 1483 | 57 | 1478 | 1483 | 65 | 1481 | 1483 | 33 |
| ρasym(CH3) | 1454 | 1441 | 79 | 1459 | 1440 | 84 | 1460 | 1445 | 81 | 1461 | 1461 | 83 |
| ν(CC)aromatic | 1435 | 1414 | 34 | 1432 | 1421 | 26 | 1433 | 1436 | 12 | 1435 | 1435 | 30 |
| ρsym(CH3) | 1358 | 1345 | 59 | 1362 | 1367 | 44 | 1363 | 1340 | 58 | 1367 | 1374 | 24 |
| νsym(NO2) | – | 1352 | 1320 | 67 | 1352 | 1315 | 56 | 1333 | 1307 | 46 | ||
| ν(CO) | 1281 | 1264 | 42 | 1285 | 1263 | 77 | 1286 | 1259 | 57 | 1279 | 1262 | 60 |
| Atom ID | Azo1 | Azo2 | Azo3 | Azo4 | ||||
|---|---|---|---|---|---|---|---|---|
| Exp | Theo 1 | Exp | Theo 2 | Exp | Theo 2 | Exp | Theo 2 | |
| H-2 | 12.58 | 12.47 | – | 12.10 | – | 12.56 | 11.87 | 12.47 |
| H-10 | 6.75 | 6.65 | 6.70 | 6.61 | 6.75 | 6.71 | 6.77 | 6.57 |
| H-tert-butyl | 1.39 | 1.72 | 1.25 | 1.61 | 1.32 | 1.64 | 1.35 | 1.60 |
| H-tert-butyl | 1.39 | 1.11 | 1.25 | 1.59 | 1.32 | 1.62 | 1.35 | 1.55 |
| H-tert-butyl | 1.39 | 1.46 | 1.25 | 1.58 | 1.32 | 1.60 | 1.35 | 1.55 |
| H-tert-butyl | 1.39 | 1.44 | 1.25 | 1.54 | 1.32 | 1.55 | 1.35 | 1.53 |
| H-tert-butyl | 1.39 | 1.31 | 1.25 | 1.31 | 1.32 | 1.39 | 1.35 | 1.36 |
| H-tert-butyl | 1.39 | 1.27 | 1.25 | 1.27 | 1.32 | 1.37 | 1.35 | 1.32 |
| H-tert-butyl | 1.39 | 1.16 | 1.25 | 1.25 | 1.32 | 1.19 | 1.35 | 1.16 |
| H-tert-butyl | 1.39 | 1.14 | 1.25 | 1.22 | 1.32 | 1.19 | 1.35 | 1.16 |
| H-tert-butyl | 1.39 | 1,12 | 1.25 | 1.07 | 1.32 | 1.16 | 1.35 | 1.14 |
| H-27 | 7.69 | 7.72 | NA | NA | 8.63 | 8.19 | 8.03 | 7.85 |
| H-29 | 7.56 | 7.65 | 8.08 | 8.29 | 7.55 | 7.79 | 8.37 | 8.65 |
| H-31 | 7.45 | 7.69 | 7.77 | 7.91 | 8.18 | 8.49 | NA | NA |
| H-33 | 7.56 | 7.87 | 7.68 | 8.16 | NA | NA | 8.37 | 8.69 |
| H-35 | 7.69 | 7.88 | 7.86 | 8.05 | 7.78 | 8.89 | 8.03 | 8.36 |
| H-37 | 9.26 | 9.20 | 9.16 | 9.07 | 9.21 | 9.25 | 9.23 | 9.20 |
| H-41 | 8.25 | 8.43 | 8.19 | 8.28 | 8.37 | 8.35 | 8.36 | 8.35 |
| H-44 | 8.06 | 8.34 | 7.93 | 8.34 | 7.96 | 8.41 | 7.99 | 8.37 |
| H-46 | 7.09 | 7.20 | 7.05 | 7.10 | 7.15 | 7.14 | 7.15 | 7.19 |
| H-49 | 7.91 | 8.28 | 7.80 | 8.43 | 7.57 | 8.33 | 7.85 | 8.34 |
| H-51 | 7.59 | 7.84 | 7.55 | 7.69 | 7.80 | 7.74 | 7.56 | 7.73 |
| H-53 | 7.54 | 7.78 | 7.52 | 7.68 | 8.21 | 7.79 | 7.53 | 7.75 |
| H-55 | 7.59 | 7.77 | 7.55 | 7.83 | 7.80 | 7.92 | 7.56 | 7.87 |
| H-57 | 7.91 | 8.30 | 7.80 | 8.29 | 7.57 | 8.27 | 7.85 | 8.33 |
| Atom ID | Azo1 | Azo2 | Azo3 | Azo4 | ||||
|---|---|---|---|---|---|---|---|---|
| Exp | Theo 1 | Exp | Theo 2 | Exp | Theo 2 | Exp | Theo 2 | |
| C-8 | 162.09 | 169.08 | 163.17 | 172.14 | 162.79 | 170.24 | 163.67 | 171.60 |
| C-9 | 91.12 | 95.52 | 91.58 | 97.91 | 92.77 | 96.94 | 93.54 | 99.21 |
| C-11 | 147.35 | 123.63 | 151.92 | 156.13 | 149.66 | 155.75 | 150.35 | 158.97 |
| C-12 | 34.68 | 38.80 | 32.40 | 39.24 | 32.87 | 39.38 | 32.87 | 39.18 |
| C-13 | 29.78 | 27.91 | 30.26 | 27,28 | 30.32 | 21.77 | 30.43 | 27.29 |
| C-17 | 29.78 | 30.39 | 30.26 | 31.32 | 30.32 | 31.01 | 30.43 | 31.10 |
| C-21 | 29.78 | 32.82 | 30.26 | 31.94 | 30.32 | 32.23 | 30.43 | 31.38 |
| C-25 | 139.33 | 146.53 | 145.38 | 139.78 | 139.76 | 147.23 | 144.44 | 152.68 |
| C-26 | 124.95 | 129.85 | 149.27 | 153.7 | 130.95 | 137.24 | 124.1 | 127.14 |
| C-28 | 129.21 | 132.82 | 125.39 | 131.70 | 131.41 | 134.17 | 125.07 | 130.11 |
| C-30 | 127.62 | 132.08 | 129.48 | 133.15 | 121.44 | 126.65 | 145.49 | 152.17 |
| C-32 | 129.21 | 134.15 | 125.68 | 141.07 | 147.97 | 155.75 | 125.07 | 131.04 |
| C-34 | 124.95 | 129.68 | 134.06 | 134.19 | 118.25 | 124.88 | 124.1 | 128.04 |
| C-36 | 164.03 | 167.11 | 161.18 | 170.19 | 162.2 | 167.77 | 161.25 | 169.36 |
| C-38 | 163.12 | 171.70 | 162.29 | 172.04 | 160.14 | 172.24 | 162.56 | 172.60 |
| C-39 | 119.17 | 124.62 | 120.59 | 123.07 | 120.96 | 123.90 | 121.41 | 123.46 |
| C-40 | 128.42 | 122.93 | 125.92 | 145.62 | 125.21 | 145.37 | 125.62 | 145.85 |
| C-42 | 145.69 | 151.65 | 145.12 | 152.22 | 145.08 | 152.04 | 145.64 | 152.17 |
| C-43 | 127.49 | 145.25 | 128.00 | 125.64 | 128.24 | 125.43 | 128.35 | 125.86 |
| C-45 | 117.76 | 121.24 | 118.09 | 122.05 | 118.23 | 122.33 | 118.21 | 122.56 |
| C-47 | 152.87 | 158.53 | 153.51 | 159.13 | 151.93 | 158.73 | 152.35 | 158.97 |
| C-48 | 122.44 | 139.40 | 122.68 | 117.85 | 129.94 | 117.32 | 122.82 | 117.48 |
| C-50 | 129.02 | 134.21 | 129.89 | 134.27 | 130.89 | 133.73 | 129.9 | 134.09 |
| C-52 | 130.54 | 137.40 | 131.53 | 137.18 | 129.84 | 137.48 | 131.55 | 137.42 |
| C-54 | 129.02 | 133.53 | 129.89 | 134.55 | 130.89 | 134.24 | 129.9 | 134.09 |
| C-56 | 122.44 | 117.47 | 122.68 | 139.78 | 129.94 | 139.21 | 122.82 | 117.48 |
| Molecule | (3,−3) | (3,−1) | (3,+1) | (3,+3) | Total | PHR |
|---|---|---|---|---|---|---|
| Azo1 | 57 | 61 | 5 | 0 | 123 | 1 |
| Azo2 | 59 | 66 | 9 | 1 | 135 | 1 |
| Azo3 | 59 | 64 | 6 | 0 | 129 | 1 |
| Azo4 | 59 | 64 | 6 | 0 | 129 | 1 |
| Molecule | C. albicans | C. neoformans | ||
|---|---|---|---|---|
| CaTMPK | CaNmT | CnFTase | CnAdSS | |
| Azo1 | −35.882 | −33.111 | −15.183 | −31.599 |
| Azo2 | −37.317 | −33.627 | −15.484 | −33.300 |
| Azo3 | −48.055 | −43.832 | −19.861 | −41.357 |
| Azo4 | −37.658 | −32.734 | −12.993 | −42.273 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandez, D.; Restrepo-Acevedo, A.; Rocha-Roa, C.; Le Lagadec, R.; Abonia, R.; Zacchino, S.A.; Gómez Castaño, J.A.; Cuenú-Cabezas, F. Synthesis, Structural Characterization, and In Vitro and In Silico Antifungal Evaluation of Azo-Azomethine Pyrazoles (PhN2(PhOH)CHN(C3N2(CH3)3)PhR, R = H or NO2). Molecules 2021, 26, 7435. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26247435
Fernandez D, Restrepo-Acevedo A, Rocha-Roa C, Le Lagadec R, Abonia R, Zacchino SA, Gómez Castaño JA, Cuenú-Cabezas F. Synthesis, Structural Characterization, and In Vitro and In Silico Antifungal Evaluation of Azo-Azomethine Pyrazoles (PhN2(PhOH)CHN(C3N2(CH3)3)PhR, R = H or NO2). Molecules. 2021; 26(24):7435. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26247435
Chicago/Turabian StyleFernandez, Dorancelly, Andrés Restrepo-Acevedo, Cristian Rocha-Roa, Ronan Le Lagadec, Rodrigo Abonia, Susana A. Zacchino, Jovanny A. Gómez Castaño, and Fernando Cuenú-Cabezas. 2021. "Synthesis, Structural Characterization, and In Vitro and In Silico Antifungal Evaluation of Azo-Azomethine Pyrazoles (PhN2(PhOH)CHN(C3N2(CH3)3)PhR, R = H or NO2)" Molecules 26, no. 24: 7435. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules26247435