
Probable Mechanisms of Doxorubicin Antitumor Activity Enhancement by Ginsenoside Rh2

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Enhanced Anti-Tumor Effects of DOX and Rh2 in a Solid Tumor Model
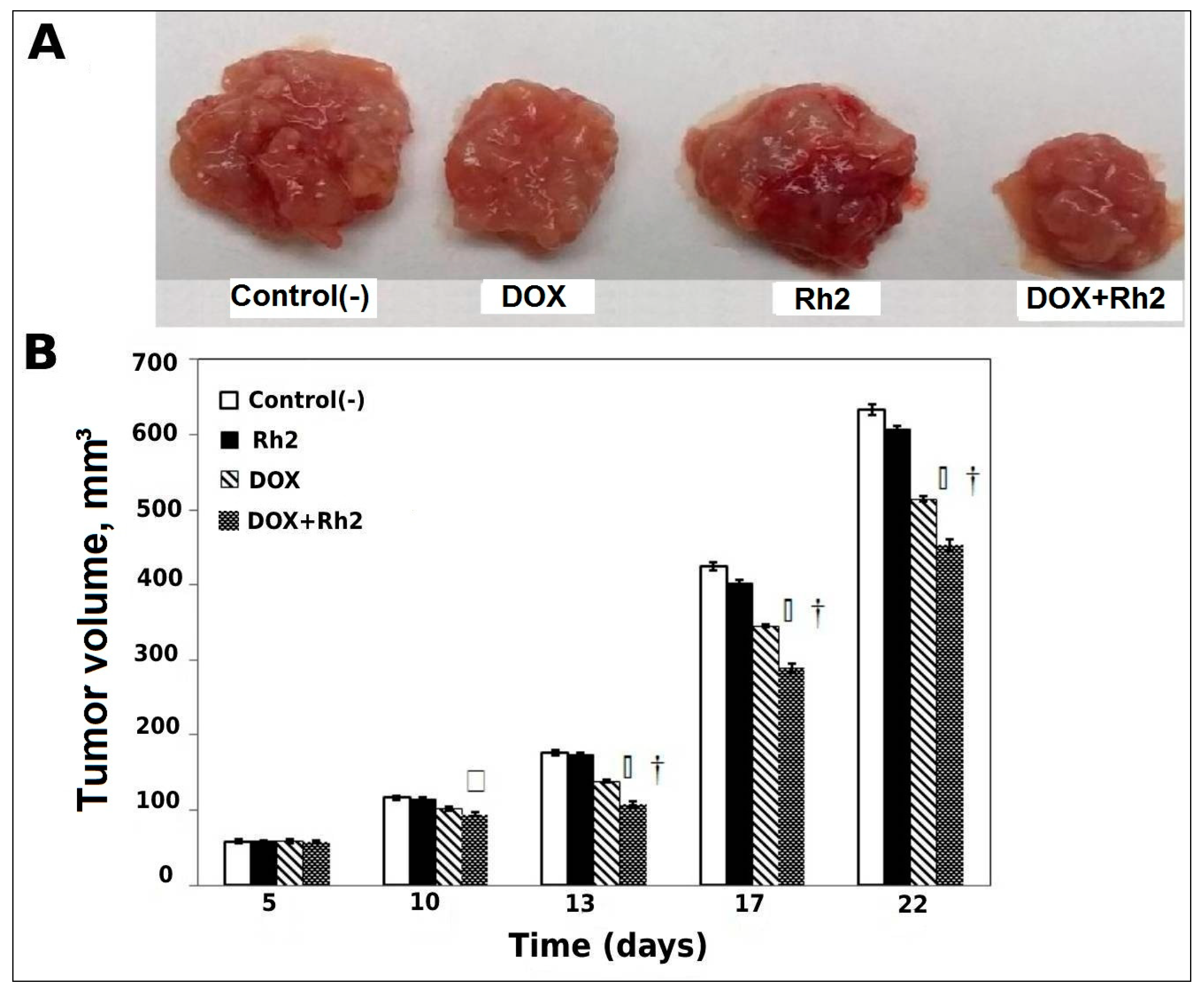
2.1.1. Solid Tumor Model—Delayed Treatment (Variant A, Post-Tumor Formation)
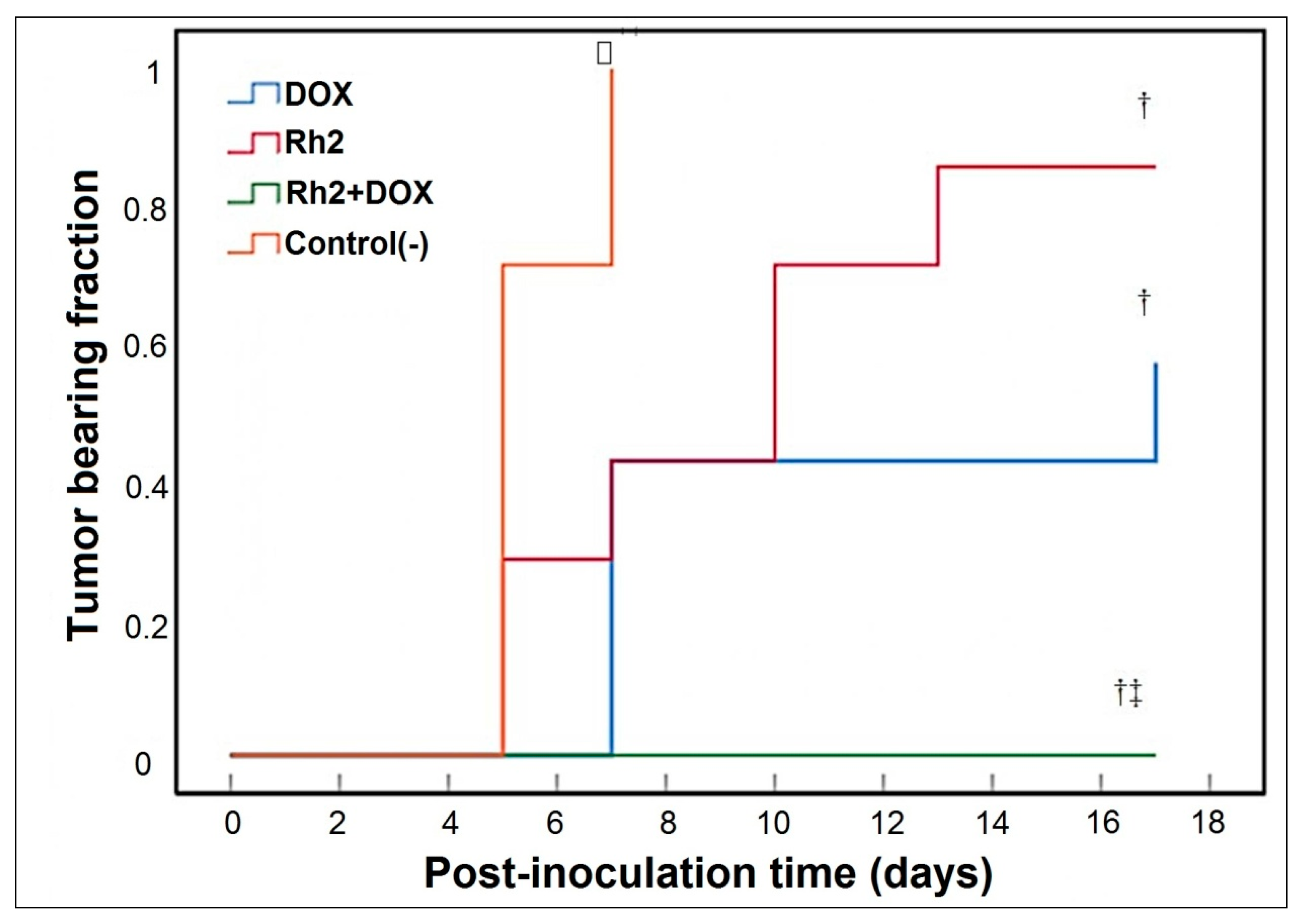
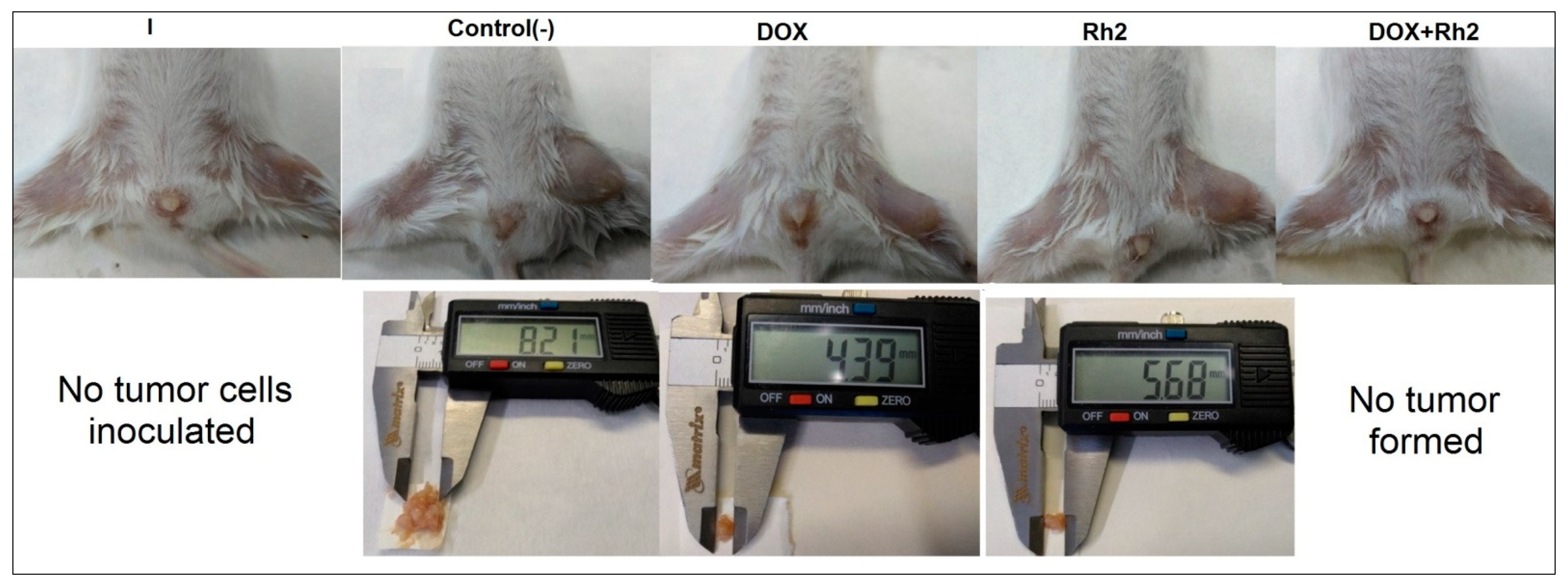
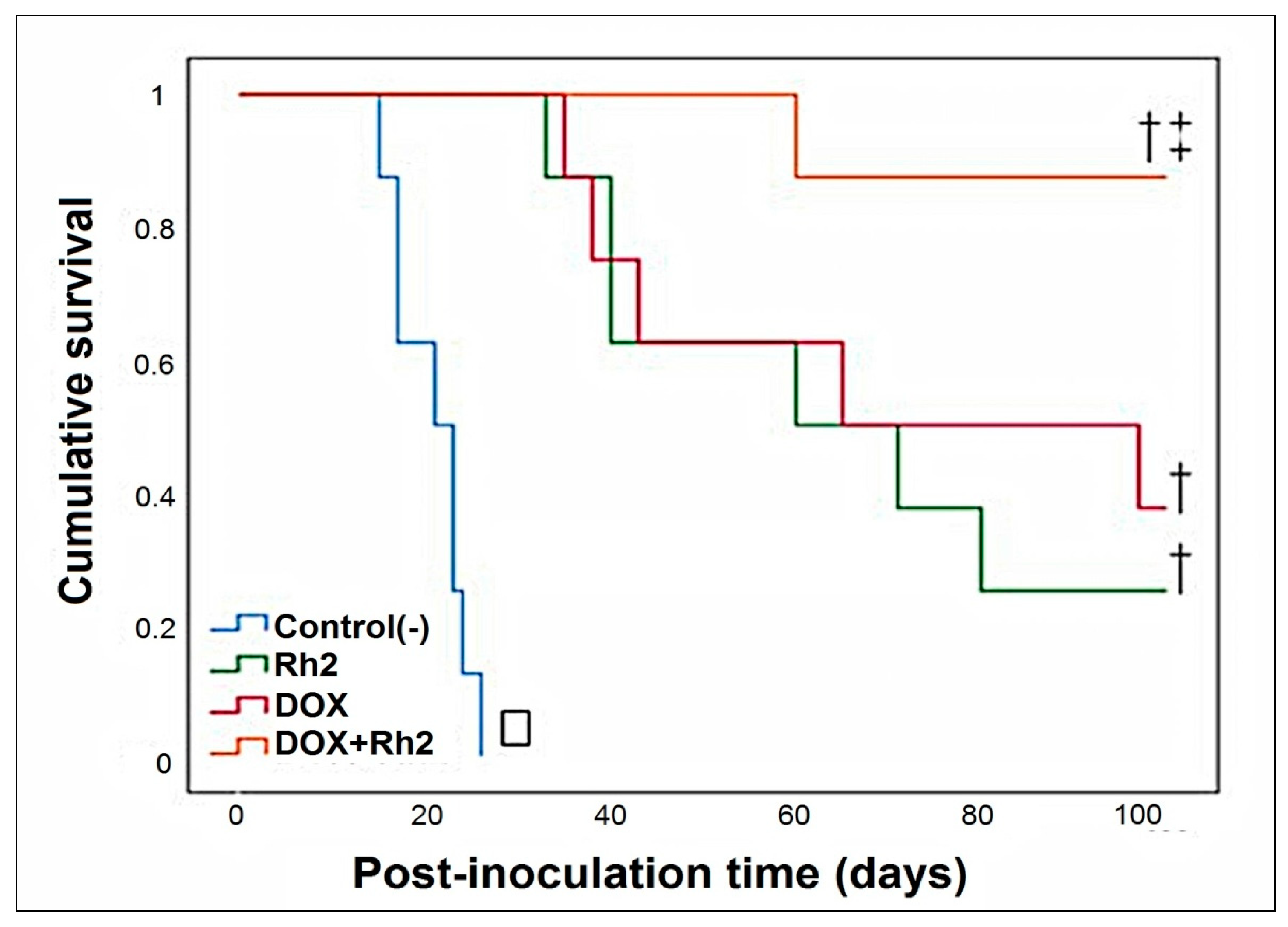
2.1.2. Solid Tumor Model—Early Treatment (Variant B; 24 h after Inoculation of Tumor Cells)
2.2. Enhanced Anti-Tumor Effects of DOX and Rh2 in the Ascites Model
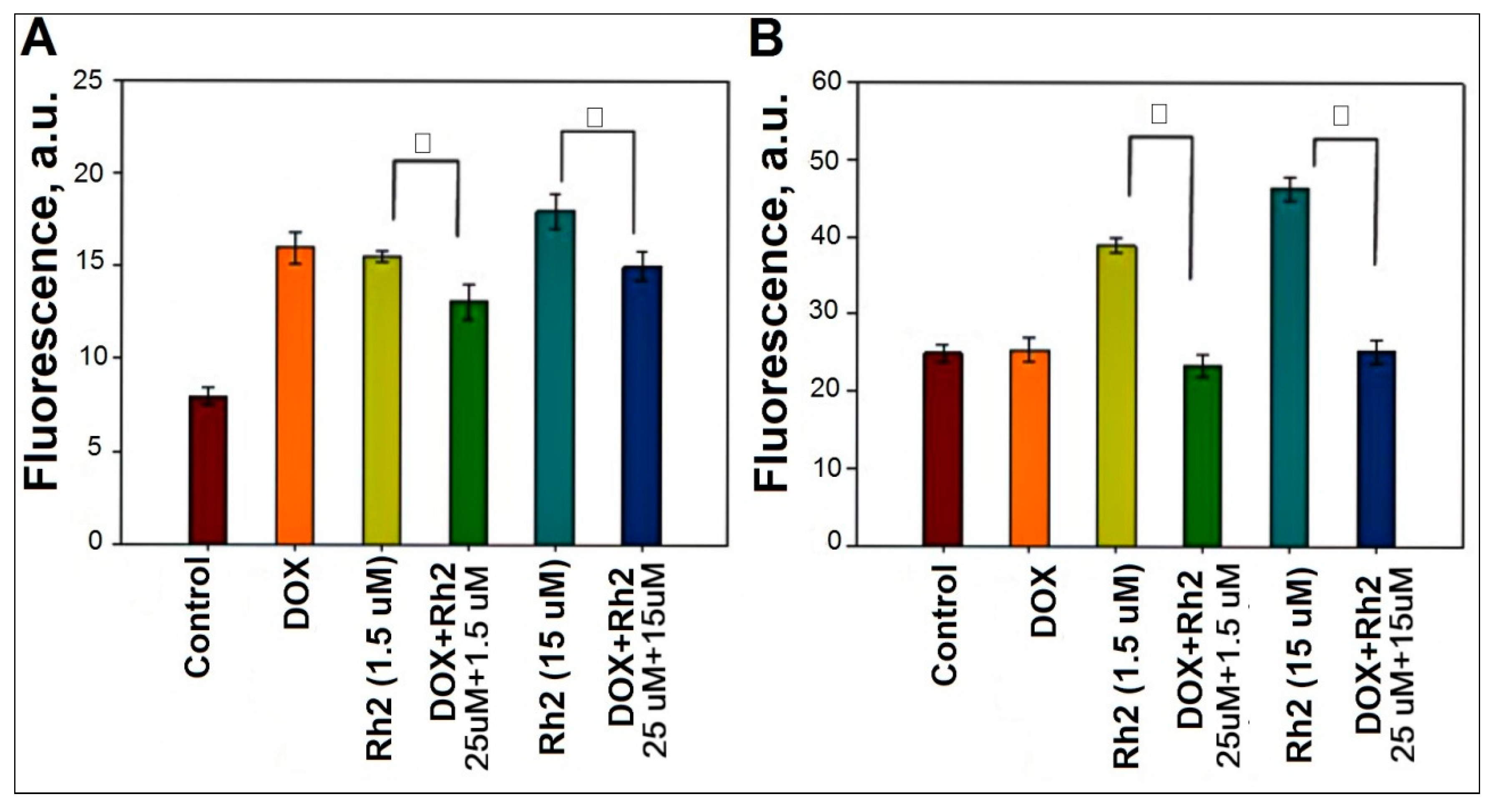
2.3. Effect of Rh2 on ROS Production in Primary Cell Cultures of Adenocarcinoma and Splenocytes
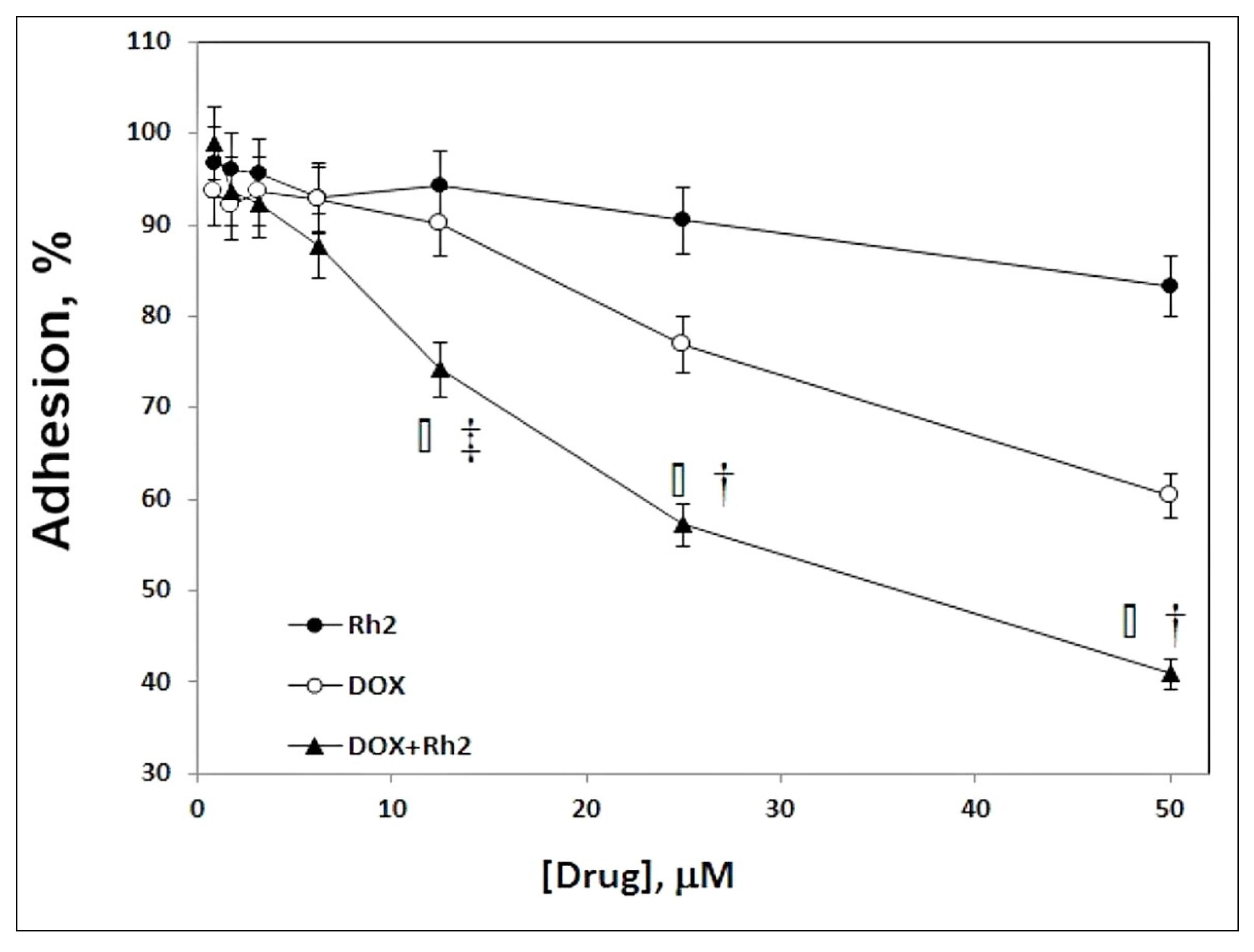
2.4. Effect of DOX + Rh2 Co-Treatment on Primary Adenocarcinoma Cell Adhesion
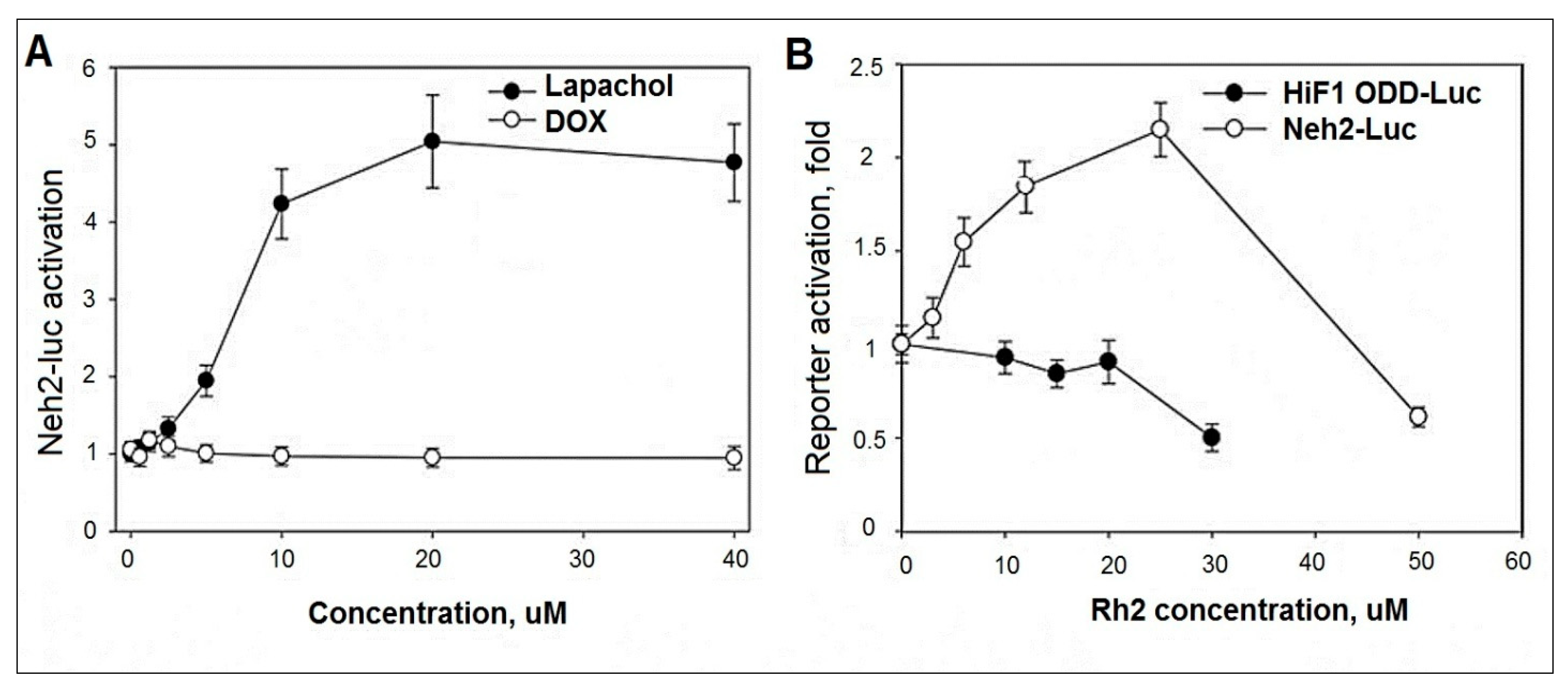
2.5. Evaluation of DOX and Rh2 Activity in Neh2-luc Reporter Assay
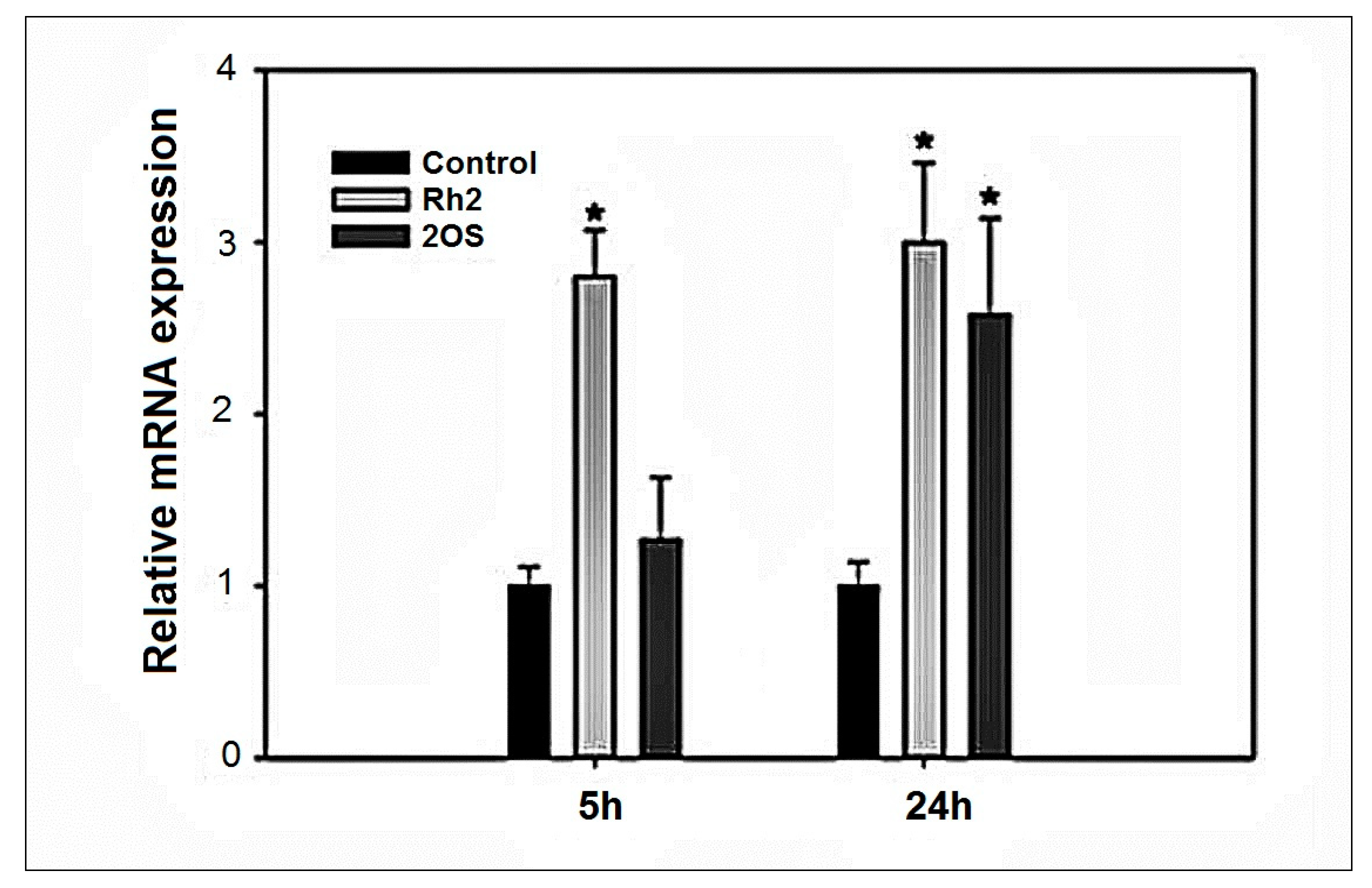
2.6. HMOX1 Expression Induction by Rh2 and 20S-Protopanaxadiol
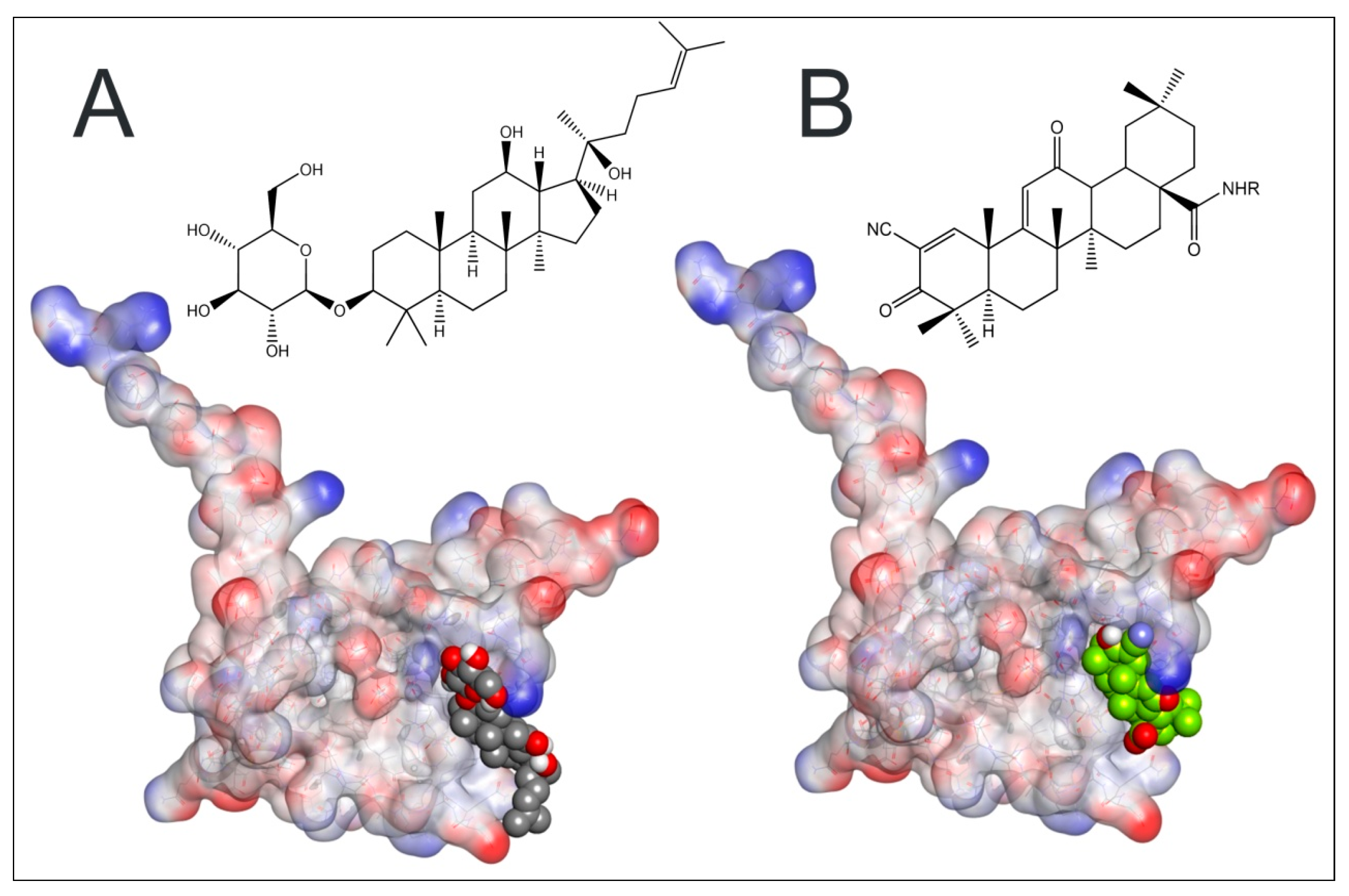
2.7. Plausible Interaction of Rh2 with BTB-Domain of Keap 1
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. HIF1 ODD-luc and Neh2-luc Reporter Assays
4.3. RT-PCR Analysis
4.4. Fluorescent Assay for ROS in the Cell Culture
4.5. Adhesion Test
4.6. Animals and Ethics Approval
4.7. Murine Model of Solid Ehrlich’s Adenocarcinoma
4.7.1. Variant A (Delayed Treatment—After Tumor Formation)
4.7.2. Variant B (Early—Next Day Treatment)
4.8. Murine Model of Ascites Ehrlich’s Adenocarcinoma
4.9. Computer Modeling
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Zhu, H.; Sarkar, S.; Scott, L.; Danelisen, I.; Trush, M.A.; Jia, Z.; Li, Y.R. Doxorubicin Redox Biology: Redox Cycling, Topoisomerase Inhibition, and Oxidative Stress. React. Oxyg. Species 2016, 1, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Ajaykumar, C. Overview on the Side Effects of Doxorubicin. In Overview of Doxorubicin-Clinical Use, Resistance, Side Effects, and Palliative Care [Working Title]; IntechOpen: London, UK, 2020; p. 20. [Google Scholar]
- Tsybulsky, A.V.; Popov, A.M.; Klimovich, A.; Artyukov, A.A.; Kostetsky, E.Y.; Veselova, M.D. Comparative study of echinochrome a, oxygenated carotenoids, ginsenoside Rh2, luteolin disulfate and metformin as a mean to potentiate antitumor effect of doxorubicin. Med. Immunol. 2018, 20, 179–192. [Google Scholar] [CrossRef] [Green Version]
- Razina, T.G.; Krylova, S.G.; Amosova, E.N.; Zueva, E.P.; Lopatina, K.A.; Popov, A.M.; Atopkina, L.N.; Kozlovskaia, E.P. Effect of gin-senoside Rh2 on the development of transferred tumors and chemotherapy efficiency. Eksp. Klin. Farmakol. 2010, 73, 27–30. [Google Scholar]
- Popov, A.M.; Atopkina, L.N.; Uvarova, N.I.; Elyakov, G.B. The antimetastatic and immunomodulating activities of ginseng minor glycosides. Dokl. Biochem. Biophys. 2001, 380, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Popov, A.M.; Atopkina, L.N.; Samoshina, N.F.; Uvarova, N.I. Immunomodulating activity of tetracyclic triterpene glycosides of the dammarane and holostane series. Antibiot. Khimioterapiia Antibiot. Chemoterapy [Sic] 1994, 39, 19–25. [Google Scholar]
- Yang, Z.; Zhao, T.; Liu, H.; Zhang, L. Ginsenoside Rh2 inhibits hepatocellular carcinoma through β-catenin and autophagy. Sci. Rep. 2016, 6, 183–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, T.; Wang, J.; Wang, Y.; Wang, Y.; Cai, J.; Wang, M.; Chen, Q.; Song, J.; Yu, Z.; Huang, W.; et al. Inhibition of autophagy potentiates anticancer property of 20(S)-ginsenoside Rh2 by promoting mitochondria-dependent apoptosis in human acute lymphoblastic leukaemia cells. Oncotarget 2016, 7, 27336–27349. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Yan, S.-J.; Zhang, H.-T.; Li, N.; Liu, T.; Zhang, Y.-L.; Li, X.-X.; Ma, Q.; Qiu, X.-C.; Fan, Q.-Y.; et al. Ginsenoside Rh2 enhances the antitumor immunological response of a melanoma mice model. Oncol. Lett. 2017, 13, 681–685. [Google Scholar] [CrossRef]
- Pink, J.J.; Planchon, S.M.; Tagliarino, C.; Varnes, M.E.; Siegel, D.; Boothman, D.A. NAD(P)H:Quinone Oxidoreductase Activity Is the Principal Determinant of β-Lapachone Cytotoxicity. J. Biol. Chem. 2000, 275, 5416–5424. [Google Scholar] [CrossRef] [Green Version]
- Sodrul, I.M.; Wang, C.; Chen, X.; Du, J.; Sun, H. Role of ginsenosides in reactive oxygen species-mediated anticancer therapy. Oncotarget 2017, 9, 2931–2950. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Chen, C. Ginseng Compounds: An Update on their Molecular Mechanisms and Medical Applications. Curr. Vasc. Pharmacol. 2009, 7, 293–302. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Muramatsu, A.; Saito, R.; Iso, T.; Shibata, T.; Kuwata, K.; Kawaguchi, S.-I.; Iwawaki, T.; Adachi, S.; Suda, H.; et al. Molecular Mechanism of Cellular Oxidative Stress Sensing by Keap1. Cell Rep. 2019, 28, 746–758.e4. [Google Scholar] [CrossRef] [Green Version]
- Saw, C.L.L.; Yang, A.Y.; Cheng, D.C.; Boyanapalli, S.S.-S.; Su, Z.-Y.; Khor, T.O.; Gao, S.; Wang, J.; Jiang, Z.-H.; Kong, A.-N.T. Pharmacodynamics of Ginsenosides: Antioxidant Activities, Activation of Nrf2, and Potential Synergistic Effects of Combinations. Chem. Res. Toxicol. 2012, 25, 1574–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, C.; Li, B.; Lai, Y.; Li, H.; Windust, A.; Hofseth, L.J.; Nagarkatti, M.; Nagarkatti, P.; Wang, X.L.; Tang, D.; et al. Identifying panaxynol, a natural activator of nuclear factor erythroid-2 related factor 2 (Nrf2) from American ginseng as a suppressor of inflamed macrophage-induced cardiomyocyte hypertrophy. J. Ethnopharmacol. 2015, 168, 326–336. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Liu, D.; He, C.; Li, X.; He, F. Inhibiting adhesion events by Panax notoginseng saponins and Ginsenoside Rb1 protecting arteries via activation of Nrf2 and suppression of p38–VCAM-1 signal pathway. J. Ethnopharmacol. 2016, 192, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, D.; Wu, J.; Zhang, D.; Cheng, B.; Zhang, Y.; Yin, Z.; Wang, Y.; Du, J.; Ling, C. Ginsenoside Rg1 attenuates ultraviolet B-induced glucocortisides resistance in keratinocytes via Nrf2/HDAC2 signalling. Sci. Rep. 2016, 6, 39336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Xiang, Y.; Chen, Y.; Tang, Y.; Zhang, Y. Ginsenoside Rg1 Protects Cardiomyocytes Against Hypoxia/Reoxygenation Injury via Activation of Nrf2/HO-1 Signaling and Inhibition of JNK. Cell. Physiol. Biochem. 2017, 44, 21–37. [Google Scholar] [CrossRef]
- Zhai, Y.; Meng, X.; Luo, Y.; Wu, Y.; Ye, T.; Zhou, P.; Ding, S.; Wang, M.; Lu, S.; Zhu, L.; et al. Notoginsenoside R1 ameliorates diabetic encephalopathy by activating the Nrf2 pathway and inhibiting NLRP3 inflammasome activation. Oncotarget 2018, 9, 9344–9363. [Google Scholar] [CrossRef] [Green Version]
- Chang, T.-L.; Huang, Y.-H.; Ou, Y.-D. The role of ginsenosides in inhibiting ubiquitin activating enzyme (E1) activity. J. Funct. Foods 2014, 7, 462–470. [Google Scholar] [CrossRef]
- Shim, S.H.; Baek, K.; Kim, Y. Inhibition of Human 20S Proteasome by Ginsenosides from Panax ginseng. Bull. Korean Chem. Soc. 2009, 30, 1385–1387. [Google Scholar] [CrossRef] [Green Version]
- Kaidery, N.A.; Banerjee, R.; Yang, L.; Smirnova, N.A.; Hushpulian, D.M.; Liby, K.; Williams, C.R.; Yamamoto, M.; Kensler, T.W.; Ratan, R.R.; et al. Targeting Nrf2-Mediated Gene Transcription by Extremely Potent Synthetic Triterpenoids Attenuate Dopaminergic Neurotoxicity in the MPTP Mouse Model of Parkinson’s Disease. Antioxid. Redox Signal. 2013, 18, 139–157. [Google Scholar] [CrossRef] [Green Version]
- Park, E.K.; Lee, E.J.; Lee, S.H.; Koo, K.H.; Sung, J.Y.; Hwang, E.H.; Park, J.H.; Kim, C.W.; Jeong, K.C.; Park, B.K.; et al. Induction of apoptosis by the ginsenoside Rh2 by internalization of lipid rafts and caveolae and inactivation of Akt. Br. J. Pharmacol. 2010, 160, 1212–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, P.M.R.; Mo, H.; McConathy, W.J.; Sabnis, N.; Lacko, A.G. The role of cholesterol metabolism and cholesterol transport in carcinogenesis: A review of scientific findings, relevant to future cancer therapeutics. Front. Pharmacol. 2013, 4, 119. [Google Scholar] [CrossRef] [Green Version]
- Verstraeten, S.L. Role of Membrane Cholesterol in the Apoptosis Induced by Ginsenoside Rh2, A Steroid Saponin. Biophys. J. 2018, 114, 271a. [Google Scholar] [CrossRef]
- Popov, A.M. Comparative study of cytotoxic and hemolytic effects of triterpenoids isolated from Ginseng and Sea cucumber. Biol. Bull. 2002, 2, 155–164. [Google Scholar]
- Likhatskaya, G.N.; Popov, A.M.; Odinokova, L.E.; Atopkina, L.I.; Uvarova, N.I.; Kuznetsova, T.A.; Anisimov, M.M. The effect of free triterpenoids on the properties of model lipid membranes. Izv. AN SSSR Ser. Biol. 1992, 6, 942–946. [Google Scholar]
- Verstraeten, S.L.; Albert, M.; Paquot, A.; Muccioli, G.G.; Tyteca, D.; Mingeot-Leclercq, M.-P. Membrane cholesterol delays cellular apoptosis induced by ginsenoside Rh2, a steroid saponin. Toxicol. Appl. Pharmacol. 2018, 352, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Verstraeten, S.L.; Deleu, M.; Janikowska-Sagan, M.; Claereboudt, E.J.S.; Lins, L.; Tyteca, D.; Mingeot-Leclercq, M.-P. The activity of the saponin ginsenoside Rh2 is enhanced by the interaction with membrane sphingomyelin but depressed by cholesterol. Sci. Rep. 2019, 9, 7285. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.-S.; Choo, H.-J.; Cho, B.-R.; Kim, H.-M.; Kim, Y.-N.; Ham, Y.-M.; Ko, Y.-G. Ginsenoside Rh2 induces ligand-independent Fas activation via lipid raft disruption. Biochem. Biophys. Res. Commun. 2009, 385, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.; Kwak, H.B.; Lee, S.K.; Na, D.S.; Rudd, C.E.; Lee, Z.H.; Kim, H.-H. Membrane Rafts Play a Crucial Role in Receptor Activator of Nuclear Factor κB Signaling and Osteoclast Function. J. Biol. Chem. 2003, 278, 18573–18580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murai, T. The Role of Lipid Rafts in Cancer Cell Adhesion and Migration. Int. J. Cell Biol. 2011, 2012, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murai, T.; Maruyama, Y.; Mio, K.; Nishiyama, H.; Suga, M.; Sato, C. Low Cholesterol Triggers Membrane Microdomain-dependent CD44 Shedding and Suppresses Tumor Cell Migration. J. Biol. Chem. 2011, 286, 1999–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zhou, F.; Wu, X.; Zhang, X.; Chen, Y.; Zha, B.S.; Niu, F.; Lu, M.; Hao, G.; Sun, Y.; et al. Cellular pharmacokinetic mechanisms of adriamycin resistance and its modulation by 20(S)-ginsenoside Rh2 in MCF-7/Adr cells. Br. J. Pharmacol. 2012, 165, 120–134. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhou, F.; Wu, X.; Gu, Y.; Ai, H.; Zheng, Y.; Li, Y.; Zhang, X.; Hao, G.; Sun, J.; et al. 20(S)-Ginsenoside Rh2 Noncompetitively Inhibits P-Glycoprotein In Vitro and In Vivo: A Case for Herb-Drug Interactions. Drug Metab. Dispos. 2010, 38, 2179–2187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Cai, Q.; Wang, W.; Lu, M.; Liu, J.; Zhou, F.; Sun, M.; Wang, G.; Zhang, J. Ginsenoside Rh2 pretreatment and withdrawal reactivated the pentose phosphate pathway to ameliorate intracellular redox disturbance and promoted intratumoral penetration of adriamycin. Redox Biol. 2020, 32, 101452. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-S.; Lin, Y.; Li, H.; Li, Y.; Song, Z.; Jin, Y.-H. The identification of molecular target of (20S) ginsenoside Rh2 for its anti-cancer activity. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.-P.; Tang, G.-D.; Fang, C.-Y.; Liang, Z.-H.; Zhang, L.-Y. Effects of ginsenoside Rh2 on growth and migration of pancreatic cancer cells. World J. Gastroenterol. 2013, 19, 1582–1592. [Google Scholar] [CrossRef]
- Christensen, M.V.; Høgdall, C.K.; Jochumsen, K.M.; Høgdall, E.V. Annexin A2 and cancer: A systematic review. Int. J. Oncol. 2017, 52, 5–18. [Google Scholar] [CrossRef]
- Wang, Y.-S.; Li, H.; Li, Y.; Zhu, H.; Jin, Y.-H. Identification of natural compounds targeting Annexin A2 with an anti-cancer effect. Protein Cell 2018, 9, 568–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Cao, Y.; Zheng, L.; Lv, D.; Chen, L.; Xing, X.; Zhu, Z.; Li, X.; Chai, Y. Identification of Annexin A2 as a target protein for plant alkaloid matrine. Chem. Commun. 2017, 53, 5020–5023. [Google Scholar] [CrossRef]
- Staquicini, D.I.; Rangel, R.; Guzman-Rojas, L.; Staquicini, F.I.; Dobroff, A.S.; Tarleton, C.A.; Ozbun, M.; Kolonin, M.G.; Gelovani, J.G.; Marchiò, S.; et al. Intracellular targeting of annexin A2 inhibits tumor cell adhesion, migration, and in vivo grafting. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Wang, L.; Liu, J.; Liu, L.; Huang, J.; Chen, X.; Luo, Z. MicroRNA-206 regulates the epithelial-mesenchymal transition and inhibits the invasion and metastasis of prostate cancer cells by targeting Annexin A2. Oncol. Lett. 2018, 15, 8295–8302. [Google Scholar] [CrossRef]
- Li, B.; Zhao, J.; Wang, C.-Z.; Searle, J.; He, T.-C.; Yuan, C.-S.; Du, W. Ginsenoside Rh2 induces apoptosis and paraptosis-like cell death in colorectal cancer cells through activation of p53. Cancer Lett. 2011, 301, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.; Li, J.; Feng, Z.; Zhao, L.; Luo, L.; You, Z.; Li, D.; Xia, J.; Zuo, G.; Chen, D. Effect of ginsenoside Rh2 on the migratory ability of HepG2 liver carcinoma cells: Recruiting histone deacetylase and inhibiting activator protein 1 transcription factors. Mol. Med. Rep. 2014, 10, 1779–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, E.-A.; Kim, E.-J.; Park, J.-S.; Kim, H.-S.; Ryu, J.; Kim, N.-H. Ginsenosides Rg3 and Rh2 Inhibit the Activation of AP-1 and Protein Kinase A Pathway in Lipopolysaccharide/Interferon-γ-Stimulated BV-2 Microglial Cells. Planta Med. 2006, 72, 627–633. [Google Scholar] [CrossRef]
- Su, S.-C.; Maxwell, S.A.; Bayless, K.J. Annexin 2 Regulates Endothelial Morphogenesis by Controlling AKT Activation and Junctional Integrity. J. Biol. Chem. 2010, 285, 40624–40634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherukuri, A.; Dykstra, M.; Pierce, S.K. Floating the Raft Hypothesis: Lipid Rafts Play a Role in Immune Cell Activation. Immunity 2001, 14, 657–660. [Google Scholar] [CrossRef] [Green Version]
- Mills, C.D. M1 and M2 Macrophages: Oracles of Health and Disease. Crit. Rev. Immunol. 2012, 32, 463–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karnevi, E.; Andersson, R.; Rosendahl, A.H. Tumour-educated macrophages display a mixed polarisation and enhance pancreatic cancer cell invasion. Immunol. Cell Biol. 2014, 92, 543–552. [Google Scholar] [CrossRef] [Green Version]
- Pollard, J.W. Tumour-educated macrophages promote tumour progression and metastasis. Nat. Cancer 2004, 4, 71–78. [Google Scholar] [CrossRef]
- Li, H.; Huang, N.; Zhu, W.; Wu, J.; Yang, X.; Teng, W.; Tian, J.; Fang, Z.; Luo, Y.; Chen, M.; et al. Modulation the crosstalk between tumor-associated macrophages and non-small cell lung cancer to inhibit tumor migration and invasion by ginsenoside Rh2. BMC Cancer 2018, 18, 579. [Google Scholar] [CrossRef] [PubMed]
- Ohradanova-Repic, A.; Machacek, C.; Charvet, C.; Lager, F.; Le Roux, D.; Platzer, R.; Leksa, V.; Mitulović, G.; Burkard, T.; Zlabinger, G.J.; et al. Extracellular Purine Metabolism Is the Switchboard of Immunosuppressive Macrophages and a Novel Target to Treat Diseases With Macrophage Imbalances. Front. Immunol. 2018, 9, 852. [Google Scholar] [CrossRef] [PubMed]
- Savio, L.E.B.; de Andrade Mello, P.; Da Silva, C.G.; Coutinho-Silva, R. The P2X7 Receptor in Inflammatory Diseases: Angel or Demon? Front. Pharmacol. 2018, 9, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Virgilio, F.; Adinolfi, E. Extracellular purines, purinergic receptors and tumor growth. Oncogene 2017, 36, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Helliwell, R.M.; ShioukHuey, C.O.; Dhuna, K.; Molero, J.C.; Ye, J.-M.; Xue, C.; Stokes, L. Selected ginsenosides of the protopanaxdiol series are novel positive allosteric modulators of P2X7 receptors. Br. J. Pharmacol. 2015, 172, 3326–3340. [Google Scholar] [CrossRef]
- Kitamura, H.; Motohashi, H. NRF2 addiction in cancer cells. Cancer Sci. 2018, 109, 900–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bose, C.; Awasthi, S.; Sharma, R.; Beneš, H.; Hauer-Jensen, M.; Boerma, M.; Singh, S.P. Sulforaphane potentiates anticancer effects of doxorubicin and attenuates its cardiotoxicity in a breast cancer model. PLoS ONE 2018, 13, e0193918. [Google Scholar] [CrossRef]
- Yang, Y.; Luo, L.; Cai, X.; Fang, Y.; Wang, J.; Chen, G.; Yang, J.; Zhou, Q.; Sun, X.; Cheng, X.; et al. Nrf2 inhibits oxaliplatin-induced peripheral neuropathy via protection of mitochondrial function. Free Radic. Biol. Med. 2018, 120, 13–24. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, Z.; Du, G.; Sun, H.; Liu, H.; Zhou, Z.; Gou, X.; Wu, X.; Yu, X.; Huang, Y. Nrf2 promotes breast cancer cell migration via up-regulation of G6PD/HIF-1α/Notch1 axis. J. Cell. Mol. Med. 2019, 23, 3451–3463. [Google Scholar] [CrossRef] [Green Version]
- Popov, A.M. Antitumor and antimetastatic activities monoglycosides of ginseng: Modern conception. Russ. J. Biopharm. 2011, 3, 3–8. [Google Scholar]
- Klimovich, A.A.; Popov, A.M.; Gazaryan, I.G.; Styshova, O.N.; Tsybulsky, A.V.; Veselova, M.D. The peculiarities of biological activity ginsenoside Rh2 and prospects of its application in the treatment oncological diseases. Russ. J. Biopharm. 2019, 11, 24–38. [Google Scholar]
- Atopkina, L.N.; Uvarova, N.I.; Elyakov, G.B. Simplified preparation of the ginsenoside-Rh2 minor saponin from ginseng. Carbohydr. Res. 1997, 303, 449–451. [Google Scholar] [CrossRef]
- Smirnova, N.A.; Haskew-Layton, R.E.; Basso, M.; Hushpulian, D.M.; Payappilly, J.B.; Speer, R.E.; Ahn, Y.-H.; Rakhman, I.; Cole, P.A.; Pinto, J.T.; et al. Development of Neh2-Luciferase Reporter and Its Application for High Throughput Screening and Real-Time Monitoring of Nrf2 Activators. Chem. Biol. 2011, 18, 752–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smirnova, N.A.; Rakhman, I.; Moroz, N.; Basso, M.; Payappilly, J.; Kazakov, S.; Hernandez-Guzman, F.; Gaisina, I.N.; Kozikowski, A.P.; Ratan, R.R.; et al. Utilization of an in vivo reporter for high throughput identification of branched small mol-ecule regulators of hypoxic adaptation. Chem. Biol. 2010, 17, 380–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smirnova, N.A.; Kaidery, N.A.; Hushpulian, D.M.; Rakhman, I.I.; Poloznikov, A.A.; Tishkov, V.I.; Karuppagounder, S.S.; Gaisina, I.N.; Pekcec, A.; Van Leyen, K.; et al. Bioactive Flavonoids and Catechols as Hif1 and Nrf2 Protein Stabilizers-Implications for Parkinson’s Disease. Aging Dis. 2016, 7, 745–762. [Google Scholar] [CrossRef] [Green Version]
- Gaisina, I.N.; Lee, S.; Kaidery, N.A.; Ben Aissa, M.; Ahuja, M.; Smirnova, N.N.; Wakade, S.; Gaisin, A.; Bourassa, M.W.; Ratan, R.R.; et al. Activation of Nrf2 and Hypoxic Adaptive Response Contribute to Neuroprotection Elicited by Phenylhydroxamic Acid Selective HDAC6 Inhibitors. ACS Chem. Neurosci. 2018, 9, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Osipyants, A.I.; Poloznikov, A.; Smirnova, N.A.; Hushpulian, D.M.; Khristichenko, A.Y.; Chubar, T.A.; Zakhariants, A.A.; Ahuja, M.; Gaisina, I.; Thomas, B.; et al. L-ascorbic acid: A true substrate for HIF prolyl hydroxylase? Biochimicals 2018, 147, 46–54. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popov, A.; Klimovich, A.; Styshova, O.; Tsybulsky, A.; Hushpulian, D.; Osipyants, A.; Khristichenko, A.; Kazakov, S.; Ahuja, M.; Kaidery, N.; et al. Probable Mechanisms of Doxorubicin Antitumor Activity Enhancement by Ginsenoside Rh2. Molecules 2022, 27, 628. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27030628
Popov A, Klimovich A, Styshova O, Tsybulsky A, Hushpulian D, Osipyants A, Khristichenko A, Kazakov S, Ahuja M, Kaidery N, et al. Probable Mechanisms of Doxorubicin Antitumor Activity Enhancement by Ginsenoside Rh2. Molecules. 2022; 27(3):628. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27030628
Chicago/Turabian StylePopov, Alexander, Anna Klimovich, Olga Styshova, Alexander Tsybulsky, Dmitry Hushpulian, Andrey Osipyants, Anna Khristichenko, Sergey Kazakov, Manuj Ahuja, Navneet Kaidery, and et al. 2022. "Probable Mechanisms of Doxorubicin Antitumor Activity Enhancement by Ginsenoside Rh2" Molecules 27, no. 3: 628. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27030628