
Spin Trapping Hydroxyl and Aryl Radicals of One-Electron Reduced Anticancer Benzotriazine 1,4-Dioxides

 , , and
, , and
Abstract
:1. Introduction
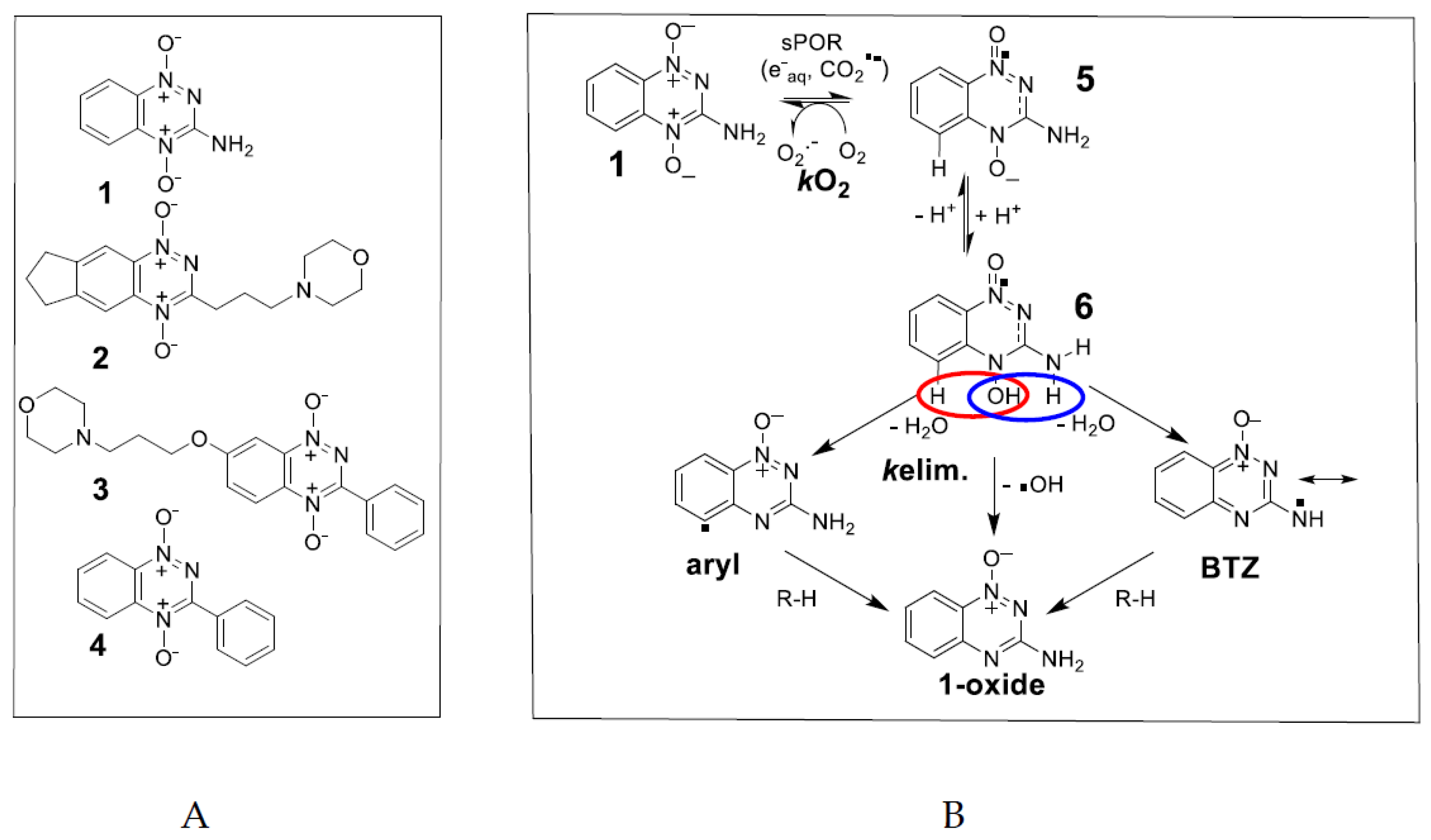
2. Results
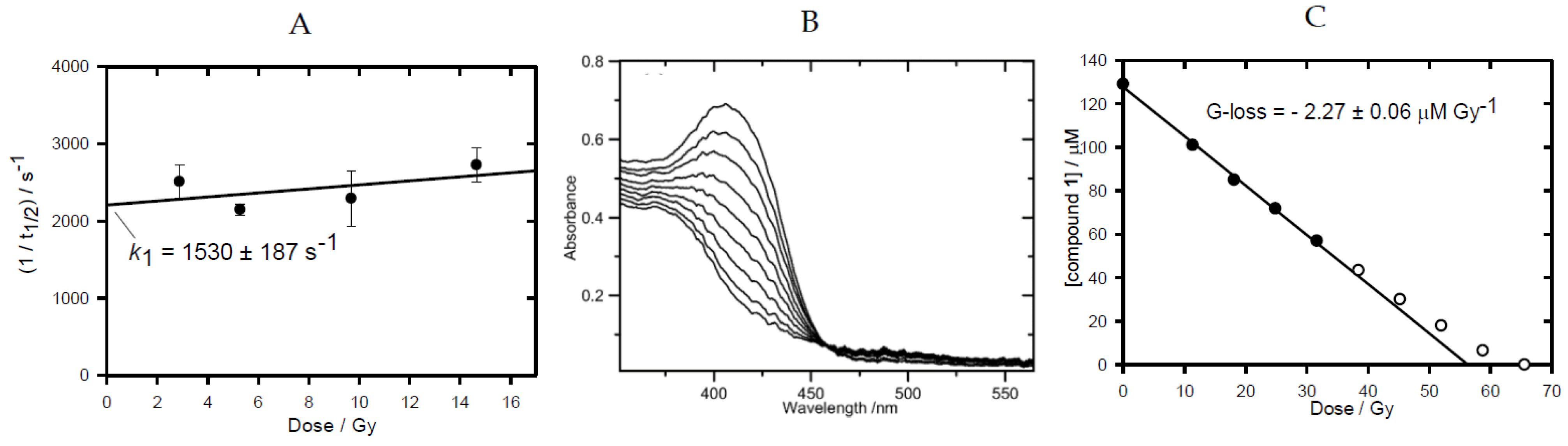
2.1. Pulse and Steady-State Radiolysis Studies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
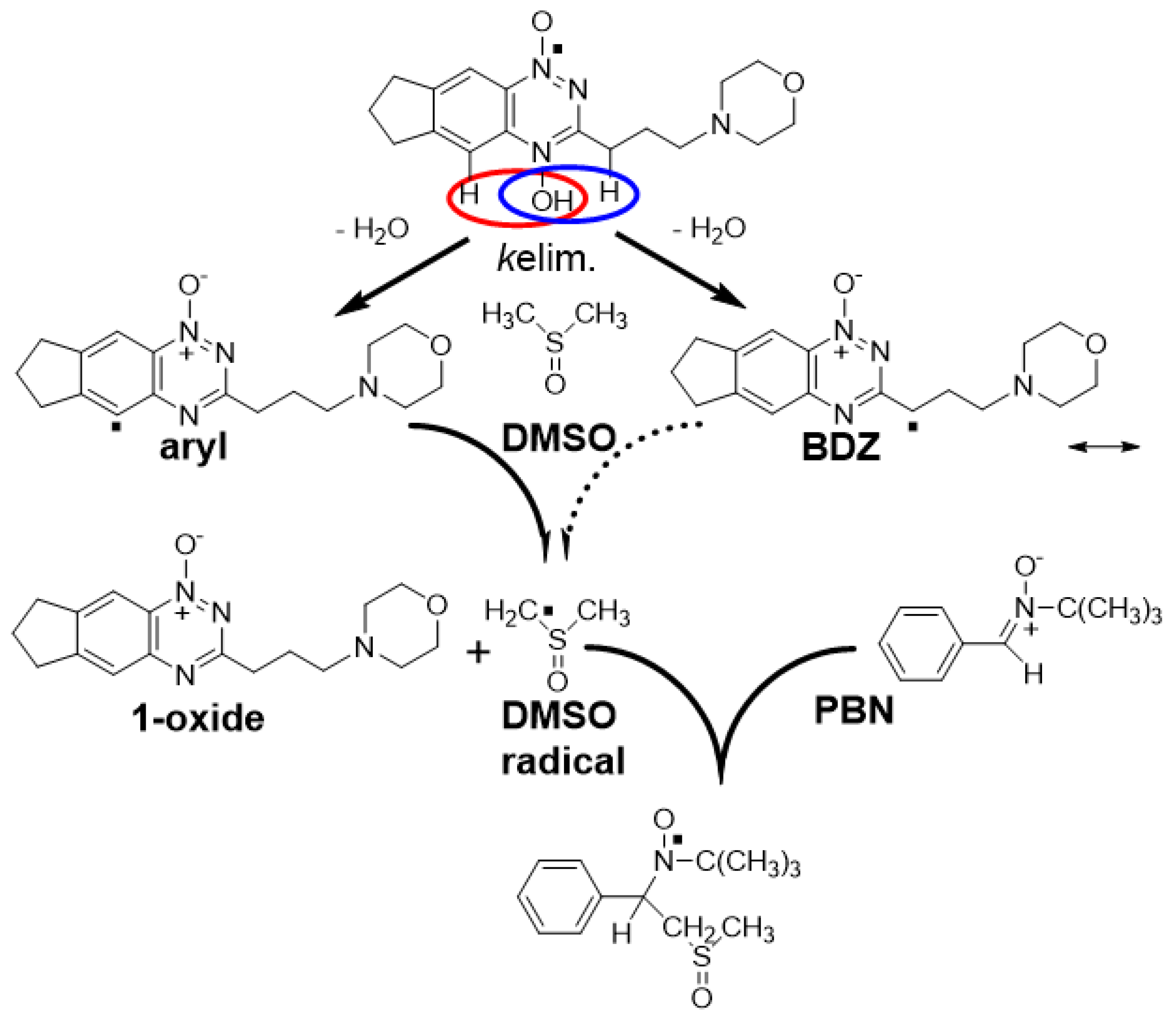{kind=link}
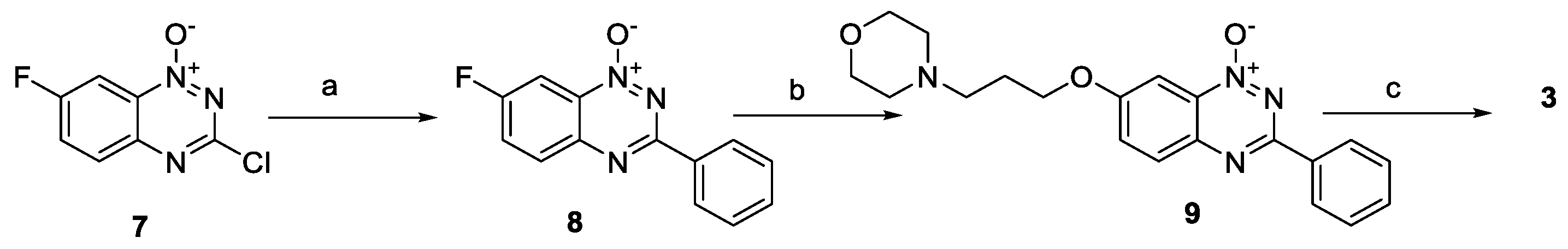{kind=link}
| Compound | E0’(A/A.−)/mV | kelim/s−1 | 106kO2/M−1 s−1 | G-loss/μM.Gy−1 |
|---|---|---|---|---|
| 1 | −456 ± 8 a,b | 83 ± 6 e | 6.20 ± 0.25 g | 1.55 ± 0.07 |
| 2 | −401 ± 8 c | 125 ± 15 c | 3.33 ± 0.03 c | 0.95 ± 0.04 |
| 3 | −387 ± 7 | 1530 ± 187 | 2.32 ± 0.17 | 1.96 ± 0.05 |
| 4 | −369 ± 8 d | 150 ± 15 f | 2.10 ± 0.06 | 2.20 ± 0.06 |
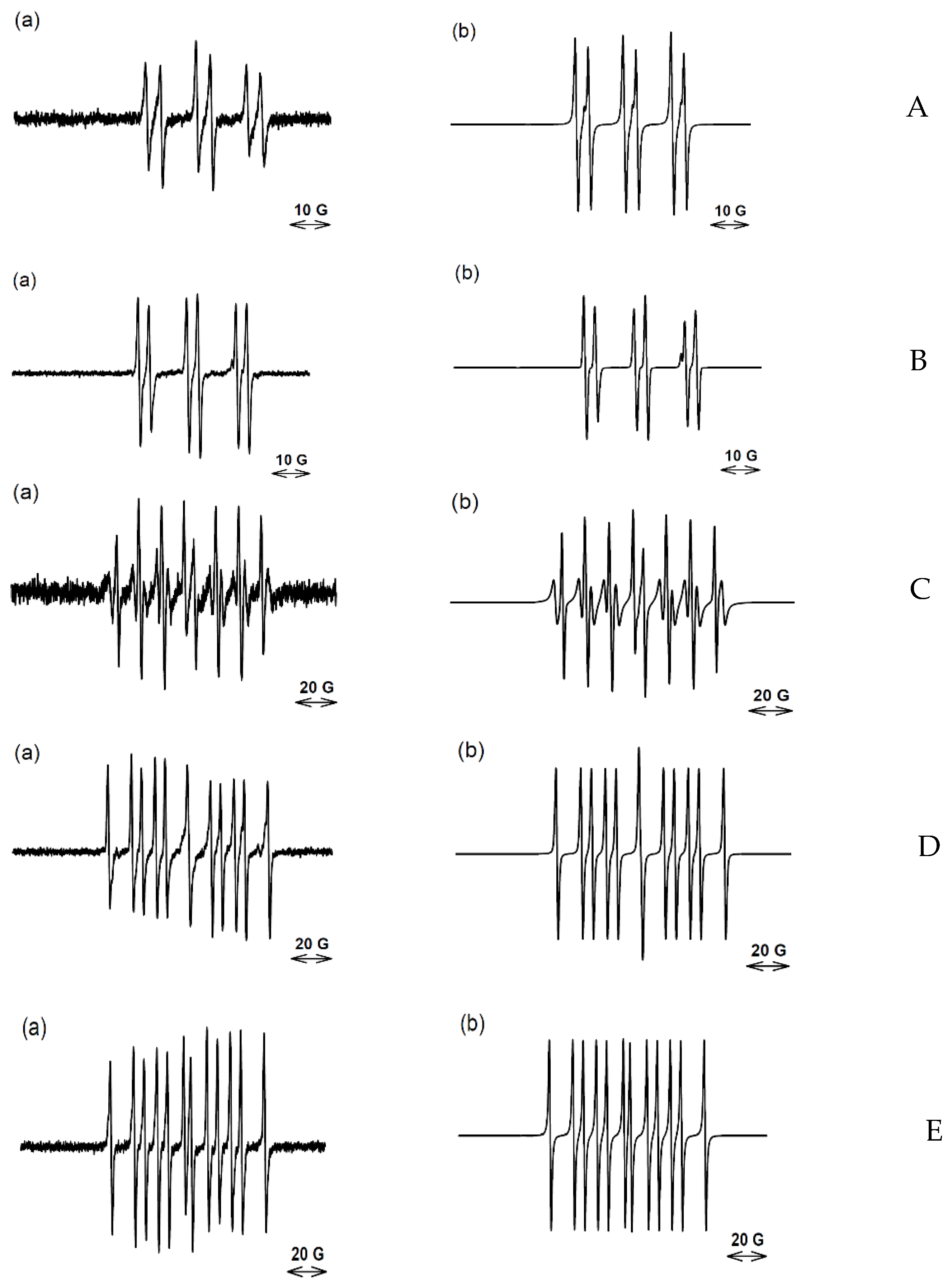
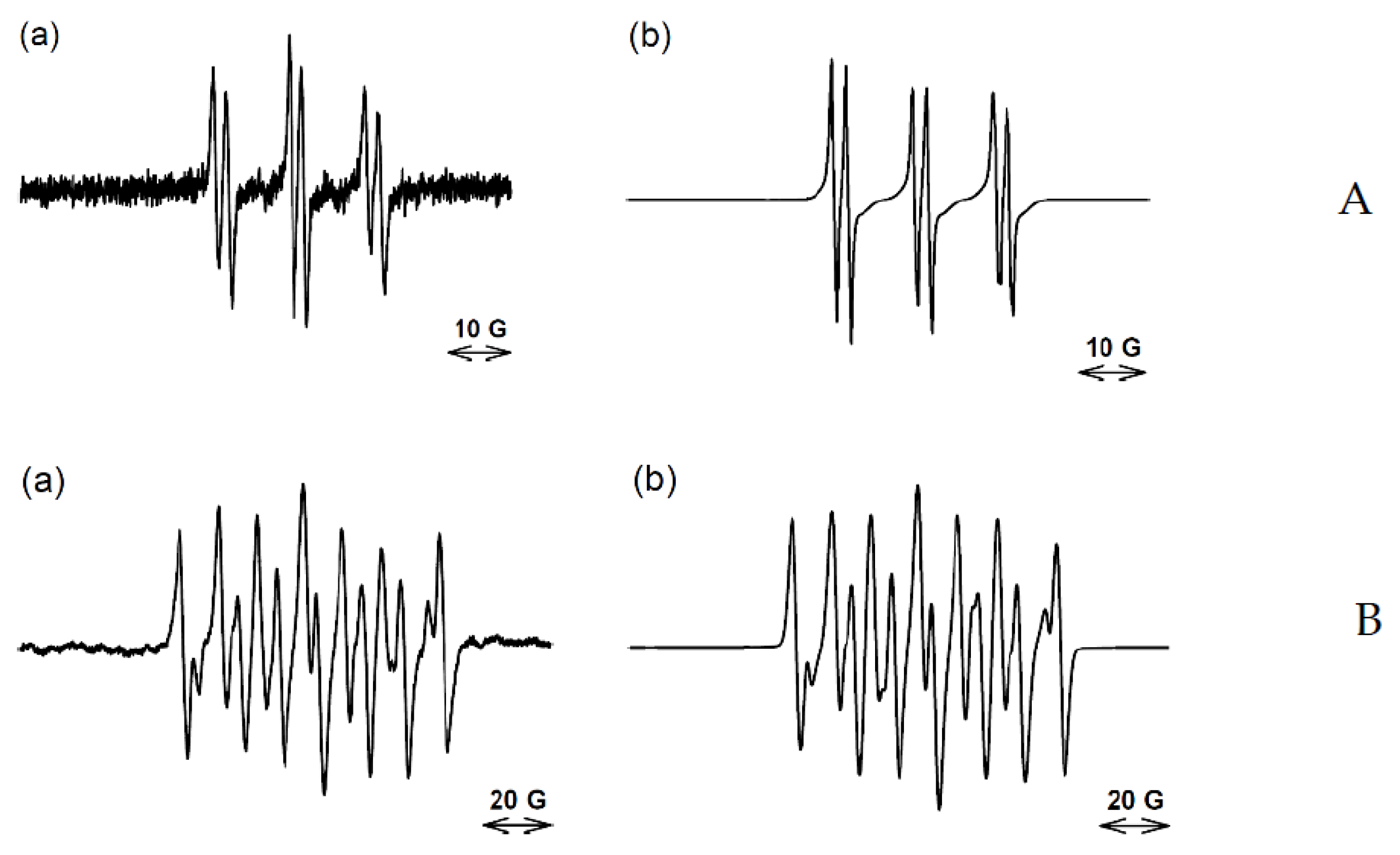
2.2. EPR Experiments
2.3. Cytotoxicity Data
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Radiation Chemistry
4.3. Methodology for EPR Experiments
4.4. In Vitro Cytotoxicity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Brown, J.M. SR 4233 (tirapazamine): A new anticancer drug exploiting hypoxia in solid tumours. Br. J. Cancer 1993, 67, 1163–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.M.; Wilson, W.R. Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer 2004, 4, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Zeman, E.M.; Brown, J.M.; Lemmon, M.J.; Hirst, V.K.; Lee, W.W. SR 4233: A new bioreductive agent with high selective toxicity for hypoxic mammalian cells. Int. J. Radiat. Oncol. Biol. Phys. 1986, 12, 1239–1242. [Google Scholar] [CrossRef]
- Baker, M.A.; Zeman, E.M.; Hirst, V.K.; Brown, J.M. Metabolism of SR 4233 by chinese hamster ovary cells: Basis of selective hypoxic cytotoxicity. Cancer Res. 1988, 48, 5947–5952. [Google Scholar]
- Anderson, R.F.; Shinde, S.S.; Hay, M.P.; Gamage, S.A.; Denny, W.A. Activation of 3-amino-1,2,4-benzotriazine 1,4-dioxide antitumor agents to oxidizing species following their one-electron reduction. J. Am. Chem. Soc. 2003, 125, 748–756. [Google Scholar] [CrossRef]
- Anderson, R.F.; Shinde, S.S.; Hay, M.P.; Gamage, S.A.; Denny, W.A. Radical properties governing the hypoxia-selective cytotoxicity of antitumor 3-amino-1,2,4-benzotriazine 1,4-dioxides. Org. Biomol. Chem. 2005, 3, 2167–2174. [Google Scholar] [CrossRef]
- Hay, M.P.; Gamage, S.A.; Kovacs, M.; Pruijn, F.B.; Anderson, R.F.; Patterson, A.V.; Wilson, W.R.; Brown, J.M.; Denny, W.A. Structure-activity relationships of 1,2,4-Benzotriazine 1,4-dioxides as hypoxia-selective analogs of tirapazamine. J. Med. Chem. 2003, 46, 169–182. [Google Scholar] [CrossRef]
- Koch, C.J. Unusual oxygen concentration dependence of toxicity of SR-4233, a hypoxic cell toxin. Cancer Res. 1993, 53, 3992–3997. [Google Scholar]
- Hicks, K.O.; Siim, B.G.; Jaiswal, J.K.; Pruijn, F.B.; Fraser, A.M.; Patel, R.; Hogg, A.; Liyanage, H.D.S.; Dorie, M.J.; Brown, J.M.; et al. Parmacokinetic/pharmacodynamic modeling identifies SN30000 and SN29751 as tirapazamine analogues with improved tissue penetration and hypoxic cell killing in tumors. Clin. Cancer Res. 2010, 16, 4946–4957. [Google Scholar] [CrossRef] [Green Version]
- Birincioglu, M.; Jaruga, P.; Chowdhury, G.; Rodriguez, H.; Dizdaroglu, M.; Gates, K.S. DNA base damage by the antitumor agent 3-amino-1,2,4-benzotriazine 1,4-dioxide (Tirapazamine). J. Am. Chem. Soc. 2003, 125, 11607–11615. [Google Scholar] [CrossRef]
- Chowdhury, G.; Junnotula, V.; Daniels, J.S.; Greenberg, M.M.; Gates, K.S. DNA strand damage product analysis provides evidence the the tumor cell-specific cytotoxin tirapazamine produces hydroxyl radical and acts as surrogate for O2. J. Am. Chem. Soc. 2007, 129, 12870–12877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniels, J.S.; Gates, K.S. DNA cleavage by the antitumor agent 3-amino-1,2,4-benzotriazine 1,4-dioxide (SR4233): Evidence for involvement of hydroxyl radical. J. Am. Chem. Soc. 1996, 118, 3380–3385. [Google Scholar] [CrossRef]
- Kotandeniya, D.; Ganley, B.; Gates, K.S. Oxidative DNA base damage by the antitumor agent 3-amino-1,2,4-benzotriazine 1,4-dioxide (tirapazamine). Bioorg. Med. Chem. Lett. 2002, 12, 2325–2329. [Google Scholar] [CrossRef]
- Junnotula, V.; Sarkar, U.; Sinha, S.; Gates, K.S. Initiation of DNA strand cleavage by 1,2,4-benzotriazine 1,4-dioxide antitumor agents: Mechanistic insight from studies of 3-methyl-1,2,4-benzotriazine 1,4-dioxide. J. Am. Chem. Soc. 2009, 131, 1015–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, L.H.; Taiwo, F.A. Electron paramagnetic resonance spectrometry evidence for bioreduction of tirapazamine to oxidising free radicals under anaerobic conditions. Biochem. Pharmacol. 2000, 60, 1933–1935. [Google Scholar] [CrossRef]
- Ranguelova, K.; Mason, R.P. The fidelity of spin trapping with DMPO in biological systems. Magn. Reson. Chem. 2011, 49, 152–158. [Google Scholar] [CrossRef] [Green Version]
- Shinde, S.S.; Hay, M.P.; Patterson, A.V.; Denny, W.A.; Anderson, R.F. Spin trapping of radicals other than the.OH radical upon reduction of the anticancer agent tirapazamine by cytochrome P450 reductase. J. Am. Chem. Soc. 2009, 131, 14220–14221. [Google Scholar] [CrossRef]
- Hay, M.P.; Hicks, K.O.; Pchalek, K.; Lee, H.H.; Blaser, A.; Pruijn, F.B.; Anderson, R.F.; Shinde, S.S.; Wilson, W.R.; Denny, W.A. Tricyclic [1,2,4]triazine 1,4-dioxides as hypoxia selective cytotoxins. J. Med. Chem. 2008, 51, 6853–6865. [Google Scholar] [CrossRef] [Green Version]
- Anderson, R.F.; Yadav, P.; Patel, D.; Reynisson, J.; Tipparaju, S.R.; Guise, C.P.; Patterson, A.V.; Denny, W.A.; Maroz, A.; Shinde, S.S.; et al. Characterisation of radicals formed by the triazine 1,4-dioxide hypoxia-activated prodrug, SN30000. Org. Biomol. Chem. 2014, 12, 3386–3392. [Google Scholar] [CrossRef]
- Shinde, S.S.; Maroz, A.; Hay, M.P.; Anderson, R.F. One-electron reduction potential of the neutral guanyl radical in the GC base pair of duplex DNA. J. Am. Chem. Soc. 2009, 131, 5203–5207. [Google Scholar] [CrossRef] [Green Version]
- Steenken, S.; Jovanovic, S.V. How easily oxidizable is DNA? One-electron reduction potentials of adenosine and guanosine radicals in aqueous solution. J. Am. Chem. Soc. 1997, 119, 617–618. [Google Scholar] [CrossRef]
- Shinde, S.S.; Maroz, A.; Hay, M.P.; Patterson, A.V.; Denny, W.A.; Anderson, R.F. Characterization of radicals formed following enzymatic reduction of 3-substituted analogues of the hypoxia-activated cytotoxin 3-amino-1,2,4-benzotriazine 1,4-dioxide (tirapazamine). J. Am. Chem. Soc. 2010, 132, 2591–2599. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, B.C.; Ionita, P.; Smith, J.R.L.; Oakes, J.; Ouwerkerk, N. Mechanistic studies on the free radical decomposition of some oxalic acid arylhydrazides: A source of aryl radicals in aqueous solution. ARKIVOC 2006, 3, 127–147. [Google Scholar] [CrossRef] [Green Version]
- Hill, H.A.O.; Thornalley, P.J. The effect of spin traps on phenylhydrazine-induced haemolysis. Biochim. Biophys. Acta 1983, 762, 44–51. [Google Scholar] [CrossRef]
- Janzen, E.G.; Coulter, G.A.; Oehler, U.M.; Bergsma, J.P. Solvent effects on the nitrogen and β-hydrogen hyperfine splitting constants of aminoxyl radicals obtained in spin trapping experiments. Can. J. Chem. 1982, 60, 2725–2733. [Google Scholar] [CrossRef]
- Lorance, E.D.; Kramer, W.H.; Gould, I.R. Kinetics of reductive N-O bond fragmentation: The role of a conical intersection. J. Am. Chem. Soc. 2002, 124, 15225–15238. [Google Scholar] [CrossRef]
- Yin, J.; Glaser, R.; Gates, K.S. On the reaction mechanism of tirapazamine reduction chemistry: Unimolecular N-OH homolysis, stepwise dehydration, of triazene ring-opening. Chem. Res. Toxicol. 2012, 25, 634–645. [Google Scholar] [CrossRef]
- Frejaville, C.; Karoui, H.; Tuccio, B.; Le Moigne, F.; Culcasi, M.; Pietri, S.; Lauricella, R.; Tordo, P. 5-(Diethoxyphosphoryl)-5-methyl-1-pyrroline N-oxide: A new efficient phosphorylated nitrone for the in vitro and in vivo spin trapping of oxygen-centred radicals. J. Med. Chem. 1995, 38, 258–265. [Google Scholar] [CrossRef]
- Janzen, E.G.; Kotake, Y.; Hinton, R.D. Stabilities of hydroxyl radical adducts of PBN-type spin traps. Free Radic. Biol. Med. 1992, 12, 169–173. [Google Scholar] [CrossRef]
- Greenstock, C.L.; Wiebe, R.H. Substituent effects in the kinetic analysis of free radical reactions with nitrone spin traps. Can. J. Chem. 1982, 60, 1560–1564. [Google Scholar] [CrossRef] [Green Version]
- Villamena, F.A.; Hadad, C.M.; Zweier, J.L. Kinetic study and theoretical analysis of hydroxyl radical trapping and spin adduct decay of alkoxycarbonyl and dialkoxyphosphoryl nitrones in aqueous media. J. Phys. Chem. A 2003, 107, 4407–4414. [Google Scholar] [CrossRef]
- Priyadarsini, K.I.; Tracy, M.; Wardman, P. The one-electron reduction potential of 3-amino-1,2,4-benzotriazine 1,4-dioxide (tirapazamine): A hypoxia-selective bioreductive drug. Free Rad. Res. 1996, 25, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Hay, M.P.; Pchalek, K.; Pruijn, F.B.; Hicks, K.O.; Siim, B.G.; Anderson, R.F.; Shinde, S.S.; Phillips, V.; Denny, W.A.; Wilson, W.R. Hypoxia-selective 3-alkyl 1,2,4-benzotriazine 1,4-dioxides: The influence of hydrogen bond donors on extravascular transport and antitumor activity. J. Med. Chem. 2007, 50, 6654–6664. [Google Scholar] [CrossRef] [PubMed]
- Laderoute, K.; Wardman, P.; Rauth, A.M. Molecular mechanisms for the hypoxia-dependent activation of 3-amino-1,2,4-benzotriazine-1,4-dioxide (SR 4233). Biochem. Pharmacol. 1988, 37, 1487–1495. [Google Scholar] [CrossRef]
- Mulazzani, Q.G.; Venturi, M.; Hoffman, M.Z.; Rodgers, M.A.J. Interaction of formate and oxalate ions with radiation-generated radicals in aqueous solution. Methylviologen as a mechanistic probe. J. Phys. Chem. 1986, 90, 5347–5352. [Google Scholar] [CrossRef]
- Dixon, W.T.; Norman, R.O.C.; Buley, A. L Electron spin resonance studies of oxidation. Part II. Aliphatic acids and substituted acids. J. Chem. Soc. 1964, 701, 3625–3634. [Google Scholar] [CrossRef]
- Kondo, T.; Riesz, P. Sonochemistry of nitrone spin traps in aqueous solutions. Evidence for pyrolysis radicals from spin traps. Free Radic. Biol. Med. 1989, 7, 259–268. [Google Scholar] [CrossRef]
- Saprin, A.; Piette, L.H. Spin trapping and its application in the study of lipid peroxidation and free radical production with liver microsomes. Arch. Biochem. Biophys. 1977, 180, 480–492. [Google Scholar] [CrossRef]
- Blanksby, S.J.; Ellison, G.B. Bond dissociation energies of organic molecules. Acc. Chem. Res. 2003, 36, 255–263. [Google Scholar] [CrossRef]
- Eberhardt, M.K.; Colina, R. The reaction of OH radicals with dimethyl sulfoxide. A comparative study of Fenton’s reagent and the radio;ysis of aqueous dimethyl sulfoxide solutions. J. Org. Chem. 1988, 53, 1071–1074. [Google Scholar] [CrossRef]
- Asmus, K.-D.; Mockel, H.; Henglein, A. Pulse radiolysis of the site of OH radical attack on aliphatic alcohols in aqueous solution. J. Phys. Chem. 1973, 77, 1218–1221. [Google Scholar] [CrossRef]
- Janzen, E.G.; Nutter, D.E.; Evans, C.A. Rate constants for the hydrogen atom abstraction by phenyl radical from methanol, ethanol, and 2-propanol as studied by electron spin resonance spin trapping techniques. J. Phys. Chem. 1975, 79, 1983–1984. [Google Scholar] [CrossRef]
- Stolze, K.; Udilova, N.; Nohl, H. Spin trapping of lipid radicals with DEPMPO-derived spin traps: Detection of superoxide, alkyl and alkoxyl radicals in aqueous and lipid phase. Free Radic. Biol. Med. 2000, 29, 1005–1014. [Google Scholar] [CrossRef]
- Karoui, H.; Chalier, F.; Finet, J.-P.; Tordo, P. DEPMPO: An efficient tool for the coupled ESR-spin trapping of alkylperoxyl radicals in water. Org. Biomol. Chem. 2011, 9, 2473–2480. [Google Scholar] [CrossRef]
- Quintero, B.; Morales, J.J.; Quiros, M.; Martinez-Puentedura, M.I.; del Carmen Cabeza, M. Dediazoniation of p-hydroxybenzenediazonium ion in a neutral aqueous medium. Free Radic. Biol. Med. 2000, 29, 464–479. [Google Scholar] [CrossRef]
- Yamamoto, K.; Kawanishi, S. Site-specific DNA damage by phenylhydrazine and phenelzine in the presence of Cu(II) ion or Fe(III) complexes: Roles of active oxygen species and carbon radicals. Chem. Res. Toxicol. 1992, 5, 440–446. [Google Scholar] [CrossRef]
- Albano, E.; Tomasi, A.; Goria-Gatti, L.; Poli, G.; Vannini, V.; Dianzani, M.U. Free radical metabolism of alcohols by rat liver microsomes. Free Rad. Res. Comm. 1987, 3, 243–249. [Google Scholar] [CrossRef]
- Augusto, O.; Du Plessis, L.R.; Weingrill, C.L.V. Spin-trapping of methyl radical in the oxidative metabolism of 12-dimethylhydrazine. Biochem. Biophys. Res. Commun. 1985, 126, 853–858. [Google Scholar] [CrossRef]
- Thomas, J.K. Pulse radiolysis of aqueous solutions of methyl iodide and methyl bromide. The reactions of iodine atoms and methyl radicals in water. J. Phys. Chem. 1967, 71, 1919–1925. [Google Scholar] [CrossRef]
- Alfassi, Z.B.; Marguet, S.; Neta, P. Formation and reactivity of phenylperoxyl radicals in aqueous solution. J. Phys. Chem. 1994, 98, 8019–8023. [Google Scholar] [CrossRef]
- Merenyi, G.; Lind, J.; Engman, E. One- and two-electron reduction potentials of peroxyl radicals and related species. J. Chem. Soc. Perkin Trans. 1994, 2, 2551–2553. [Google Scholar] [CrossRef]
- Mason, J.C.; Tennant, G. Heterocyclic N-oxides. Part VI. Synthesis and nuclear magnetic resonance spectra of 3-aminobenzo-1,2,4-triazines and their mono- and di-N-oxides. J. Chem. Soc. B 1970, 911–916. [Google Scholar] [CrossRef]
- Pchalek, K.; Hay, M.P. Stille coupling reactions in the synthesis of hypoxia-selective 3-alkyl-1,2,4-benzotriazine 1,4-dioxide anticancer agents. J. Org. Chem. 2006, 71, 6530–6535. [Google Scholar] [CrossRef] [PubMed]
- Plé, P.A.; Green, T.P.; Hennequin, L.F.; Curwen, J.; Fennell, M.; Allen, J.; Lambert-van der Brempt, C.; Costello, G. Discovery of a new class of anilinoquinazoline inhibitors with high affinity and specificity for the tyrosine kinase domain of c-src. J. Med. Chem. 2004, 47, 871–887. [Google Scholar] [CrossRef]
- Anderson, R.F.; Denny, W.A.; Li, W.; Packer, J.E.; Tercel, M.; Wilson, W.R. Pulse radiolysis studies on the fragmentation of arylmethyl quaternary nitrogen mustards by one-electron reduction in aqueous solution. J. Phys. Chem. A 1997, 101, 9704–9709. [Google Scholar] [CrossRef]
- Schwarz, H.A.; Dodson, R.W. Reduction potentials of CO2− and the alcohol radicals. J. Phys. Chem. 1989, 93, 409–414. [Google Scholar] [CrossRef]
- Wardman, P. Reduction potentials of one-electron couples involving free radicals in aqueous solution. J. Phys. Chem. Ref. Data 1989, 18, 1637–1755. [Google Scholar] [CrossRef] [Green Version]
- Hunter, F.W.; Young, R.J.; Shalev, Z.; Vellanki, R.N.; Wang, J.; Gu, Y.N.; Joshi, N.; Sreebhavan, S.; Weinreb, I.; Goldstein, G.D.; et al. Identification of P450 oxidoreductase as a major determinant of sensitivity to hypoxia-activated prodrugs. Cancer Res. 2015, 75, 4211–4223. [Google Scholar] [CrossRef] [Green Version]






| Compd | Spin Trap | Scans | Radical Adduct | aN /G | aHβ/G | aP/G | % | r |
|---|---|---|---|---|---|---|---|---|
| 1 | PBN 50 mM a | 10 | -aryl | 16.50 | 4.18 | 20 | 0.96 | |
| (4 mM) | -carbon | 16.18 | 3.45 | 80 | ||||
| DEPMPO 25 mM b | 20 | -carbon | 14.70 | 21.40 | 47.40 | 100 | 0.92 | |
| DEPMPO 105 mM | 20 | -carbon -OH | 15.01 14.15 | 22.30 13,22 | 47.90 47.56 | 89.8 10.2 | 0.88 | |
| 2 | PBN 50 mM c | 100 | -aryl | 16.0 | 4.20 | 43 | 0.96 | |
| (12 mM) | -carbon | 16.2 | 3.44 | 57 | ||||
| PBN 250 mM | 100 | -aryl | 16.03 | 4.27 | 91 | 0.94 | ||
| (16 mM) | -carbon | 16.13 | 3.44 | 7 | ||||
| PBN 100 mM + DMSO 2 M | 70 | -CH2(CH3)SO -aryl | 16.50 15.88 | 3.62 4.18 | 74 26 | 0.99 | ||
| PBN 100 mM + DMSO 2 M | 137 | -CH2(CH3)SO | 16.50 | 3.62 | 100 | 0.99 | ||
| DEPMPO 25 mM | 30 | -carbon c | 15.2 | 22.1 | 48.8 | 100 | 0.92 | |
| DEPMPO 250 mM | 50 | -aryl | 14.64 | 22.47 | 45.75 | 62 | 0.96 | |
| -carbon | 15.10 | 21.78 | 47.50 | 10 | ||||
| -OH | 14.12 | 13.25 | 47.53 | 28 | ||||
| DEPMPO 250 mM + methanol 2.5 M | 149 | -CH2OH | 14.80 | 21.19 | 49.59 | 100 | 0.98 | |
| DEPMPO 25 mM + DMSO 2 M | 110 | -carbon | 15.25 | 22.00 | 48.14 | 100 | 0.98 | |
| 3 | PBN 100 mM | 150 | -aryl | 15.07 | 4.02 | 79 | 0.81 | |
| (10 mM) | -carbon | 16.05 | 3.57 | 21 | ||||
| 4-POBN 100 mM | 150 | -aryl | 15.41 | 3.20 | 75 | 0.96 | ||
| -carbon | 15.65 | 2.69 | 25 | |||||
| DEPMPO 100 mM | 125 | -carbon | 14.62 | 22.02 | 47.18 | 65 | 0.99 | |
| -OH | 14.08 | 13.58 | 47.62 | 35 | ||||
| # | HT29 Oxic | HT29 Anoxic | HT29 HCR | SiHa Oxic | SiHa Anoxic | SiHa HCR | HCT116 Oxic | HCT116 Anoxic | HCT116 HCR |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 411 ± 18 | 5.69 ± 0.58 | 72.2 | 140 ± 8.0 | 2.80 ± 0.27 | 50 | 91.7 ± 4.4 | 1.69 ± 0.66 | 54 |
| 2 | 375 ± 89 | 2.88 ± 0.22 | 130 | 245 ± 27 | 1.42 ± 0.32 | 173 | 126 ± 11 | 2.05 ± 0.66 | 61 |
| 3 | 4.07 ± 1.04 | 2.83 ± 0.50 | 1.44 | 1.93 ± 0.22 | 1.09 ± 0.27 | 1.77 | 1.36 ± 0.23 | 1.64 ± 0.06 | 0.83 |
| 4 | 6.9 a | 4.4 a | 1.56 | 2.1 a | 2.2 a | 1.0 | 0.84 ± 0.12 | 1.67 ± 0.52 | 0.5 |
| Compound | G-loss μM.Gy−1 | x | y | Fraction x of G-Value |
|---|---|---|---|---|
| 1 | 1.55 ± 0.07 | 0.60 | 0.40 | 0.93 |
| 2 | 0.95 ± 0.04 | 0.35 | 0.65 | 0.33 |
| 3 | 2.20 ± 0.06 | 0.68 | 0.32 | 1.33 |
| 4 | 1.96 ± 0.05 | 0.72 | 0.28 | 1.58 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qi, W.; Yadav, P.; Hong, C.R.; Stevenson, R.J.; Hay, M.P.; Anderson, R.F. Spin Trapping Hydroxyl and Aryl Radicals of One-Electron Reduced Anticancer Benzotriazine 1,4-Dioxides. Molecules 2022, 27, 812. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27030812
Qi W, Yadav P, Hong CR, Stevenson RJ, Hay MP, Anderson RF. Spin Trapping Hydroxyl and Aryl Radicals of One-Electron Reduced Anticancer Benzotriazine 1,4-Dioxides. Molecules. 2022; 27(3):812. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27030812
Chicago/Turabian StyleQi, Wen, Pooja Yadav, Cho R. Hong, Ralph J. Stevenson, Michael P. Hay, and Robert F. Anderson. 2022. "Spin Trapping Hydroxyl and Aryl Radicals of One-Electron Reduced Anticancer Benzotriazine 1,4-Dioxides" Molecules 27, no. 3: 812. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27030812