
Investigating the Broad Matrix-Gate Network in the Mitochondrial ADP/ATP Carrier through Molecular Dynamics Simulations


Abstract
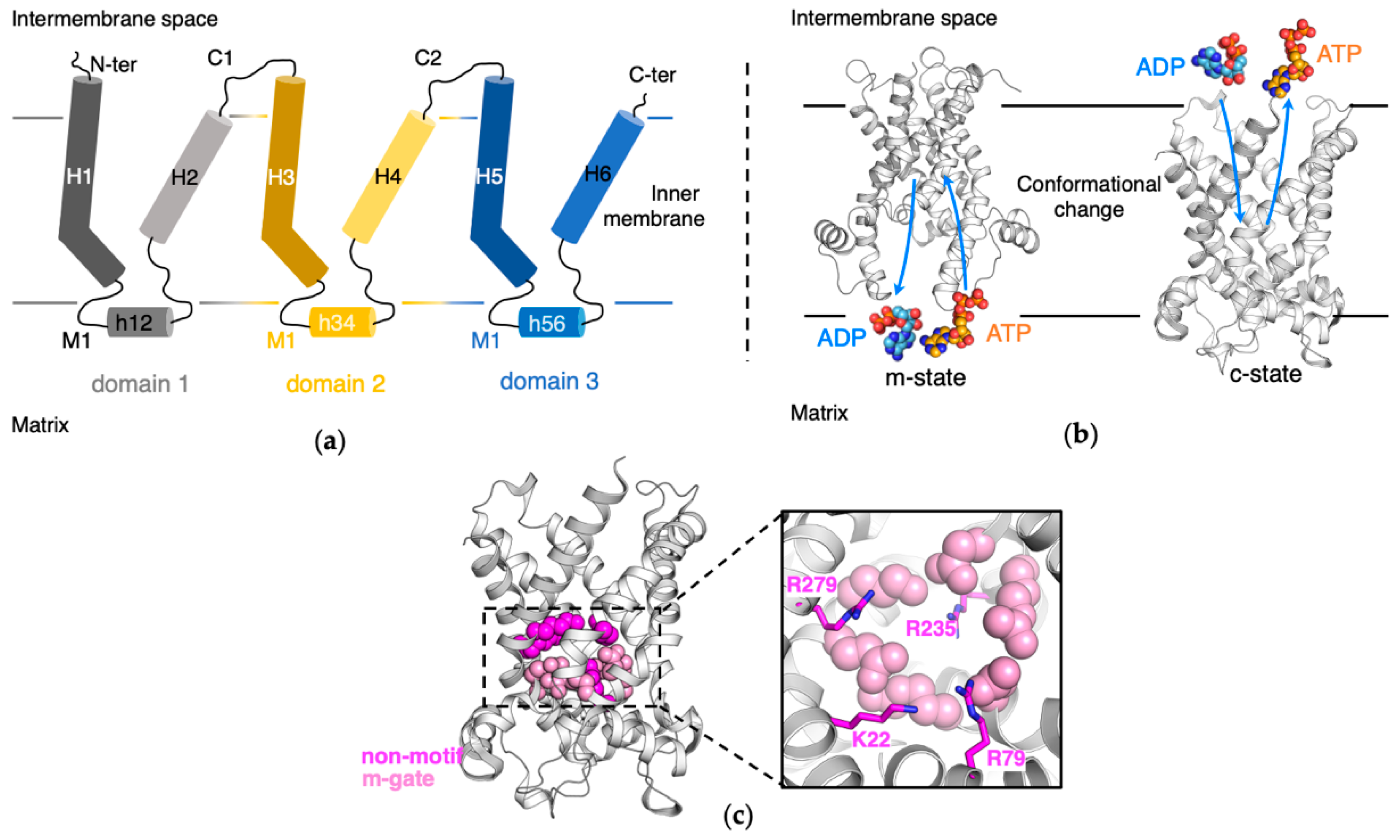
:1. Introduction

2. Results
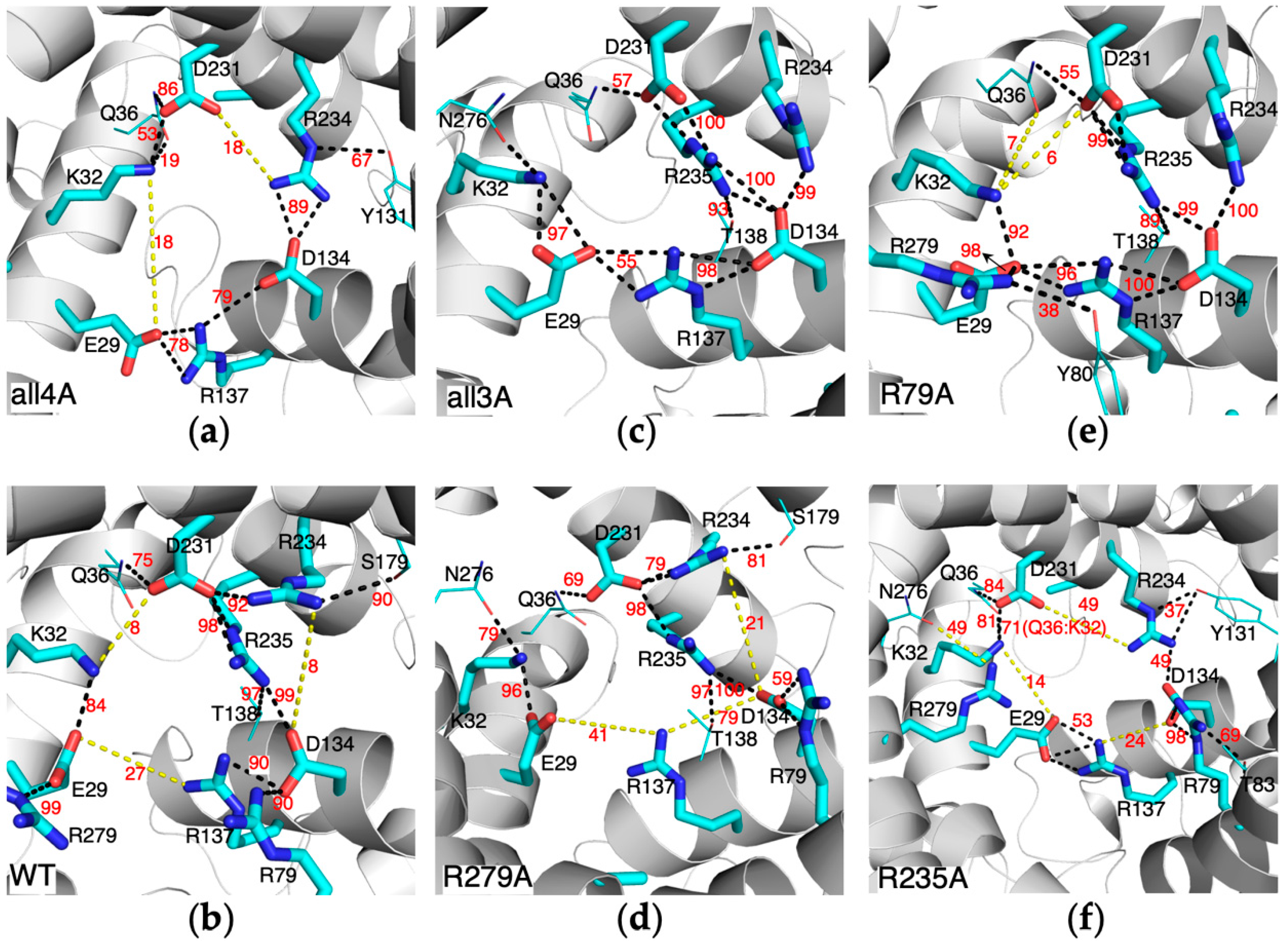
2.1. Impact of Mutations of the Non-Motif Positive Residues on the m-Gate Network
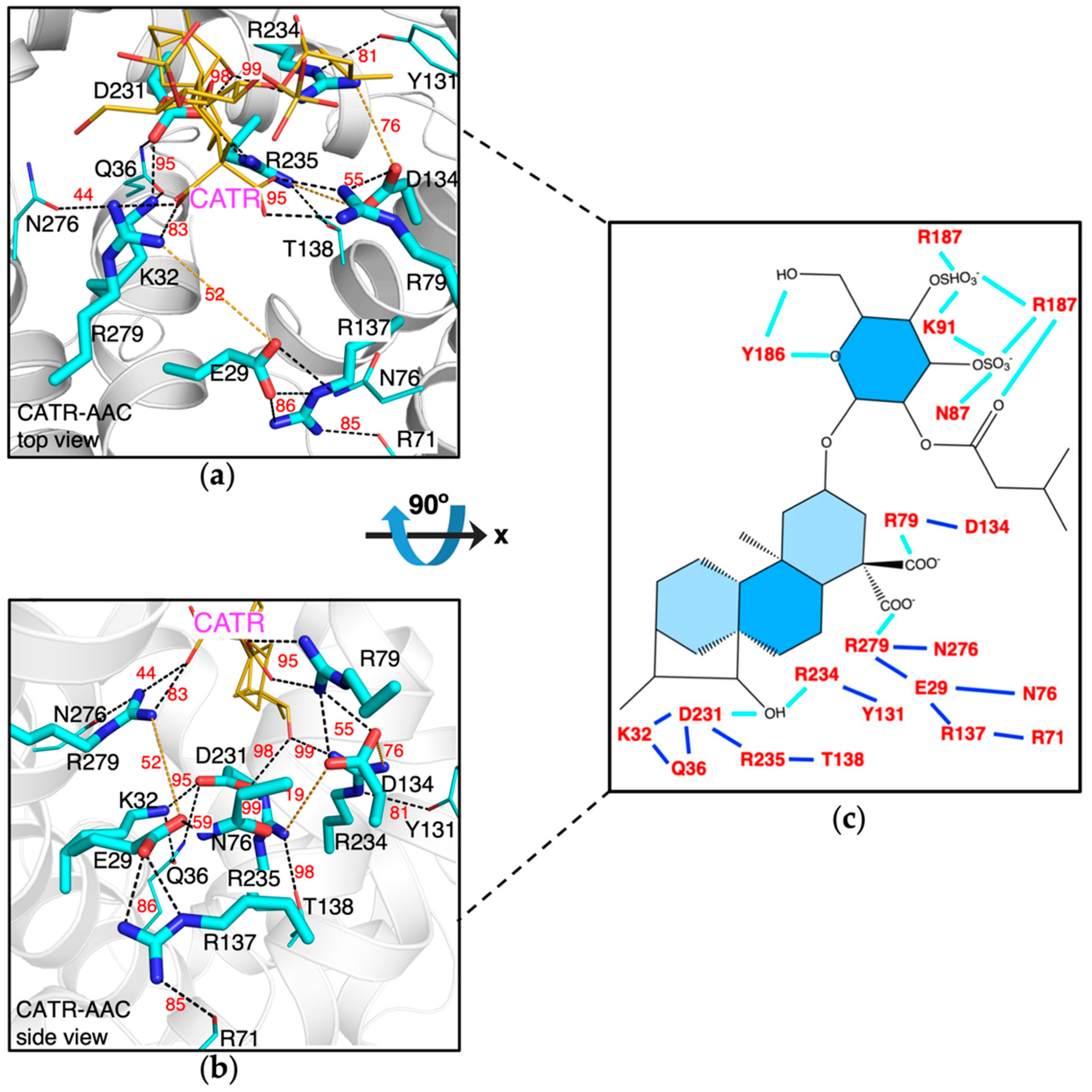
2.2. Impact of CATR Binding on the m-Gate Network
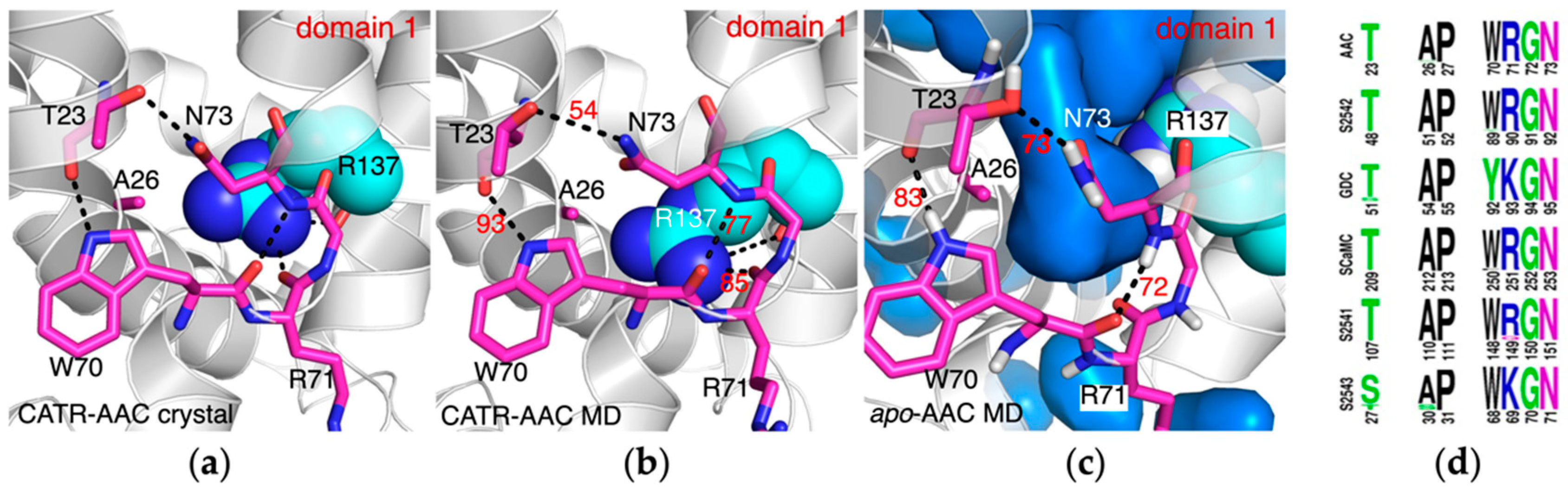
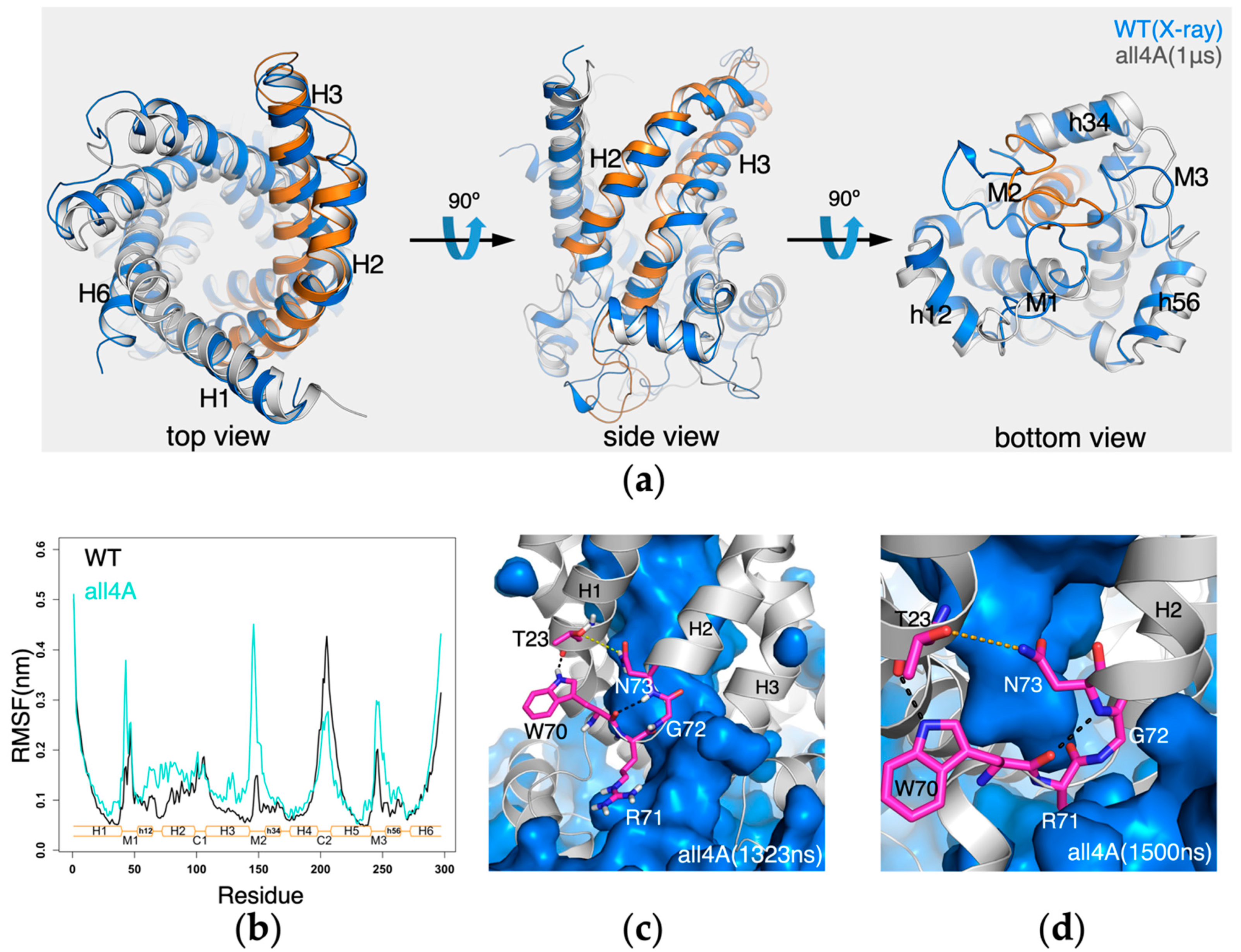
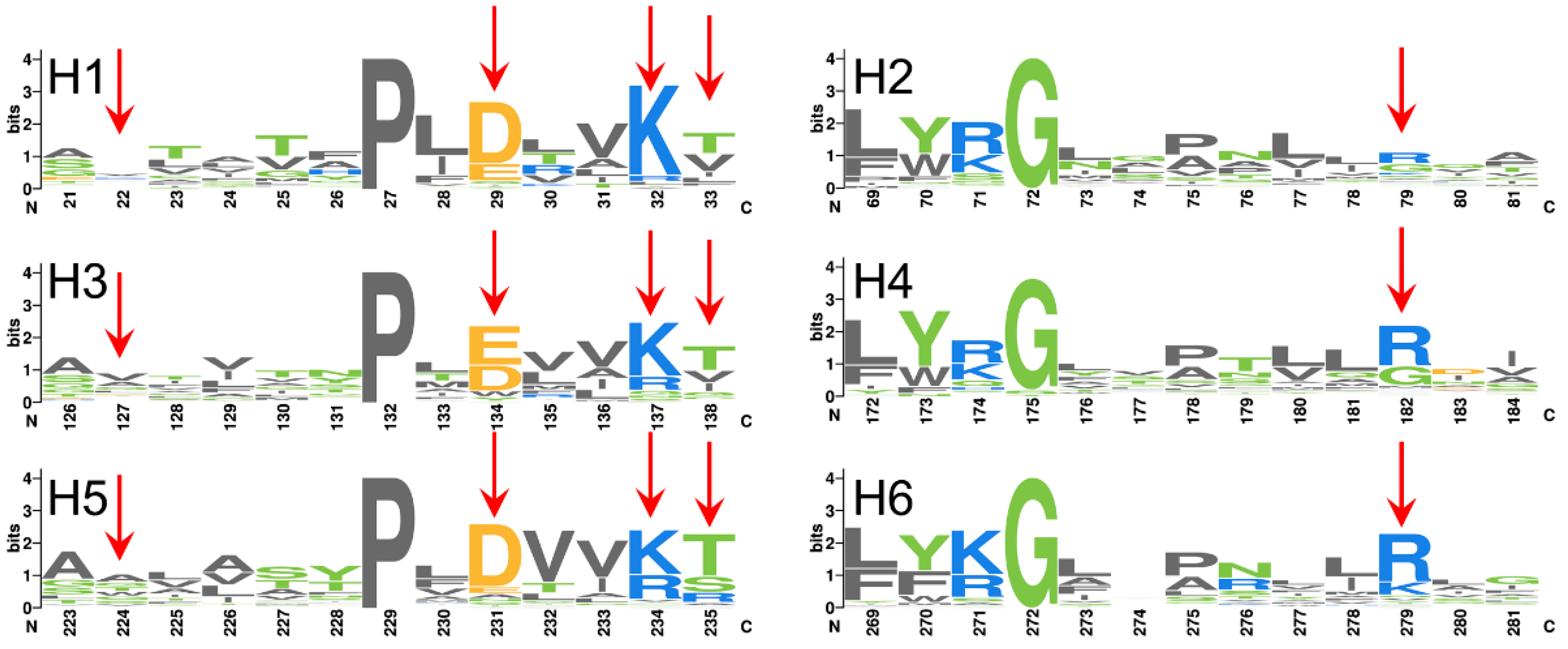
2.3. The Conserved Gap between A26 and the [YWF][KR]G Motif in Domain 1
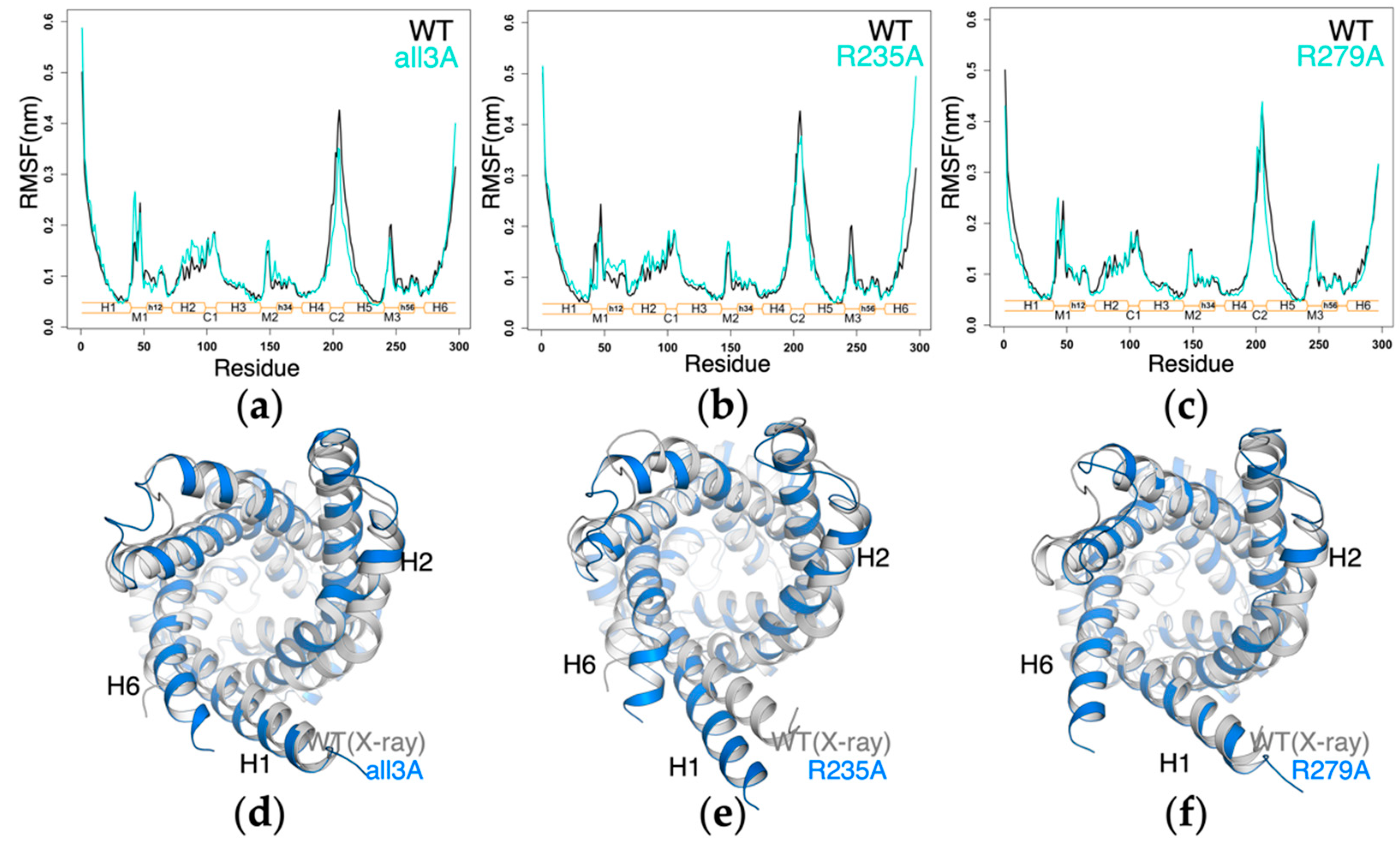
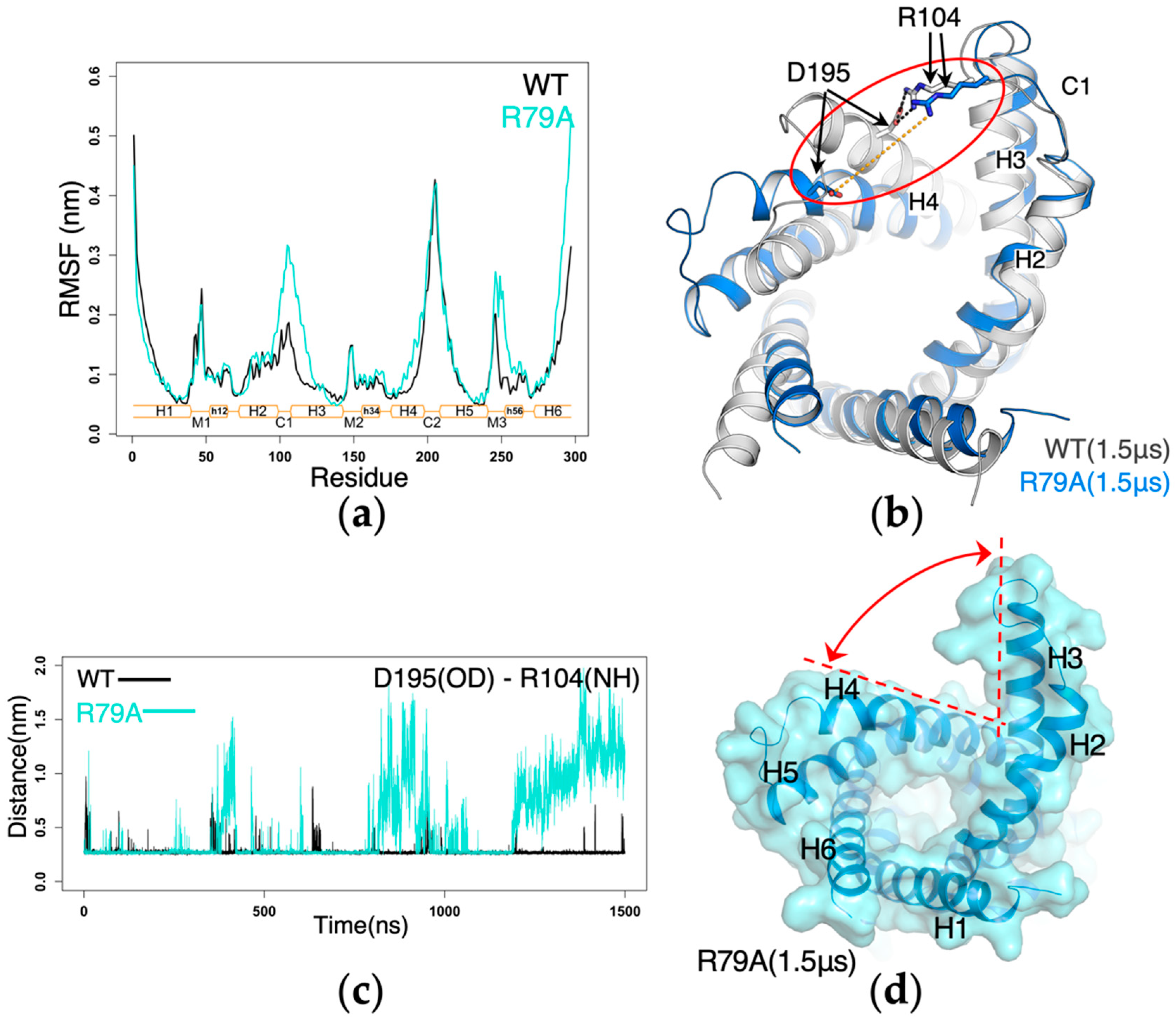
2.4. Impact of Mutations of the Non-Motif Positive Residues on the Structural Dynamics of AAC
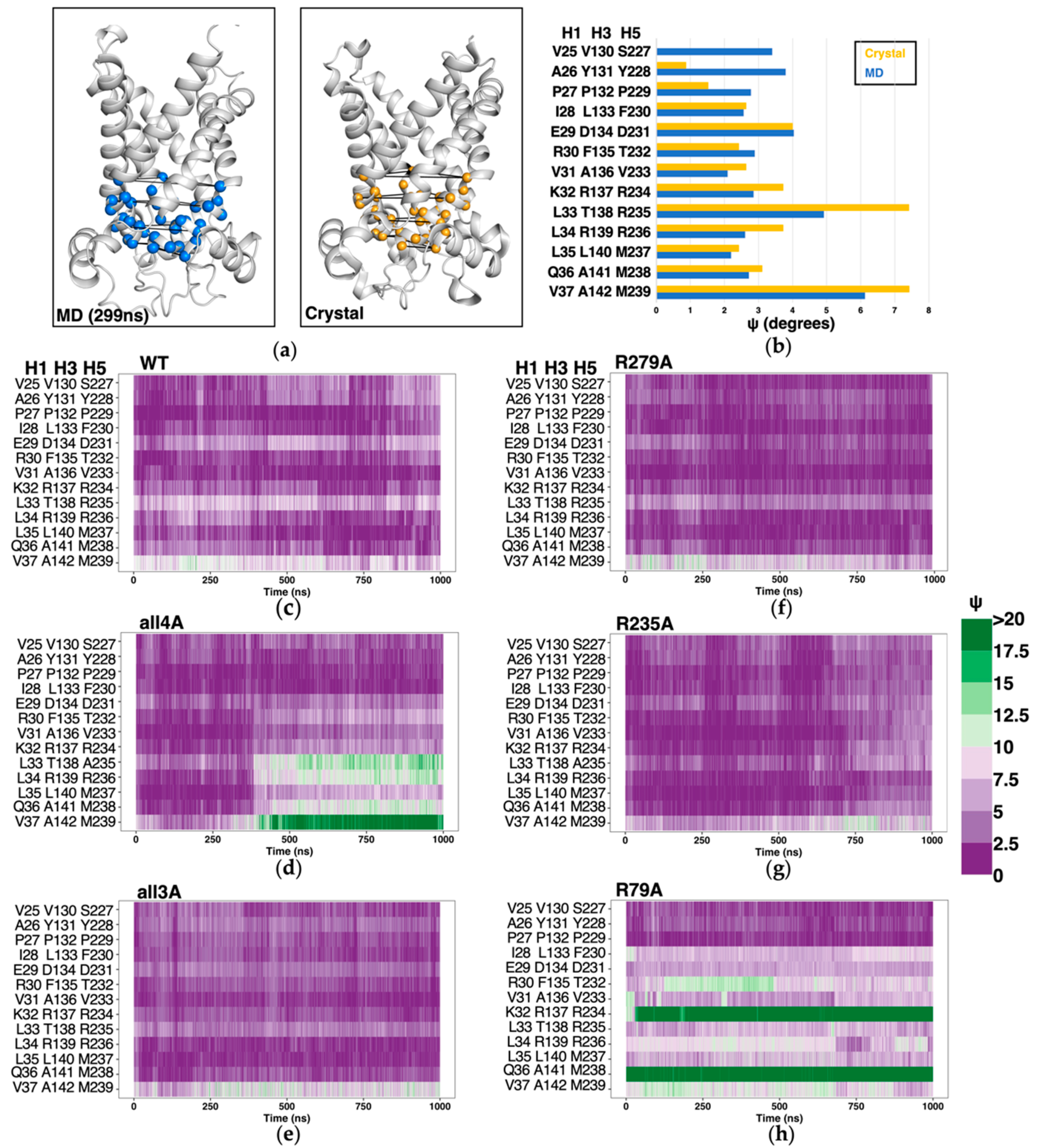
2.5. Structure-Based Symmetry Analysis near the m-Gate Level
3. Discussion
4. Materials and Methods
4.1. System Setup
4.2. MD Simulation Protocol
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| AAC | ADP/ATP carrier |
| ATR | atractyloside |
| BKA | bongkrekic acid |
| CATR | carboxyatractyloside |
| GDC | graves disease carrier |
| hAAC1 | human ADP/ATP carrier 1 |
| IMM | the inner mitochondrial membrane |
| m-gate | matrix gate |
| MCF | mitochondrial carrier family |
| MCs | mitochondrial carriers |
| MD | molecular dynamics |
| OXPHOS | oxidative phosphorylation |
| POPC | palmitoyl-oleoyl-phosphatidylcholine |
| RMSF | root-mean square fluctuation |
| SCaMC | calcium-binding mitochondrial carrier protein |
| SLC | solute carrier |
| SLC25A16 | Solute carrier family 25, member 16 |
References
- Aquila, H.; Link, T.A.; Klingenberg, M. Solute carriers involved in energy transfer of mitochondria form a homologous protein family. FEBS Lett. 1987, 212, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, F. Mitochondrial carrier proteins. FEBS Lett. 1994, 346, 48–54. [Google Scholar] [CrossRef] [Green Version]
- Ruprecht, J.J.; Kunji, E.R.S. Structural Mechanism of Transport of Mitochondrial Carriers. Annu. Rev. Biochem. 2021, 90, 535–558. [Google Scholar] [CrossRef] [PubMed]
- Yi, Q.; Li, Q.; Yao, S.; Chen, Y.; Guan, M.X.; Cang, X. Molecular dynamics simulations on apo ADP/ATP carrier shed new lights on the featured motif of the mitochondrial carriers. Mitochondrion 2019, 47, 94–102. [Google Scholar] [CrossRef]
- Robinson, A.J.; Overy, C.; Kunji, E.R. The mechanism of transport by mitochondrial carriers based on analysis of symmetry. Proc. Natl. Acad. Sci. USA 2008, 105, 17766–17771. [Google Scholar] [CrossRef] [Green Version]
- Pierri, C.L.; Palmieri, F.; De Grassi, A. Single-nucleotide evolution quantifies the importance of each site along the structure of mitochondrial carriers. Cell. Mol. Life Sci. 2014, 71, 349–364. [Google Scholar] [CrossRef]
- Klingenberg, M. The ADP and ATP transport in mitochondria and its carrier. Biochim. Biophys. Acta 2008, 1778, 1978–2021. [Google Scholar] [CrossRef] [Green Version]
- Pebay-Peyroula, E.; Dahout-Gonzalez, C.; Kahn, R.; Trezeguet, V.; Lauquin, G.J.; Brandolin, G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 2003, 426, 39–44. [Google Scholar] [CrossRef]
- Ruprecht, J.J.; Hellawell, A.M.; Harding, M.; Crichton, P.G.; McCoy, A.J.; Kunji, E.R. Structures of yeast mitochondrial ADP/ATP carriers support a domain-based alternating-access transport mechanism. Proc. Natl. Acad. Sci. USA 2014, 111, E426–E434. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, F.; Pierri, C.L. Structure and function of mitochondrial carriers—Role of the transmembrane helix P and G residues in the gating and transport mechanism. FEBS Lett. 2010, 584, 1931–1939. [Google Scholar] [CrossRef] [Green Version]
- Kunji, E.R.; Robinson, A.J. Coupling of proton and substrate translocation in the transport cycle of mitochondrial carriers. Curr. Opin. Struct. Biol. 2010, 20, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tajkhorshid, E. Electrostatic funneling of substrate in mitochondrial inner membrane carriers. Proc. Natl. Acad. Sci. USA 2008, 105, 9598–9603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, J.M.; Khalid, S.; Sansom, M.S. Conformational dynamics of the mitochondrial ADP/ATP carrier: A simulation study. Mol. Membr. Biol. 2008, 25, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Dehez, F.; Pebay-Peyroula, E.; Chipot, C. Binding of ADP in the mitochondrial ADP/ATP carrier is driven by an electrostatic funnel. J. Am. Chem. Soc. 2008, 130, 12725–12733. [Google Scholar] [CrossRef] [PubMed]
- Ruprecht, J.J.; King, M.S.; Zogg, T.; Aleksandrova, A.A.; Pardon, E.; Crichton, P.G.; Steyaert, J.; Kunji, E.R.S. The Molecular Mechanism of Transport by the Mitochondrial ADP/ATP Carrier. Cell 2019, 176, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Mao, X.; Yao, S.; Yi, Q.; Xu, Z.M.; Cang, X. Function-related asymmetry of the specific cardiolipin binding sites on the mitochondrial ADP/ATP carrier. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183466. [Google Scholar] [CrossRef]
- Yao, S.; Yi, Q.; Ma, B.; Mao, X.; Chen, Y.; Guan, M.-X.; Cang, X. Structural Basis of Substrate Recognition by the Mitochondrial ADP/ATP Transporter. BioRxiv 2021, preprint. [Google Scholar] [CrossRef]
- Montalvo-Acosta, J.J.; Kunji, E.R.S.; Ruprecht, J.J.; Dehez, F.; Chipot, C. Structure, substrate binding, and symmetry of the mitochondrial ADP/ATP carrier in its matrix-open state. Biophys. J. 2021, 120, 5187–5195. [Google Scholar] [CrossRef]
- Falconi, M.; Chillemi, G.; Di Marino, D.; D’Annessa, I.; Morozzo della Rocca, B.; Palmieri, L.; Desideri, A. Structural dynamics of the mitochondrial ADP/ATP carrier revealed by molecular dynamics simulation studies. Proteins 2006, 65, 681–691. [Google Scholar] [CrossRef]
- Ardalan, A.; Sowlati-Hashjin, S.; Oduwoye, H.; Uwumarenogie, S.O.; Karttunen, M.; Smith, M.D.; Jelokhani-Niaraki, M. Biphasic Proton Transport Mechanism for Uncoupling Proteins. J. Phys. Chem. 2021, 125, 9130–9144. [Google Scholar] [CrossRef]
- Nelson, D.R.; Lawson, J.E.; Klingenberg, M.; Douglas, M.G. Site-directed mutagenesis of the yeast mitochondrial ADP/ATP translocator. Six arginines and one lysine are essential. J. Mol. Biol. 1993, 230, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Muller, V.; Basset, G.; Nelson, D.R.; Klingenberg, M. Probing the role of positive residues in the ADP/ATP carrier from yeast. The effect of six arginine mutations of oxidative phosphorylation and AAC expression. Biochemistry 1996, 35, 16132–16143. [Google Scholar] [CrossRef] [PubMed]
- Thompson, K.; Majd, H.; Dallabona, C.; Reinson, K.; King, M.S.; Alston, C.L.; He, L.; Lodi, T.; Jones, S.A.; Fattal-Valevski, A.; et al. Recurrent De Novo Dominant Mutations in SLC25A4 Cause Severe Early-Onset Mitochondrial Disease and Loss of Mitochondrial DNA Copy Number. Am. J. Hum. Genet. 2016, 99, 1405. [Google Scholar] [CrossRef]
- Mielke, C.; Lefort, N.; McLean, C.G.; Cordova, J.M.; Langlais, P.R.; Bordner, A.J.; Te, J.A.; Ozkan, S.B.; Willis, W.T.; Mandarino, L.J. Adenine Nucleotide Translocase Is Acetylated in Vivo in Human Muscle: Modeling Predicts a Decreased ADP Affinity and Altered Control of Oxidative Phosphorylation. Biochemistry 2014, 53, 3817–3829. [Google Scholar] [CrossRef] [Green Version]
- Finlayson, J.; Barakati, N.; Langlais, P.R.; Funk, J.; Zapata Bustos, R.; Coletta, D.K.; Luo, M.; Willis, W.T.; Mandarino, L.J. Site-specific acetylation of adenine nucleotide translocase 1 at lysine 23 in human muscle. Anal. Biochem. 2021, 630, 114319. [Google Scholar] [CrossRef] [PubMed]
- Bertholet, A.M.; Chouchani, E.T.; Kazak, L.; Angelin, A.; Fedorenko, A.; Long, J.Z.; Vidoni, S.; Garrity, R.; Cho, J.; Terada, N.; et al. H(+) transport is an integral function of the mitochondrial ADP/ATP carrier. Nature 2019, 571, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Delano, W. Pymol Molecular Graphics System: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [Green Version]
- MacKerell, A., Jr.; Feig, M.; Rd, B.C. Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D., Jr.; Pastor, R.W. Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J. Phys. Chem. 2010, 114, 7830–7843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 2384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; Vangunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular-Dynamics with Coupling To an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, S.; Kollman, P.A. Settle—An Analytical Version of the Shake and Rattle Algorithm for Rigid Water Models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.F.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Mutations | Simulation Time |
|---|---|---|
| all4A-AAC | K22A + R79A + R235A + R279A | 1.5 μs |
| all3A-AAC | K22A + R79A + R279A | 1 μs |
| R279A-AAC | K22A + R279A | 1 μs |
| R235A-AAC | K22A + R235A | 1 μs |
| R79A-AAC | K22A + R79A | 1.5 μs |
| CATR-AAC | / | 1 μs |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yao, S.; Ma, B.; Yi, Q.; Guan, M.-X.; Cang, X. Investigating the Broad Matrix-Gate Network in the Mitochondrial ADP/ATP Carrier through Molecular Dynamics Simulations. Molecules 2022, 27, 1071. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27031071
Yao S, Ma B, Yi Q, Guan M-X, Cang X. Investigating the Broad Matrix-Gate Network in the Mitochondrial ADP/ATP Carrier through Molecular Dynamics Simulations. Molecules. 2022; 27(3):1071. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27031071
Chicago/Turabian StyleYao, Shihao, Boyuan Ma, Qiuzi Yi, Min-Xin Guan, and Xiaohui Cang. 2022. "Investigating the Broad Matrix-Gate Network in the Mitochondrial ADP/ATP Carrier through Molecular Dynamics Simulations" Molecules 27, no. 3: 1071. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27031071