
Resolution of a Configurationally Stable Hetero[4]helicene
by
 , , , and
, , , and
Michela Lupi
1 ,
,
Martina Onori
1,
Stefano Menichetti
1,
Sergio Abbate
2,
Giovanna Longhi
2 and
Caterina Viglianisi
1,*
1
Department of Chemistry “Ugo Schiff” (DICUS), University of Florence, Via della Lastruccia 13, Sesto Fiorentino (FI), 50019 Florence, Italy
2
Department of Molecular and Translational Medicine (DMMT), University of Brescia, V.le Europa 11 Brescia (BS), 25121 Brescia, Italy
*
Author to whom correspondence should be addressed.
Molecules 2022, 27(4), 1160; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27041160
Submission received: 31 December 2021
/
Revised: 2 February 2022
/
Accepted: 4 February 2022
/
Published: 9 February 2022
(This article belongs to the Special Issue Women in Bioorganic Chemistry)
Abstract
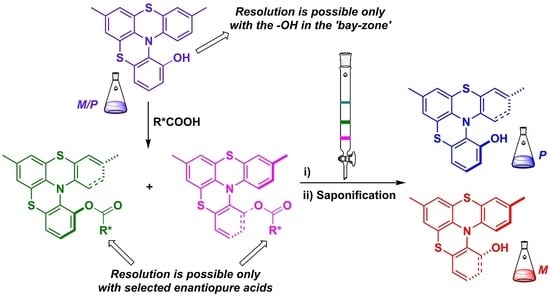
:We have developed an efficient chemical resolution of racemic hydroxy substituted dithia-aza[4]helicenes (DTA[4]H) 1(OH) using enantiopure acids as resolving agents. The better diastereomeric separation was achieved on esters prepared with (1S)-(−)-camphanic acid. Subsequent simple manipulations produced highly optically pure (≥ 99% enantiomeric excess) (P) and (M)-1(OH) in good yields. The role of the position where the chiral auxiliary is inserted (cape- vs. bay-zone) and the structure of the enantiopure acid used on successful resolution are discussed.
1. Introduction
Chirality is one of the most crucial assets of nature and is of paramount importance in several areas of science, technology and medicine. Molecular chirality has been recognized for a long time and has provided guidance in the design of drugs and functional materials. Furthermore, a smart combination of chiral phenomena and supramolecular chemistry resulted in an emerging interdisciplinary field called supramolecular chirality [1].
Helicenes are compounds with a screw-shaped skeleton formed by ortho-condensed (hetero)aromatic rings with a non-planar structure due to the steric superimposition of terminal rings or/and the substituents on these rings [2], which force the molecule to adopt a helical conformation (Figure 1). This important class of axially chiral compounds has a barrier of interconversion between M and P enantiomers increasing with the increase of the ring number forming the helicene backbone.
Circular dichroism (CD) and circularly polarized luminescence (CPL) are just a few of the chiroptical proprieties that make helicenes valuable in potential applications such as advanced optical information storage, circularly polarized organic light-emitting diodes (CP-OLEDs), circularly polarized light detecting organic field effect transistors (CP-OFETs), chirality-induced spin selectivity (CISS) devices and stereoselective sensing chiroptical probes in biological processes [3,4,5,6,7,8,9,10,11,12,13,14].
Recently, increased attention has focused on the binding of small molecules to specific DNA structures to inhibit the biological functions in which these structures participate. Indeed, helicenes enantioselectivity offered a way to rationally design Z-DNA-depending inhibitors of biological functions [11].
Over the years, a variety of heterohelicenes and helical-shaped molecules, containing nitrogen, oxygen, sulfur, and other hetero-elements, have been synthesized and their unique properties studied. Great effort has been devoted to the setting of new simple and multi-gram synthetic procedures that allow for the isolation of helicenes in enantiopure form as required for various practical applications, including chirogenesis [7,15,16].
Dithia-aza[4]helicenes (DTA[4]H) 1 (Scheme 1) can be described as bis-phenothiazines with an aryl ring and a nitrogen atom in common forced into a helical shaped structure by the four long carbon–sulfur bonds. These [1,4]benzothiazino[2,3,4-kl]phenothiazines represent one of the attractive rare examples of geometrically stable [4]helicenes with racemization barriers higher than those measured for all carbon [5]helicenes.
DTA[4]H are obtained [17,18,19,20,21], as racemic mixture, from properly substituted triarylamines (TAA) 2 or N-aryl phenothiazines (PTZ) 3 (Scheme 1, pathway A and B respectively), through regioselective sulfenylation(s) with two or one equivalents of phthalimidesulfenyl chloride (PhtNSCl (4), Pht = phthaloyl). Reacting the resulting sulfenylated derivatives 5 or 6 with a Lewis Acid (L.A.), typically BF3OEt2 or AlCl3, causes two or one electrophilic intramolecular cyclization with formation of helicenes 1.
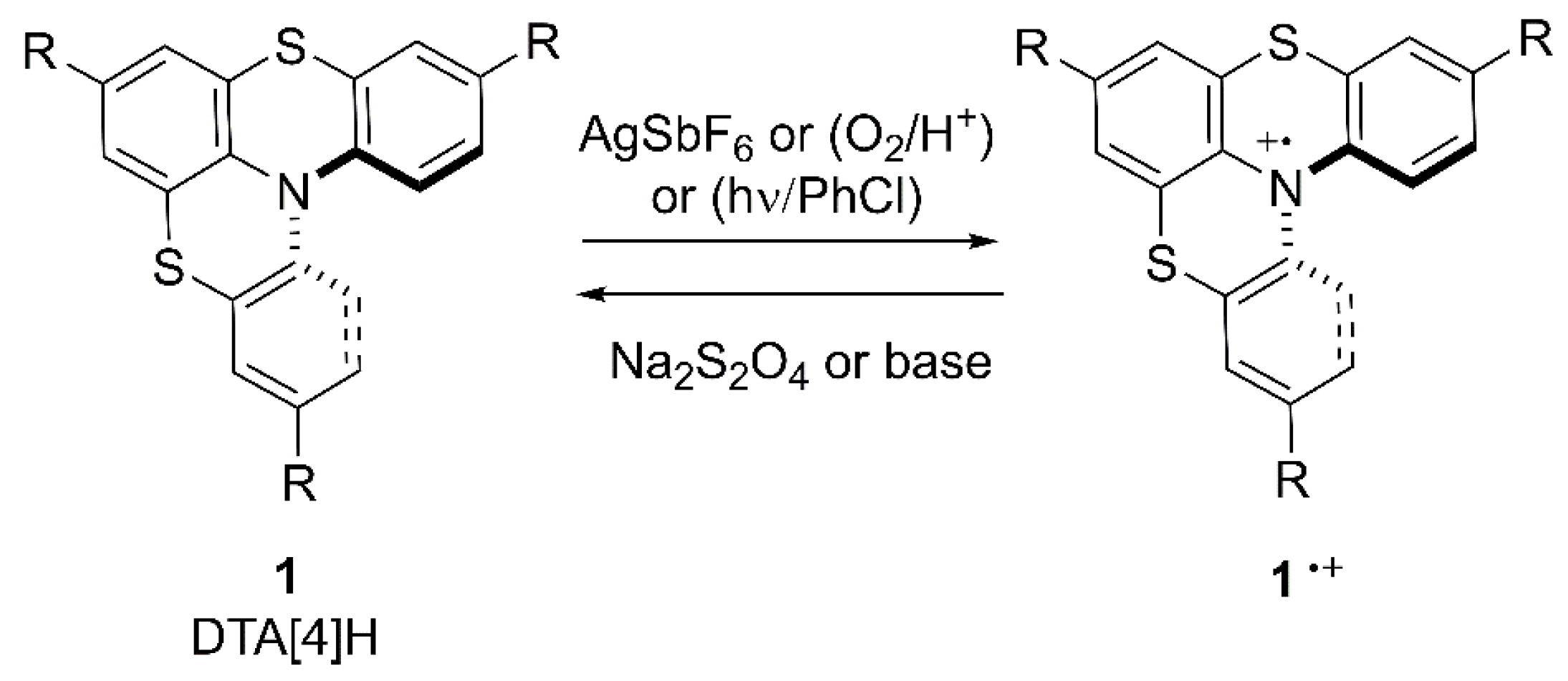
Along with their peculiar helical-shaped structure, derivatives 1 show a very good one-electron donor ability, being easily, and reversibly, oxidized to the corresponding exceptionally stable crystalline chiral radical cations 1•+ [18,21] (Scheme 2). We have also demonstrated that the oxidative process is extremely sensitive to the medium, and under acidic conditions, molecular oxygen becomes an efficient single electron transfer (SET) oxidant, giving rise to the formation of 1•+. Furthermore, radical cations 1•+ can be generated also via irradiation at 240–400 nm of helicenes in the presence of PhCl [21] (Scheme 2).
Indeed, we have also prepared, via ring-opening metathesis polymerization, dithia-aza[4]helicene functionalized polynorbornenes showing a pH depending reversible redox behavior as a new class of tunable material reversibly switchable by pH-triggered redox processes [22].
The availability of differently functionalized enantiomerically pure helicenes, avoiding the limitation associated with chiral HPLC resolution [17,23] is mandatory for the development of the appealing applications of these peculiar systems [21,22,24]. Therefore, in recent years we tried to set off regio-, stereo- and enantioselective synthetic approaches for the preparations of 1.
Actually, the synthetic procedure depicted in Scheme 1, while failing to control the absolute stereochemistry of the process, allowed for the control of the regiochemistry during ring closure as well as the possibility of inserting different substituents in specific positions. Thus, we have studied the chemical resolutions of 1 using the classical temporary insertion of chiral auxiliaries.
The DTA[4]H 1 derivatives requested for the above described applications require the insertion, as an anchoring unit, of a hydroxyl group in different positions of the helical backbone. Thus, we decided to take advantage of these phenolic groups for the introduction of chiral auxiliaries through esterification with enantiopure acids 7 and in order to verify whether the diastereoisomeric mixture of esters 8 obtained can be separated. Herein we report how the helicene topology and chiral auxiliary structure could be matched to allow the resolution of phenolic DTA[4]H 1(OH) (Scheme 3).
2. Results and Discussion
Selected DTA[4]H 1 can be resolved through HPLC in the chiral stationary phase as we previously reported [17,23]; however, this method is unsuitable to obtain enantiopure DTA[4]H in multigram scale. Instead, the diastereomeric process-based resolution has advantages in the the viewpoint of cost, generality and the amount of enantiopure products achieved. Thus, we decided to study the insertion of chiral auxiliaries, for example through esterification reactions, to verify whether the mixture of diastereomers obtained can be separated by flash chromatography allowing isolation of pure M and P DTA[4]H in relevant quantity.
We have demonstrated that N-arylphenothiazines PTZ 3 are suitable substrates for the synthesis of unsymmetrically hydroxy substituted helicenes 1(OH) [20]. We selected helicene 1a(OH) and 1b(OH) (Scheme 4) to prepare diastereoisomeric esters using enantiopure acid 7 (Scheme 5).
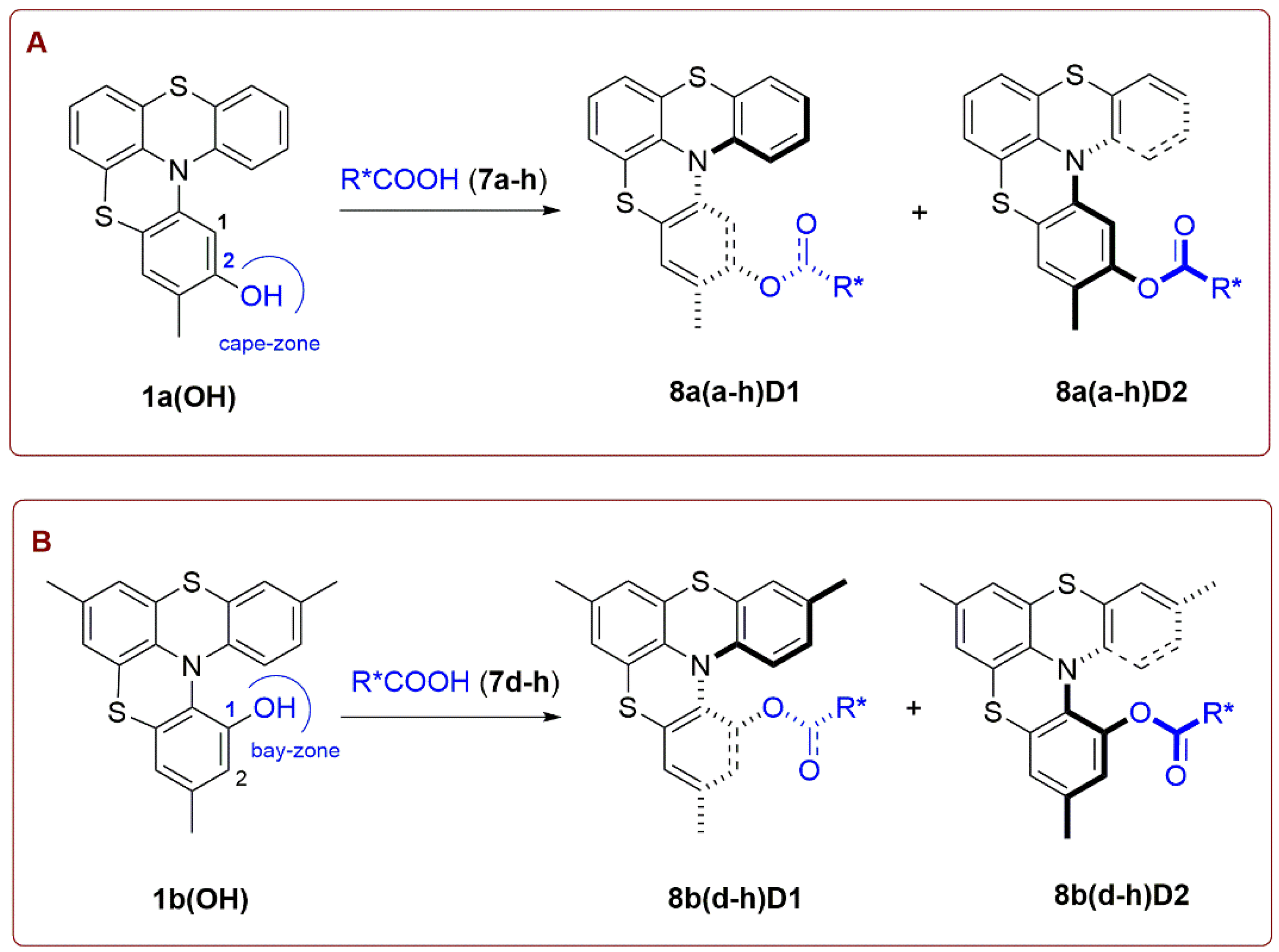
Firstly, we planned the introduction of chiral auxiliaries in 2-hydroxy-substituted ADT[4]H 1a(OH) presenting a hydroxyl group in the 2-position (that we indicate as cape-zone) of the helicene, which was relatively easy to prepare [20].
Racemic 1a(OH) was esterified with different enantiopure acids 7a–h yielding a diastereomeric mixture (D1+D2) of esters 8a(a–h). Esterification reactions were carried out in presence of diisopropylcarbodiimide (DIC) and 4-dimethylaminopyridine (DMAP) as catalysts, in dry CH2Cl2 at room temperature (Scheme 5A).
Regardless, all chiral acids 7a–h (Scheme 5 panel A and Table 1) allowed the formation of diastereomeric esters 8a(a–h) (Scheme 5 panel A and Table 1) in good yields, in none of these cases it was possible to separate the diastereoisomeric mixtures by flash chromatography or crystallization.
We thought, as suggested by literature data [25,26,27,28,29,30,31,32,33,34,35,36,37,38], that functionalization of the cape-zone position keeps the chiral auxiliaries too far from the superimposition area of the terminal aryl rings vanishing separation. Thus, we moved to helicene 1b(OH), which allowed for the insertion of chiral auxiliaries on 1-position, ortho to the nitrogen atom, which we indicated as bay-zone, i.e., exactly in the area of terminal ring superimposition (Scheme 5B).
Using chiral acids 7d–h (Scheme 5 panel B and Table 1), the corresponding diastereomeric esters 8b(d–h) (Scheme 5 panel B and Table 1)were obtained in moderate yields generally lower than those of the corresponding esters prepared using phenol 1a(OH), indicating, as expected, a more difficult access to the OH group of 1b(OH) laying the bay-zone (Table 1). For several esters 8b it was possible to identify the presence of the two diastereomers (D1 and D2, chromatographic elution order) by 1H and 13C NMR, and, eventually, to reveal a slightly different chromatographic behavior on TLC (Figure 2).
Despite the introduction of the chiral auxiliary in the bay-zone diastereoisomeric esters D1 and D2 of 8b(d–f) were not separable by flash chromatography in spite of an accurate selection of eluent mixtures. However, esterification of 1b(OH) with N-boc pipecolic acid 7g allowed for the partial separation by flash chromatography on silica gel of diastereomers 8bgD1 and 8bgD2, which were characterized by 1H and 13C NMR. Optical rotation of 8bgD1 was: −157 (c 0.1, CH2Cl2), while for 8bgD2 was: +49 (c 0.1, CH2Cl2). 1H NMR spectra show that the product 8bgD1 was isolated as single diastereomer, while the product 8bgD2 was isolated as a roughly 3:1 mixture of the two diastereomers.
Esters 8bgD1 and 8bgD2 were hydrolysed with 3 eq. of NaOH in CH2Cl2/MeOH to give enantiomeric phenols (M)-1b(OH) and (P)-1b(OH). Phenols were analysed by HPLC in the chiral stationary phase in order to calculate the enantiomeric ratio. Chromatograms showed that product (M)-1b(OH) −161 (c 0.1, CH2Cl2) was obtained as single enantiomer (e.e. ≥ 99%), while helicene (P)-1b(OH) +75 (c 0.1, CH2Cl2) exhibits the enantiomeric ratio 72:28 (e.e. = 44%).
Esterification of 1b(OH) with (1S)-(−)-camphanic acid 7h provided, with our great satisfaction, diastereomers 8bhD1 and 8bhD2 that were successfully separated by flash chromatography and characterized by 1H and 13C NMR spectroscopy (Figure 3 and Supplementary Materials).
Optical rotation was measured and gave −129 (c 0.1, CH2Cl2) for 8bhD1 and + 126, (c 0.1, CH2Cl2) for 8bhD2. Hydrolysis of diastereomeric esters provided helicenes (M)-1b(OH) and (P)-1b(OH), respectively. HPLC analysis with a chiral stationary phase showed that the first eluted product, (P)-1b(OH) +166 (c 0.1, CH2Cl2), exhibits an enantiomeric ratio = 98:2 (e.e. = 96%), while the second eluted product, (M)-1b(OH) −167 (c 0.1, CH2Cl2), was obtained as a single enantiomer (e.e. ≥ 99%), (Scheme 6).
The assignment of the absolute configuration of DTA[4]H 1 derivatives has been established as P-(+) and M-(−), which is typical for helicene systems; opposite assignment is quite unusual, as we have established in ref [17,19]. In this work the absolute configuration of 1b(OH) was validated by comparison of the electronic circular dichroism (ECD) spectra of the two optical enantiomers-(+)-1b(OH) and (−)-1b(OH), assigned to (P)-1b(OH) and (M)-1b(OH), with the calculated spectrum of the M structure.
In order to assign the configuration, DFT and TD-DFT calculations have been conducted with the Gaussian16 package [39]. Two orientations are possible for the hydroxyl-group; the two optimized structures in the M configuration are reported in Figure 4 with their Boltzmann populations. Two functionals have been considered; the two differing in the amount of the exact exchange included M06 with 27% HF exchange and M06-2X with 54% HF exchange [40]. The solvent has been treated at the iefpcm level. Structural results are similar for the two functionals.
CD and absorption spectra have been calculated at the same level of theory, a constant Gaussian 0.2 eV bandwidth was applied to each transition. The experimental CD and absorption spectra have been recorded for the two enantiomers in 4.2 mM dichloromethane solution in a 0.1 mm quartz cuvette using a JASCO-815SE instrument.
The comparison of experimental data with calculations are presented in Figure 5. In order to compare with data, +4 nm shift has been applied to the results obtained with M06, +26 nm with M06-2X; calculation of similarity index between experimental and calculated spectra suggested the best shift for the best correspondence, as reported in the Supplementary Materials paragraph. It is also shown that the two conformers give very similar spectra, while in Figure 5 the Boltzmann weighed average is presented. Both functionals permit confirmation of the configuration as M-(-) (correspondingly P-(+)). This conclusion agrees with what was obtained for the parent molecule triarylamine hetero[4]helicene examined in reference [19].
Overall, our results confirm that, on chemical resolution of helicenes, the position where the chiral auxiliaries are inserted is extremely important, being the bay-zone that allows for the higher effect on enantiomeric discrimination. At the same time we have confirmed previous studies reporting chromatographic resolutions of [7]carbo- and [7]hetero-helicenes by means of tetra- and monocamphanate esters [25,26,27,28,29,30,31]. In each of these cases, the (1S)-camphanate of the (P)-helicenol moves more slowly upon chromatography on silica gel than the (1S)-camphanate of the (M)-helicenol [27].
3. Materials and Methods
1H and 13C NMR spectra were recorded with Varian Mercury Plus 400, Varian Inova 400, using CDCl3 as solvent. Residual CHCl3 at δ = 7.26 ppm and central line of CDCl3 at δ = 77.16 ppm were used as the reference of 1H-NMR spectra and 13C NMR spectra, respectively. FT-IR spectra were recorded with a Spectrum Two FT-IR Spectrometer. ESI-MS spectra were recorded with a JEOL MStation JMS700. Melting points were measured with a Stuart SMP50 Automatic Melting Point Apparatus. Optical rotation measurements were performed on a JASCO DIP-370 polarimeter (JASCO, Easton, MD, USA) and the specific rotation of compounds was reported [41].
All the reactions were monitored by TLC on commercially available precoated plates (silica gel 60 F 254) and the products were visualized with acidic vanillin solution. Silica gel 60 (230–400 mesh) was used for column chromatography. Dry solvents were obtained by The PureSolv Micro Solvent Purification System. Chloroform was washed with water several times and stored over calcium chloride. Pyridine and TEA were freshly distilled from KOH. Phthalimide sulfenyl chloride was prepared from the corresponding disulfide (purchased from Chemper snc) as reported elsewhere. Helicenes 1a and 1b were described elsewhere [17].
General Procedure for the synthesis of diastereomeric esters from 1 by Steglich esterification: To a solution of 1 in dry CH2Cl2 (roughly 0.03–0.04 M), the enantiopure acid 7 (1.2 eq), DMAP (0.1 eq) and DIC (1–1.2 eq) were added at 0 °C. The solution was stirred at room temperature under a nitrogen atmosphere for 2–29 h, then was diluted with CH2Cl2 (60 mL), washed with a saturated solution of NH4Cl (2 × 40 mL), with a saturated solution of NaHCO3 (3 × 40) then with NH4Cl (3 × 40 mL). The organic layer was dried over Na2SO4, filtered and evaporated under reduced pressure. The crude was purified by flash chromatography on silica gel.
Diastereoisomers 8aaD1 and 8aaD2. (M/P)-3-methyl[1,4]benzothiazino[2,3,4-kl]phenothiazine-2-yl (S)-perillate. Following the general Steglich esterification procedure from 1a(OH) (60 mg, 0.18 mmol) and (S)-(−)-perillic acid 7a (36 mg, 0.22 mmol), kept for 22 h at rt. The crude was purified by flash chromatography on silica gel (petroleum ether/ CH2Cl2 1:3, Rf 0.86) to afford the mixture of the two diastereomeric compounds 8aaD1 and 8aaD2 (52 mg, 60% yield) as a white solid (mp 105–115 °C). 1H NMR (400 MHz, CDCl3)* δ: 1.45- 1.55 (m, 2H), 1.76 (s, 6H), 1.89–1.95 (m, 2H), 2.12 (s, 6H), 2.16–2.41 (m, 8H), 2.51–2.59 (m, 2H), 4.74 (bs, 2H), 4.78 (bs, 2H), 6.89 (bs, 2H) 6.92–7.05 (m, 8H), 7.06 (bs, 2H), 7.12–7.25 (m, 8H) ppm. 13C NMR (100 MHz, CDCl3)* δ: 15.8, 20.9, 24.76, 24.80, 27.1, 31.4, 40.1, 108.63, 108, 64, 110.0, 110.6, 114.7, 120.6, 124.1, 124.8, 125.0, 125.7, 125.75, 125.83, 126.0, 127.0, 127.3, 127.7, 128.0, 129.3, 129.5, 139.5, 141.2, 141.8, 142.5, 148.7, 149.2, 165.2, 165.3 ppm (34 signals for 58 different carbons). Elem. Anal. for C29H25NO2S2: Calcd. C 72.02, H 5.21, N 2.90; found C 71.80; H 5.21, N 2.89. *Et3N was added to neutralize CHCl3 acidity.
Diastereoisomers 8abD1 and 8abD2. (M/P)-3-methyl[1,4]benzothiazino[2,3,4-kl]phenothiazine-2-yl (1S)-10-camphorsulfonate. To a solution of 1a(OH) (60 mg; 0.18 mmol) and TEA (22 mg, 0.22 mmol) in 4 mL of dry CH2Cl2, (1S)-(+)-10-camphorsulfonyl chloride 7b (51 mg, 0.20 mmol) is added at 0 °C. After 10 min the solution was allowed to warm at room temperature and was stirred for 18 h under a nitrogen atmosphere. The mixture was diluted with AcOEt (25 mL) and washed with water (3 × 20 mL). The organic layer was dried over Na2SO4, filtered, and then evaporated under reduced pressure. The crude was purified by flash chromatography on silica gel (petroleum ether/AcOEt: 5/1, Rf 0.38) to afford the mixture of the two diastereomeric compounds 8abD1 and 8abD2 (55 mg, 56% yield) as a white solid (mp 190–195 °C). 1H NMR (400 MHz, CDCl3) δ: 0.87 (s, 3H), 0.88 (s, 3H), 1.11 (s, 6H), 1.40–1.47 (m, 2H), 1.64–1.72 (m, 2H), 1.92–1.98 (m, 2H), 2.00–2.13 (m, 4H), 2.31 (s, 3H), 2.32 (s, 3H), 2.36–2.57 (m, 4H), 3.13–3.18 (m, 2H), 3.73–3.78 (m, 2H), 6.93–7.25 (m, 18H) ppm. 13C NMR (100 MHz, CDCl3) δ: 11.6, 16.4, 16.5, 19.8, 20.02, 20.03, 25.3, 26.9, 27.0, 42.5, 43.0, 43.1, 46.3, 48.01, 48.03, 48.4, 48.6, 58.19, 58.24, 114.7, 114.8, 120.45, 120.50, 125.1, 125.2, 125.4, 125.5, 125.7, 125.8, 125.9, 126.0, 126.1, 126.2, 127.1, 127.8, 127.9, 128.11, 128.13, 128.2, 128.4, 130.0, 130.1, 139.2, 141.29, 141.32, 142.26, 142.29, 147.0, 147.1, 213.8, 213.9 ppm. Elem. Anal. for C29H27NO4S3: Calcd. C 63.36, H 4.95, N 2.55; found C 63.38, H 4.95, N 2. 54.
Diastereoisomers 8acD1 and 8acD2. (M/P)-3-methyl[1,4]benzothiazino[2,3,4-kl]phenothiazine-2-yl o,o’-dibenzoyl-L-tartrate. Following the general procedure from 1a(OH) (85 mg, 0.25 mmol) and (-)-o,o’-dibenzoyl-L-tartaric acid mono(dimethyl amide) 7c (117 mg, 0.30 mmol), kept for 2 h at room temperature. The crude was purified by flash chromatography on silica gel (AcOEt/CH2Cl2 1/20, Rf 0.65) to afford the mixture of the two diastereomeric compounds 8acD1 and 8acD2 (99 mg, 56% yield) as a white solid (mp 102–106 °C). 1H NMR (400 MHz, CDCl3) δ: 2.09 (s, 3H), 2.10 (s, 3H), 2.92 (s, 3H), 2.93 (s, 3H), 3.17 (s, 3H), 3.30 (s, 3H), 6.16 (d, 1H, J = 4.8 Hz), 6.20 (d, 1H, J = 5.2 Hz), 6.31 (d, 1H, J = 4.8 Hz), 6.33 (d, 1H, J = 5.2 Hz), 6.83–7.20 (m, 18H), 7.44–7.54 (m, 8H), 7.54–7.58 (m, 4H), 7.99–8.08 (m, 8H) ppm. 13C NMR (100 MHz, CDCl3) δ: 15.61, 15.63, 36.27, 36.31, 37.2, 69.5, 69.6, 70.9, 113.8, 114.0, 120.35, 120.41, 124.8, 124.9, 125.0, 125.1, 125.5, 125.6, 125.72, 125.74, 125.86, 125.92, 126.75, 126.84, 126.9, 127.6, 127.7, 127.9, 128.0, 128.35, 128.43, 128.52, 128.53, 128.58, 128.59, 128.60, 129.58, 129.59, 130.0, 130.09, 130.12, 130.15 133.82, 133.86, 139.12, 139.13, 141.1, 141.2, 142.1, 142.3, 148.09, 148.10, 165.0, 165.1, 165.3, 165.43, 165.44, 165.45, 165.50 ppm (59 signals for 78 carbons). IR (ATR solid) n: 3063, 2929, 1763, 1724, 1664, 1477, 1432, 1240 cm−1. Elem. Anal. for C39H30N2O7S2: Calcd. C 66.65, H 4.30, N 3.99; found C 66.61, H 4.28, N 4.00.
Diastereoisomers 8adD1 and 8adD2. (M/P)-3-methyl[1,4]benzothiazino[2,3,4-kl]phenothiazine-2-yl (1S)-ketopinate. Following the general Steglich esterification procedure from 1a(OH) (60 mg, 0.18 mmol) and (1S)-(+)-ketopinic acid 7d (56 mg, 0.31 mmol), kept for 29 h at room temperature. The crude was purified by flash chromatography on silica gel (petroleum ether/CH2Cl2 1/3, Rf 0.45) to afford the mixture of the two diastereomeric compounds 8adD1 and 8adD2 (71 mg, 79% yield) as a white solid (mp 130–133 °C). 1H NMR (400 MHz, CDCl3)* δ: 1.14 (s, 6H), 1.21 (s, 3H), 1.22 (s, 3H), 1.42–1.48 (m, 2H), 1.88–1.97 (m, 3H), 2.01–2.10 (m, 3H), 2.13–2.15 (m, 2H), 2.18 (s, 3H), 2.19 (s, 3H), 2.40–2.48 (m, 2H), 2.54–2.60 (m, 2H), 6.85 (s, 1H), 6.87 (s, 1H), 6.94–7.06 (m, 10H), 7.12–7.23 (m, 6H) ppm. 13C NMR (100 MHz, CDCl3)* δ: 16.2, 20.0, 21.50, 21.52, 26.49, 26.52, 26.7, 26.8, 44.02, 44.58, 44.61, 49.51, 49.56, 68.20, 68.24, 110.6, 114.70, 114.72, 120.41, 120.44, 124.6, 124.8, 124.9, 125.0, 125.70, 125.72, 125.8, 126.1, 126.8, 127.2, 127.3, 127.75, 127.82, 128.1, 129.59, 129.61, 139.4, 140.9, 141.0, 142.66, 142.71, 148.8, 168.1, 210.3 ppm (44 signals for 58 different carbons). Elem. Anal. for C29H25NO3S2: Calcd. C 69.71, H 5.04, N 2.80; found: C 69.73, H 5.01, N 2.77. *Et3N was added to neutralize CHCl3 acidity.
Diastereoisomers 8aeD1 and 8aeD2. (M/P)-3-methyl[1,4]benzothiazino[2,3,4-kl]phenothiazine-2-yl (S)-2-(6-methoxy-2-naphthyl) propionate. Following the general Steglich esterification procedure from 1a(OH) (60 mg, 0.18 mmol) and (S)-(+)-2-(6-methoxy-2-naphthyl) propionic acid 7e (50 mg, 0.22 mmol), kept for 20 h at room temperature. The crude was purified by flash chromatography on silica gel (petroleum ether/CH2Cl2 1/3, Rf 0.80) to afford the mixture of the two diastereomeric compounds 8aeD1 and 8aeD2 (94 mg, 94% yield) as an orange solid (mp 132–136 °C). 1H NMR (400 MHz, CDCl3)* δ: 1.39 (d, 6H, J = 6.5 Hz), 1.77 (s, 3H), 1.79 (s, 3H), 3.39 (s, 6H), 3.84 (q, 1H, J = 6.6 Hz), 4.12 (q, 1H, J = 6.7 Hz), 6.81 (bs, 2H), 6.91–7.05 (m, 10H), 7.10–7.20 (m, 10H), 7.44–7.47 (m, 2H), 7.67–7.73 (m, 6H) ppm. 13C NMR (100 MHz, CDCl3)* δ: 15.46, 15.48, 18.5, 18.6, 20.6, 21.2, 22.7, 23.6, 42.3, 45.56, 45.59, 55.4, 105.7, 108.6, 110.6, 114.4, 114.5, 119.3, 120.5, 124.3, 124.8, 125.0, 125.7, 125.8, 125.96, 126.01, 126.3, 126.4, 126.9, 127.1, 127.2, 127.4, 127.7, 127.8, 127.98, 128.01, 129.0, 129.4, 129.5, 133.9, 134.95, 134.99, 139.4, 141.0, 142.50, 142.54, 148.9, 157.9, 172.7 ppm (49 signals for 66 different carbons). Elem. Anal. for C33H25NO3S2: Calcd. C 72.37, H 4.60, N 2.56; found C 72.29; H 4.58, N 2.57. *Et3N was added to neutralize CHCl3 acidity.
Diastereoisomers 8afD1 and 8afD2. (M/P)-3-methyl[1,4]benzothiazino[2,3,4-kl]phenothiazine-2-yl mono-(1R)-menthylphthalate. Following the general procedure from 1a(OH) (70 mg, 0.21 mmol) and (-)mono(1R)-menthylphthalate 7f (76 mg, 0.25 mmol), kept for 24 h at room temperature. The crude was purified by flash chromatography on silica gel (petroleum ether/ CH2Cl2 2/1, Rf 0.78)) to afford the mixture of the two diastereomeric compounds 8afD1 and 8afD2 (70 mg, 54% yield) as a white solid (mp 140.8–146.8 °C). 1HNMR (400 MHz, CDCl3) δ: 0.73–0.69 (m, 6H), 0.83–0.89 (m, 14H), 0.97–1.16 (m, 4H), 1.36–1.51 (m, 4H), 1.64–1.71 (m, 4H), 1.85–1.98 (m, 2H), 2.05–2.11 (m, 2H), 2.21 (s, 3H), 2.22 (s, 3H), 4.82–4.89 (m, 2H), 6.93–7.00 (m, 6H), 7.03 (dd, 2H, J = 1.2 Hz, J = 7.5 Hz), 7.08 (bs, 1H), 7.09 (bs, 1H), 7.11–7.16 (m, 2H), 7.18 (d, 2H, J = 7.7 Hz), 7.24–7.29 (m, 2H), 7.54–7.60 (m, 4H), 7.68–7.73 (m, 2H), 7.85–7.90 (m, 2H) ppm. 13C NMR (100 MHz, CDCl3) δ: 15.19, 16.0, 16.3, 16.5, 20.89, 20.93, 22.15, 22.18, 23.5, 23.6, 26.3, 26.4, 31.55, 31.57, 34.37, 34.41, 40.6, 40.7, 47.23, 47.25, 75.95, 75.02, 114.47, 114.49, 120.60, 120.64, 124.5, 124.6, 124.9, 125.0, 125.71, 125.75, 125.77, 125.79, 126.07, 126.08, 126.95, 126.99, 127.33, 127.34, 127.71, 127.75, 127.98, 128.00, 128.8, 129.0, 129.51, 129.52, 129.56, 129.60, 130.6, 130.89, 130.94, 131.0, 131.8, 131.9, 133.5, 133.7, 139.5, 141.2, 142.50, 142.52, 148.97, 148.99, 165.3, 165.4, 166.8, 166.9 ppm (68 signals for 74 different carbons). IR (ATR solid) n: 2953, 2923, 2867, 1749, 1715, 1477, 1431, 1276, 1236 cm−1. Elem. Anal. for C37H35NO4S2: Calcd. C 71.47, H 5.67, N 2.25; found C 71.36, H 5.65, N 2.26.
Diastereoisomers 8agD1 and 8agD2. (M/P)-3-methyl[1,4]benzothiazino[2,3,4-kl]phenothiazine-2-yl (S)-N-Boc pipecolinate. Following the general procedure from 1a(OH) (61 mg, 0.18 mmol) and (S)-N-Boc pipecolic acid 7g (50 mg, 0.22 mmol) kept for 22 h at room temperature. The crude was purified by flash chromatography (CH2Cl2, Rf 0.51) on silica gel to afford the mixture of the two diastereomeric compounds 8agD1 and 8agD2 (70 mg, 69%yield) as a white solid (mp 106–117 °C). 1H NMR (400 MHz, CDCl3)* δ: 1.37–1.44 (m, 22H), 1.65–1.80 (m, 8H), 2.13 (s, 6H), 2.29–2.33 (m, 2H), 2.93–3.09 (m, 2H), 3.89–3.95 (m, 1H), 4.04–4.09 (m, 2H), 4.92 (bs, 1H), 5.07 (bs, 1H), 7.21–6.77 (m, 18H) ppm. Elem. Anal. For C30H30N2O4S2: Calcd. C 65.91, H 5.53, N 5.12; found: C 65.35, H 5.31, N 4.98. *Et3N was added to neutralize CHCl3 acidity.
Diastereoisomers 8ahD1 and 8ahD2. (M/P)-3-methyl[1,4]benzothiazino[2,3,4-kl]phenothiazine-2-yl (1S)-camphanate. Following a Steglich esterification procedure from 1a(OH) (60 mg, 0.18 mmol) and (1S)-(−)-camphanic acid 7h (43 mg, 0.22 mmol), kept for 17 h at room temperature. The crude was purified by flash chromatography on silica gel (petroleum ether/AcOEt/diethyl ether: 10:1:3, Rf 0.43)) to afford the mixture of the two diastereomeric compounds 8ahD1 and 8ahD2 (52 mg, 57% yield) as a white solid (mp 170 °C dec). 1H NMR (400 MHz, CDCl3)* δ: 1.04 (s, 3H), 1.06 (s, 3H), 1.11 (s, 6H), 1.13 (s, 3H), 1.14 (s, 3H), 1.69–1.77 (m, 2H), 1.92–1.99 (m, 2H), 2.11–2.19 (m, 8H), 2.45–2.54 (m, 2H), 6.84 (s, 1H), 6.86 (s, 1H), 6.93–7.00 (m, 6H), 7.02–7.08 (m, 4H), 7.12–7.21 (m, 6H) ppm. 13C NMR (100 MHz, CDCl3)* δ: 9.78, 9.81, 16.0, 16.2, 16.94, 16.96, 17.01, 17.04, 29.0, 31.2, 31.3. 53.6, 54.6, 54.97, 55.00, 90.89, 90.94, 114.16, 114.21, 120.4, 120.5, 125.0, 125.1, 125.24, 125.27, 125.6, 125.8, 125.9, 126.0, 126.1, 126.50, 126.52, 126.9, 127.0, 127.8, 128.1, 129.8, 139.3, 141.2, 141.3, 142.4, 142.5, 148.1, 148.2, 165.8, 165.9, 177.8 ppm (47 signals for 58 different carbons). Elem. Anal. for C29H25NO4S2: Calcd. C 67.55, H 4.89, N 2.72; found C 67.49, H 4.88, N 2.72. *Et3N was added to neutralize CHCl3 acidity.
Diastereoisomers 8bdD1, 8bdD2. (M/P)-1,3,7-trimethyl[1,4]benzothiazino[2,3,4-kl]phenothiazine-2-yl (1S)-ketopinate. Following procedure from 1b(OH) (40 mg, 0.11 mmol) and (1S)-(+)-ketopinic acid 7d (20 mg, 0.11 mmol), kept for 18 h at room temperature. The crude was purified by flash chromatography on silica gel (petroleum eter/CH2Cl2 1/2, Rf 0.38)) to afford the mixture of the two diastereomeric compounds 8bdD1 and 8bdD2 (32 mg, 55% yield) as a white solid (mp 190–199 °C). 1H NMR (400 MHz, CDCl3) δ: 0.76 (s,3H), 1.01 (s, 3H), 1.06 (s, 3H), 1.11 (s, 3H), 1.18–1.36 (m, 5H), 1.63–1.70 (m, 1H), 1.73–1.90 (m, 4H), 1.94–1.99 (m, 2H), 2.21–2.31 (m, 18H), 2.42–2.49 (m, 2H), 6.77–6.84 (m, 7H), 6.88–7.01 (m, 7H) ppm. 13C NMR (100 MHz, CDCl3) δ: 19.61, 19.64, 20.51, 20.54, 20.61, 20.7, 20.78, 20.82, 20.9, 24.9, 25.3, 26.16, 26.24, 43.7, 43.8, 43.9, 44.0, 48.2, 48.5, 67.5, 67.6, 118.1, 118.6, 123.4, 123.8, 125.45, 125.55, 125.6, 125.78, 125.83, 125.9, 126.0, 126.3, 126.4, 127.0, 127.2, 127.5, 127.8, 128.3, 128.5, 129.4, 130.0, 130.7, 130.9, 133.6, 133.7, 134.8, 135.70, 135.74, 138.0, 138.1, 142.1, 142.4, 142.5, 167.2, 167.8, 210.54, 210.55 ppm (58 signals for 62 carbons). IR (ATR solid) n: 2961, 2920, 2888, 1762, 1737, 1480, 1448, 1313, 1270 cm−1. Elem. Anal. for C31H29NO3S2: Calcd. C 70.56, H 5.54, N 2.65; found C 70.48, H 5.55, N 2.64.
Diastereoisomers 8beD1 and 8beD2. (M/P)-1,3,7-trimethyl[1,4]benzothiazino[2,3,4-kl]phenothiazine-2-yl (S)-2-(6-methoxy-2-naphthyl) propionate. Following the general procedure from 1b(OH) (48 mg, 0.13 mmol) and (S)-(+)-2-(6-methoxy-2-naphthyl) propionic acid 7e (28 mg, 0.13 mmol), kept for 12 h at room temperature. The crude was purified by flash chromatography on silica gel (petroleum ether/CH2Cl2 1/2, Rf 0.74)) to afford the mixture of the two diastereomeric compounds 8beD1 and 8beD2 (43 mg, 58% yield) as a white solid (mp 250 °C dec). 1H NMR (400 MHz, CDCl3) δ: 1.23 (d, 3H, J = 7.2 Hz), 1.39 (d, 3H, J = 7.2 Hz) 2.208 (s, 3H), 2.214 (s, 3H), 2.22 (s, 3H), 2.23 (s, 3H), 2.29 (s, 3H), 2.33 (s, 3H), 3.10 (q, 1H, J = 7.3 Hz), 3.20 (q, 1H, J = 7.2 Hz), 3.91 (s, 3H), 3.92 (s, 3H), 6.52 (bs, 1H), 6.65 (bs, 1H), 6.75–6.94 (m, 10H), 6.99 (bs, 1H), 7.04 (bs, 1H), 7.09–7.17 (m, 5H), 7.27–7.30 (m, 1H), 7.39 (bs, 1H), 7.53 (bs, 1H), 7.64–7.71 (m, 4H) ppm. Elem Anal. for C35H29NO3S2: Calcd. C 73.02, H 5.08, N 2.43; found C 73.03, H 5.09, N 2.43.
Diastereoisomers 8bfD1 and 8bfD2. (M/P)-1,3,7-trimethyl[1,4]benzothiazino[2,3,4-kl]phenothiazine-2-yl mono(1R)-menthylphthalate. Following the general procedure from 1b(OH) (40 mg, 0.11 mmol) and (-)mono(1R)-menthylphthalate 7f (33 mg, 0.11 mmol), kept for 18 h at room temperature. The crude was purified by flash chromatography on silica gel (petroleum ether/CH2Cl2 1/1, Rf 0.63)) to afford the mixture of the two diastereomeric compounds 8bfD1 and 8bfD2 (32 mg, 45% yield) as a white solid (mp 180–190 °C). 1HNMR (400 MHz, CDCl3) δ: 0.66 (d, 3H, J = 6.9 Hz), 0.78–0.90 (m, 17H), 0.98–1.11 (m, 4H), 1.40–1.53 (m, 4H), 1.65–1.71 (m, 4H), 1.90–1.99 (m, 2H), 2.05–2.26 (m, 14H), 2.53 (s, 6H), 4.85–4.93 (m, 2H), 6.70–6.85 (m, 10H), 6.95–7.00 (m, 4H), 7.07 (bs, 2H), 7.26–7.30 (m, 2H), 7.44–7.54 (m, 4H) ppm. 13CNMR (100 MHz, CDCl3) δ: 16.1, 16.4, 20.5, 20.6, 20.90, 20.92, 21.02, 22.1, 22.2, 23.4, 23.5, 26.2, 26.3, 31.5, 31.6, 34.4, 34.5, 40.5, 40.7, 47.3, 47.4, 75.7, 118.45, 118.50, 123.35, 123.40, 125.7, 125.76, 125.77, 125.79, 125.9, 126.26, 126.34, 126.4, 127.0, 127.46, 127.48, 128.05, 128.12. 128.16, 128.3, 129.50, 129.53, 129.6, 129.75, 129.77, 130.15, 130.18, 130.7, 130.8, 131.4, 131.5, 133.6, 133.9, 134.1, 134.7, 134.8, 135.36, 135.40, 138.1, 138.2, 141.45, 141.52, 141.98, 142.01, 163.82, 163.84, 167.28, 167.33 ppm (56 signals for 78 different carbons). Elem. Anal. for C39H39NO4S2: Calcd. C 72.08, H 6.05, N 2.16; found C 71.99, H 6.06, N 2.15.
Diastereoisomers 8bgD1 and 8bgD2. (M/P)-1,3,7-trimethyl[1,4]benzothiazino[2,3,4-kl]phenothiazine-2-yl (S)-N-Boc pipecolate. Following the general procedure from 1b(OH) (40 mg, 0.11 mmol) and (S)-N-Boc pipecolic acid 7g (25 mg, 0.11 mmol), kept for 3 h at room temperature. The crude was purified by flash chromatography on silica gel (petroleum ether/CH2Cl2 1/3, D1 Rf 0.27, D2 Rf 0.20) to afford the product 8bgD1 (17 mg, 28% yield) as a white solid (mp 79–82 °C) and the product 8bgD2 (9 mg, 15% yield) as a white solid (mp 121–125 °C). 8bgD1: 1HNMR (400Mz, CDCl3) δ: 1.10–1.21 (m, 1H), 1.26–1.53 (m, 14H), 2.21 (s, 3H), 2.28 (s, 3H), 2.29 (s, 3H), 2.29 (s, 3H), 2.80–2.95 (m, 1H), 3.79–3.96 (m, 1H), 4.40–4.47 (m, 1H), 6.78–6.91 (m, 6H), 7.00 (bs, 1H) ppm. 13C NMR (100 MHz, CDCl3) δ: 20.6, 20.7, 21.1, 21.4, 24.7, 25.0, 26.0, 28.5, 29.8, 41.2, 42.2, 54.3, 55.3, 77.2, 80.0, 80.1, 118.2, 122.6, 123.2, 125.5, 125.8, 126.3, 127.2, 127.7, 127.9, 128.1, 128.5, 129.5, 130.9, 131.3, 133.8, 134.2, 134.8, 134.9, 135.5, 138.1, 141.9, 142.4, 155.4, 155.7, 169.7, ppm. IR (ATR solid) n: 2973, 2924, 2860, 1764, 1689, 1480, 1448, 1364, 1252 cm−1. −157, (c 0.1, CH2Cl2). 8bgD2: 1HNMR (400Mz, CDCl3) δ: 0.79–0.99 (m, 1H), 1.26–1.45 (m, 14H), 2.14–2.29 (m, 9H), 2.46–2.94 (m, 1H), 3.65–3.92 (m, 1H), 4.39–4.63 (m, 1H), 6.78–6.91 (m, 6H), 7.00–7.02 (m, 1H) ppm. 13C NMR (100 MHz, CDCl3) δ: 20.1, 20.2, 20.5, 20.6, 20.70, 20.74, 20.8, 21.0, 24.7, 24.9, 26.0, 26.1, 28.4, 28.6, 41.0, 42.0, 54.4, 55.3, 80.0, 80.3, 115.2, 118.1, 118.2, 122.7, 122.9, 125.5, 125.8, 126.4, 127.5, 128.1, 128.3, 128.9, 129.5, 131.5, 133.8, 134.9, 135.6, 138.1, 142.5, 155.7, 169.7, 169.9, ppm. +49 (c 0.1, CH2Cl2).
Diastereoisomers 8bhD1 and 8bhD2. (M/P)-1,3,7-trimethyl[1,4]benzothiazino[2,3,4-kl]phenothiazine-2-yl (1S)-camphanate. Following the general procedure from 1b(OH) (259 mg, 0.71 mmol) and (1S)-(−)-camphanic acid 7h (170 mg, 0.86 mmol), kept for 22 h at room temperature. The crude was purified by flash chromatography on silica gel (petroleum ether/CH2Cl2 1/3, D1 Rf 0.37, D2 Rf 0.26) to afford product 8bhD1 (143 mg, 37% yield) as a white solid (mp 70–72 °C) and product 8bhD2 (126 mg, 33% yield) as a white solid (mp 86–88 °C). 8bhD1: 1HNMR (400 MHz, CDCl3) δ: 0.97 (s, 3H), 0.98 (s, 3H), 1.06 (s, 3H), 1.23–1.30 (m, 1H), 1.50–1.55 (m, 1H), 1.77–1.84 (m, 1H), 1.98–2.05 (m, 1H), 2.22 (s, 3H), 2.26 (s, 3H), 2.32 (s, 3H), 6.73 (bs, 1H), 6.79–6.81 (m, 2H), 6.85 (bs, 1H), 6.91–6.96 (m, 2H), 7.00 (bs, 1H) ppm. 13C NMR (100 MHz, CDCl3) δ: 9.8, 16.9, 17.0, 20.6, 20.7, 20.8, 29.0, 29.7, 54.5, 54.8, 90.8, 118.2, 122.7, 125.46, 125.49, 126.0, 126.2, 126.5, 127.1, 127.9, 128.7, 129.8, 131.6, 134.5, 135.0, 135.7, 137.9, 141.3, 141.6, 164.5, 177.9. IR (ATR solid) n: 2970, 2922, 2867, 1787, 1776, 1481, 1449, 1309, 1250 cm−1. −129 (c 0.1, CH2Cl2). 8bhD2 1HNMR (400 MHz, CDCl3) δ: 0.89 (s, 3H), 0.93 (s, 3H), 1.06 (s, 3H), 1.51–1.56 (m, 3H), 1.65–1.71 (m, 1H), 2.21 (s, 3H), 2.25 (s, 3H), 2.31 (s, 3H), 6.68 (bs, 1H), 6.77–6.79 (m, 2H), 6.84–6.89 (m, 2H), 6.96 (bs, 1H), 7.02 (bs, 1H) ppm. 13C NMR (100 MHz, CDCl3) δ: 9.8, 16.9, 17.0, 20.6, 20.7, 20.8, 28.9 29.6, 54.3, 54.8, 90.8, 118.1, 122.7, 125.2, 125.9, 126.3, 126.6 (2C), 127.5, 128.0, 128.2, 129.9, 132.0, 133.9, 135.0, 135.7, 137.8, 141.6, 142.1, 164.5, 177.7 ppm. IR (ATR solid) n: 2967, 2922, 2865, 1776, 1482, 1449, 1309, 1250 cm−1. + 126 (c 0.1, CH2Cl2).
General Procedure for the hydrolysis: To a solution of ester 8 in CH2Cl2/MeOH: 10/1 (roughly 0.05 M) 3 eq. of NaOH was added, and the solution was stirred for 4–6 h at room temperature. The solution was diluted with water and HCl (1M) was added until the pH was neutral, then the mixture was extracted with CH2Cl2 (3 × 5 mL). The organic layer was dried over Na2SO4, filtered, and then evaporated under reduced pressure. The crude was purified by flash chromatography on silica gel (Petroleum Ether/CH2Cl2 1/2) to afford the products (M)-1b(OH) or (P)-1b(OH) as a white solid in quantitative yield. (M)-1b(OH) −161 (c 0.1, CH2Cl2) and (P)-1b(OH) +166 (c 0.1, CH2Cl2).
Experimental HPLC Analytical (250 × 4.6 mm) column packed with Chiralpak IA chiral stationary phase was purchased from Chiral Technologies Europe. The HPLC resolution of products was performed on a HPLC Waters Alliance 2695 equipped with a 200 μL loop injector and a spectrophotometer UV Waters PDA 2996. The mobile phase, delivered at a flow rate of 1.2 mL/min, was hexane/CH2Cl2 70/30 v/v + 1% MeOH.
4. Conclusions
In this paper we have reported that the fine matching of the structures of the chiral auxiliaries used and, above all, the topology of their insertion on the helical skeleton, bay-zone vs cape-zone, allow for the chemical resolution of DTA[4]H) 1. Helicene 1b(OH) allowed for the insertion of the chiral auxiliary on the 1-position, the area of terminal ring superimposition that we indicated as the bay-zone. Esterification of 1b(OH) with (1S)-(−)-camphanic acid 7h provided diastereomers 8bhD1 and 8bhD2 which were successfully separated by flash chromatography and hydrolyzed providing enantiopure helicenes (M)-1b(OH) and (P)-1b(OH), respectively.
Supplementary Materials
The following supporting information is available online: HPLC Analysis, NMR spectra, DFT calculations of compound 1b(OH), Optimized structures’ coordinates.
Author Contributions
Conceptualization, S.M. and C.V.; methodology, validation and investigation, M.O. and M.L.; data curation, M.L.; formal analysis, S.A. and G.L.; writing—original draft preparation, C.V.; writing—review and editing, C.V., S.M. and M.L.; supervision and project administration, C.V. and S.M.; funding acquisition, S.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank MUR-Italy ‘Progetto Dipartimenti di Eccellenza 2018–2022′ allocated to the Department of Chemistry ‘Ugo Schiff’, University of Florence, Italy.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are available from the authors.
References
- Liu, M.; Zhang, L.; Wang, T. Supramolecular chirality in self-assembled systems. Chem. Rev. 2015, 115, 7304–7397. [Google Scholar] [CrossRef]
- Shen, Y.; Chen, C.-F. Helicenes: Synthesis and applications. Chem. Rev. 2012, 112, 1463–1535. [Google Scholar] [CrossRef] [PubMed]
- Mori, T. Chiroptical properties of symmetric double, triple, and multiple helicenes. Chem. Rev. 2021, 121, 2373–2412. [Google Scholar] [CrossRef] [PubMed]
- Reiné, P.; Ortuño, A.M.; Resa, S.; de Cienfuegos, L.Á.; Ribagorda, M.; Mota, A.J.; Abbate, S.; Longhi, G.; Miguel, D.; Cuerva, J.M. Enantiopure double ortho oligophenylethynylene-based helical structures with circularly polarized luminescence activity. ChemPhotoChem 2021, 6, e202100160. [Google Scholar] [CrossRef]
- Hong, J.; Xiao, X.; Liu, H.; Fu, L.; Wang, X.-C.; Zhou, L.; Wang, X.-Y.; Qiu, Z.; Cao, X.-Y.; Narita, A.; et al. X-shaped thiadiazole-containing double [7]heterohelicene with strong chiroptical response and π-stacked homochiral assembly. Chem. Com. 2021, 57, 5566–5569. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Huang, Z.; Huang, Z.; Cheng, R.; Yang, Y.; You, J. Triple Oxa[7]helicene with circularly polarized luminescence: Enhancing the dissymmetry factors via helicene subunit multiplication. Org. Lett. 2021, 23, 4559–4563. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.; Borovkov, V. Helicene-based chiral auxiliaries and chirogenesis. Symmetry 2018, 10, 10. [Google Scholar] [CrossRef] [Green Version]
- Neidle, S.; Waring, M. Molecular Aspects of Anticancer Drug-DNA Interactions; CRC Press: Boca Raton, FL, USA, 1993. [Google Scholar]
- D’Incalci, M.; Sessa, C. DNA minor groove binding ligands: A new class of anticancer agents. Expert Opin. Invest. Drugs 1997, 6, 875–884. [Google Scholar] [CrossRef]
- Honzawa, S.; Okubo, H.; Anzai, S.; Yamaguchi, M.; Tsumoto, K.; Kumagai, I. Chiral recognition in the binding of helicenediamine to double strand DNA: Interactions between low molecular weight helical compounds and a helical polymer. Bioorg Med. Chem. 2002, 10, 3213–3218. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, Y.X.; Sugiyama, H.; Umano, T.; Osuga, H.; Tanaka, K. (P)-helicene displays chiral selection in binding to Z-DNA. J. Am. Chem Soc. 2004, 126, 6566–6567. [Google Scholar] [CrossRef]
- Shinohara, K.; Sannohe, Y.; Kaieda, S.; Tanaka, K.; Osuga, H.; Tahara, H.; Xu, Y.; Kawase, T.; Bando, T.; Sugiyama, H. A chiral wedge molecule inhibits telomerase activity. J. Am. Chem. Soc. 2010, 132, 3778–3782. [Google Scholar] [CrossRef] [PubMed]
- Leydecker, T.; Wang, Z.M.; Torricelli, F.; Orgiu, E. Organic-based inverters: Basic concepts, materials, novel architectures and applications Chem. Soc. Rev. 2020, 49, 7627–7670. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Y.; Xie, X.; Ren, Y.; Zhang, B.; He, L.; Zhang, J.; Wang, L.-D.; Wang, P. A helicene-based molecular semiconductor enables 85 °C stable perovskite solar cells. ACS Energy Lett. 2021, 6, 1764–1772. [Google Scholar] [CrossRef]
- Gingras, M. One hundred years of helicene chemistry. Part 1: Non-stereoselective syntheses of carbohelicenes. Chem. Soc. Rev. 2013, 42, 968–1006. [Google Scholar] [CrossRef]
- Gingras, M.; Félix, G.; Peresutti, R. One hundred years of helicene chemistry. Part 2: Stereoselective syntheses and chiral separations of carbohelicenes. Chem. Soc. Rev. 2013, 42, 1007–1050. [Google Scholar] [CrossRef]
- Lamanna, G.; Faggi, C.; Gasparrini, F.; Ciogli, A.; Villani, C.; Stephens, P.J.; Devlin, F.J.; Menichetti, S. Efficient thia-bridged triarylamine heterohelicenes: Synthesis, resolution, and absolute configuration determination. Chem. Eur. J. 2008, 14, 5747–5750. [Google Scholar] [CrossRef]
- Menichetti, S.; Cecchi, S.; Procacci, P.; Innocenti, M.; Becucci, L.; Franco, L.; Viglianisi, C. Thia-bridged triarylamine heterohelicene radical cations as redox-driven molecular switches. Chem. Commun. 2015, 51, 11452–11454. [Google Scholar] [CrossRef]
- Longhi, G.; Castiglioni, E.; Villani, C.; Sabia, R.; Menichetti, S.; Viglianisi, C.; Devlin, F.; Abbate, S. Chiroptical properties of the ground and excited states of two thia-bridged triarylamine heterohelicenes. J. Photochem. Photobiol. A Chem. 2016, 331, 138–145. [Google Scholar] [CrossRef]
- Menichetti, S.; Faggi, C.; Onori, M.; Piantini, S.; Ferreira, M.; Rocchi, S.; Lupi, M.; Marin, I.; Maggini, M.; Viglianisi, C. Thia-bridged triarylamine hetero[4]helicenes: Regioselective synthesis and functionalization C. Eur. J. Org. Chem. 2019, 2019, 168–175. [Google Scholar] [CrossRef]
- Amorati, R.; Valgimigli, L.; Baschieri, A.; Guo, Y.; Mollica, F.; Menichetti, S.; Lupi, M.; Viglianisi, C. SET and HAT/PCET acid-mediated oxidation processes in helical shaped fused bis-phenothiazines. ChemPhysChem 2021, 22, 1446–1454. [Google Scholar] [CrossRef]
- Lupi, M.; Menichetti, S.; Stagnaro, P.; Utzeri, R.; Viglianisi, C. Thia-bridged triarylamine[4]helicene-functionalized polynorbor nenes as redox-active pH-sensitive polymers. Synthesis 2021, 53, 2602–2611. [Google Scholar] [CrossRef]
- Gliemann, B.D.; Petrovic, A.G.; Zolnhofer, E.M.; Dral, P.O.; Hampel, F.; Breitenbruch, G.; Schulze, P.; Raghavan, V.; Meyer, K.; Polavarapu, P.L.; et al. Configurationally stable chiral dithia-bridged hetero[4]helicene radical cation: Electronic structure and absolute configuration. Chem. Asian J. 2017, 12, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Giaconi, N.; Sorrentino, A.L.; Poggini, L.; Lupi, M.; Polewczyk, V.; Vinai, G.; Torelli, P.; Magnani, A.; Sessoli, R.; Menichetti, S.; et al. Stabilization of an enantiopure sub-monolayer of helicene radical cations on a Au(111) surface through noncovalent interactions. Angew. Chem. 2021, 133, 15404–15408. [Google Scholar] [CrossRef]
- Nuckolls, C.; Katz, T.J.; Katz, G.; Collings, P.J.; Castellanos, L. Synthesis and aggregation of a conjugated helical molecule. J. Am. Chem. Soc. 1999, 121, 79–88. [Google Scholar] [CrossRef]
- Nuckolls, C.; Katz, T.J.; Castellanos, L. Aggregation of conjugated helical molecules. J. Am. Chem. Soc. 1996, 118, 3767–3768. [Google Scholar] [CrossRef]
- Thongpanchang, T.; Paruch, K.; Katz, T.J.; Rheingold, A.L.; Lam, K.-C.; Liable-Sands, L. Why (1S)-camphanates are excellent resolving agents for helicen-1-ols and why they can be used to analyze absolute configurations. J. Org. Chem. 2000, 65, 1850–1856. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.M.; Goldberg, N.M.; Katz, T.J. Efficient synthesis of functionalized [7] helicenes. J. Org. Chem. 1998, 63, 7456–7462. [Google Scholar] [CrossRef]
- Dreher, S.D.; Weix, D.J.; Katz, T.J. Easy synthesis of functionalized hetero [7] helicenes. J. Org. Chem. 1999, 64, 3671–3678. [Google Scholar] [CrossRef]
- Dreher, S.D.; Paruch, K.; Katz, T.J. Application of the Russig-Laatsch reaction to synthesize a bis [5] helicene chiral pocket for asymmetric catalysis. J. Org. Chem. 2000, 65, 815–822. [Google Scholar] [CrossRef]
- Dreher, S.D.; Katz, T.J.; Lam, K.-C.; Rheingold, A.L. First Friedel-Crafts diacylation of a phenanthrene as the basis for an efficient synthesis of nonracemic [7]helicenes. J. Org. Chem. 2000, 65, 7602–7608. [Google Scholar] [CrossRef]
- Li, H.-Y.; Nehira, T.; Hagiwara, M.; Harada, N. Total synthesis and absolute stereochemistry of the natural atropisomer of the biflavone 4′,4‴,7,7″-tetra-O-methylcupressuflavone. J. Org. Chem. 1997, 62, 7222–7227. [Google Scholar] [CrossRef] [PubMed]
- Fuji, K.; Sakurai, M.; Kinoshita, T.; Kawabata, T. Palladium-catalyzed asymmetric reduction of allylic esters with a new chiral monodentate ligand, 8-diphenylphosphino-8′-methoxy-1, 1′-binaphthyl. Tetrahedron Lett. 1998, 39, 6323–6326. [Google Scholar] [CrossRef]
- Ohmori, K.; Kitamura, M.; Suzuki, K. From axial chirality to central chiralities: Pinacol cyclization of 2,2′-biaryldicarbaldehyde to trans-9,10-dihydrophenanthrene-9,10-diol. Angew. Chem. Int. Ed. Engl. 1999, 38, 1226–1229. [Google Scholar] [CrossRef]
- Schaefer, T.; Penner, G.H. The conformational properties of some phenyl esters. Molecular orbital and nuclear magnetic resonance studies. Can. J. Chem. 1987, 65, 2175–2178. [Google Scholar] [CrossRef]
- Schaefer, T.; Sebastian, R.; Penner, G.H. Long-range formyl proton coupling constants of 4-X-phenyl formats (X=H, F, CH3, NO2) and 2,6-dichlorophenyl formate. Conformations in solution. Can. J. Chem. 1988, 66, 1787–1793. [Google Scholar] [CrossRef]
- Mohamadi, F.; Richards, N.G.J.; Guida, W.C.; Liskamp, R.; Lipton, M.; Caufield, C.; Chang, G.; Hendrickson, T.; Still, W.C. Macromodel—An integrated software system for modeling organic and bioorganic molecules using molecular mechanics. J. Comput. Chem. 1990, 11, 440–467. [Google Scholar] [CrossRef]
- Allinger, N.L.; Yuh, Y.H.; Lii, J.-H. Molecular mechanics. The MM3 force field for hydrocarbons. 1. J. Am. Chem. Soc. 1989, 111, 8551–8566. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01. Gaussian, Inc.: Wallingford, CT, USA, 2016. Available online: https://gaussian.com/citation/ (accessed on 28 December 2021).
- Zhao, Y.; Truhlar, D.G. Truhlar The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215. [Google Scholar]
- Coghill, A.M.; Garson, L.R. The ACS Style Guide, 3rd ed.; American Chemical Society: Washington, DC, USA, 2006; p. 274. [Google Scholar] [CrossRef]
Figure 1.
M and P enantiomers for a [6]carbohelicene.

Scheme 1.
Synthetic pathways (A or B) to dithia-aza[4]helicenes (DTA[4]H) 1.

Scheme 2.
Red-ox behavior of DTA[4]H 1.

Scheme 3.
Esterification with enantiopure acids 7.

Scheme 4.
Synthesis of hydroxy substituted helicenes 1a(OH) and 1b(OH).

Scheme 5.
Esterification of helicenes 1a(OH) and 1b(OH), panels (A) and (B), respectively, with enantiopure acids 7.
Scheme 5.
Esterification of helicenes 1a(OH) and 1b(OH), panels (A) and (B), respectively, with enantiopure acids 7.

Figure 2.
1H NMR spectrum of diastereomers (D1 and D2) 8bh.

Figure 3.
1H-NMR spectra of diastereomers 8bhD1 and 8bhD2.

Scheme 6.
Chemical resolution of helicene (rac)-1b(OH).

Figure 4.
Optimized 3D-structures for the two possible conformers of (M)-1b(OH). Percent population factors calculated at M06/cc-pvtz, iefpcm level; in parenthesis population factors calculated at M062X/cc-pvtz, iefpcm level.
Figure 4.
Optimized 3D-structures for the two possible conformers of (M)-1b(OH). Percent population factors calculated at M06/cc-pvtz, iefpcm level; in parenthesis population factors calculated at M062X/cc-pvtz, iefpcm level.

Figure 5.
CD (top) and absorption (bottom) experimental and calculated spectra with two choices for the DFT functional (see text); calculation performed on M-1b(OH). The calculated spectra are Boltzmann weighed averages of the two conformers A and B.
Figure 5.
CD (top) and absorption (bottom) experimental and calculated spectra with two choices for the DFT functional (see text); calculation performed on M-1b(OH). The calculated spectra are Boltzmann weighed averages of the two conformers A and B.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Diastereomeric esters 8 obtained reaction racemic phenols 1a(OH) or 1b(OH) with enantiopure acids 7a–h.
Table 1.
Diastereomeric esters 8 obtained reaction racemic phenols 1a(OH) or 1b(OH) with enantiopure acids 7a–h.
| Chiral Auxiliary | Product | Yield | Resolution |
|---|---|---|---|
 7a (S)-(−)-Perillic acid | 8aaD1/8aaD2 | 60% | No resolution |
 7b Camphorsulfonyl chloride | 8abD1/8abD2 | 56% | No resolution |
 7c (−)-o,o’-Dibenzoyl-L-tartaric acid mono(dimethyl amide) | 8acD1/8acD2 | 56% | No resolution |
 7d (1S)-(+)-Ketopinic acid | 8adD1/8adD2 8bdD1/8bdD2 | 79% 55% | No resolution No resolution |
 7e (S)-(+)-2-(6-Methoxy-2-naphthyl)propionic acid | 8aeD1/8aeD2 8beD1/8beD2 | 94% 58% | No resolution No resolution |
 7f (-)mono-(1R)-Menthylphthalate | 8afD1/8afD2 8bfD1/8bfD2 | 54% 45% | No resolution No resolution |
 7g (S)-N-Boc pipecolic acid | 8agD1/8agD2 8bgD1/8bgD2 | 69% 28%15% | No resolution Chromatographic resolution is possible |
 7h (1S)-(−)-Camphanic acid | 8ahD1/8ahD2 8bhD1/8bhD2 | 57% 37% 33% | No resolution Chromatographic resolution is possible |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lupi, M.; Onori, M.; Menichetti, S.; Abbate, S.; Longhi, G.; Viglianisi, C. Resolution of a Configurationally Stable Hetero[4]helicene. Molecules 2022, 27, 1160. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27041160
AMA Style
Lupi M, Onori M, Menichetti S, Abbate S, Longhi G, Viglianisi C. Resolution of a Configurationally Stable Hetero[4]helicene. Molecules. 2022; 27(4):1160. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27041160
Chicago/Turabian StyleLupi, Michela, Martina Onori, Stefano Menichetti, Sergio Abbate, Giovanna Longhi, and Caterina Viglianisi. 2022. "Resolution of a Configurationally Stable Hetero[4]helicene" Molecules 27, no. 4: 1160. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27041160