
Photoionization Observables from Multi-Reference Dyson Orbitals Coupled to B-Spline DFT and TD-DFT Continuum


Abstract
:1. Introduction
2. Theory
2.1. Dyson Orbitals and Their Norms for a Biorthonormal Orbital Basis
2.2. The Continuum and the Calculation of the Photoionization Observables
3. Computational Details
4. Results
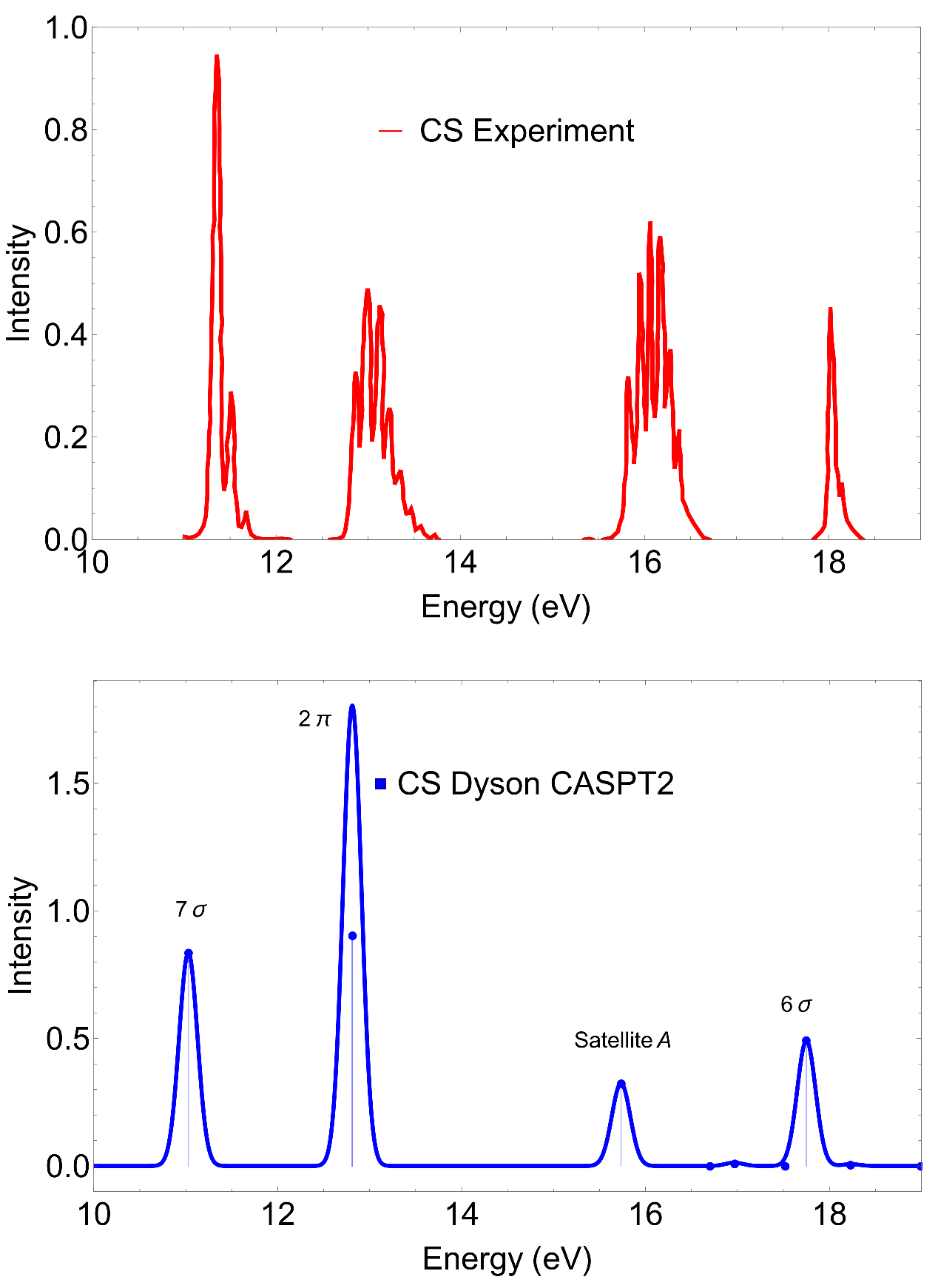
4.1. CS
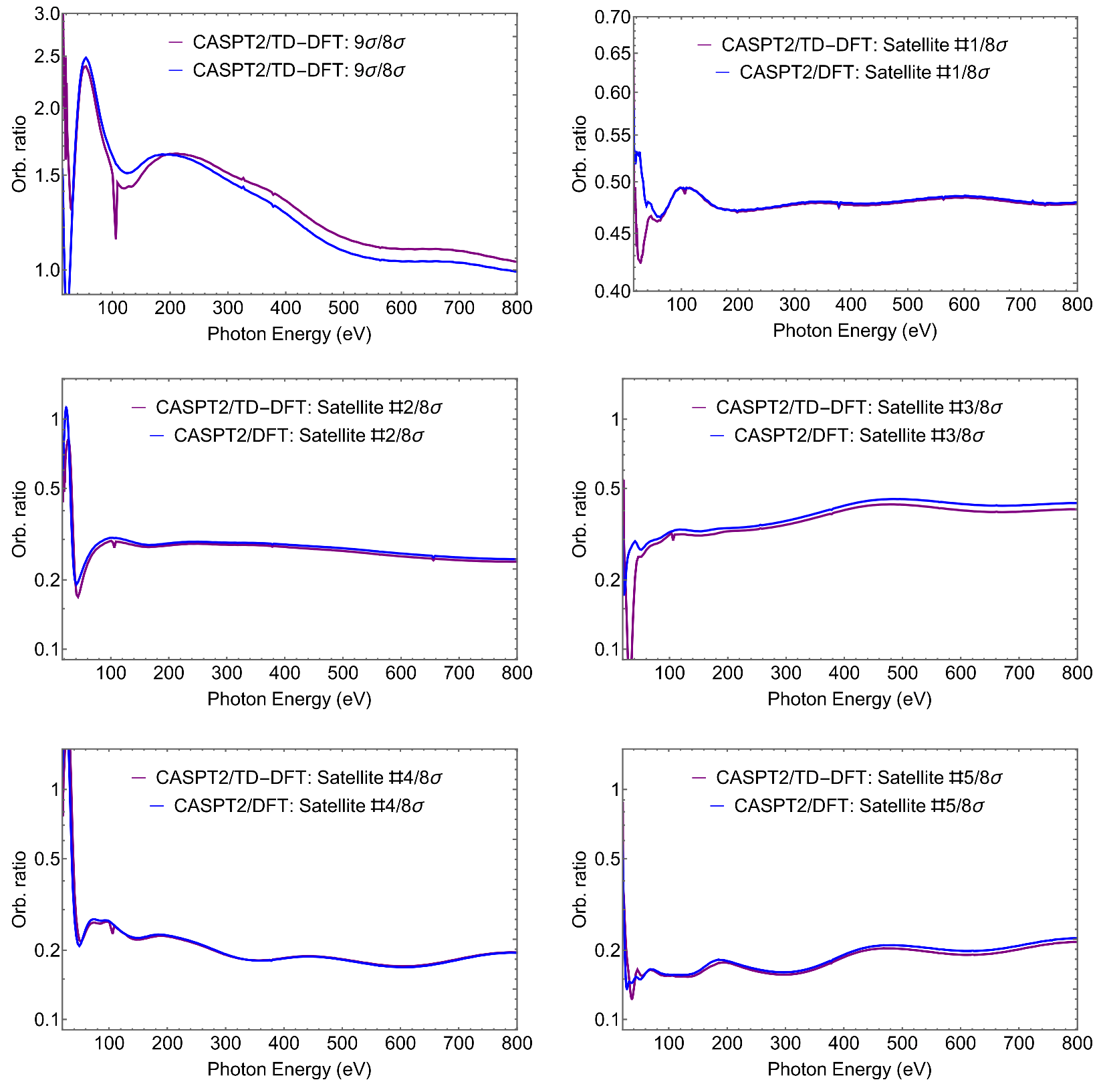
4.2. SiS
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Abbreviations
| MS-CASPT2 | Multi-State Complete Active Space Perturbation Theory to Second Order |
| CC | Coupled Cluster |
| EOM-CC | Equation of Motion Coupled Cluster |
| CCSD | Coupled Cluster Singles and Doubles |
| CC3 | Coupled Cluster Singles, Doubles, and perturbative triples |
| ADC | Algebraic Diagrammatic Construction |
| DFT | Density Functional Theory |
| TD-DFT | Time-Dependent DFT |
| I.E. | Ionization Energy |
| PES | Photoelectron Spectroscopy |
| HF | Hartree-Fock |
| FCI | Full Configuration Interaction |
| MFPADS | Molecular Frame Photoelectron Angular Distributions |
| LCAO | Linear Combination of Atomic Orbitals |
| RASSI | Restricted Active Space State-Interaction |
| HOMO | Highest Occupied Molecular Orbital |
| KT | Koopmans’ Theorem |
References
- Öksüz, İ.; Sinanoğlu, O. Theory of Atomic Structure Including Electron Correlation. I. Three Kinds of Correlation in Ground and Excited Configurations. Phys. Rev. 1969, 181, 42–53. [Google Scholar] [CrossRef]
- Beck, D.; Nicolaides, C. (Eds.) Excited States in Quantum Chemistry; Springer: Dordrecht, The Netherlands, 1979. [Google Scholar] [CrossRef]
- Roos, B. (Ed.) Lecture Notes in Quantum Chemistry; Springer: Berlin/Heidelberg, Germany, 1992. [Google Scholar] [CrossRef]
- Bartlett, R.J.; Musiał, M. Coupled-cluster theory in quantum chemistry. Rev. Mod. Phys. 2007, 79, 291–352. [Google Scholar] [CrossRef] [Green Version]
- Martin, R.L.; Shirley, D.A. Theory of the neon 1s correlation-peak intensities. Phys. Rev. A 1976, 13, 1475–1483. [Google Scholar] [CrossRef] [Green Version]
- Cederbaum, L.S.; Domcke, W. Theoretical Aspects of Ionization Potentials and Photoelectron Spectroscopy: A Green’s Function Approach. In Advances in Chemical Physics; Prigogine, I., Rice, S.A., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 1977; Volume XXXVI, pp. 205–344. [Google Scholar] [CrossRef]
- Schirmer, J. Beyond the random-phase approximation: A new approximation scheme for the polarization propagator. Phys. Rev. A 1982, 26, 2395–2416. [Google Scholar] [CrossRef]
- Stanton, J.F.; Bartlett, R.J. The equation of motion coupled-cluster method. A systematic biorthogonal approach to molecular excitation energies, transition probabilities, and excited state properties. J. Chem. Phys. 1993, 98, 7029–7039. [Google Scholar] [CrossRef]
- Jahnke, T.; Titze, J.; Foucar, L.; Wallauer, R.; Osipov, T.; Benis, E.; Jagutzki, O.; Arnold, W.; Czasch, A.; Staudte, A.; et al. Carbon K-shell photoionization of CO: Molecular frame angular distributions of normal and conjugate shakeup satellites. J. Electron Spectros. Relat. Phenom. 2011, 183, 48–52. [Google Scholar] [CrossRef] [Green Version]
- Nakano, M.; Selles, P.; Lablanquie, P.; Hikosaka, Y.; Penent, F.; Shigemasa, E.; Ito, K.; Carniato, S. Near-Edge X-ray Absorption Fine Structures Revealed in Core Ionization Photoelectron Spectroscopy. Phys. Rev. Lett. 2013, 111, 123001. [Google Scholar] [CrossRef]
- Lebech, M.; Houver, J.C.; Raseev, G.; dos Santos, A.S.; Dowek, D.; Lucchese, R.R. Valence and inner-valence shell dissociative photoionization of CO in the 26–33 eV range. II. Molecular-frame and recoil-frame photoelectron angular distributions. J. Chem. Phys. 2012, 136, 094303. [Google Scholar] [CrossRef]
- Waitz, M.; Bello, R.Y.; Metz, D.; Lower, J.; Trinter, F.; Schober, C.; Keiling, M.; Lenz, U.; Pitzer, M.; Mertens, K.; et al. Imaging the square of the correlated two-electron wave function of a hydrogen molecule. Nat. Commun. 2017, 8, 2266, Correction in Nat. Commun. 2018, 9, 2259. [Google Scholar] [CrossRef]
- Arneberg, R.; Müller, J.; Manne, R. Configuration interaction calculations of satellite structure in photoelectron spectra of H2O. Chem. Phys. 1982, 64, 249–258. [Google Scholar] [CrossRef]
- Martin, R.L.; Shirley, D.A. Theory of core-level photoemission correlation state spectra. J. Chem. Phys. 1976, 64, 3685–3689. [Google Scholar] [CrossRef] [Green Version]
- Ortiz, J.V. Dyson-orbital concepts for description of electrons in molecules. J. Chem. Phys. 2020, 153, 070902. [Google Scholar] [CrossRef]
- Krylov, A.I. From orbitals to observables and back. J. Chem. Phys. 2020, 153, 080901. [Google Scholar] [CrossRef]
- Pomogaev, V.; Lee, S.; Shaik, S.; Filatov, M.; Choi, C.H. Exploring Dyson’s Orbitals and Their Electron Binding Energies for Conceptualizing Excited States from Response Methodology. J. Phys. Chem. Lett. 2021, 12, 9963–9972. [Google Scholar] [CrossRef]
- Ponzi, A.; Angeli, C.; Cimiraglia, R.; Coriani, S.; Decleva, P. Dynamical photoionization observables of the CS molecule: The role of electron correlation. J. Chem. Phys. 2014, 140, 204304. [Google Scholar] [CrossRef]
- Moitra, T.; Ponzi, A.; Koch, H.; Coriani, S.; Decleva, P. Accurate Description of Photoionization Dynamical Parameters. J. Phys. Chem. Lett. 2020, 11, 5330–5337. [Google Scholar] [CrossRef]
- Shao, Y.; Gan, Z.; Epifanovsky, E.; Gilbert, A.T.B.; Wormit, M.; Kussmann, J.; Lange, A.W.; Behn, A.; Deng, J.; Feng, X.; et al. Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Mol. Phys. 2015, 113, 184–215. [Google Scholar] [CrossRef] [Green Version]
- Oana, C.M.; Krylov, A.I. Dyson orbitals for ionization from the ground and electronically excited states within equation-of-motion coupled-cluster formalism: Theory, implementation, and examples. J. Chem. Phys. 2007, 127, 234106. [Google Scholar] [CrossRef]
- Folkestad, S.D.; Kjønstad, E.F.; Myhre, R.H.; Andersen, J.H.; Balbi, A.; Coriani, S.; Giovannini, T.; Goletto, L.; Haugland, T.S.; Hutcheson, A.; et al. eT 1.0: An open source electronic structure program with emphasis on coupled cluster and multilevel methods. J. Chem. Phys. 2020, 152, 184103. [Google Scholar] [CrossRef]
- Moitra, T.; Paul, A.; Decleva, P.; Koch, H.; Coriani, S. Multi-electron excitation contributions towards primary and satellite states in the photoelectron spectrum. ChemRxiv 2021. [Google Scholar] [CrossRef]
- Grell, G.; Bokarev, S.; Winter, B.; Seidel, R.; Aziz, E.; Aziz, S.; Kühn, O. Multi-reference approach to the calculation of photoelectron spectra including spin-orbit coupling. J. Chem. Phys. 2015, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fdez Galván, I.; Vacher, M.; Alavi, A.; Angeli, C.; Aquilante, F.; Autschbach, J.; Bao, J.J.; Bokarev, S.I.; Bogdanov, N.A.; Carlson, R.K.; et al. OpenMolcas: From Source Code to Insight. J. Chem. Theory Comput. 2019, 15, 5925–5964. [Google Scholar] [CrossRef] [PubMed]
- Aquilante, F.; Autschbach, J.; Baiardi, A.; Battaglia, S.; Borin, V.A.; Chibotaru, L.F.; Conti, I.; De Vico, L.; Delcey, M.; Fdez Galván, I.; et al. Modern quantum chemistry with [Open]Molcas. J. Chem. Phys. 2020, 152, 214117. [Google Scholar] [CrossRef] [PubMed]
- Norell, J.; Grell, G.; Kühn, O.; Odelius, M.; Bokarev, S.I. Photoelectron shake-ups as a probe of molecular symmetry: 4d XPS analysis of in solution. Phys. Chem. Chem. Phys. 2018, 20, 19916–19921. [Google Scholar] [CrossRef] [Green Version]
- Toffoli, D.; Stener, M.; Fronzoni, G.; Decleva, P. Convergence of the multicenter B-spline DFT approach for the continuum. Chem. Phys. 2002, 276, 25–43. [Google Scholar] [CrossRef]
- Stener, M.; Toffoli, D.; Fronzoni, G.; Decleva, P. Time dependent density functional study of the photoionization dynamics of SF6. J. Chem. Phys. 2006, 124, 114306. [Google Scholar] [CrossRef]
- Jonathan, N.; Morris, A.; Okuda, M.; Ross, K.J.; Smith, D.J. Photoelectron spectroscopy of transient species. The CS molecule. Faraday Discuss. Chem. Soc. 1972, 54, 48–55. [Google Scholar] [CrossRef]
- Cockett, M.C.R.; Dyke, J.M.; Morris, A.; Niavaran, M.H.Z. High-temperature photoelectron spectroscopy. A study of SiS(X 1Σ+). J. Chem. Soc. Faraday Trans. 1989, 85, 75–83. [Google Scholar] [CrossRef]
- Ponzi, A.; Quadri, N.; Angeli, C.; Decleva, P. Electron correlation effects in the photoionization of CO and isoelectronic diatomic molecules. Phys. Chem. Chem. Phys. 2019, 21, 1937–1951. [Google Scholar] [CrossRef]
- Malmqvist, P.Å.; Roos, B.O. The CASSCF state interaction method. Chem. Phys. Lett. 1989, 155, 189–194. [Google Scholar] [CrossRef]
- Malmqvist, P.Å.; Roos, B.O.; Schimmelpfennig, B. The restricted active space (RAS) state interaction approach with spin–orbit coupling. Chem. Phys. Lett. 2002, 357, 230–240. [Google Scholar] [CrossRef]
- OpenMolcas Is a Quantum Chemistry Package. Available online: https://gitlab.com/Molcas/OpenMolcas (accessed on 6 January 2022).
- Moitra, T.; Coriani, S.; Decleva, P. Capturing Correlation Effects on Photoionization Dynamics. J. Chem. Theory Comput. 2021, 17, 5064–5079. [Google Scholar] [CrossRef]
- Bachau, H.; Cormier, E.; Decleva, P.; Hansen, J.E.; Martín, F. Applications of B-splines in atomic and molecular physics. Rep. Prog. Phys. 2001, 64, 1815–1943. [Google Scholar] [CrossRef]
- Fischer, C.F.; Guo, W. Spline algorithms for the Hartree-Fock equation for the helium ground state. J. Comput. Phys. 1990, 90, 486–496. [Google Scholar] [CrossRef]
- Brosolo, M.; Decleva, P.; Lisini, A. Accurate variational determination of continuum wavefunctions by a one-centre expansion in a spline basis. An application to and HeH2+ photoionization. J. Phys. B At. Mol. Opt. Phys. 1992, 25, 3345–3356. [Google Scholar] [CrossRef]
- Andersson, K.; Malmqvist, P.Å.; Roos, B.O.; Sadlej, A.J.; Wolinski, K. Second-order perturbation theory with a CASSCF reference function. J. Phys. Chem. 1990, 94, 5483–5488. [Google Scholar] [CrossRef]
- Andersson, K.; Malmqvist, P.; Roos, B.O. Second-order perturbation theory with a complete active space self-consistent field reference function. J. Chem. Phys. 1992, 96, 1218–1226. [Google Scholar] [CrossRef]
- Malmqvist, P.Å.; Pierloot, K.; Shahi, A.R.M.; Cramer, C.J.; Gagliardi, L. The restricted active space followed by second-order perturbation theory method: Theory and application to the study of CuO2 and Cu2O2 systems. J. Chem. Phys. 2008, 128, 204109. [Google Scholar] [CrossRef]
- Sauri, V.; Serrano-Andrés, L.; Shahi, A.R.M.; Gagliardi, L.; Vancoillie, S.; Pierloot, K. Multiconfigurational Second-Order Perturbation Theory Restricted Active Space (RASPT2) Method for Electronic Excited States: A Benchmark Study. J. Chem. Theory Comput. 2011, 7, 153–168. [Google Scholar] [CrossRef]
- Johnson, R.D., III (Ed.) NIST Computational Chemistry Comparison and Benchmark Database, NIST Standard Reference Database Number 101. Available online: https://cccbdb.nist.gov/ (accessed on 21 August 2020).
- Balabanov, N.B.; Peterson, K.A. Systematically convergent basis sets for transition metals. I. All-electron correlation consistent basis sets for the 3d elements Sc–Zn. J. Phys. Chem. 2005, 123, 064107. [Google Scholar] [CrossRef] [Green Version]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. IV. Calculation of static electrical response properties. J. Phys. Chem. 1994, 100, 2975–2988. [Google Scholar] [CrossRef] [Green Version]
- te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Stener, M.; Furlan, S.; Decleva, P. Density functional calculations of photoionization with an exchange-correlation potential with the correct asymptotic behaviour. J. Phys. B-At. Mol. Opt. 2000, 33, 1081–1102. [Google Scholar] [CrossRef]
- van Leeuwen, R.; Baerends, E.J. Exchange-correlation potential with correct asymptotic behavior. Phys. Rev. A 1994, 49, 2421–2431. [Google Scholar] [CrossRef] [Green Version]
- Trofimov, A.B.; Schirmer, J. Molecular ionization energies and ground- and ionic-state properties using a non-Dyson electron propagator approach. J. Chem. Phys. 2005, 123, 144115. [Google Scholar] [CrossRef]
- Kushawaha, R.K.; Patanen, M.; Guillemin, R.; Journel, L.; Miron, C.; Simon, M.; Piancastelli, M.N.; Skates, C.; Decleva, P. From double-slit interference to structural information in simple hydrocarbons. Proc. Natl. Acad. Sci. USA 2013, 110, 15201–15206. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, I.E.; Weigold, E. Electron momentum spectroscopy of atoms and molecules. Rep. Prog. Phys. 1991, 54, 789–879. [Google Scholar] [CrossRef]
- Chattopadhyaya, S.; Chattopadhyay, A.; Das, K.K. Electronic Spectrum of Silicon Monosulfide: Configuration Interaction Study. J. Phys. Chem. A 2002, 106, 833–841. [Google Scholar] [CrossRef]
- Chattopadhyaya, S.; Das, K.K. Multireference configuration interaction study of the low-lying electronic states of SiS+. J. Phys. B At. Mol. Opt. Phys. 2004, 37, 3355–3367. [Google Scholar] [CrossRef]
- Von Niessen, W.; Cederbaum, L.; Schirmer, J.; Diercksen, G.; Kraemer, W. Ionization energies of some molecules found in interstellar clouds calculated by a Green’s function method. J. Electron Spectros. Relat. Phenom. 1982, 28, 45–78. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ionic State (I) | MS-CASPT2 State Character | Exp. | KT | MS-CASPT2 | EOM-CCSD a | ||
|---|---|---|---|---|---|---|---|
| I.E. | I.E. | I.E. | I.E. | ||||
| 7 | 7(0.79) | 11.33 | 12.81 | 11.03 | 0.8354 | 11.52 | 0.8672 |
| 2 | 2(0.87) | 12.79 | 12.58 | 12.81 | 0.9019 | 13.06 | 0.9091 |
| Satellite A | 723(0.50) + 6(0.28) | 16.05 | 15.73 | 0.3242 | 20.32 | 0.1056 | |
| 6 | 6(0.47) + 723(0.24) | 18.00 | 18.85 | 17.75 | 0.4915 | 17.26 | 0.7683 |
| Satellite B | 5(0.17) + 623(0.22) | 22.85 | 0.1810 | 24.83 b | 0.468 | ||
| Ionic States | Orbital Ratio () | ||||
|---|---|---|---|---|---|
| I/J | 30 eV | 60 eV | 400 eV | 800 eV | |
| 6/7 | 0.59 | 0.50 | 0.47 | 1.16 | 1.10 |
| Sat.A/7 | 0.39 | 0.30 | 0.35 | 0.64 | 0.56 |
| Sat.B/7 | 0.21 | 0.43 | 0.16 | 0.29 | 0.32 |
| Sat.A/6 | 0.66 | 0.76 | 0.70 | 0.55 | 0.55 |
| Sat.B/6 | 0.37 | 0.71 | 0.35 | 0.25 | 0.32 |
| Sat.B/Sat.A | 0.55 | 0.91 | 0.50 | 0.45 | 0.57 |
| Ionic State (I) | MS-CASPT2 State Character | Exp. | KT | CASPT2 | EOM-CCSD a | ||
|---|---|---|---|---|---|---|---|
| I.E. | I.E. | I.E. | I.E. | ||||
| 9 | 9(0.74) | 10.53 | 10.61 | 10.20 | 0.8882 | 10.49 | 0.8993 (9) |
| 3 | 3(0.90) | 10.56 | 10.57 | 10.50 | 0.8674 | 10.61 | 0.8939 (3) |
| 8 | 8(0.45) + 934(0.40) | 13.88 | 15.65 | 13.54 | 0.4845 | 14.49 | 0.7776 (8) |
| Satellite #1 | 934(0.62) + 8(0.21) | - | 15.30 | 0.2241 | |||
| Satellite #2 | 934(0.34) + 910(0.14) | 16.9 | 16.68 | 0.1122 | |||
| Satellite #3 | 7(0.16) + 934(0.22) | 18.37 | 19.56 | 0.1597 | |||
| Satellite #4 | 910(0.28) | 19.88 | 0.1065 | ||||
| Satellite #5 | 7(0.10) + 934(0.23) | 20.92 | 0.0959 | ||||
| Ionic States | Orbital Ratio () | ||||
|---|---|---|---|---|---|
| I/J | 30 eV | 60 eV | 400 eV | 800 eV | |
| 9/8 | 1.83 | 1.13 | 2.4 | 1.2 | 1.04 |
| Sat.1/8 | 0.46 | 0.43 | 0.46 | 0.47 | 0.48 |
| Sat.2/8 | 0.23 | 0.68 | 0.24 | 0.28 | 0.24 |
| Sat.3/8 | 0.33 | 0.09 | 0.27 | 0.40 | 0.41 |
| Sat.4/8 | 0.22 | 1.48 | 0.24 | 0.18 | 0.20 |
| Sat.5/8 | 0.20 | 0.14 | 0.15 | 0.19 | 0.22 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tenorio, B.N.C.; Ponzi, A.; Coriani, S.; Decleva, P. Photoionization Observables from Multi-Reference Dyson Orbitals Coupled to B-Spline DFT and TD-DFT Continuum. Molecules 2022, 27, 1203. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27041203
Tenorio BNC, Ponzi A, Coriani S, Decleva P. Photoionization Observables from Multi-Reference Dyson Orbitals Coupled to B-Spline DFT and TD-DFT Continuum. Molecules. 2022; 27(4):1203. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27041203
Chicago/Turabian StyleTenorio, Bruno Nunes Cabral, Aurora Ponzi, Sonia Coriani, and Piero Decleva. 2022. "Photoionization Observables from Multi-Reference Dyson Orbitals Coupled to B-Spline DFT and TD-DFT Continuum" Molecules 27, no. 4: 1203. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27041203