3. Materials and Methods
3.1. General
Analytical grade solvents and commercially available reagents were used without further purification. Reactions requiring an inert atmosphere were carried out under a nitrogen atmosphere.
1H-NMR and
13C-NMR spectra were recorded on a Mercury 400 (
1H: 400 MHz,
13C: 100 MHz), or a Mercury 200 (
1H: 200 MHz,
13C: 50 MHz) spectrometer (Varian, Palo Alto, CA, USA). The chemical shifts (
δ) and coupling constants (
J) are expressed in parts per million (ppm) and Hertz (Hz), respectively. Copies of
1H- and
13C-NMR spectra of compounds
3–21 are provided as
Supplementary Material. Flash column chromatography (FCC) purifications were performed manually using glass columns filled with silica gel (40–63 μm, Merck KGaA, Darmstadt, Germany) or using an Isolera automatic system (Biotage Sweden AB, Uppsala, Sweden) and SNAP silica cartridges. TLC analyses were performed on Merck silica gel 60 F254 plates. Optical rotations were measured with DIP-360 digital polarimeter (JASCO, Easton, MD, USA). ESI-MS spectra were recorded on a LCQ Fleet ion-trap double quadrupole mass spectrometer (Thermo Scientific, Waltham, MA, USA) using electrospray (ES
+) ionization techniques. Dichloromethane (DCM) was dried by distillation over CaH2 and THF was dried by distillation over Na/benzophenone. Reactions conducted with microwave irradiation were performed with an automatic single-mode Biotage Initiator Sixty microwave synthesizer (Biotage Sweden AB, Uppsala, Sweden) equipped with temperature and pressure monitoring sensors, using sealed reaction tubes.
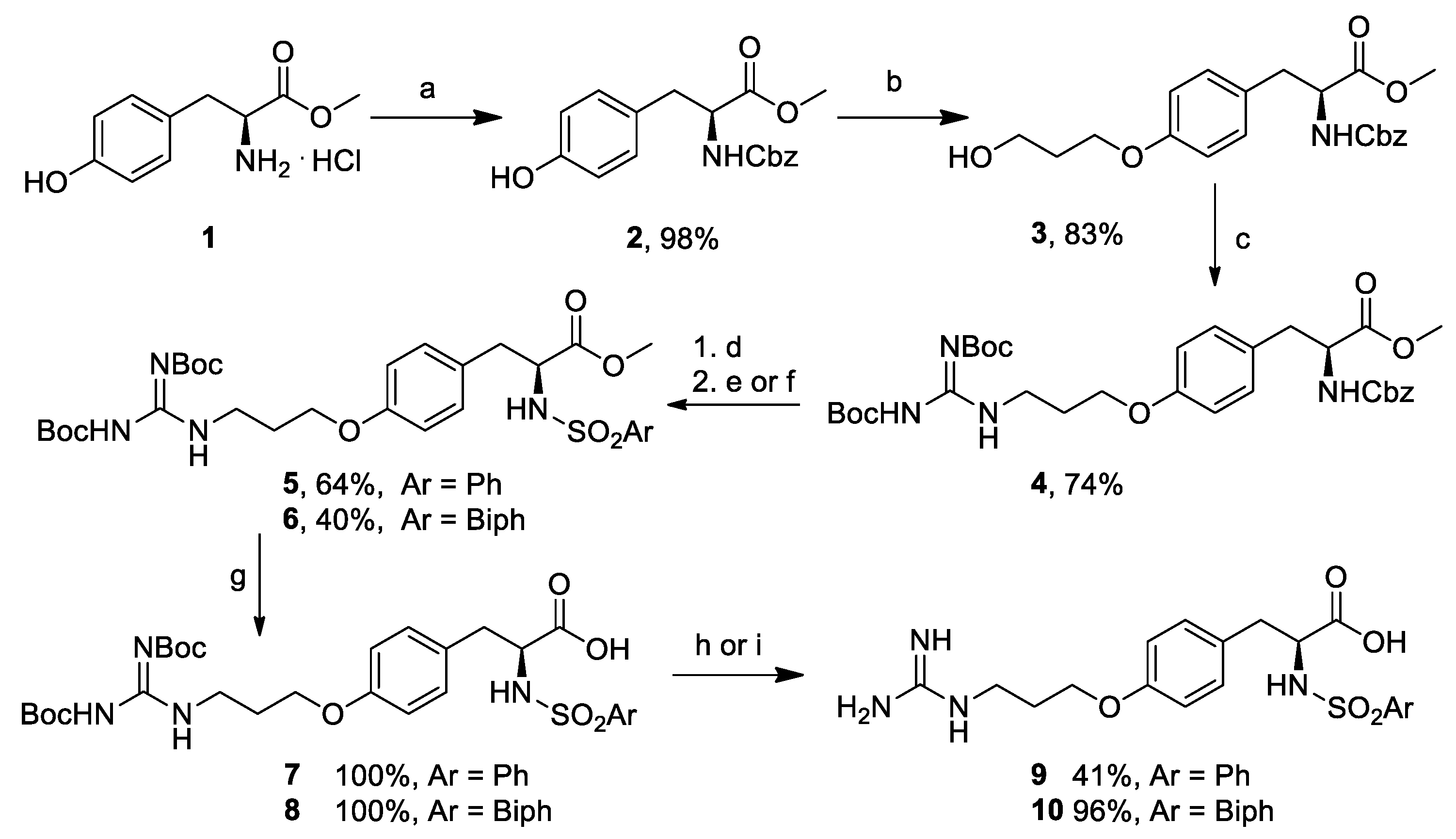
3.2. (S)-Methyl 2-(((Benzyloxy)carbonyl)amino)-3-(4-hydroxyphenyl)propanoate (2)
To a 250 mL round bottom flask containing a solution of L-Tyr-OMe
.HCl (1) (3.0 g, 12.9 mmol) in EtOAc (28.0 mL), a solution of NaHCO
3 (2.7 g, 32.0 mmol) in H
2O (30.0 mL) was added. Benzyl chloroformate (3.30 g, 19.4 mmol) was then added dropwise at 0 °C, and the resulting reaction mixture was warmed to room temperature and stirred overnight. After a TLC check (PE/EtOAc 2.5:1, R
f = 0.53), the organic phase was diluted with 80 mL of a mixture of EtOAc/H
2O (1:1). The resulting organic phase was separated, washed with H
2O and brine, dried over Na
2SO
4, and concentrated in vacuo. The title compound was obtained as a grainy white solid (4.20 g, 98% yield), that was used without further purification. Spectroscopic data were in agreement with those reported in the literature [
27].
3.3. (S)-Methyl 2-(((Benzyloxy)carbonyl)amino)-3-(4-(3-hydroxypropoxy)phenyl)propanoate (3)
Under an atmosphere of N2, a 250 mL round bottom flask was charged with PPh3 (954 mg, 3.64 mmol) followed by compound 2 (1.0 g, 3.04 mmol) in dry THF (30 mL). DIAD (716 μL, 3.64 mmol) was added dropwise at −10 °C, and the reaction mixture was stirred for 10 min. Propanediol (263 μL, 3.64 mmol) was added dropwise at −10 °C. The reaction solution was warmed to room temperature and reacted for 1 h at 65 °C under microwave irradiation. The solvent was removed under reduced pressure, and the residue was purified by Biotage Isolera system to provide the title compound as a colorless oil (960 mg, 2.48 mmol, 83% yield). = 27.2 (c 1.0, CHCl3). 1H-NMR (400 MHz, CDCl3) δ (ppm) = 7.35–7.31 (m, 5H), 6.99 (d, J = 8.5 Hz, 2H), 6.80 (d, J = 8.5 Hz, 2H), 5.23 (d, J = 7.9 Hz, 1H), 5.09 (s, 2H), 4.61 (dd, J = 13.3, 5.8 Hz, 1H), 4.08 (t, J = 5.9 Hz, 2H), 3.85 (t, J = 5.4 Hz, 2H), 3.71 (s, 3H), 3.04 (dd, J = 8.6, 6.2 Hz, 2H), 2.08–1.98 (m, 2H). 13C-NMR (100 MHz, CDCl3) δ 172.0, 157.9, 155.6, 136.2, 131.9, 130.2, 128.5, 128.1, 128.0, 127.8, 114.6, 66.9, 65.6, 60.4, 54.9, 52.3, 37.3, 32.0. MS (ESI) m/z (%) = 410.25 (100, [M + Na]+).
3.4. (S)-Methyl 2-(((Benzyloxy)carbonyl)amino)-3-(4-(3-(2,3-bis(tert-butoxycarbonyl)guanidino)propoxy)phenyl)propanoate (4)
Under an atmosphere of N2, a 100 mL round bottom flask was charged with PPh3 (776 mg, 2.96 mmol) and compound 3 (1.04 g, 2.69 mmol) in dry THF (27 mL). DIAD (582 μL, 2.96 mmol) was added dropwise at −10 °C, and the reaction mixture was stirred for 10 min. A solution of bis-N-Boc guanidine (766 mg, 2.96 mmol) in dry THF (2 mL) was added dropwise at −10 °C. The reaction solution was warmed to room temperature, divided between two 20 mL microwave vials, which were reacted for 30 min at 110 °C under microwave irradiation. After TLC check (PE/EtOAc 2:1, Rf = 0.44), the solvent was removed under reduced pressure. The reaction was purified by Biotage Isolera system to provide the title compound as a colorless oil (1.25 g, 2.69 mmol, 74% yield). = 27.9 (c 1.0, CHCl3). 1H- NMR (400 MHz, CDCl3) δ 9.30 (br s, 2H), 7.42–7.26 (m, 5H), 6.97 (d, J = 8.5 Hz, 2H), 6.76 (d, J = 8.5 Hz, 2H), 5.21 (d, J = 7.9 Hz, 1H), 5.09 (s, 2H), 4.60 (dd, J = 13.3, 5.7 Hz, 1H), 4.10 (t, J = 6.9 Hz, 2H), 3.97 (t, J = 6.1 Hz, 2H), 3.70 (s, 3H), 3.10–2.98 (m, 2H), 2.06 (dd, J = 14.2, 7.6 Hz, 2H), 1.48 (s, 18H). 13C-NMR (100 MHz, CDCl3) δ 172.0, 163.8, 160.6, 158.1, 155.6, 155.0, 136.2, 132.1, 130.2, 128.5, 128.2, 128.1, 127.5, 114.4, 83.8, 78.7, 66.9, 65.6, 54.9, 52.3, 42.3, 37.3, 28.7, 28.3, 28.0. MS (ESI) m/z (%) = 629.01 (100, [M + H]+).
3.5. (S)-Methyl 3-(4-(3-(2,3-bis(tert-Butoxycarbonyl)guanidino)propoxy)phenyl)-2-(phenylsulfonamido)propanoate (5)
To a two neck 25 mL round bottom flask a solution of compound 4 (950 mg, 1.5 mmol) in MeOH (7.5 mL), Pd/C (75 mg), and catalytic AcOH were added. H2 gas was let to stream inside the flask for 15 min, and the reaction solution was overnight stirred at room temperature under a H2 atmosphere. After TLC check (EtOAc/PE 1:3, Rf = 0.0), the reaction solution was filtered over Celite, and concentrated in vacuo. The so obtained free amine (720 mg, 1.5 mmol, quantitative yield) was used in the next synthetic step without further purification. 1H-NMR (200 MHz, CDCl3) δ (ppm) = 9.40 (br s, 2H), 7.13 (d, J = 10 Hz, 2H), 6.87 (d, J = 8 Hz, 2H), 4.17 (t, J = 6 Hz, 2H), 4.05 (t, J = 6 Hz, 1H), 3.78 (s, 3H), 3.13 (d, J = 6 Hz, 2H), 2.14 (t, J = 6 Hz, 2H), 1.56 (s, 18H). In a 10 mL round bottom flask containing a solution of the free amine (100 mg, 0.2 mmol) and DMAP (5 mg, 0.04 mmol) in dry DCM (2 mL), TEA (56 μL, 0.4 mmol) and then phenylsulfonyl chloride (28 μL, 0.2 mmol) were added dropwise at 0 °C. The reaction solution was stirred at room temperature overnight, and after TLC check (EtOAc/PE 1:2, Rf = 0.23), it was washed with 1M HCl, satd. NaHCO3 and Brine. The organic phase was dried over Na2SO4, and the solvent was removed under reduced pressure. The crude was purified by FCC (Et2O/PE 1:1) to provide the title compound (81 mg, 0.1 mmol, 64% yield). = 1.4 (c 0.4, CHCl3). 1H-NMR (400 MHz, CDCl3) δ (ppm) = 9.26 (br s, 2H), 7.76 (d, J = 7.4 Hz, 2H), 7.54 (t, J = 7.4 Hz, 1H), 7.45 (t, J = 7.7 Hz, 2H), 6.95 (d, J = 8.5 Hz, 2H), 6.73 (d, J = 8.5 Hz, 2H), 5.06 (d, J = 8.9 Hz, 1H), 4.18 (dd, J1 = 14.5 Hz, J2 = 5.8 Hz, 1H), 4.10 (t, J = 7.0 Hz, 2H), 3.97 (t, J = 6.2 Hz, 2H), 3.46 (s, 3H), 2.97 (d, J = 5.8 Hz, 2H), 2.17–1.98 (m, 2H), 1.50 (s, 9H), 1.48 (s, 9H). 13C-NMR (100 MHz, CDCl3) δ (ppm) = 171.1, 163.8, 160.6, 158.2, 155.0, 139.6, 132.7, 130.3, 128.9, 127.1, 126.6, 114.4, 83.8, 78.7, 65.6, 56.7, 52.3, 42.2, 38.5, 28.7, 28.3, 28.0. MS (ESI) m/z (%) = 657.12 (100, [M + Na]+).
3.6. (S)-Methyl 2-([1,1′-Biphenyl]-4-ylsulfonamido)-3-(4-(3-(2,3-bis(tert-butoxycarbonyl)guanidino)propoxy)phenyl)propanoate (6)
To a 50 mL round bottom flask containing a solution of the free amine obtained from
4 as in
Section 3.5 (720 mg, 1.5 mmol), DMAP (40 mg, 0.3 mmol) in dry DCM (15 mL), and TEA (606 μL, 4.4 mmol), biphenylsulfonyl chloride (731 mg, 2.9 mmol) was added dropwise at 0 °C. The solution was stirred at room temperature overnight, and after TLC check (EtOAc/PE 1:3, R
f = 0.50), it was washed with 1M HCl, satd. NaHCO
3 and brine. The organic phase was dried over Na
2SO
4 and concentrated in vacuo. The crude was purified by FCC (Et
2O/PE 2:3) to provide the title compound (412 mg, 0.6 mmol, 40% yield).
= −42.2 (c 0.7, CHCl
3).
1H-NMR (200 MHz, CDCl
3) δ 9.28 (s, 2H), 7.80 (d,
J = 8.1 Hz, 2H), 7.72–7.54 (m, 5H), 7.46 (d,
J = 7.3 Hz, 3H), 6.96 (d,
J = 8.0 Hz, 2H), 6.73 (d,
J = 8.1 Hz, 2H), 5.09 (d,
J = 8.9 Hz, 1H), 4.20 (d,
J = 8.8 Hz, 1H), 4.08 (t,
J = 6.5 Hz, 2H), 3.94 (d,
J = 5.5 Hz, 2H), 3.48 (s, 3H), 2.99 (d,
J = 5.2 Hz, 2H), 2.06 (d,
J = 6.0 Hz, 2H), 1.50 (s, 9H), 1.49 (s, 9H).
13C-NMR (50 MHz, CDCl
3) δ (ppm) = 171.3, 163.8, 160.7, 158.3, 155.0, 145.7, 139.2, 138.3, 130.4, 129.1, 128.6, 127.7, 127.6, 127.3, 114.5, 83.9, 78.7, 65.8, 56.9, 52.4, 42.3, 38.6, 28.8, 28.4, 28.1. MS (ESI)
m/
z (%) = 733.92 (100, [M + Na]
+).
3.7. (S)-3-(4-(3-(2,3-bis(tert-Butoxycarbonyl)guanidino)propoxy)phenyl)-2-(phenylsulfonamido)propanoic Acid (7)
In a 5 mL round bottom flask containing a solution of compound 5 (50 mg, 0.08 mmol) in MeOH (0.6 mL), 1M LiOH (0.2 mL) was added and stirred at room temperature overnight. The solution was then diluted with EtOAc, acidified to pH 1 with 1N HCl and extracted with EtOAc. The organic phase was dried over Na2SO4, and the solvent was removed under reduced pressure to provide the title compound (48 mg, 0.08 mmol, quantitative yield) which was used in the next synthetic step without further purification. = 10.3 (c 0.9, CHCl3). 1H-NMR (400 MHz, CDCl3) δ (ppm) = 9.03 (s, 1H), 7.77 (d, J = 7.6 Hz, 2H), 7.57–7.38 (m, 3H), 7.05 (d, J = 8.0 Hz, 2H), 6.72 (d, J = 7.8 Hz, 2H), 5.64 (br s, 1H), 4.12 (br s, 1H), 3.94 (br s, 2H), 3.44 (d, J = 13.5, 2H), 3.08 (dd, J1 =19.2 Hz, J2 = 14.5 Hz, 1H), 2.99 (d, J = 4.3 Hz, 2H), 2.00 (br s, 2H), 1.51 (s, 18H). 13C-NMR (100 MHz, CDCl3) δ (ppm) = 173.0, 157.3, 154.3, 152.3, 132.8, 132.2, 131.0, 129.1, 127.0, 114.4, 85.3, 64.9, 56.8, 46.0, 39.3, 29.7, 27.9. MS (ESI) m/z (%) = 619.09 (100, [M − H]−).
3.8. (S)-2-([1,1′-Biphenyl]-4-ylsulfonamido)-3-(4-(3-(2,3-bis(tert-butoxycarbonyl)guanidino)propoxy)phenyl)propanoic Acid (8)
In a 5 mL round bottom flask containing a solution of compound 6 (200 mg, 0.28 mmol) in MeOH (1.0 mL), 1M LiOH (1.0 mL) was added and stirred at room temperature overnight. The reaction solution was then diluted with EtOAc, acidified to pH 1 with 1N HCl and extracted with EtOAc. The organic phase was dried over Na2SO4 and concentrated in vacuo to provide the title compound (195 mg, 0.28 mmol, quantitative yield) which was used in the next step without further purification. = −9.5 (c 1.0, MeOH). 1H-NMR (200 MHz, CD3OD) δ 7.79–7.44 (m, 9H), 7.11 (d, J = 8 Hz, 2H), 6.81 (d, J = 8 Hz, 2H), 4.14 (s, 1H), 3.99 (s, 2H), 3.75 (d, J = 4 Hz, 1H), 3.25–2.68 (m, 4H), 2.05 (s, 2H), 1.60 (s, 9H). 13C-NMR (100 MHz, CDCl3) δ 166.0, 155.3, 145.9, 136.5, 131.7, 129.3, 128.8, 128.4, 128.3, 127.5, 122.2, 87.8, 84.8, 62.1, 58.0, 57.3, 54.6, 45.2, 30.8, 21.9. MS (ESI) m/z (%) = 695.73 (100, [M − H]−).
3.9. (S)-3-(4-(3-Guanidinopropoxy)phenyl)-2-(phenylsulfonamido)propanoic Acid (9)
In a 5 mL round bottom flask, compound 7 (19 mg, 0.03 mmol) was dissolved in 3M HCl (150 μL). The reaction solution was stirred at room temperature overnight. After TLC check (EtOAc, Rf = 0.06), the solvent was removed under reduced pressure to provide the title compound (56 mg, 0.13 mmol, 41% yield). = 14.2 (c 0.5, H2O). 1H-NMR (400 MHz, DMSO) δ (ppm) = 8.22 (d, J = 9.0 Hz, 1H), 7.82 (br s, 1H), 7.64–7.34 (m, 5H), 7.01 (d, J = 8.6 Hz, 2H), 6.74 (dd, J1 = 12.0 Hz, J2 = 5.3 Hz, 2H), 3.97 (m, 2H), 3.80 (td, J1 = 8.9 Hz, J2 = 5.7 Hz, 1H), 3.35-3.32 (m, 1H), 3.04 (d, J = 3.5 Hz, 1H), 2.84 (dt, J1 = 15.8 Hz, J2 = 7.9 Hz, 1H), 2.61 (dd, J1 = 13.7 Hz, J2 = 9.1 Hz, 1H), 2.03–1.83 (m, 2H). 13C-NMR (100 MHz, DMSO) δ (ppm) = 172.7, 158.2, 157.5, 141.5, 132.5, 132.3, 130.6, 129.2, 126.6, 114.6, 64.7, 58.1, 38.1, 28.7, 28.0. MS (ESI) m/z (%) = 443.21 (100, [M + Na]+).
3.10. (S)-2-([1,1′-Biphenyl]-4-ylsulfonamido)-3-(4-(3-guanidinopropoxy)phenyl)propanoic Acid (10)
In a 5 mL round bottom flask, compound 8 (33 mg, 0.05 mmol) was dissolved in 2 mL of a mixture of TFA (95%), water (2.5%) and TES (2.5%). The reaction solution was stirred at room temperature for 2 h. After TLC check (EtOAc, Rf = 0.0), the solvent was removed under reduced pressure to provide the title compound (24 mg, 0.05 mmol, 96% yield). = −27.1 (c 1.0, MeOH). 1H-NMR (400 MHz, CD3OD) δ 7.70–7.58 (m, 6H), 7.48 (t, J = 7.5 Hz, 2H), 7.40 (t, J = 7.3 Hz, 1H), 7.02 (d, J = 8.5 Hz, 2H), 6.71 (d, J = 8.5 Hz, 2H), 4.00 (dd, J = 9.1, 5.0 Hz, 1H), 3.86 (tt, J = 9.1, 4.6 Hz, 2H), 3.28 (d, J = 6.9 Hz, 2H), 3.01 (dd, J = 13.8, 4.9 Hz, 1H), 2.75 (dd, J = 13.8, 9.2 Hz, 1H), 1.95–1.84 (m, 2H). 13C-NMR (100 MHz, CD3OD) δ (ppm) = 174.7, 159.0, 158.7, 146.2, 140.8, 140.7, 131.4, 130.3, 130.1, 129.4, 128.5, 128.3, 115.3, 65.8, 59.2, 39.6, 39.0, 29.6. MS (ESI) m/z (%) = 519.36 (100, [M+Na]+).
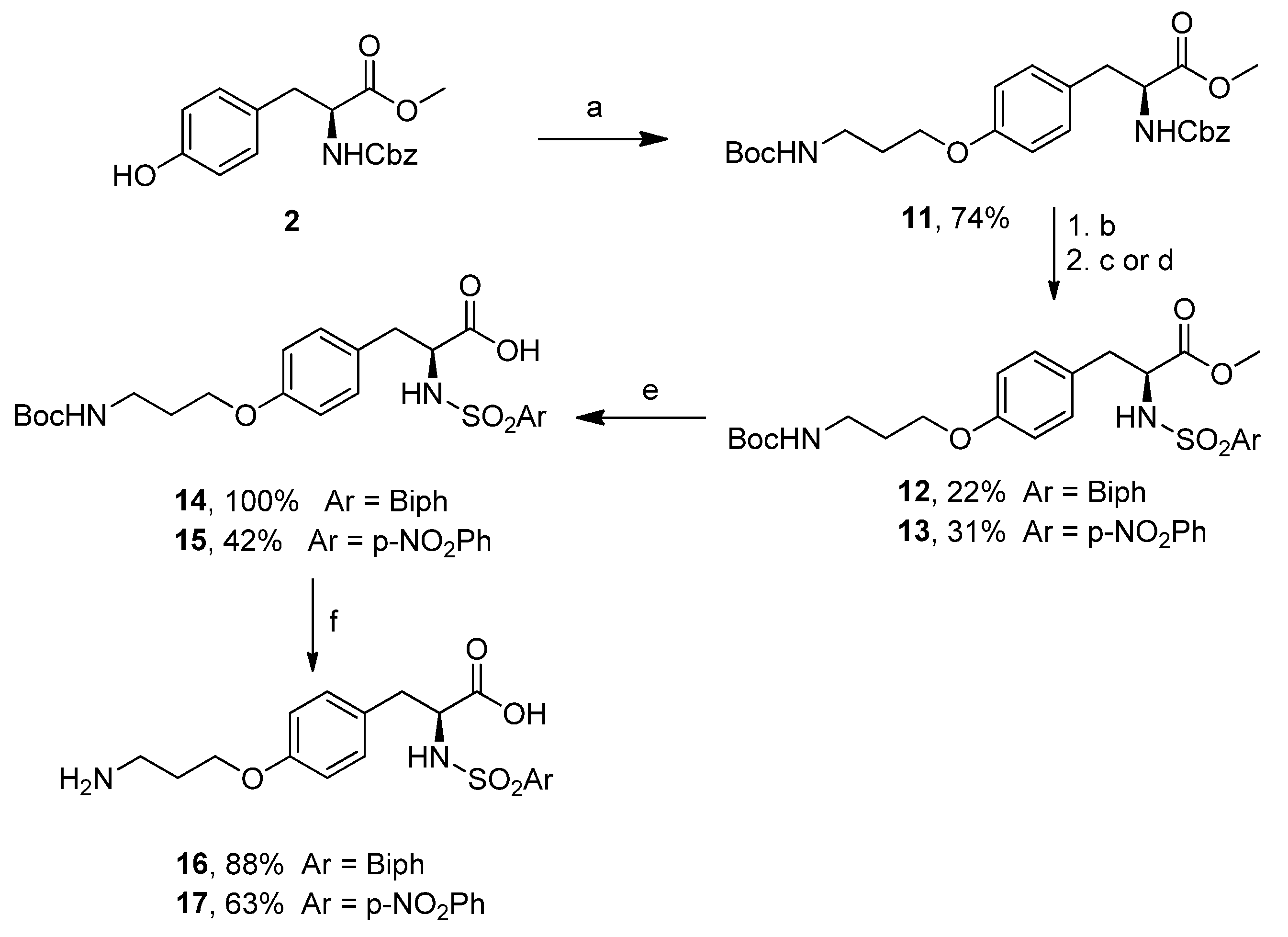
3.11. (S)-Methyl 2-(((Benzyloxy)carbonyl)amino)-3-(4-(3-((tert-butoxycarbonyl)amino)propoxy)phenyl)propanoate (11)
Under an atmosphere of N2, a 250 mL round bottom flask was charged with PPh3 (2.8 g, 10.6 mmol) and a solution of compound 2 (3.2 g, 9.7 mmol) in dry THF (90 mL). DIAD (2.1 g, 10.6 mmol) was then added dropwise at −10 °C, and the reaction mixture was stirred for 10 min. A solution of tert-butyl (3-hydroxypropyl) carbamate (1.6 g, 10.6 mmol) in dry THF (4 mL) was added dropwise at −10 °C. The reaction solution was warmed to room temperature, partitioned between 5 microwave 20 mL vials, and reacted at 110 °C for 30 min under microwave irradiation. After TLC check (PE/EtOAc 2:1, Rf = 0.19), the solvent was removed under reduced pressure, and the residue was purified by FCC (Et2O/PE 2:1) to provide the title compound as a colorless oil (3.5 g, 7.1 mmol, 74% yield). = 39.6 (c 1.0, CHCl3). 1H-NMR (200 MHz, CDCl3) δ (ppm) = 7.34 (s, 5H), 6.98 (d, J = 4 Hz, 2H), 6.78 (d, J = 4 Hz, 2H), 5.22 (d, J = 4 Hz, 1H), 5.09 (s, 2H), 4.79 (m, 1H), 4.66-4.56 (m, 1H), 3.97 (t, J = 6 Hz, 2H), 3.72 (s, 3H), 3.32 (q, J = 6 Hz, 2H), 3.05–3.03 (m, 2H), 1.96 (quint, J = 5.5 Hz, 2H), 1.43 (s, 9H). 13C-NMR (50 MHz, CDCl3) δ (ppm) = 172.1, 157.9, 156.1, 155.7, 136.3, 130.2, 128.5, 128.1, 128.0, 127.8, 115.6, 114.6, 79.2, 66.9, 65.7, 55.0, 52.3, 38.0, 37.3, 29.5, 28.4. MS (ESI) m/z (%) = 509.04 (100, [M + Na]+).
3.12. (S)-Methyl 2-([1,1′-Biphenyl]-4-ylsulfonamido)-3-(4-(3-((tert-butoxycarbonyl)amino)propoxy)phenyl)propanoate (12)
To a 100 mL round bottom two neck flask a solution of compound 11 (3.3 g, 6.9 mmol) in MeOH (34 mL), Pd/C (340 mg), and catalytic AcOH were added. H2 gas was let to stream inside the flask for 15 min. The reaction solution was then stirred at room temperature, under H2 atmosphere, overnight. The reaction solution was filtered over Celite, and the solvent was removed under reduced pressure. The obtained free amine (2.2 g, 6.4 mmol, 93% yield) was used in the next step without further purification. 1H-NMR (200 MHz, CDCl3) δ (ppm) = 7.30 (d, J = 7 Hz, 2H), 6.82 (d, J = 5 Hz, 2H), 4.83 (m, 1H), 3.97 (t, J = 3 Hz, 2H), 3.71 (s, 3H), 3.30 (m, 2H), 3.10–3.00 (m, 1H), 2.90-2.80 (m, 1H), 2.59 (br s, 2H), 1.95 (m, 2H), 1.42 (s, 9H). In a 100 mL round bottom flask containing a solution of the free amine (1.1 g, 3.2 mmol), DMAP (78 mg, 0.64 mmol) in dry DCM (32 mL), and TEA (1.33 mL, 9.5 mmol) biphenylsulfonyl chloride (882 mg, 3.5 mmol) was added dropwise at 0 °C. The reaction solution was stirred at room temperature overnight, and after TLC check (Et2O/PE 2:1, Rf = 0.34), it was washed with 1M HCl, satd. NaHCO3 and brine. The organic phase was then dried over Na2SO4 and concentrated in vacuo. The crude was purified by FCC (Et2O/PE 1:1) to provide the title compound (320 mg, 0.56 mmol, 22% yield). = 1.3 (c 2.0, CHCl3). 1H-NMR (200 MHz, CDCl3) δ (ppm) = 7.80–7.41 (m, 9H), 6.97 (d, J = 4 Hz, 2H), 6.74 (d, J = 4 Hz, 2H), 5.07 (d, J = 6 Hz, 1H), 4.72 (br, 1H), 4.21–4.17 (m, 1H), 3.91 (t, J = 3 Hz, 2H), 3.50 (s, 3H), 3.28 (m, 2H), 3.04-2.93 (m, 2H), 1.92 (m, 2H), 1.44 (s, 9H). 13C-NMR (100 MHz, CDCl3) δ (ppm) = 171.3, 157.9, 155.9, 145.3, 139.1, 138.2, 132.0, 130.3, 129.0, 128.4, 127.4, 127.1, 114.4, 79.0, 65.6, 56.9, 52.3, 38.3, 37.9, 29.4, 28.3. MS (ESI) m/z (%) = 591.03 (100, [M + Na]+).
3.13. (S)-Methyl 3-(4-(3-((tert-Butoxycarbonyl)amino)propoxy)phenyl)-2-(4-nitrophenylsulfonamido)propanoate (13)
In a 50 mL round bottom flask containing a solution of free amine (1.1 g, 3.2 mmol), prepared from 11 as in (12), DMAP (78 mg, 0.64 mmol) in dry DCM (32 mL), and TEA (1.3 mL, 9.5 mmol) 4-nitrophenylsulfonyl chloride (773 mg, 3.5 mmol) was added dropwise at 0 °C. The reaction solution was stirred at room temperature overnight. After TLC check (EtOAc/PE 2:3, Rf = 0.75), it was washed with 1M HCl, satd. NaHCO3 and brine. The organic phase was dried over Na2SO4 and concentrated in vacuo. The crude was purified by FCC (Et2O/PE (3:2)) to provide the title compound (530 mg, 1.0 mmol, 31% yield). = −6.6 (c 1.0, CHCl3). 1H-NMR (400 MHz, CDCl3) δ 8.22 (d, J = 8.8 Hz, 2H), 7.84 (d, J = 8.8 Hz, 2H), 6.93 (d, J = 8.5 Hz, 2H), 6.71 (d, J = 8.5 Hz, 2H), 5.27 (d, J = 9.3 Hz, 1H), 4.77 (br s, 1H), 4.21 (ddd, J = 9.3, 7.1, 5.2 Hz, 1H), 3.94 (t, J = 6.0 Hz, 2H), 3.62 (s, 3H), 3.32 (d, J = 6.3 Hz, 2H), 3.05 (dd, J = 14.0, 5.1 Hz, 1H), 2.90 (dd, J = 14.0, 7.2 Hz, 1H), 2.01–1.90 (m, 2H), 1.44 (s, 9H). 13C-NMR (100 MHz, CDCl3) δ 171.1, 158.2, 149.8, 145.6, 130.4, 128.2, 126.7, 124.1, 114.6, 110.5, 110.0, 108.5, 65.7, 57.2, 52.8, 38.4, 28.4. MS (ESI) m/z (%) = 559.99 (100, [M + Na]+).
3.14. (S)-2-([1,1′-Biphenyl]-4-ylsulfonamido)-3-(4-(3-((tert-butoxycarbonyl)amino)propoxy)phenyl)propanoic Acid (14)
In a 5 mL round bottom flask containing the solution of compound 12 (74 mg, 0.13 mmol) in MeOH (580 μL), 1M LiOH (580 μL) was added and stirred at room temperature overnight. The reaction solution was then diluted with EtOAc, acidified to pH 1 with 1N HCl and extracted with further EtOAc. The organic phase was dried over Na2SO4 and concentrated in vacuo to provide the title compound (72 mg, 0.13 mmol, quantitative yield) which was used in the next step without further purification. = −4.0 (c 1.0, CHCl3). 1H-NMR (200 MHz, CDCl3) δ (ppm) = 7.77–7.33 (m, 9H), 7.01 (d, J = 8 Hz, 2H), 6.70 (d, J = 8 Hz, 2H), 5.36 (d, J = 8 Hz, 1H), 4.24–4.10 (m, 1H), 3.85 (m, 2H), 3.22–3.02 (m, 4H), 1.85 (m, 2H), 1.42 (s, 9H). 13C-NMR (50 MHz, CDCl3) δ (ppm) = 157.9, 145.4, 139.2, 138.1, 130.6, 129.0, 128.5, 127.6, 127.5, 127.3, 114.4, 77.2, 65.6, 56.9, 38.0, 29.7, 29.4, 28.4. MS (ESI) m/z (%) = 553.04 (100, [M − H]−).
3.15. (S)-3-(4-(3-((tert-Butoxycarbonyl)amino)propoxy)phenyl)-2-(4-nitrophenylsulfonamido)propanoic Acid (15)
In a 5 mL round bottom flask containing the solution of compound 13 (209 mg, 0.38 mmol) in MeOH (1.0 mL), an aqueous solution of 1M LiOH (1.0 mL) was added and stirred at room temperature overnight. The reaction solution was then diluted with EtOAc, acidified to pH 1 with 1N HCl and extracted with further EtOAc. The organic phase was dried over Na2SO4 and concentrated in vacuo to provide the title compound (83 mg, 0.16 mmol, 42% yield) which was used the next synthetic step without further purification. = −15.8 (c 0.95, CHCl3). 1H-NMR (200 MHz, CDCl3) δ (ppm) = 8.14 (d, J = 8 Hz, 2H), 7.78 (m, 2H), 6.96 (d, J = 8 Hz, 2H), 6.64 (d, J = 10 Hz, 2H), 5.76 (m, 1H), 4.15 (m, 1H), 3.88 (m, 1H), 3.26 (m, 2H), 2.89 (m, 1H), 1.89 (m, 2H), 1.43 (s, 9H). 13C-NMR (50 MHz, CDCl3) δ (ppm) = 174.3, 158.0, 149.7, 145.6, 130.5, 128.1, 127.3, 124.0, 114.5, 65.6, 57.4, 53.4, 37.9, 29.4, 28.3. MS (ESI) m/z (%) = 545.95 (100, [M + H]+).
3.16. (S)-2-([1,1′-Biphenyl]-4-ylsulfonamido)-3-(4-(3-aminopropoxy)phenyl)propanoic Acid (16)
In a 5 mL round bottom flask, compound 14 (44 mg, 0.08 mmol) was dissolved in a solution of 3M HCl in 1,4-dioxane (400 μL). The reaction solution was stirred at room temperature overnight. After TLC check (DCM/MeOH (5:1), Rf = 0.0), the solvent was removed under reduced pressure to provide the title compound (34 mg, 0.07 mmol, 88% yield). = −13.7 (c 1.0, MeOH). 1H-NMR (400 MHz, CD3OD) δ (ppm) = 7.60 (s, 6H), 7.47–7.36 (m, 3H), 6.98 (d, J = 8 Hz, 2H), 6.67 (d, J = 8 Hz, 2H), 3.92–3.85 (m, 3H), 3.60-3.54 (m, 1H), 2.97–2.93 (m, 3H), 2.74–2.63 (m, 1H), 1.95 (m, 2H). 13C-NMR (101 MHz, CDCl3) δ (ppm) = 164.0, 157.4, 144.8, 139.3, 139.2, 130.0, 129.0, 128.8, 128.1, 127.8, 127.1, 126.9, 113.9, 64.6, 37.6, 37.2, 29.3, 27.0. MS (ESI) m/z (%) = 553.17 (100, [M − H]−).
3.17. (S)-3-(4-(3-Aminopropoxy)phenyl)-2-(4-nitrophenylsulfonamido)propanoic Acid (17)
In a 5 mL round bottom flask, compound 15 (80 mg, 0.15 mmol) was dissolved in 3M HCl (800 μL). The reaction solution was stirred at room temperature overnight. After TLC check (EtOAc, Rf = 0.0), the solvent was removed under reduced pressure to provide the title compound (43 mg, 0.09 mmol, 63% yield). = −7.8 (c 1.0, MeOH). 1H-NMR (200 MHz, D2O) δ (ppm) = 7.89 (d, J = 10 Hz, 2H), 7.40 (d, J = 8 Hz, 2H), 6.70 (d, J = 10 Hz, 2H), 6.35 (d, J = 8 Hz, 2H), 3.92–3.74 (m, 4H), 3.03–2.87 (m, 4H), 2.47–2.34 (m, 1H), 1.91 (m, 2H). 13C-NMR (50 MHz, CD3OD) δ (ppm) = 171.8, 157.4, 149.3, 146.8, 130.1, 129.0, 127.7, 123.8, 114.0, 65.1, 58.2, 37.7, 37.3, 27.2. MS (ESI) m/z (%) = 424.04 (100, [M + H]+).
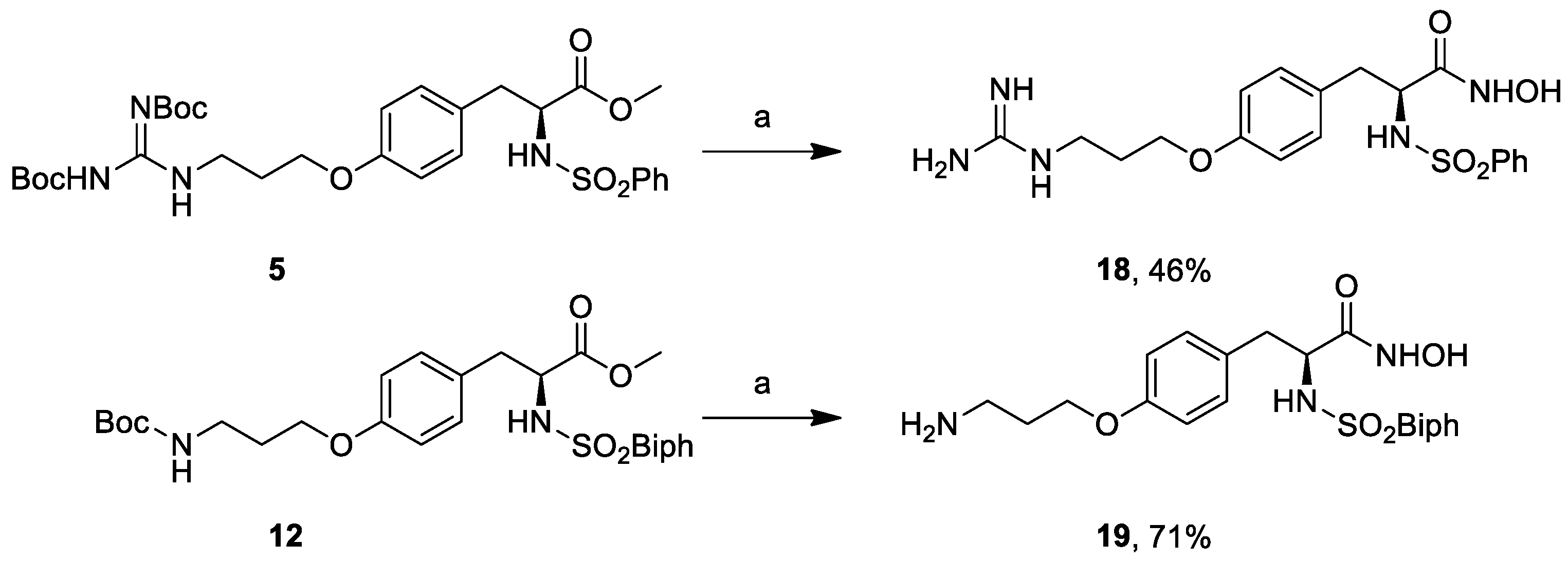
3.18. (S)-3-(4-(3-Guanidinopropoxy)phenyl)-N-hydroxy-2-(phenylsulfonamido)propanamide (18)
NH2OH.HCl (70 mg, 1.01 mmol) was solubilized in MeOH (0.4 mL) by heating to reflux until most of the salt was dissolved. The solution was cooled to <40 °C, and a solution of KOH (84 mg, 1.51 mmol) in MeOH (0.2 mL) was added in one portion. The resulting suspension was cooled to room temperature and was added without prior removal of precipitated material to compound 5 (80 mg, 0.13 mmol) and stirred at room temperature for 16 h. The reaction mixture was taken up in 1M HCl, extracted with EtOAc, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by FCC (EtOAc) to provide the pure compound that was subsequently dissolved in 3M HCl (300 μL). The reaction solution was stirred at room temperature overnight and concentrated under reduced pressure to give the title compound (26 mg, 0.06 mmol, 46% yield). = 7.5 (c 1.0, H2O). 1H-NMR (400 MHz, DMSO) δ (ppm) = 7.91 (d, J = 7.3 Hz, 1H), 7.55–7.47 (m, 2H), 7.48–7.43 (m, 5H), 6.96 (d, J = 8.4 Hz, 2H), 6.66 (d, J = 8.4 Hz, 2H), 3.98 (d, J = 21.0 Hz, 2H), 3.46 (d, J = 21.4 Hz, 2H), 3.27-3.24 (m, 1H), 2.89 (d, J = 16.6 Hz, 1H), 2.73 (d, J = 16.6 Hz, 1H), 2.07–1.79 (m, 2H). 13C-NMR (100 MHz, DMSO) δ (ppm) = 167.4, 157.8, 154.0, 132.6, 130.6, 129.3, 128.8, 126.8, 114.6, 65.1, 58.2, 38.3, 28.8, 28.1. MS (ESI) m/z (%) = 436.28 (100, [M + H]+).
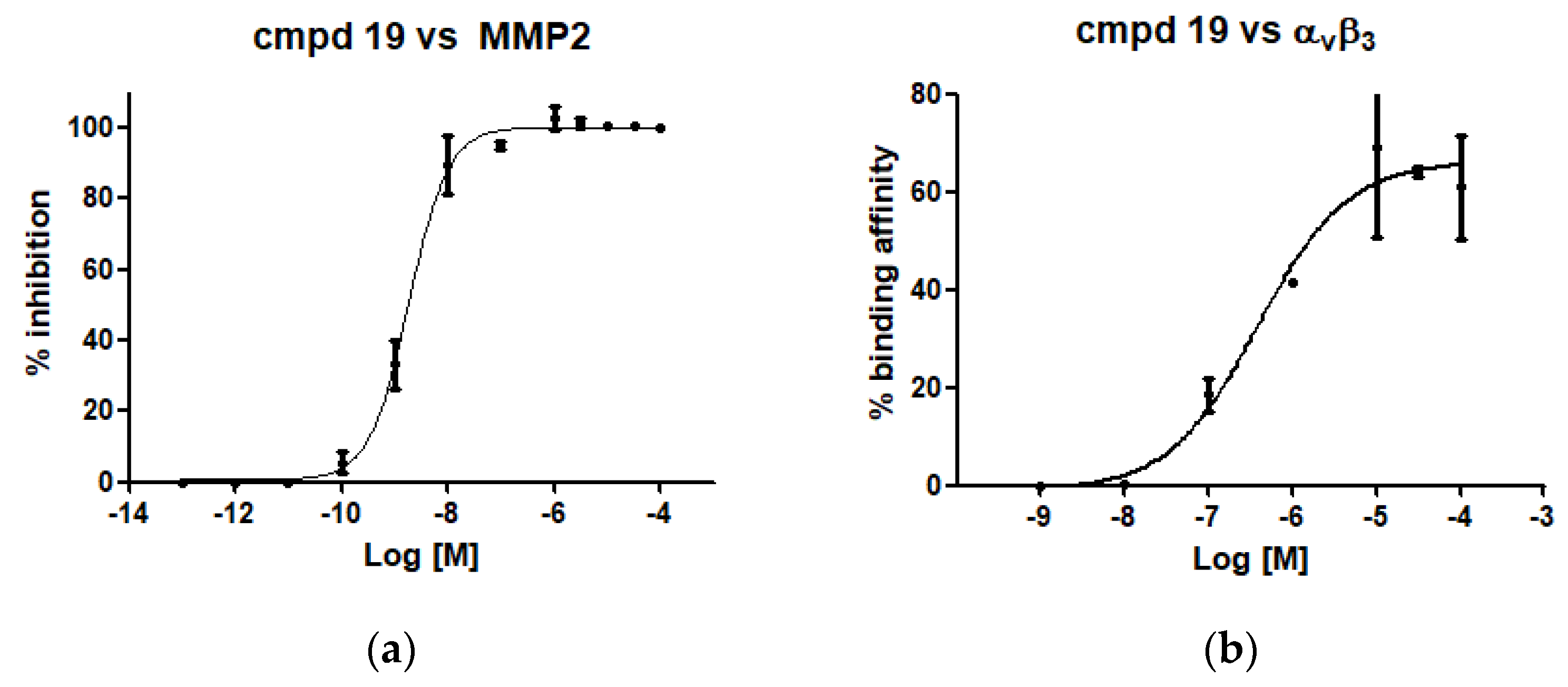
3.19. (S)-2-([1,1′-Biphenyl]-4-ylsulfonamido)-3-(4-(3-aminopropoxy)phenyl)-N-hydroxypropanamide (19)
NH2OH.HCl (39 mg, 0.56 mmol) was solubilized in MeOH (220 μL) by heating to reflux until most of the salt was dissolved. The solution was cooled to <40 °C, and a solution of KOH (47 mg, 0.84 mmol) in MeOH (120 μL) was added in one portion. The resulting suspension was cooled to room temperature and was added without prior removal of precipitated material to the methyl ester 12 (39 mg, 0.07 mmol) and stirred at room temperature for 16 h. The reaction mixture was taken up in 1M HCl, extracted with EtOAc, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by FCC (EtOAc) to provide the pure compound that was subsequently dissolved in 3M HCl (350 μL). The reaction solution was stirred at room temperature overnight and concentrated under reduced pressure to give the title compound (25 mg, 0.05 mmol, 71% yield). = −1.8 (c 0.9, MeOH). 1H-NMR (400 MHz, DMSO) δ 10.69 (s, 1H), 8.86 (s, 1H), 8.08 (s, 3H), 7.74–7.34 (m, 9H), 6.96 (d, J = 8.4 Hz, 2H), 6.66 (d, J = 8.4 Hz, 2H), 3.86 (d, J = 6.6 Hz, 2H), 3.75 (dd, J = 8.7, 5.9 Hz, 1H), 2.99 (s, 1H), 2.84 (s, 2H), 2.71 (dd, J = 13.7, 5.5 Hz, 1H), 2.57–2.49 (m, 1H), 1.92 (s, 2H), 1.29–1.11 (m, 2H). 13C-NMR (50 MHz, CD3OD) δ 168.8, 157.4 144.8, 139.3, 139.1, 129.9, 128.8, 128.7, 128.1, 127.1, 127.0, 126.9, 114.0, 64.6, 56.5, 37.7, 37.2, 27.0. MS (ESI) m/z (%) = 467.99 (100, [M − H]−).
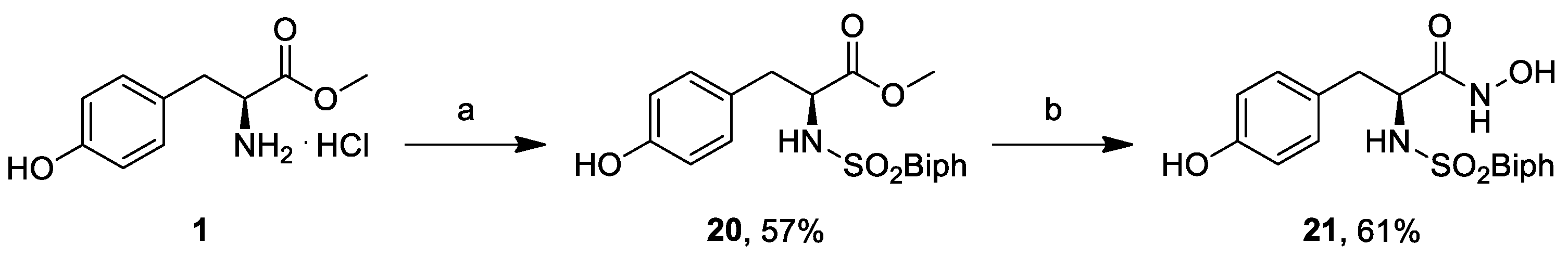
3.20. (S)-Methyl 2-([1,1′-Biphenyl]-4-ylsulfonamido)-3-(4-hydroxyphenyl)propanoate (20)
In a 5 mL round bottom flask L-tyrosine methyl ester (1, 218 mg, 1.37 mmol) was dissolved in dry THF (1.37 mL) and dry DMF (440 μL), and a solution of biphenylsulfonyl chloride (346 mg, 1.37 mmol) in dry THF (850 μL) was added dropwise at 0 °C. The solution was warmed to room temperature and stirred for 1 h, and dry Na2CO3 (145 mg, 1.37 mmol) was then added. The consumption of biphenylsulfonyl chloride was monitored by TLC (EtOAc/PE 1:2), and the solvent was removed under reduced pressure. The solid was dissolved in 2M HCl (2 mL) and brine (2 mL), and extracted three times with EtOAc. The organic phase was washed with brine. The crude was purified by FCC (EtOAc/PE 1:2) to provide the title compound (320 mg, 0.78 mmol, 57% yield). = −13.3 (c 1.0, MeOH). 1H-NMR (400 MHz, CD3OD) δ 7.70–7.62 (m, 6H), 7.46 (t, J = 8 Hz, 2H), 7.39 (d, J = 8 Hz, 1H), 6.90 (d, J = 8 Hz, 2H), 6.61 (d, J = 8 Hz, 2H), 4.00 (dd, J = 8, 6 Hz, 1H), 3.43 (s, 3H), 3.92–3.87 (m, 1H), 2.90 (A part of a second order ABX system, 1H), 2.75 (B part of a second order ABX system, 1H). 13C-NMR (50 MHz, CDCl3) δ 172.0, 156.0, 145.0, 139.2, 139.0, 130.0, 128.7, 128.0, 127.2, 127.0, 126.7, 114.8, 57.9, 51.1, 37.6. MS (ESI) m/z (%) = 434.18 (100, [M + Na]+).
3.21. (S)-2-([1,1′-Biphenyl]-4-ylsulfonamido)-N-hydroxy-3-(4-hydroxyphenyl)propanamide (21)
NH2OH.HCl (428 mg, 6.2 mmol) was solubilized in MeOH (2.5 mL) by heating to reflux until most of the salt was dissolved. The solution was cooled to <40 °C, and a solution of KOH (518 mg, 9.2 mmol) in MeOH (1.3 mL) was added in one portion. The resulting suspension was cooled to room temperature and was added without prior removal of precipitated material to the methyl ester 20 (320 mg, 0.77 mmol) and stirred at room temperature for 16 h. The reaction mixture was taken up in 1M HCl, extracted with EtOAc, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The residue was purified by FCC (DCM/MeOH 30:1) to provide, after concentration under reduced pressure, the title compound (193 mg, 0.47 mmol, 61% yield). = −3.4 (c 1.0, MeOH). 1H-NMR (400 MHz, DMSO) δ 10.59 (s, 1H), 8.20 (s, 1H), 7.77–7.35 (m, 9H), 6.83 (d, J = 8.0 Hz, 2H), 6.53 (d, J = 8.1 Hz, 2H), 4.23 (t, J = 7.0 Hz, 1H), 3.68 (s, 2H). 13C-NMR (50 MHz, CDCl3) δ 168.0, 155.8, 145.5, 139.1, 137.8, 130.1, 128.9, 128.3, 127.4, 127.2 126.3, 115.4, 56.5, 37.7. MS (ESI) m/z (%) = 410.95 (100, [M − H]−).
3.22. Enzyme Inhibition Assays
The inhibition potency of hydroxamic acid derivatives against MMP2 and MMP9 was assayed through a fluorometric assay using the fluorogenic substrate Mca-Lys-Pro-Leu-Gly-Leu-Dpa-Ala-Arg-NH2 (Enzo life science). All the measurements were performed in 96-well plates with a Fluostar Optima microplate reader (BMG Labtech, Ortenberg, Germany). Excitation and emission wavelengths were 320 and 420 nm, respectively. All incubations were performed at 28 °C in 50 mM TrisHCl, 150 mM NaCl, 10 mM CaCl2, 0.05% Brij 35, 1% DMSO at pH 7.5. The inhibitors were pre-incubated with enzymes (1 nM) for 5 min at room temperature before the reaction was started by the addition of the fluorogenic substrate (3 µM). The decrease of fluorescence was monitored over 30 min (λex = 320 nm, λem = 420 nm) at 28 °C. The percentages of inhibition for the test compounds were determined through the equation (1 − Vs/Vo) × 100, where vs. is the initial velocity in the presence of the inhibitor and V0 is the initial velocity of the uninhibited reaction. The IC50 values were obtained by dose-response measurements using inhibitor range of concentrations 0.00001–20 μM and enzyme concentration equal to 1 nM. A detergent-based assay was used to determine the presence of promiscuous inhibitors. All the experiments were performed in triplicates and data collected were analyzed using Graphpad 5.0 Software Package (GraphPad Prism Inc., San Diego, CA, USA).
3.23. Solid-Phase Integrin Binding Assay
Purified αvβ3 receptors (Sino Biological Europe GmbH, Eschborn, Germany) were diluted to 0.5 μg/mL in coating buffer containing Tris-HCl (20 mmol/L; pH 7.4), NaCl (150 mmol/L), MnCl2 (1 mmol/L), CaCl2 (2 mmol/L) and MgCl2 (1 mmol/L). An aliquot of diluted receptors (100 μL per well) was added to 96-well microtiter plates (MW 96F Medisorp Straight Nunc, Thermo Fischer Scientific, Waltham, MA, USA) and incubated, overnight, at 4 °C. The plates were then incubated with blocking solution (coating buffer plus 5% bovine serum albumin) for an additional 2 h at room temperature to block nonspecific binding followed by 3 h incubation at room temperature with various concentrations of test compounds in the presence of vitronectine (1 μg/mL, Sigma Aldrich, Merck KGaA, Darmstadt, Germany) biotinylated by using EZ-Link Sulfo-NHS-Biotinylation kit (Thermo Fisher, Waltham, MA, USA). After washing, the plates were incubated for 1 h at room temperature with streptavidin–biotinylated peroxidase complex (GE Healthcare, Chicago, IL, USA), followed by 30 min incubation with substrate reagent solution (100 μL; R&D Systems) before stopping the reaction by addition of H2SO4 (2 N, 50 μL). Absorbance at 415 nm was read with a BMG Labtech Fluostar Optima microplate reader. All the experiments were performed in triplicates and data collected were analyzed using the GraphPad 5.0 Software Package.
3.24. Inhibition of Cell Adhesion Assay
The expression levels of α
Vβ
3 integrin receptors on M21 melanoma cell lines were reported previously [
26]. Next, 96 wells plates were coated overnight, at 4 °C, with vitronectin (5 μg/mL in PBS) (V8379 Sigma). Plates were, then washed with phosphate buffered saline (PBS) solution and incubated at 37 °C for 1 h with PBS containing 1% bovine serum albumin (BSA). M21 cells were centrifugated (RT, at 700×
g) in PBS, to remove serum. Cells were counted and suspended in serum-free medium at 5.0 × 10
5 cells/mL. Melanoma cell suspensions were pre-incubated with different amounts of the compounds. The final concentration ranged from 30 uM to 0.3 uM, using cilengitide as a control. The incubation was performed at 37 °C for 30 min to allow the ligand-receptor equilibrium to be reached. Next, cells were plated on VN substrata (5–6 × 10
4 cells/well) and incubated at 37 °C for 1 h. The assays were conducted in the presence of 2 mmol/L MnCl
2 [
26]. At the end of the incubation, plates were washed with PBS to remove the non-adherent cells, and 200 μL of 0.5% crystal violet solution in 20% methanol were added. After 2 h of static incubation at 4 °C, plates were examined at 540 nm in a counter ELX800 (Bio TEK Instruments, Winooski, VT, USA). Experiments were done in triplicate and repeated at least three times. The values are expressed as % inhibition of cell adhesion relative to cells exposed to vehicle alone (PBS).
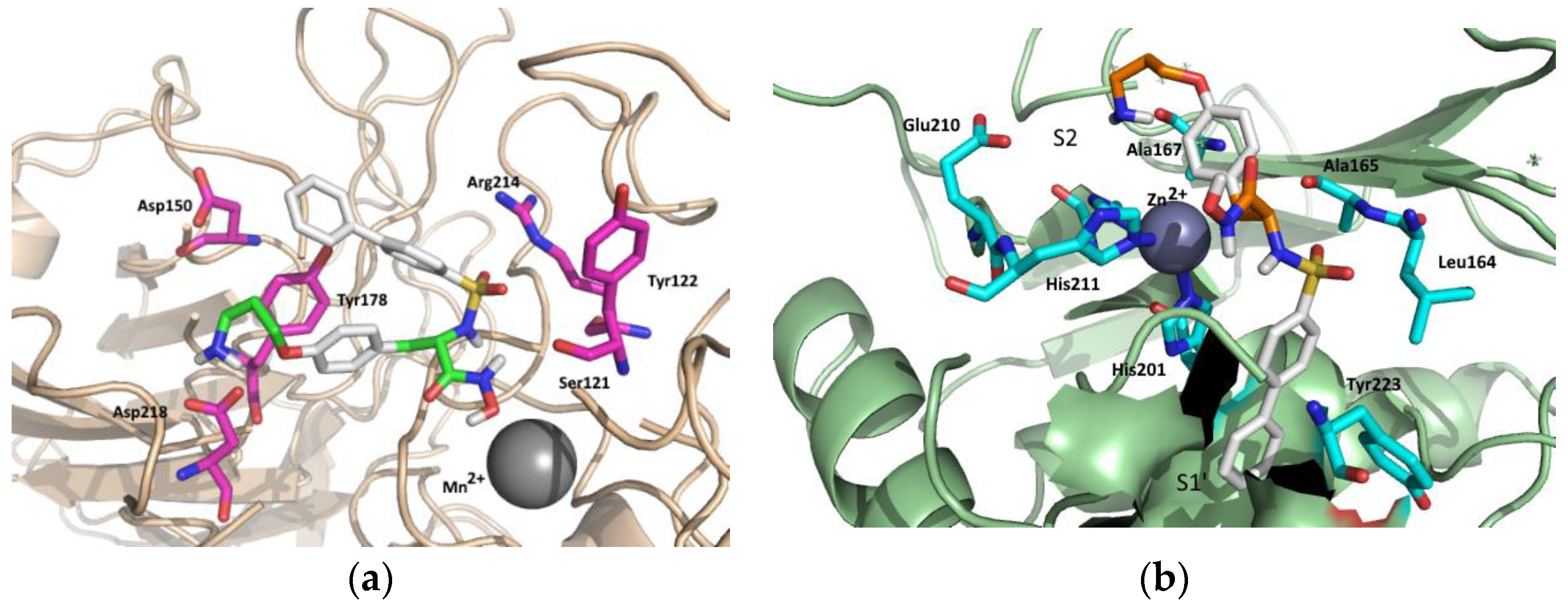
3.25. Molecular Modeling
Automated docking studies were carried out using the Lamarckian Genetic Algorithm (LGA) as a search engine implemented in the Autodock 4.0.1 program [
28]. The AutoDockTools 1.4.5 (ADT) graphical interface [
29] was used to prepare integrin and ligands PDBQT files. Coordinates of compound 19 were generated using Spartan (version 5.147, Wavefunction, Inc. Irvine, CA, USA) using Monte Carlo method within MMFF94 force field, and then energy-minimized through the AM1 semi-empirical method [
30] to calculate the equilibrium geometry. The coordinates of α
vβ
3 receptor and MMP2 were retrieved from the Protein Data Bank (PDB code: 1L5G and 1QIB for α
vβ
3 and MMP2, respectively), and ligand–protein complex was unmerged for achieving free protein structure. Water molecules were removed. For protein structures and compound 19, all hydrogen atoms were added, Gasteiger charges were computed, and nonpolar hydrogen atoms were merged. A charge value of +2.0 was successively added to each Mn atoms of α
vβ
3 receptor and to Zn atom of MMP2. Three-dimensional energy scoring grids of 0.375 Å resolution and 40 × 40 × 40 Å dimensions were computed. The center of the grid was set to be coincident with mass center of ligands preliminary fitted on the X-ray structure of c[RGDf(Me)V] in the α
vβ
3 complex (1L5G) and on Zn atom for MMP2 enzyme (1QIB). A total of 50 runs with a maximum of 2,500,000 energy evaluations were carried out for each ligand, using the default parameters for LGA. Low energy ligand-protein complexes were subjected to AMMP energy minimization using VegaZZ [
31], then cluster analysis was performed on docked results using a root-mean-square (rms) tolerance of 1.5 Å. Analysis of the binding mode, calculation of the binding energy, and prediction of the binding activity of docked conformations were carried out using Autodock plugin within the PyMol software v0.99 [
32].














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



















