
Targeted Drug Delivery for the Treatment of Blood Cancers


1
Nuffield Division of Clinical Laboratory Sciences, Radcliffe Department of Medicine, University of Oxford, John Radcliffe Hospital, Oxford OX3 9DU, UK
2
Institute of Clinical Sciences, University of Birmingham, Birmingham B15 2TT, UK
3
Department of Molecular Chemistry and Materials Science, Weizmann Institute of Sciences, Rehovot 761001, Israel
4
Arthritis Research UK Centre for Osteoarthritis Pathogenesis, Kennedy Institute of Rheumatology, University of Oxford, Oxford OX3 7FY, UK
*
Author to whom correspondence should be addressed.
Molecules 2022, 27(4), 1310; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27041310
Submission received: 23 December 2021
/
Revised: 2 February 2022
/
Accepted: 8 February 2022
/
Published: 15 February 2022
(This article belongs to the Special Issue Recent Advances in Biomedical Engineering and Molecular Medicine)
Abstract
:Blood cancers are a type of liquid tumor which means cancer is present in the body fluid. Multiple myeloma, leukemia, and lymphoma are the three common types of blood cancers. Chemotherapy is the major therapy of blood cancers by systemic administration of anticancer agents into the blood. However, a high incidence of relapse often happens, due to the low efficiency of the anticancer agents that accumulate in the tumor site, and therefore lead to a low survival rate of patients. This indicates an urgent need for a targeted drug delivery system to improve the safety and efficacy of therapeutics for blood cancers. In this review, we describe the current targeting strategies for blood cancers and recently investigated and approved drug delivery system formulations for blood cancers. In addition, we also discuss current challenges in the application of drug delivery systems for treating blood cancers.
1. Introduction
Cancers are one of the leading causes of death in the world [1]. Unlike solid tumors such as those in organs, blood cancers (including multiple myeloma, leukemia, and lymphoma) form in the bone marrow or in the lymphatic system [2,3]. Table 1 provides an overview of different types of blood cancers. Current treatments for blood cancers consist of chemotherapy, radiotherapy, immunotherapy, and transplantation [4]. Although, many chemotherapeutic drugs are clinically available for the treatment of blood cancers, there are no curative treatment approaches in clinical practice for these types of cancers due to the inevitable aggravation of blood cancers and bone metastasis [5]. Furthermore, it is difficult to achieve a sufficient therapeutic dose of anticancer agents at tumor sites inside bone marrow or the lymphatic system to suppress tumor growth via systematic administration [6]. To maintain therapeutic levels in bone marrow or the lymphatic system, chemotherapeutics require high dosage and/or more frequent administration which can result in increased side effects [7]. In addition, the bone marrow microenvironment contains a huge number of hematopoietic stem/progenitor cells which are resistance to chemotherapy and mediate disease refractory/relapse [5]. Therefore, a targeted drug delivery system for blood cancers is a significant challenge for chemotherapy.
In recent years, different types of nanoparticles have received considerable attention for the treatment of various types of solid tumors, leading to several successful drug delivery systems that have entered clinical practice. However, most of them have been done in solid tumors and few works have been done to develop drug delivery systems for the treatment of blood cancers. In this review, we summarized currently available strategies for drug delivery systems for treating blood cancers.
2. Targeting Delivery Strategy
2.1. Targeting Bone Marrow and Its Microenvironment
The bone marrow microenvironment plays a critical role in the maintenance of cell renewal and differentiation, especially for cancer cells. The bone marrow contains numerous blood vessels and capillaries. It is considered to be one of the most complex systems comprising various cell types including endothelia cells, stromal cells, osteocytes, fibroblasts, mesenchymal stem cells, macrophages, osteoclasts, and osteoblasts. Moreover, the non-cellular component including the extracellular matrix, oxygen tension, cytokines, and mechanical forces are also essential for cancer cell proliferation and are related to resistance. Targeting the bone marrow microenvironment can be improved by drug delivery systems and can be achieved passively or actively.
2.1.1. Passive Targeting Strategy
Potentially beneficial properties of nanotherapeutics include improved bioavailability, reduced toxicity, greater dose response, and enhanced solubility as compared with conventional medicines [8]. Passive targeting depends on accumulation of the drug delivery system at a specific end organ or tumor site, through leaky vasculatures which mainly require a delivery system for its own characteristics including the size, shape, surface zeta-potential, and other properties. In the bone marrow, the drug accumulation amounts in the bone are related to the reticulo-endothelial cells in vessels [9]. The enhanced permeability and retention (EPR) effect in solid tumors was described more than 30 years ago, however, it has been less appreciated in blood cancers in contrast to solid tumors [10]. Particle size plays a critical role since the transcellular route takes place through the fenestrae between the endothelial cells in the bone marrow [11]. It has been reported that the sizes of the fenestrae in the endothelial wall are less than 150 nm which means the particles larger than 150 nm would be less likely to pass through [12]. Furthermore, nanoparticles smaller than 60 nm can penetrate and distribute into the bone marrow interstitial space, since reticuloendothelial sinusoidal blood capillaries consist of pores as large as 60 nm in diameter [13]. In addition, to achieve high efficiency for drug delivery in bone marrow, a long circulation time in blood vessels is critical for nanomedicine. Liposomes less than 100 nm in diameter circulate longer in the blood and have less interaction with plasma proteins. However, there is also a limitation of nanoparticles with a small size, since nanoparticles less than 50 nm limit the drug encapsulation efficiency [14]. In addition to the role of diameter size in nanomedicine, surface charge also plays a major role in bone marrow uptake. It has been reported that negatively charged liposomes increased the efficiency of bone marrow uptake rate by the macrophages [11]. Therefore, the ideal size of nanoparticles for blood cancers should be between 50 nm and 100 nm.
2.1.2. Targeting Bone Surface-Mediated Bone Marrow
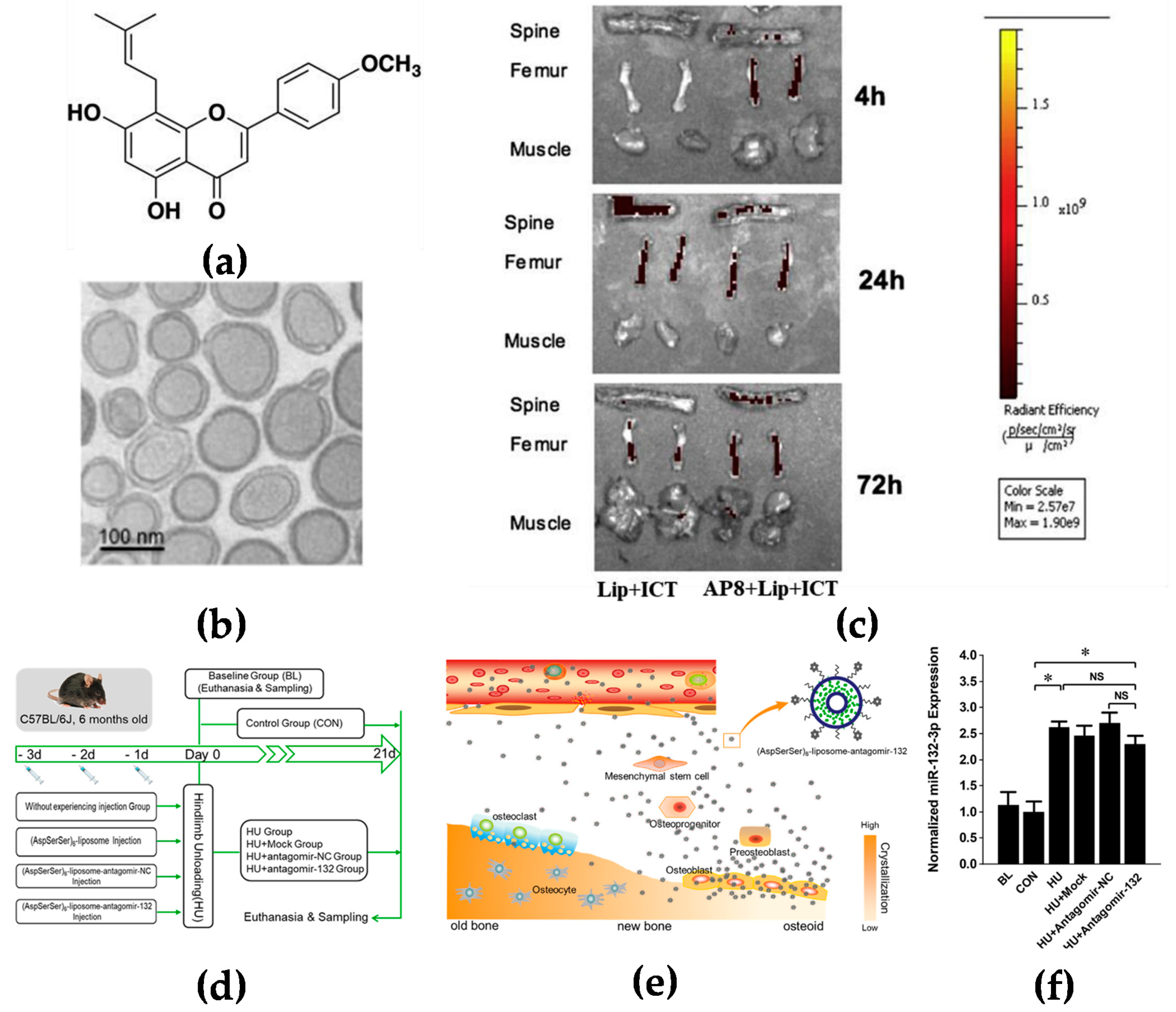
Bone is rich in hydroxyapatite which has a high affinity with glutamic acid or aspartic acid [15]. It has been reported that several oligopeptides have demonstrated their particular interactions with bone tissues. Eight repetitive aspartic acids, also known as Asp8, is one of the successful examples which has been reported to bind to bone-resorption surfaces [16]. Asp8 has been demonstrated to mainly bind to the highly crystallized hydroxyapatite of bone-resorption surfaces. It was found that Asp8-icaritin-liposome enhanced bone formation in ovariectomized mice as compared with an icaritin-liposome control lacking the Asp8 moiety (Figure 1a–c) [17]. (AspSerSer)6 is another type of oligopeptide used for the bone-surface delivery that mainly binds to calcium phosphate which is mainly distributed in the mantle dentin in the bone [18]. Hu et al. used the (AspSerSer)6-cationic liposome system to deliver miRNA-132-3p in bone, resulting in prevention and treatment of osteoporosis (Figure 1d–f) [19].
Another type of stable bone targeting by high affinity to hydroxyapatite is bisphosphonates [16]. Bisphosphonates as radionuclides have been reported for delivery of antineoplastic compounds into the bone marrow [20]. Furthermore, some studies have also demonstrated that bisphosphonates can reduce bone metastasis by preventing osteoclast differentiation [21]. Tian et al. reported that antibody conjugated bisphosphonates could deliver therapeutic antibodies directly into the bone for treatment of bone metastatic cancers and other bone diseases [22]. Therefore, bone-targeting molecules that are capable of binding to bone-formation surfaces may precisely deliver therapeutic agents to the bone marrow niche for blood cancer therapy.
2.1.3. Active Targeting
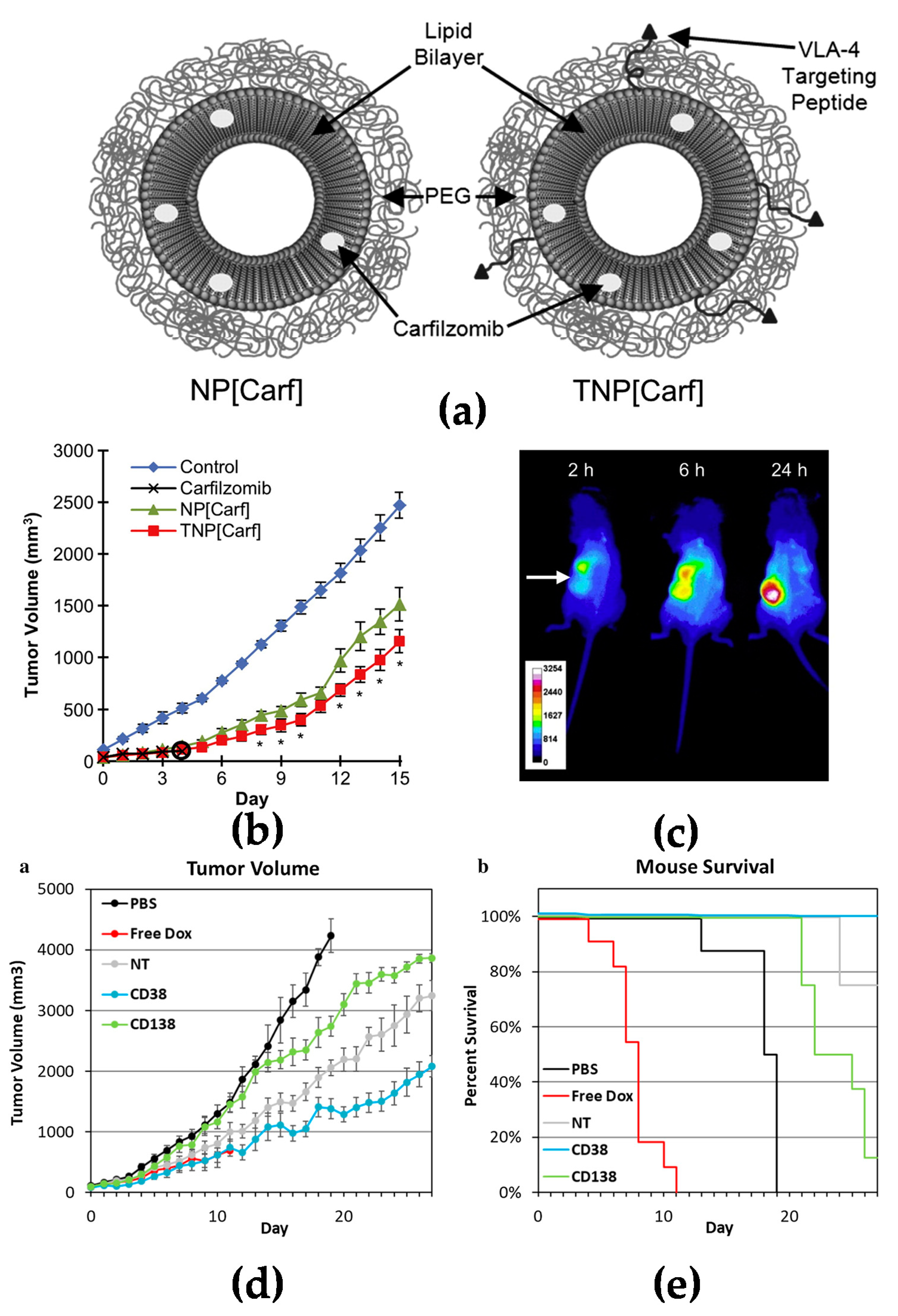
Active targeting increases specific delivery to tumor tissues. Many blood cancer cells express specific surface biomarkers which can be specifically targeted by coupling peptides/antibodies/ligands to the surface of nanomedicines for drug delivery [23,24]. Targeting nanomedicines modified with peptides is a common strategy investigated extensively in drug delivery research. Liposomes modified with RGD peptide (Arg-Gly-Asp) have been widely used to target angiogenic endothelial cells in tumors [25]. It has also been reported that liposomes conjugated with a cyclic pentamer peptide (VLA-4, very late antigen-4) can be used to target multiple myeloma (Figure 2a–c) [26]. Targeting via antibodies is another approach for an active targeting strategy [27]. It is known that CD38 and CD138 are widely expressed on multiple myeloma cells. Liposomes modified with anti-CD38 or anti-CD138 monoclonal antibody could be a new approach for a targeted delivery system with both targeting myeloma cells and also delivery anticancer agents to cancer cells (Figure 2d,e) [28]. Similar to CD38 and CD138, CD19 is one of the markers expressed in most of the lymphoma diseases [29]; an anti-CD19 targeted liposome encapsuled rapamycin showed promising lymphoma cell-specific treatment inducing autophagic cell death [30,31].
2.2. Targeting Spleen and Lymphoid Nodes
Spleen and lymph nodes provide a distinct microenvironment for tumor cells in blood cancers. The spleen is considered to be involved in many blood cancers, especially in lymphomas. It has been reported that the spleen also plays a key role in tumor immunity by recruiting monocytes and macrophages to the tumor tissues [32]. Spleen involvement is found in approximately one third of lymphomas and can also upstage the disease, especially in Hodgkin lymphoma [33]. Intravenously administered nanoparticles tend to target the spleen because of the phagocytic activity of monocytes and macrophages [34]. In vivo experiments have shown that siRNA encapsuled nanoparticles can reduce tumor growth [35]. Enhanced drug concentration in the spleen has also provided therapeutic benefits in spleen resident infections and hematological disorders including malaria, hairy cell leukemia, idiopathic thrombocytopenic purpura, and autoimmune hemolytic anemia [36].
Lymph nodes initiate most immune responses which can prevent malignant transformation [37]. Antitumor immune responses are still active in some malignancies, impacting progression and outcome. In addition, the cytokines in lymphoid nodes also provide a proinflammatory microenvironment which can also support proliferation of malignant cells [38].
2.3. Targeting Vascular System
Neovascularization is always associated with poor prognosis in most blood cancers including acute myeloid leukemia, multiple myeloma, acute lymphatic leukemia, chronic lymphatic leukemia, and Burkett’s lymphoma [39]. Endothelial surface receptors are highly expressed on the inner lining of blood vessels. Shamay et al. reported that vascular endothelial growth factor receptor 1 (VEGFR1)-targeted polymer drug conjugates showed efficient antitumor effect by targeting tumor vasculature [40]. Another strategy is to utilize tumor-homing immunocytokines such as interleukin-2 (IL-2) [41]. The antibody-based delivery of IL-2 to extracellular targets expressed in the easily accessible tumor-associated vasculature showed therapeutic potential for acute myeloid leukemia and other solid tumors [42]. E-selectin is mainly expressed on inflamed endothelial cells which always exist in the vasculature of inflammatory and tumor sites [43]. Gholizadeh et al. reported that E-selectin targeted immunoliposomes could delivery rapamycin, which specifically inhibited inflammatory responses in inflamed endothelial cells [44]. Targeting the vascular system can direct antiangiogenic agents to the blood vessels to suppress angiogenesis, and can also contribute released chemotherapeutic drugs to inhibit cell proliferation near the vascular in the bone marrow. A vascular targeting co-delivery strategy can maximize the combination therapeutic efficacy for the treatment of blood cancers.
3. Nanomedicines for Blood Cancers
3.1. Multiple Myeloma
Multiple myeloma (MM) is a B cell malignancy disease which is characterized by the accumulation of malignant plasma cells in the bone marrow. Although the new treatment and transplant has been utilized in recent decades and has prolonged the overall survival for patients, multiple myeloma is still not curable since it is difficult to remove the tumor cells from the bone marrow. Swami et al. reported that PEG-PLGA encapsuled bortezomib nanoparticles inhibited myeloma growth in a mouse model [5]. Ashley et al. reported that carfilzomib-loaded liposomal nanoparticles targeted myeloma cells [26]. A doxorubicin liposome combined with bortezomib for the treatment of relapsed or refractory multiple myeloma has already been approved by FDA for clinical use [45]. The outcome was based on a phase III clinical trial which showed that liposomal doxorubicin was superior to bortezomib monotherapy [46].
In recent years, protease inhibitors have been widely used in the treatment of multiple myeloma [47]. Nanoparticles encapsuled with protease inhibitors have also been investigated. Lee et al. reported on an injectable nanomedicine for MM therapy by encapsulating bortezomib (class I protease inhibitor) in nanoparticles that possessed a catechol-functionalized polycarbonate core through a pH-sensitive covalent bond between the biodegradable phenylboronic acid in bortezomib and catechol [48]. An in vitro release study showed that, at pH 7.4, the bortezomib release from the composite remained low at 7%, whereas in an acidic environment, ∼85% of bortezomib was released gradually over 9 days. In vivo studies showed that tumor progression of mice treated with the bortezomib-loaded micelle/hydrogel composite resulted in significant delay in tumor progression in a xenograft mouse model, thus, demonstrating the potential of the hydrogel for subcutaneous administration and sustained drug delivery. In addition, an antibody-based delivery system has also been investigated in the myeloma treatment. Huang et al. reported the development of monoclonal anti-CD38 antibody conjugated nanoparticles encapsulated with S3I-1757 (a STAT3 inhibitor) in MM therapy [49]. In this study, they generated two nanoparticle delivery systems with or without the anti-CD38 antibody. The in vitro release study showed two formulations with a comparable drug release (about 68%) property. However, the anti-CD38 antibody coated nanoparticles showed increased drug uptake in two different MM cell lines. In vivo studies performed on a xenograft mouse model demonstrated that the anti-CD38 antibody coated nanoparticles were able to significantly reduce tumor size by four-fold as compared with non-coated nanoparticles, after 12 days drug administration, which indicated that the anti-CD38 antibody on nanoparticles loaded with STAT3 inhibitors can further improve their therapeutic effects against MM.
3.2. Acute Myeloid Leukemia
Acute myeloid leukemia (AML) is another common type of hematological malignancy which is characterized by high proliferation of abnormal myeloblasts in the bone marrow [50,51]. Chemotherapy is still the primary choice for AML treatment. However, the overall survival of single chemotherapy for AML patients is still very low [52]. A combination of two or more anticancer reagents is often used for AML therapy to increase the treatment outcome, but various adverse effects can happen during the treatments [53]. Recently, there are some drug delivery systems that have been investigated to increase the anti-AML effect. Roboz et al. reported on a lipid-drug conjugate encapsuled cytarabine that has been put into a phase III clinical trial [54]. Alakhova et al. reported on a pluronic-based micelle which could increase the anti-AML efficacy of doxorubicin and was also in a phase III clinical trial [55]. Tardi et al. reported on a cytarabine liposome which could increase accumulation in leukemia cells inside the bone marrow and enhance efficacy in AML xenograft model [56].
In 2017, CPX-351 (trade name Vyxeos) was approved by the FDA and EMA for treating newly diagnosed therapy-related AML and/or AML with myelodysplasia-related changes [57]. CPX-351 was initially synthesized and evaluated in in vitro and in vivo studies with leukemia cell lines. The results indicated that the liposomal-encapsulated cytarabine and daunorubicin could display the best synergistic effect and minimum antagonism at a ratio of 5:1, with higher proportions of response rates, more durable remissions, and longer maintenances in bone marrows as compared with a free drug cocktail of cytarabine and daunorubicin with their maximum tolerated doses. Lancet et al. analyzed the data from the clinical trials and found that CPX-351 indicated a significant improvement in survival over standard induction chemotherapy for high-risk patients with AML, older patients with sAML, and a poor-risk subgroup of patients with AML [58].
AZD2811 polymeric nanoparticles are loaded with aurora kinase B inhibitor. AZD2811 has been assessed in AML xenograft models and has shown improved efficacy in inhibiting tumor growth and inducing apoptosis as compared with free aurora kinase B inhibitor (AZD1152). Moreover, this formulation has also demonstrated transient cellular reduction in bone marrow, and may be a potential agent for targeting residual disease. There are two clinical trials ongoing for evaluating the safety, tolerability, and pharmacokinetics of AZD2811 (NCT02579226, NCT03217838) [59].
3.3. B Cell Lymphomas
Lymphoma is a type of cancer which often happens in lymph nodes. The majority arise from B cells, and therefore, are called B cell lymphomas which include both Hodgkin’s lymphomas and most non-Hodgkin lymphomas [60]. Chemotherapy and stem cell transplantation are two main treatments for B cell lymphomas; however, relapse is often inevitable [61]. Antibody conjugates provided a new way for targeting therapy for B cell lymphomas. Brentuximab vedotin (Adcetris®, Seattle Genetics, Bothell, WA, USA) and ibritumomab tiuxetan (Zevalin®, IDEC, Cambridge, MA /Spectrum, Irvine, CA, USA) are two commercially available antibody-drug conjugates for Hodgkin lymphoma and non-Hodgkin lymphoma which have already been approved by the FDA [62,63]. Furthermore, new technology provides the possibility to selectively deliver anticancer agents to malignant cells without damaging healthy cells or systemic toxicity, allowing them to reach the lymph nodes. Nevala et al. reported on a nano-antibody targeted chemotherapy delivery system that used a slight modification of existing cancer drugs with significantly improved treatment efficacy in CD20+ B-cell lymphoma [64]. Martucci et al. reported on siRNA targeting Bcl2-loaded diatomite nanoparticles that demonstrated significant biological improvements for personalized treatment of lymphomas [65]. Choi et al. reported on dually targeted siRNA nanoformulation constructed using layer-by-layer nanoparticles (LbL-NP) for the treatment of non-Hodgkin lymphoma. The LbL-NP protects siRNA from nucleases in the bloodstream by embedding within polyelectrolyte layers that coat a polymeric core. The outermost layer consists of hyaluronic acid (a CD44 ligand) covalently conjugated to CD20 antibodies. The CD20/CD44 dual-targeting outer layer provides precise binding to blood cancer cells, followed by receptor-mediated endocytosis of the LbL-NP. The dual-targeting approach significantly enhances internalization of BCL-2 siRNA in lymphoma and leukemia cells, which leads to significant downregulation of BCL-2 expression. An in vivo study has demonstrated that systemic administration of the dual-targeted, siRNA-loaded nanoparticles induced apoptosis and hampered proliferation of blood cancer cells, both in cell culture and in orthotopic non-Hodgkin’s lymphoma animal models [66].
4. Challenges in Drug Delivery Systems for Treating Blood Cancers
To date, only a few targeted nano-based drug delivery systems are available in clinical practice. There are still many challenges and issues that need to be resolved. Although nano-based medicines provide a significant advantage in delivery strategies, it is still difficult to develop successful formulations that can be put into clinical use. There are biological challenges and non-biological challenges which are described below.
4.1. Biological Challenges
4.1.1. Characterization of Nano-Based Medicines
One of the key points in the development of nano-based medicines is the stability and the efficiency of the loading drugs inside the drug delivery systems. In the design of drug delivery systems such as nanoparticles or liposomes, we can tune the polymer/lipid properties and introduce specific side groups, for instance, to increase the compatibility between the materials and drugs to be loaded. It is possible in the lab to change the properties of a nanocarrier such as molecular weight, ratio of hydrophobic/hydrophilic block, and concentration of drug carrier in relation to the drug to optimize the best property (the most anti-tumor efficiency and/or the lowest side effect) for the nano-based delivery system. However, it is completely different to translate a small-scale formulation into a large-scale production to satisfy industrial demands [67]. To overcome this issue, microfluidics technology and particle replication in non-wetting template (PRINT) technology can control chemical composition, drug loading, and surface properties of nanoparticles with precision [67,68,69,70,71].
4.1.2. Toxicity and Side Effects of Nano-Based Medicines
Another major challenge associated with the translation of nano-based medicines to clinical practice is the nano–bio interactions which may cause toxicity and severe side effects [72]. The potential toxicity of nano-based medicines is usually caused by interactions with biological material which can generate immunoreaction, inflammation, or related disorders in the human beings. The toxic effect is greatly dependent upon various parameters such as size, zeta potential, and solubility of the formulations [73]. When nanoparticles or liposomes enter a biological system, they interact with and are absorbed by proteins [74]. This adsorption of protein onto the surface of nanoparticles or liposomes results in altering their size, surface charge, stability, dispersibility, pharmacokinetics, biodistribution, and toxicity profile [75,76]. Furthermore, it has been reported that many nano-based medicines have caused severe acute adverse immune reactions in vivo [77]. Szebeni et al. reported that liposomes were known to activate the complement (C) system, which could lead, in vivo, to a hypersensitivity syndrome called C activation-related pseudoallergy (CARPA) which has received increased attention as a safety risk of i.v. therapy with liposomes [78]. Furthermore, long circulation properties may also result in non-specific accumulation in the skin causing serious side effects such as hand-foot syndrome, or palmar-plantar erythrodysesthesia, as reported for PEGylated liposomal doxorubicin [79,80]. In addition, there are off-target risks, due to some of the antibody nanoparticles ligands that are also targets in normal cells [81]. Additionally, nanoparticles may deactivate immune responses and can be cleared by immune cells which become a further burden for the immune system and lead to even worsen the blood cancer [82]. Therefore, it is quite critical to investigate the physicochemical characteristics of nano-based medicines with respect to pathophysiology and heterogeneity of human diseases.
4.1.3. Circulation and Clearance
The liver and spleen are the two major organs of nano-based medicine uptake and clearance, as well as kidney, lung, and bone marrow that are also involved in this process [83,84]. Macrophages play a critical role during the clearance of nanoparticles or liposomes in these organs [85,86]. To maintain a long circulation profile and to decrease recognition by host cells, surface coatings have been developed to increase circulation times in blood [87,88]. Polyethylene glycol (PEG) and polyglycerol (PG) are highly hydrophilic molecules which can reduce the protein absorption and affect the composition of the proteins absorbed on the surface of nanoparticles. It is one of the commonly used materials for coating in nanoparticle preparation which can evade immune cells [89,90,91]. It has been reported that anti-PEG antibodies identified in patients could accelerate clearance of PEG-modified nanomedicines. They may also increase the rate of adverse events, such as allergic reactions [92]. Depletion of phagocytes is another strategy for enhancing the circulation time of nano-based medicines by using clodronate-loaded liposomes [93,94,95,96]. Furthermore, macrophage depletion by clodronate liposomes supports prevention of nanoparticle clearance from the peripheral blood and is also a tool to study the role of macrophages and other phagocytes in health and disease [97,98,99]. In addition, clodronate is a first-generation bisphosphonate that has been approved for the prevention and treatment of osteoporosis [100,101] and it has also shown potential therapeutic efficacy for treating chronic lymphocytic leukemia [102,103,104].
4.1.4. Translational Study in the In Vivo Model
The first step of a preclinical study to evaluate novel pharmaceuticals including nano-based medicines is in vitro testing in order to identify the biocompatibility and efficacy and it is usually performed on cancer cell lines. Some novel in vitro culture systems have been established recently to simulate the microenvironment of tumors such as 3D culture systems or organoid culture systems which allow evaluation of therapeutics in a microenvironment that is somewhat closer to the actual disease situation [105,106]. Although these novel in vitro systems can mimic the microenvironment of a tumor or the cell–cell interactions, an in vivo animal model is still required for the development of circulation, biodistribution, safety and efficacy profiles before the clinical trial process [107]. There are lots of factors that should be considered to improve in vivo animal models to study drug delivery and therapeutic efficacy in blood cancers. First of all, the cell line in the in vitro culture has been clonal selected for lots of passages which may have already changed the original profile of the disease. Therefore, using a primary tumor derived animal model rather than a cell line-based model should be considered in in vivo animal models. Secondly, different patient-derived models may response completely different to the same nanoparticles or liposomes. The difference in the sensitivity may also provide personalized precise medicine according to the patient’s personal genomic profile which could be further investigated. Last but not least, it is realistic that tumor growth in animal models is still different than in humans. There are many new medicines that have shown promising antitumor activities in animal models but are still less effective in clinical trials [108]. One more thing specifically applicable in blood cancer studies is that the immunodeficient strains are widely used to allow xenograft models to grow in animals. The lack of an immune system may alter the pharmacokinetics and the pharmacodynamics profile in vivo, especially for blood cancers that often involve the immune system [109].
4.1.5. Interfere with the Bone Marrow for Blood Cancer Drug Delivery Systems
Unlike solid tumors, blood cancers are usually the malignance in bone marrow. Nano-based medicines overcome a series of barriers to deliver an antitumor agent into the bone marrow or the tumor site. In addition, targeting malignancies inside the bone marrow is still a complex issue since it involves cancer stem/progenitor cell existence as well as bone marrow microenvironment induced resistances [110,111]. Therefore, disruption of the interactions between tumor cells and the bone marrow microenvironment via two or three different agents/drugs may achieve greater clinical responses to increase sensitization and overcome the resistance. However, the stable co-encapsulation of multiple agents into a single nano-based targeted drug delivery system might change the pharmacokinetic or pharmacodynamic profiles of combined drugs to achieve the additive or synergistic effect for tumor therapy for blood cancers [112,113]. In addition, the accumulation of antitumor drug inside the bone marrow could enhance the antitumor efficacy but also lead to cumulative toxicity to the normal hematopoiesis cells [114]. Therefore, the development of elaborate bone marrow-targeted systems is essential for specific delivery of antitumor drugs to tumor cells and to minimize the capture by healthy bone marrow cells in the bone marrow.
4.2. Non-Biological Challenges
4.2.1. Commercialized Challenges
One of the major commercialized challenges associated with the clinical translation of nano-based medicines is the difficulty in formulating a controllable and reproducible synthesis process. Working in a laboratory on a small-scale formulation is much easier and highly dependent on the operator’s experience, which are not suitable for reproducible large-scale production. In addition, the formulation must be stable to allow long term storage and shipment which also makes the situation more complex [115]. It has been noticed that nanoplatforms with laborious and complex manufacturing processes rarely find their way into clinical practice due to the inconvenience caused to the pharmaceutical industrial.
4.2.2. Policy/Regulation Challenges
Another urgent and major challenge that needs to be addressed is the huge gap between scientific research and the regulatory authorities. In most of the countries, the authorization of a new medicine is monitored by the government according to a series of policies and laws regarding safety profiles, industrial manufacturing practices, intellectual property protections, quality controls, etc. In the United States, the approval process for nano-based medicines is essentially the same as that for other drugs or biological medicines which are regulated by the Food and Drug Administration [116,117,118]. Unless there is specific consideration for a particular nano-based medicine, the development of nano-based medicines follows the typical drug-development process [119]. This regulation of guidelines for nano-based medicines has been questioned [120]. Timely and effective translation to market is highly affected due to the deficiency of clear regulatory and safety guidelines [121]. Currently, commercially available nano-based medicines on the market have passed the general regulatory standards for approval. However, these standards may not be sufficient and need further revision to confirm quality, safety, and efficacy for human use [76,114]. Nowadays, the rapid development of nanotechnology has contributed to its potential use in nanomedicine. There is an urgent need for more integrated regulatory policies which should be done by country governments. The new guidelines should benefit nanomedicine for patients by addressing any concerns delaying the releasing process for nano-based medicines [121].
5. Summary
Targeted delivery of therapeutic agents plays a pivotal role in the effective and safe treatment of blood cancers. Targeting B cell malignancies inside the bone marrow is still a biological issue due to cancer stem/initiating cell existence and bone marrow microenvironment-induced resistances. It is also notable that the accumulation of therapeutic agents inside the bone marrow or lymph nodes might also lead to cumulative toxicity to normal hematopoiesis stem cells or inhibit the immune response. Further investigations should focus on the specific delivery of therapeutic agents to tumor cells and on minimizing the capture by healthy cells in the bone marrow or lymph node. Recently, several new nanomedicines and drug delivery formulations have been successfully developed and clinically approved for treatment of many types of cancers, which indicates that effective and safe targeted formulations are expected to benefit the treatment of patients in the future. Scientists, industry bodies, and governments should work together to overcome the biological and non-biological challenges to make the translational research smoother for patients.
Funding
This research received no external funding.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are not available from the authors.
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Rowe, J.M. Perspectives on current survival and new developments in AML. Best Pract. Res. Clin. Haematol. 2021, 34, 101248. [Google Scholar] [CrossRef]
- Tuazon, S.A.; Holmberg, L.A.; Nadeem, O.; Richardson, P.G. A clinical perspective on plasma cell leukemia; current status and future directions. Blood Cancer J. 2021, 11, 23. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.; June, C.H. Going viral: Chimeric antigen receptor T-cell therapy for hematological malignancies. Immunol Rev. 2015, 263, 68–89. [Google Scholar] [CrossRef] [PubMed]
- Swami, A.; Reagan, M.R.; Basto, P.; Mishima, Y.; Kamaly, N.; Glavey, S.; Zhang, S.; Moschetta, M.; Seevaratnam, D.; Zhang, Y.; et al. Engineered nanomedicine for myeloma and bone microenvironment targeting. Proc. Natl. Acad. Sci. USA 2014, 111, 10287–10292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adjei, I.M.; Sharma, B.; Peetla, C.; Labhasetwar, V. Inhibition of bone loss with surface-modulated, drug-loaded nanoparticles in an intraosseous model of prostate cancer. J. Control. Release 2016, 232, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Yu, X.; Carbone, E.J.; Nelson, C.; Kan, H.M.; Lo, K.W. Poly aspartic acid peptide-linked PLGA based nanoscale particles: Potential for bone-targeting drug delivery applications. Int. J. Pharm. 2014, 475, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Iannazzo, D.; Ettari, R.; Giofrè, S.; Eid, A.H.; Bitto, A. Recent Advances in Nanotherapeutics for Multiple Myeloma. Cancers 2020, 12, 3144. [Google Scholar] [CrossRef]
- Beloqui, A.; Solinís, M.A.; Delgado, A.; Evora, C.; del Pozo-Rodríguez, A.; Rodríguez-Gascón, A. Biodistribution of Nanostructured Lipid Carriers (NLCs) after intravenous administration to rats: Influence of technological factors. Eur. J. Pharm. Biopharm. 2013, 84, 309–314. [Google Scholar] [CrossRef]
- Vinhas, R.; Mendes, R.; Fernandes, A.R.; Baptista, P.V. Nanoparticles-Emerging Potential for Managing Leukemia and Lymphoma. Front. Bioeng. Biotechnol. 2017, 5, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sou, K.; Goins, B.; Oyajobi, B.O.; Travi, B.L.; Phillips, W.T. Bone marrow-targeted liposomal carriers. Expert Opin. Drug Deliv. 2011, 8, 317–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moghimi, S.M. Exploiting bone marrow microvascular structure for drug delivery and future therapies. Adv. Drug Deliv. Rev. 1995, 17, 61–73. [Google Scholar] [CrossRef]
- Sarin, H. Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J. Angiogenes Res. 2010, 2, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bozzuto, G.; Molinari, A. Liposomes as nanomedical devices. Int. J. Nanomed. 2015, 10, 975–999. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, H.A.; Warner, K.J.; Li, M.C.; Hunter, G.K. Binding of bone sialoprotein, osteopontin and synthetic polypeptides to hydroxyapatite. Connect. Tissue Res. 2001, 42, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Miller, S.C.; Shlyakhtenko, L.S.; Portillo, A.M.; Liu, X.M.; Papangkorn, K.; Kopecková, P.; Lyubchenko, Y.; Higuchi, W.I.; Kopecek, J. Osteotropic Peptide that differentiates functional domains of the skeleton. Bioconjug. Chem. 2007, 18, 1375–1378. [Google Scholar] [CrossRef]
- Huang, L.; Wang, X.; Cao, H.; Li, L.; Chow, D.H.; Tian, L.; Wu, H.; Zhang, J.; Wang, N.; Zheng, L.; et al. A bone-targeting delivery system carrying osteogenic phytomolecule icaritin prevents osteoporosis in mice. Biomaterials 2018, 182, 58–71. [Google Scholar] [CrossRef]
- Yang, Y.S.; Xie, J.; Chaugule, S.; Wang, D.; Kim, J.M.; Kim, J.; Tai, P.W.L.; Seo, S.K.; Gravallese, E.; Gao, G.; et al. Bone-Targeting AAV-Mediated Gene Silencing in Osteoclasts for Osteoporosis Therapy. Mol. Ther. Methods Clin. Dev. 2020, 17, 922–935. [Google Scholar] [CrossRef]
- Hu, Z.; Zhang, L.; Wang, H.; Wang, Y.; Tan, Y.; Dang, L.; Wang, K.; Sun, Z.; Li, G.; Cao, X.; et al. Targeted silencing of miRNA-132-3p expression rescues disuse osteopenia by promoting mesenchymal stem cell osteogenic differentiation and osteogenesis in mice. Stem Cell Res. Ther. 2020, 11, 58. [Google Scholar] [CrossRef]
- Shi, Y.; Su, Z.; Li, S.; Chen, Y.; Chen, X.; Xiao, Y.; Sun, M.; Ping, Q.; Zong, L. Multistep targeted nano drug delivery system aiming at leukemic stem cells and minimal residual disease. Mol. Pharm. 2013, 10, 2479–2489. [Google Scholar] [CrossRef]
- Santini, D.; Caraglia, M.; Vincenzi, B.; Holen, I.; Scarpa, S.; Budillon, A.; Tonini, G. Mechanisms of disease: Preclinical reports of antineoplastic synergistic action of bisphosphonates. Nat. Clin. Pract. Oncol. 2006, 3, 325–338. [Google Scholar] [CrossRef]
- Tian, Z.; Wu, L.; Yu, C.; Chen, Y.; Xu, Z.; Bado, I.; Loredo, A.; Wang, L.; Wang, H.; Wu, K.L.; et al. Harnessing the power of antibodies to fight bone metastasis. Sci. Adv. 2021, 7, 26. [Google Scholar] [CrossRef]
- Jiang, Y.; Yang, N.; Zhang, H.; Sun, B.; Hou, C.; Ji, C.; Zheng, J.; Liu, Y.; Zuo, P. Enhanced in vivo antitumor efficacy of dual-functional peptide-modified docetaxel nanoparticles through tumor targeting and Hsp90 inhibition. J. Control. Release 2016, 221, 26–36. [Google Scholar] [CrossRef]
- Yang, N.; Jiang, Y.; Zhang, H.; Sun, B.; Hou, C.; Zheng, J.; Liu, Y.; Zuo, P. Active targeting docetaxel-PLA nanoparticles eradicate circulating lung cancer stem-like cells and inhibit liver metastasis. Mol. Pharm. 2015, 12, 232–239. [Google Scholar] [CrossRef]
- Mahmoudi, R.; Ashraf Mirahmadi-Babaheidri, S.; Delaviz, H.; Fouani, M.H.; Alipour, M.; Jafari Barmak, M.; Christiansen, G.; Bardania, H. RGD peptide-mediated liposomal curcumin targeted delivery to breast cancer cells. J. Biomater. Appl. 2021, 35, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Ashley, J.D.; Stefanick, J.F.; Schroeder, V.A.; Suckow, M.A.; Alves, N.J.; Suzuki, R.; Kikuchi, S.; Hideshima, T.; Anderson, K.C.; Kiziltepe, T.; et al. Liposomal carfilzomib nanoparticles effectively target multiple myeloma cells and demonstrate enhanced efficacy in vivo. J. Control. Release 2014, 196, 113–121. [Google Scholar] [CrossRef]
- Kim, D.; Park, C.Y.; Medeiros, B.C.; Weissman, I.L. CD19-CD45 low/-CD38 high/CD138+ plasma cells enrich for human tumorigenic myeloma cells. Leukemia 2012, 26, 2530–2537. [Google Scholar] [CrossRef]
- Omstead, D.T.; Mejia, F.; Sjoerdsma, J.; Kim, B.; Shin, J.; Khan, S.; Wu, J.; Kiziltepe, T.; Littlepage, L.E.; Bilgicer, B. In vivo evaluation of CD38 and CD138 as targets for nanoparticle-based drug delivery in multiple myeloma. J. Hematol. Oncol. 2020, 13, 145. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wei, G.; Liu, D. CD19: A biomarker for B cell development, lymphoma diagnosis and therapy. Exp. Hematol. Oncol. 2012, 1, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, K.; Sato, T.; Iyama, S.; Tatekoshi, A.; Hashimoto, A.; Kamihara, Y.; Horiguchi, H.; Kikuchi, S.; Kawano, Y.; Takada, K.; et al. A novel strategy inducing autophagic cell death in Burkitt’s lymphoma cells with anti-CD19-targeted liposomal rapamycin. Blood Cancer J. 2014, 4, e180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Shen, D.; Jia, M.; Zhao, H.; Tang, Y. The targeting effect of Hm2E8b-NCTD-liposomes on B-lineage leukaemia stem cells is associated with the HLF-SLUG axis. J. Drug Target. 2018, 26, 55–65. [Google Scholar] [CrossRef]
- Bronte, V.; Pittet, M.J. The spleen in local and systemic regulation of immunity. Immunity 2013, 39, 806–818. [Google Scholar] [CrossRef] [Green Version]
- Saboo, S.S.; Krajewski, K.M.; O’Regan, K.N.; Giardino, A.; Brown, J.R.; Ramaiya, N.; Jagannathan, J.P. Spleen in haematological malignancies: Spectrum of imaging findings. Br. J. Radiol. 2012, 85, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Balasubramanian, S.K.; Jittiwat, J.; Manikandan, J.; Ong, C.N.; Yu, L.E.; Ong, W.Y. Biodistribution of gold nanoparticles and gene expression changes in the liver and spleen after intravenous administration in rats. Biomaterials 2010, 31, 2034–2042. [Google Scholar] [CrossRef]
- Leuschner, F.; Dutta, P.; Gorbatov, R.; Novobrantseva, T.I.; Donahoe, J.S.; Courties, G.; Lee, K.M.; Kim, J.I.; Markmann, J.F.; Marinelli, B.; et al. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat. Biotechnol. 2011, 29, 1005–1010. [Google Scholar] [CrossRef]
- Jindal, A.B. Nanocarriers for spleen targeting: Anatomo-physiological considerations, formulation strategies and therapeutic potential. Drug Deliv. Transl. Res. 2016, 6, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Mu, C.F.; Shen, J.; Liang, J.; Zheng, H.S.; Xiong, Y.; Wei, Y.H.; Li, F. Targeted drug delivery for tumor therapy inside the bone marrow. Biomaterials 2018, 155, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Park, C.S.; Choi, Y.S. How do follicular dendritic cells interact intimately with B cells in the germinal centre? Immunology 2005, 114, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef] [Green Version]
- Shamay, Y.; Golan, M.; Tyomkin, D.; David, A. Assessing the therapeutic efficacy of VEGFR-1-targeted polymer drug conjugates in mouse tumor models. J. Control. Release 2016, 229, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Runbeck, E.; Crescioli, S.; Karagiannis, S.N.; Papa, S. Utilizing Immunocytokines for Cancer Therapy. Antibodies 2021, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Schliemann, C.; Gutbrodt, K.L.; Kerkhoff, A.; Pohlen, M.; Wiebe, S.; Silling, G.; Angenendt, L.; Kessler, T.; Mesters, R.M.; Giovannoni, L.; et al. Targeting interleukin-2 to the bone marrow stroma for therapy of acute myeloid leukemia relapsing after allogeneic hematopoietic stem cell transplantation. Cancer Immunol. Res. 2015, 3, 547–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, M.; Videira, P.A.; Sackstein, R. E-Selectin Ligands in the Human Mononuclear Phagocyte System: Implications for Infection, Inflammation, and Immunotherapy. Front. Immunol. 2017, 8, 1878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gholizadeh, S.; Visweswaran, G.R.R.; Storm, G.; Hennink, W.E.; Kamps, J.; Kok, R.J. E-selectin targeted immunoliposomes for rapamycin delivery to activated endothelial cells. Int. J. Pharm. 2018, 548, 759–770. [Google Scholar] [CrossRef]
- Ning, Y.M.; He, K.; Dagher, R.; Sridhara, R.; Farrell, A.T.; Justice, R.; Pazdur, R. Liposomal doxorubicin in combination with bortezomib for relapsed or refractory multiple myeloma. Oncology 2007, 21, 1503–1508. [Google Scholar]
- Orlowski, R.Z.; Nagler, A.; Sonneveld, P.; Bladé, J.; Hajek, R.; Spencer, A.; Robak, T.; Dmoszynska, A.; Horvath, N.; Spicka, I.; et al. Final overall survival results of a randomized trial comparing bortezomib plus pegylated liposomal doxorubicin with bortezomib alone in patients with relapsed or refractory multiple myeloma. Cancer 2016, 122, 2050–2056. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Zhuang, J. Major advances in the treatment of multiple myeloma in American Society of Hematology annual meeting 2020. Chronic Dis. Transl. Med. 2021, 7, 220–226. [Google Scholar] [CrossRef]
- Lee, A.L.Z.; Voo, Z.X.; Chin, W.; Ono, R.J.; Yang, C.; Gao, S.; Hedrick, J.L.; Yang, Y.Y. Injectable Coacervate Hydrogel for Delivery of Anticancer Drug-Loaded Nanoparticles in vivo. ACS Appl Mater. Interfaces 2018, 10, 13274–13282. [Google Scholar] [CrossRef]
- Huang, Y.H.; Vakili, M.R.; Molavi, O.; Morrissey, Y.; Wu, C.; Paiva, I.; Soleimani, A.H.; Sanaee, F.; Lavasanifar, A.; Lai, R. Decoration of Anti-CD38 on Nanoparticles Carrying a STAT3 Inhibitor Can Improve the Therapeutic Efficacy Against Myeloma. Cancers 2019, 11, 248. [Google Scholar] [CrossRef] [Green Version]
- Löwenberg, B.; Downing, J.R.; Burnett, A. Acute myeloid leukemia. N. Engl. J. Med. 1999, 341, 1051–1062. [Google Scholar] [CrossRef]
- Huang, X.; Lin, H.; Huang, F.; Xie, Y.; Wong, K.H.; Chen, X.; Wu, D.; Lu, A.; Yang, Z. Targeting Approaches of Nanomedicines in Acute Myeloid Leukemia. Dose Response 2019, 17, 1559325819887048. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef]
- Dombret, H.; Gardin, C. An update of current treatments for adult acute myeloid leukemia. Blood 2016, 127, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Roboz, G.J.; Rosenblat, T.; Arellano, M.; Gobbi, M.; Altman, J.K.; Montesinos, P.; O’Connell, C.; Solomon, S.R.; Pigneux, A.; Vey, N.; et al. International randomized phase III study of elacytarabine versus investigator choice in patients with relapsed/refractory acute myeloid leukemia. J. Clin. Oncol 2014, 32, 1919–1926. [Google Scholar] [CrossRef]
- Alakhova, D.Y.; Zhao, Y.; Li, S.; Kabanov, A.V. Effect of doxorubicin/pluronic SP1049C on tumorigenicity, aggressiveness, DNA methylation and stem cell markers in murine leukemia. PLoS ONE 2013, 8, e72238. [Google Scholar] [CrossRef]
- Tardi, P.; Wan, C.P.; Mayer, L. Passive and semi-active targeting of bone marrow and leukemia cells using anionic low cholesterol liposomes. J. Drug Target. 2016, 24, 797–804. [Google Scholar] [CrossRef]
- Krauss, A.C.; Gao, X.; Li, L.; Manning, M.L.; Patel, P.; Fu, W.; Janoria, K.G.; Gieser, G.; Bateman, D.A.; Przepiorka, D.; et al. FDA Approval Summary: (Daunorubicin and Cytarabine) Liposome for Injection for the Treatment of Adults with High-Risk Acute Myeloid Leukemia. Clin. Cancer Res. 2019, 25, 2685–2690. [Google Scholar] [CrossRef]
- Lancet, J.E.; Uy, G.L.; Cortes, J.E.; Newell, L.F.; Lin, T.L.; Ritchie, E.K.; Stuart, R.K.; Strickland, S.A.; Hogge, D.; Solomon, S.R.; et al. CPX-351 (cytarabine and daunorubicin) Liposome for Injection Versus Conventional Cytarabine Plus Daunorubicin in Older Patients With Newly Diagnosed Secondary Acute Myeloid Leukemia. J. Clin. Oncol 2018, 36, 2684–2692. [Google Scholar] [CrossRef]
- Floc’h, N.; Ashton, S.; Taylor, P.; Trueman, D.; Harris, E.; Odedra, R.; Maratea, K.; Derbyshire, N.; Caddy, J.; Jacobs, V.N.; et al. Optimizing Therapeutic Effect of Aurora B Inhibition in Acute Myeloid Leukemia with AZD2811 Nanoparticles. Mol. Cancer Ther. 2017, 16, 1031–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novo, M.; Castellino, A.; Nicolosi, M.; Santambrogio, E.; Vassallo, F.; Chiappella, A.; Vitolo, U. High-grade B-cell lymphoma: How to diagnose and treat. Expert Rev. Hematol. 2019, 12, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Deshantri, A.K.; Varela Moreira, A.; Ecker, V.; Mandhane, S.N.; Schiffelers, R.M.; Buchner, M.; Fens, M. Nanomedicines for the treatment of hematological malignancies. J. Control. Release 2018, 287, 194–215. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Gopal, A.K.; Smith, S.E.; Ansell, S.M.; Rosenblatt, J.D.; Savage, K.J.; Ramchandren, R.; Bartlett, N.L.; Cheson, B.D.; de Vos, S.; et al. Results of a pivotal phase II study of brentuximab vedotin for patients with relapsed or refractory Hodgkin’s lymphoma. J. Clin. Oncol. 2012, 30, 2183–2189. [Google Scholar] [CrossRef] [PubMed]
- Morschhauser, F.; Radford, J.; Van Hoof, A.; Vitolo, U.; Soubeyran, P.; Tilly, H.; Huijgens, P.C.; Kolstad, A.; d’Amore, F.; Gonzalez Diaz, M.; et al. Phase III trial of consolidation therapy with yttrium-90-ibritumomab tiuxetan compared with no additional therapy after first remission in advanced follicular lymphoma. J. Clin. Oncol. 2008, 26, 5156–5164. [Google Scholar] [CrossRef]
- Nevala, W.K.; Butterfield, J.T.; Sutor, S.L.; Knauer, D.J.; Markovic, S.N. Antibody-targeted paclitaxel loaded nanoparticles for the treatment of CD20(+) B-cell lymphoma. Sci. Rep. 2017, 7, 45682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martucci, N.M.; Migliaccio, N.; Ruggiero, I.; Albano, F.; Calì, G.; Romano, S.; Terracciano, M.; Rea, I.; Arcari, P.; Lamberti, A. Nanoparticle-based strategy for personalized B-cell lymphoma therapy. Int. J. Nanomed. 2016, 11, 6089–6101. [Google Scholar] [CrossRef] [Green Version]
- Choi, K.Y.; Correa, S.; Min, J.; Li, J.; Roy, S.; Laccetti, K.H.; Dreaden, E.; Kong, S.; Heo, R.; Roh, Y.H.; et al. Binary Targeting of siRNA to Hematologic Cancer Cells In Vivo using Layer-by-Layer Nanoparticles. Adv. Funct. Mater. 2019, 29, 1900018. [Google Scholar] [CrossRef]
- Roscigno, R.F.; Vaughn, T.; Parsley, E.; Hunt, T.; Eldon, M.A.; Rubin, L.J. Comparative bioavailability of inhaled treprostinil administered as LIQ861 and Tyvaso® in healthy subjects. Vascul. Pharmacol. 2021, 138, 106840. [Google Scholar] [CrossRef]
- Petersen, R.S.; Boisen, A.; Keller, S.S. Micromechanical Punching: A Versatile Method for Non-Spherical Microparticle Fabrication. Polymers 2020, 13, 83. [Google Scholar] [CrossRef]
- Perry, J.L.; Tian, S.; Sengottuvel, N.; Harrison, E.B.; Gorentla, B.K.; Kapadia, C.H.; Cheng, N.; Luft, J.C.; Ting, J.P.; DeSimone, J.M.; et al. Pulmonary Delivery of Nanoparticle-Bound Toll-like Receptor 9 Agonist for the Treatment of Metastatic Lung Cancer. ACS Nano 2020, 14, 7200–7215. [Google Scholar] [CrossRef]
- Dumont, E.F.; Oliver, A.J.; Ioannou, C.; Billiard, J.; Dennison, J.; van den Berg, F.; Yang, S.; Chandrasekaran, V.; Young, G.C.; Lahiry, A.; et al. A Novel Inhaled Dry-Powder Formulation of Ribavirin Allows for Efficient Lung Delivery in Healthy Participants and Those with Chronic Obstructive Pulmonary Disease in a Phase 1 Study. Antimicrob. Agents Chemother. 2020, 64, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, E.M.; Luft, J.C.; DeSimone, J.M. Formulation of High-Performance Dry Powder Aerosols for Pulmonary Protein Delivery. Pharm. Res. 2018, 35, 195. [Google Scholar] [CrossRef]
- Metselaar, J.M.; Lammers, T. Challenges in nanomedicine clinical translation. Drug Deliv. Transl. Res. 2020, 10, 721–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dilnawaz, F.; Acharya, S.; Sahoo, S.K. Recent trends of nanomedicinal approaches in clinics. Int. J. Pharm. 2018, 538, 263–278. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Dilnawaz, F.; Sahoo, S.K. Challenges of moving theranostic nanomedicine into the clinic. Nanomedicine 2020, 15, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef]
- Hua, S.; de Matos, M.B.C.; Metselaar, J.M.; Storm, G. Current Trends and Challenges in the Clinical Translation of Nanoparticulate Nanomedicines: Pathways for Translational Development and Commercialization. Front. Pharmacol. 2018, 9, 790. [Google Scholar] [CrossRef] [PubMed]
- Jackman, J.A.; Mészáros, T.; Fülöp, T.; Urbanics, R.; Szebeni, J.; Cho, N.J. Comparison of complement activation-related pseudoallergy in miniature and domestic pigs: Foundation of a validatable immune toxicity model. Nanomedicine 2016, 12, 933–943. [Google Scholar] [CrossRef]
- Szebeni, J.; Storm, G. Complement activation as a bioequivalence issue relevant to the development of generic liposomes and other nanoparticulate drugs. Biochem. Biophys. Res. Commun. 2015, 468, 490–497. [Google Scholar] [CrossRef]
- Hu, X.; Dong, M.; Liang, X.; Liu, Z.; Li, Q. Reactive Oxygen Species-Mediated Inflammation and Apoptosis in Hand-Foot Syndrome Induced by PEGylated Liposomal Doxorubicin. Int. J. Nanomed. 2021, 16, 471–480. [Google Scholar] [CrossRef]
- Ni, C.; Fang, J.; Qian, H.; Xu, Q.; Shen, F. Liposomal doxorubicin-related palmar-plantar erythrodysesthesia (hand-foot syndrome): A case report. J. Int. Med. Res. 2020, 48, 300060520974854. [Google Scholar] [CrossRef]
- Konopleva, M.; Pollyea, D.A.; Potluri, J.; Chyla, B.; Hogdal, L.; Busman, T.; McKeegan, E.; Salem, A.H.; Zhu, M.; Ricker, J.L.; et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer Discov. 2016, 6, 1106–1117. [Google Scholar] [CrossRef] [Green Version]
- Butcher, N.J.; Mortimer, G.M.; Minchin, R.F. Drug delivery: Unravelling the stealth effect. Nat. Nanotechnol. 2016, 11, 310–311. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, H.H.; Holt-Casper, D.; Grainger, D.W.; Ghandehari, H. Nanoparticle Uptake: The Phagocyte Problem. Nano Today 2015, 10, 487–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jantawong, C.; Priprem, A.; Intuyod, K.; Pairojkul, C.; Pinlaor, P.; Waraasawapati, S.; Mongkon, I.; Chamgramol, Y.; Pinlaor, S. Curcumin-loaded nanocomplexes: Acute and chronic toxicity studies in mice and hamsters. Toxicol. Rep. 2021, 8, 1346–1357. [Google Scholar] [CrossRef] [PubMed]
- Cicuéndez, M.; Casarrubios, L.; Barroca, N.; Silva, D.; Feito, M.J.; Diez-Orejas, R.; Marques, P.; Portolés, M.T. Benefits in the Macrophage Response Due to Graphene Oxide Reduction by Thermal Treatment. Int. J. Mol. Sci. 2021, 22, 6701. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Ma, J.; Ji, Z.; Shen, J.; Wang, Q. Recent Advances of Cell Membrane Coated Nanoparticles in Treating Cardiovascular Disorders. Molecules 2021, 26, 3428. [Google Scholar] [CrossRef]
- Chen, T.Y.; Chen, M.R.; Liu, S.W.; Lin, J.Y.; Yang, Y.T.; Huang, H.Y.; Chen, J.K.; Yang, C.S.; Lin, K.M. Assessment of Polyethylene Glycol-Coated Gold Nanoparticle Toxicity and Inflammation In Vivo Using NF-κB Reporter Mice. Int. J. Mol. Sci. 2020, 21, 8158. [Google Scholar] [CrossRef] [PubMed]
- Pandita, D.; Munjal, A.; Poonia, N.; Awasthi, R.; Kalonia, H.; Lather, V. Albumin-Coated Mesoporous Silica Nanoparticles of Docetaxel: Preparation, Characterization, and Pharmacokinetic Evaluation. Assay Drug Dev. Technol. 2021, 19, 226–236. [Google Scholar] [CrossRef]
- Zou, Y.; Ito, S.; Yoshino, F.; Suzuki, Y.; Zhao, L.; Komatsu, N. Polyglycerol Grafting Shields Nanoparticles from Protein Corona Formation to Avoid Macrophage Uptake. ACS Nano 2020, 14, 7216–7226. [Google Scholar] [CrossRef]
- Tavano, R.; Gabrielli, L.; Lubian, E.; Fedeli, C.; Visentin, S.; Polverino De Laureto, P.; Arrigoni, G.; Geffner-Smith, A.; Chen, F.; Simberg, D.; et al. C1q-Mediated Complement Activation and C3 Opsonization Trigger Recognition of Stealth Poly(2-methyl-2-oxazoline)-Coated Silica Nanoparticles by Human Phagocytes. ACS Nano 2018, 12, 5834–5847. [Google Scholar] [CrossRef]
- Lassenberger, A.; Scheberl, A.; Stadlbauer, A.; Stiglbauer, A.; Helbich, T.; Reimhult, E. Individually Stabilized, Superparamagnetic Nanoparticles with Controlled Shell and Size Leading to Exceptional Stealth Properties and High Relaxivities. ACS Appl. Mater. Interfaces 2017, 9, 3343–3353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, C.E.; Wang, Y.Y.; Yang, Q.; Hoang, T.; Chattopadhyay, S.; Hoen, T.; Ensign, L.M.; Nunn, K.L.; Schroeder, H.; McCallen, J.; et al. Anti-PEG antibodies alter the mobility and biodistribution of densely PEGylated nanoparticles in mucus. Acta Biomater. 2016, 43, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Li, M.; Qi, W.; Bai, H.; Nie, Z.; Hu, Z.; Xiao, Y.; de Bruijn, J.D.; Bao, C.; Yuan, H. Serial cellular events in bone formation initiated by calcium phosphate ceramics. Acta Biomater. 2021, 134, 730–743. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Yang, J.; Sun, G.; Hu, J.; Zhang, Q.; Cai, J.; Yuan, D.; Li, H.; Hei, Z.; Yao, W. Macrophage extracellular traps aggravate iron overload-related liver ischaemia/reperfusion injury. Br. J. Pharmacol. 2021, 178, 3783–3796. [Google Scholar] [CrossRef] [PubMed]
- Le, K.; Sun, J.; Khawaja, H.; Shibata, M.; Maggirwar, S.B.; Smith, M.R.; Gupta, M. Mantle cell lymphoma polarizes tumor-associated macrophages into M2-like macrophages, which in turn promote tumorigenesis. Blood Adv. 2021, 5, 2863–2878. [Google Scholar] [CrossRef]
- Li, J.; Sun, Z.; Luo, G.; Wang, S.; Cui, H.; Yao, Z.; Xiong, H.; He, Y.; Qian, Y.; Fan, C. Quercetin Attenuates Trauma-Induced Heterotopic Ossification by Tuning Immune Cell Infiltration and Related Inflammatory Insult. Front. Immunol. 2021, 12, 649285. [Google Scholar] [CrossRef]
- Dora, D.; Ferenczi, S.; Stavely, R.; Toth, V.E.; Varga, Z.V.; Kovacs, T.; Bodi, I.; Hotta, R.; Kovacs, K.J.; Goldstein, A.M.; et al. Evidence of a Myenteric Plexus Barrier and its Macrophage-Dependent Degradation during Murine Colitis: Implications in Enteric Neuroinflammation. Cell Mol. Gastroenterol. Hepatol. 2021, 12, 1617–1641. [Google Scholar] [CrossRef]
- Schlundt, C.; El Khassawna, T.; Serra, A.; Dienelt, A.; Wendler, S.; Schell, H.; van Rooijen, N.; Radbruch, A.; Lucius, R.; Hartmann, S. Macrophages in bone fracture healing: Their essential role in endochondral ossification. Bone 2018, 106, 78–89. [Google Scholar] [CrossRef]
- Narla, R.K.; Modi, H.; Bauer, D.; Abbasian, M.; Leisten, J.; Piccotti, J.R.; Kopytek, S.; Eckelman, B.P.; Deveraux, Q.; Timmer, J.; et al. Modulation of CD47-SIRPα innate immune checkpoint axis with Fc-function detuned anti-CD47 therapeutic antibody. Cancer Immunol. Immunother. 2021, 71, 473–489. [Google Scholar] [CrossRef]
- Alaranji, G.; Goyal, A.; Bansal, P. Bisphosphonate Toxicity. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2022. [Google Scholar]
- McCloskey, E.; Paterson, A.H.; Powles, T.; Kanis, J.A. Clodronate. Bone 2021, 143, 115715. [Google Scholar] [CrossRef]
- Hanna, B.; McClanahan, F.; Yazdanparast, H.; Zaborsky, N.; Kalter, V.; Rößner, P.; Benner, A.; Dürr, C.; Egle, A.; Gribben, J. Depletion of CLL-associated patrolling monocytes and macrophages controls disease development and repairs immune dysfunction in vivo. Leukemia 2016, 30, 570–579. [Google Scholar] [CrossRef]
- Galletti, G.; Scielzo, C.; Barbaglio, F.; Rodriguez, T.V.; Riba, M.; Lazarevic, D.; Cittaro, D.; Simonetti, G.; Ranghetti, P.; Scarfò, L.; et al. Targeting Macrophages Sensitizes Chronic Lymphocytic Leukemia to Apoptosis and Inhibits Disease Progression. Cell Rep. 2016, 14, 1748–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piaggio, F.; Kondylis, V.; Pastorino, F.; Di Paolo, D.; Perri, P.; Cossu, I.; Schorn, F.; Marinaccio, C.; Murgia, D.; Daga, A.; et al. A novel liposomal Clodronate depletes tumor-associated macrophages in primary and metastatic melanoma: Anti-angiogenic and anti-tumor effects. J. Control. Release 2016, 223, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Braham, M.V.; Minnema, M.C.; Aarts, T.; Sebestyen, Z.; Straetemans, T.; Vyborova, A.; Kuball, J.; Öner, F.C.; Robin, C.; Alblas, J. Cellular immunotherapy on primary multiple myeloma expanded in a 3D bone marrow niche model. Oncoimmunology 2018, 7, e1434465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanz, H.L.; Saleh, A.; Kramer, B.; Cairns, J.; Ng, C.P.; Yu, J.; Trietsch, S.J.; Hankemeier, T.; Joore, J.; Vulto, P. Therapy response testing of breast cancer in a 3D high-throughput perfused microfluidic platform. BMC Cancer 2017, 17, 709. [Google Scholar] [CrossRef] [Green Version]
- Gould, S.E.; Junttila, M.R.; de Sauvage, F.J. Translational value of mouse models in oncology drug development. Nat. Med. 2015, 21, 431–439. [Google Scholar] [CrossRef]
- Van Norman, G.A. Limitations of Animal Studies for Predicting Toxicity in Clinical Trials: Is it Time to Rethink Our Current Approach? JACC Basic Transl. Sci. 2019, 4, 845–854. [Google Scholar] [CrossRef]
- Song, G.; Suzuki, O.T.; Santos, C.M.; Lucas, A.T.; Wiltshire, T.; Zamboni, W.C. Gulp1 is associated with the pharmacokinetics of PEGylated liposomal doxorubicin (PLD) in inbred mouse strains. Nanomedicine 2016, 12, 2007–2017. [Google Scholar] [CrossRef]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal. Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Fairfield, H.; Falank, C.; Avery, L.; Reagan, M.R. Multiple myeloma in the marrow: Pathogenesis and treatments. Ann. N. Y. Acad. Sci. 2016, 1364, 32–51. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.; Sun, W.; Wang, C.; Gu, Z. Recent advances of cocktail chemotherapy by combination drug delivery systems. Adv. Drug Deliv. Rev. 2016, 98, 19–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miao, L.; Guo, S.; Lin, C.M.; Liu, Q.; Huang, L. Nanoformulations for combination or cascade anticancer therapy. Adv. Drug Deliv. Rev. 2017, 115, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; Rodriguez-Torres, M.D.P.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2018, 16, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, S.; Wu, S.Y. Editorial: Advances and Challenges in Nanomedicine. Front. Pharmacol. 2018, 9, 1397. [Google Scholar] [CrossRef]
- Bobo, D.; Robinson, K.J.; Islam, J.; Thurecht, K.J.; Corrie, S.R. Nanoparticle-Based Medicines: A Review of FDA-Approved Materials and Clinical Trials to Date. Pharm. Res. 2016, 33, 2373–2387. [Google Scholar] [CrossRef]
- Havel, H.; Finch, G.; Strode, P.; Wolfgang, M.; Zale, S.; Bobe, I.; Youssoufian, H.; Peterson, M.; Liu, M. Nanomedicines: From Bench to Bedside and Beyond. AAPS J. 2016, 18, 1373–1378. [Google Scholar] [CrossRef]
- Sainz, V.; Conniot, J.; Matos, A.I.; Peres, C.; Zupancic, E.; Moura, L.; Silva, L.C.; Florindo, H.F.; Gaspar, R.S. Regulatory aspects on nanomedicines. Biochem. Biophys. Res. Commun. 2015, 468, 504–510. [Google Scholar] [CrossRef]
- Grossman, J.H.; Crist, R.M.; Clogston, J.D. Early Development Challenges for Drug Products Containing Nanomaterials. AAPS J. 2017, 19, 92–102. [Google Scholar] [CrossRef]
- Ventola, C.L. Progress in Nanomedicine: Approved and Investigational Nanodrugs. Pharm. Ther. 2017, 42, 742–755. [Google Scholar]
- Tinkle, S.; McNeil, S.E.; Mühlebach, S.; Bawa, R.; Borchard, G.; Barenholz, Y.C.; Tamarkin, L.; Desai, N. Nanomedicines: Addressing the scientific and regulatory gap. Ann. N. Y. Acad. Sci. 2014, 1313, 35–56. [Google Scholar] [CrossRef]
Figure 1.
Targeting the bone surface-mediated bone marrow: (a) Characterization of the bone-targeting delivery system in vitro and in vivo. Chemical structure of icaritin (ICT); (b) the morphology of Asp8-liposome-icaritin taken by cryo-transmission electron microscope (Cryo-TEM) (scale bar = 100 nm, magnification = 105×); (c) localization of fluorescent-labeled liposome delivery system with or without Asp8 targeting peptide in mice by optical imaging IVIS analysis, 72 h after administration; (d) a schematic diagram to illustrate the experimental design for targeted delivery of antagomir-132 to specifically decrease miRNA-132-3p levels in bone; (e) a schematic diagram to illustrate how antagomir-132 is selectively delivered to bone formation region by (AspSerSer)6; (f) analysis of miRNA-132-3p expression in the femur bone tissues of mice after hindlimb unloading for 21 days. BL—baseline group, mice were euthanatized and sampled at the beginning of experiment; CON—control group mice were raised in normal conditions during the experiment; HU—hindlimb unloading group, mice were submitted to a hindlimb unloading experiment; HU + Mock—hindlimb unloading plus (AspSerSer)6-liposome injection group, mice were injected with the (AspSerSer)6-liposome before HU; HU + antagomir-NC—hindlimb unloading plus (AspSerSer)6-liposome-antagomir-NC injection group, mice were injected with the (AspSerSer)6-liposome-antagomir-NC before HU; HU + antagomir-132—hindlimb unloading plus (AspSerSer)6-liposome-antagomir-132 injection group, mice were injected with the (AspSerSer)6-liposome-antagomir-132 before HU. Values are shown as mean ± SD, n = 6. * p < 0.05. NS, not significant. (a–c) Adapted with permission from [17] and (d–f) adapted with permission from [19].
Figure 1.
Targeting the bone surface-mediated bone marrow: (a) Characterization of the bone-targeting delivery system in vitro and in vivo. Chemical structure of icaritin (ICT); (b) the morphology of Asp8-liposome-icaritin taken by cryo-transmission electron microscope (Cryo-TEM) (scale bar = 100 nm, magnification = 105×); (c) localization of fluorescent-labeled liposome delivery system with or without Asp8 targeting peptide in mice by optical imaging IVIS analysis, 72 h after administration; (d) a schematic diagram to illustrate the experimental design for targeted delivery of antagomir-132 to specifically decrease miRNA-132-3p levels in bone; (e) a schematic diagram to illustrate how antagomir-132 is selectively delivered to bone formation region by (AspSerSer)6; (f) analysis of miRNA-132-3p expression in the femur bone tissues of mice after hindlimb unloading for 21 days. BL—baseline group, mice were euthanatized and sampled at the beginning of experiment; CON—control group mice were raised in normal conditions during the experiment; HU—hindlimb unloading group, mice were submitted to a hindlimb unloading experiment; HU + Mock—hindlimb unloading plus (AspSerSer)6-liposome injection group, mice were injected with the (AspSerSer)6-liposome before HU; HU + antagomir-NC—hindlimb unloading plus (AspSerSer)6-liposome-antagomir-NC injection group, mice were injected with the (AspSerSer)6-liposome-antagomir-NC before HU; HU + antagomir-132—hindlimb unloading plus (AspSerSer)6-liposome-antagomir-132 injection group, mice were injected with the (AspSerSer)6-liposome-antagomir-132 before HU. Values are shown as mean ± SD, n = 6. * p < 0.05. NS, not significant. (a–c) Adapted with permission from [17] and (d–f) adapted with permission from [19].

Figure 2.
Active targeting strategy by peptide or antibody mediated nanomedicines: (a) Illustration of PEGylated non-targeted liposomal carfilzomib nanoparticles (NP[Carf], left) and VLA-4 targeted liposomal carfilzomib nanoparticles (TNP[Carf], right); (b) liposomal carfilzomib nanoparticles preferentially accumulate in the tumor, inhibit tumor growth, and reduce systemic toxicities in vivo. Tumor bearing SCID mice were injected intravenously on Days 1, 2, 8, and 9 with NP[Carf], TNP[Carf], free carfilzomib, and PBS at a dose of 5 mg/kg carfilzomib equivalence. Tumor growth inhibition was measured via calipers; (c) in vivo images of near infrared dye loaded targeted nanoparticles in tumor bearing mice. Images were taken for all mice at t = 2, 6, and 24 h using non-invasive methods. The representative images show the accumulation of the nanoparticles in the tumor (white arrow) over time; (d,e) in vivo efficacy of CD38pep- and CD138pep-targeted nanoparticles loaded with prodrug doxorubicin. Nanoparticles targeted with CD38pep or CD138pep were prepared loaded with a doxorubicin prodrug and their in vivo efficacy was tested against that of free doxorubicin in a subcutaneous xenograft mouse model. Mice were injected with H929 cells and tumors were allowed to grow to a predetermined size before i.v. injection of nanoparticle formulations began on Day 1. Mice were injected with 3 mg/kg of doxorubicin or nanoparticle prodrug equivalent on Days 1, 3, 5, 7, and 9. Tumor volume (d) and survival (e) were tracked with mice being killed when tumor volume grew too large or mouse weight was too low. n = 6 for all groups and data represent means (± s.e.m.). (a–c) Adapted with permission from [26] and (d–e) adapted with permission from [28]).
Figure 2.
Active targeting strategy by peptide or antibody mediated nanomedicines: (a) Illustration of PEGylated non-targeted liposomal carfilzomib nanoparticles (NP[Carf], left) and VLA-4 targeted liposomal carfilzomib nanoparticles (TNP[Carf], right); (b) liposomal carfilzomib nanoparticles preferentially accumulate in the tumor, inhibit tumor growth, and reduce systemic toxicities in vivo. Tumor bearing SCID mice were injected intravenously on Days 1, 2, 8, and 9 with NP[Carf], TNP[Carf], free carfilzomib, and PBS at a dose of 5 mg/kg carfilzomib equivalence. Tumor growth inhibition was measured via calipers; (c) in vivo images of near infrared dye loaded targeted nanoparticles in tumor bearing mice. Images were taken for all mice at t = 2, 6, and 24 h using non-invasive methods. The representative images show the accumulation of the nanoparticles in the tumor (white arrow) over time; (d,e) in vivo efficacy of CD38pep- and CD138pep-targeted nanoparticles loaded with prodrug doxorubicin. Nanoparticles targeted with CD38pep or CD138pep were prepared loaded with a doxorubicin prodrug and their in vivo efficacy was tested against that of free doxorubicin in a subcutaneous xenograft mouse model. Mice were injected with H929 cells and tumors were allowed to grow to a predetermined size before i.v. injection of nanoparticle formulations began on Day 1. Mice were injected with 3 mg/kg of doxorubicin or nanoparticle prodrug equivalent on Days 1, 3, 5, 7, and 9. Tumor volume (d) and survival (e) were tracked with mice being killed when tumor volume grew too large or mouse weight was too low. n = 6 for all groups and data represent means (± s.e.m.). (a–c) Adapted with permission from [26] and (d–e) adapted with permission from [28]).


{kind=link}
{kind=link}
Table 1.
An overview of different types of blood cancers. Due to the scope of this review the somewhat rarer forms are not discussed here. Data were adopted from Cancer Statistics 2022 (USA), adapted with permission from ref. [1]. Copyright 2022 John Wiley & sons.
Table 1.
An overview of different types of blood cancers. Due to the scope of this review the somewhat rarer forms are not discussed here. Data were adopted from Cancer Statistics 2022 (USA), adapted with permission from ref. [1]. Copyright 2022 John Wiley & sons.
| Types of Blood Cancers | Origin | New Cases | Deaths |
|---|---|---|---|
| Est. in 2022 | Est. in 2022 | ||
| Multiple myeloma (MM) | B cells (plasma cells) | 34,470 | 12,640 |
| Acute myeloid leukaemia (AML) | Myeloid lineage hematopoietic precursors | 20,050 | 11,540 |
| B cell lymphoma (BL) | B cells and lymphocytes | 80,910 | 21,170 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Jiang, Y.; Lin, W.; Zhu, L. Targeted Drug Delivery for the Treatment of Blood Cancers. Molecules 2022, 27, 1310. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27041310
AMA Style
Jiang Y, Lin W, Zhu L. Targeted Drug Delivery for the Treatment of Blood Cancers. Molecules. 2022; 27(4):1310. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27041310
Chicago/Turabian StyleJiang, Yao, Weifeng Lin, and Linyi Zhu. 2022. "Targeted Drug Delivery for the Treatment of Blood Cancers" Molecules 27, no. 4: 1310. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27041310