
Non-Peptide Opioids Differ in Effects on Mu-Opioid (MOP) and Serotonin 1A (5-HT1A) Receptors Heterodimerization and Cellular Effectors (Ca2+, ERK1/2 and p38) Activation

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
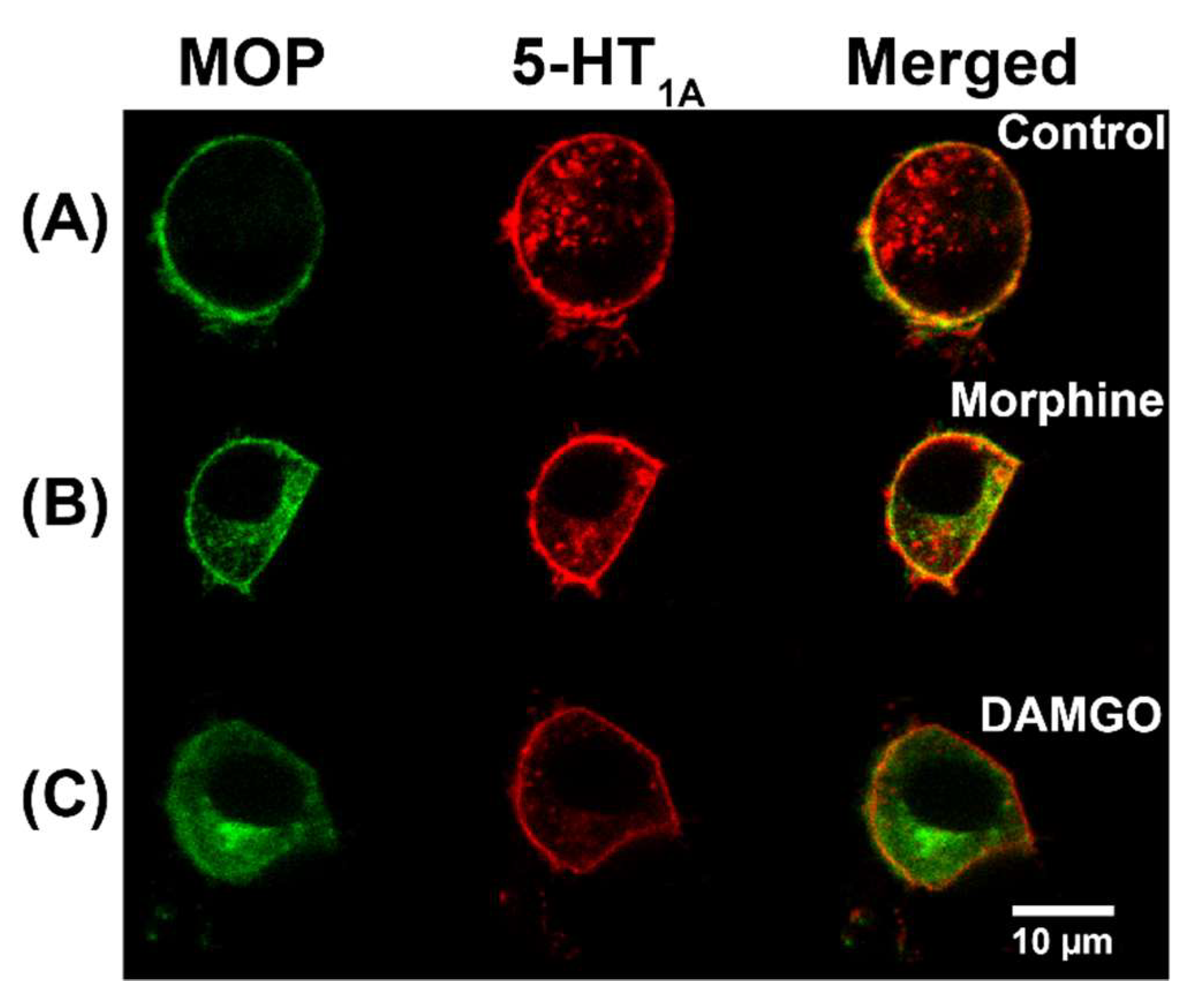
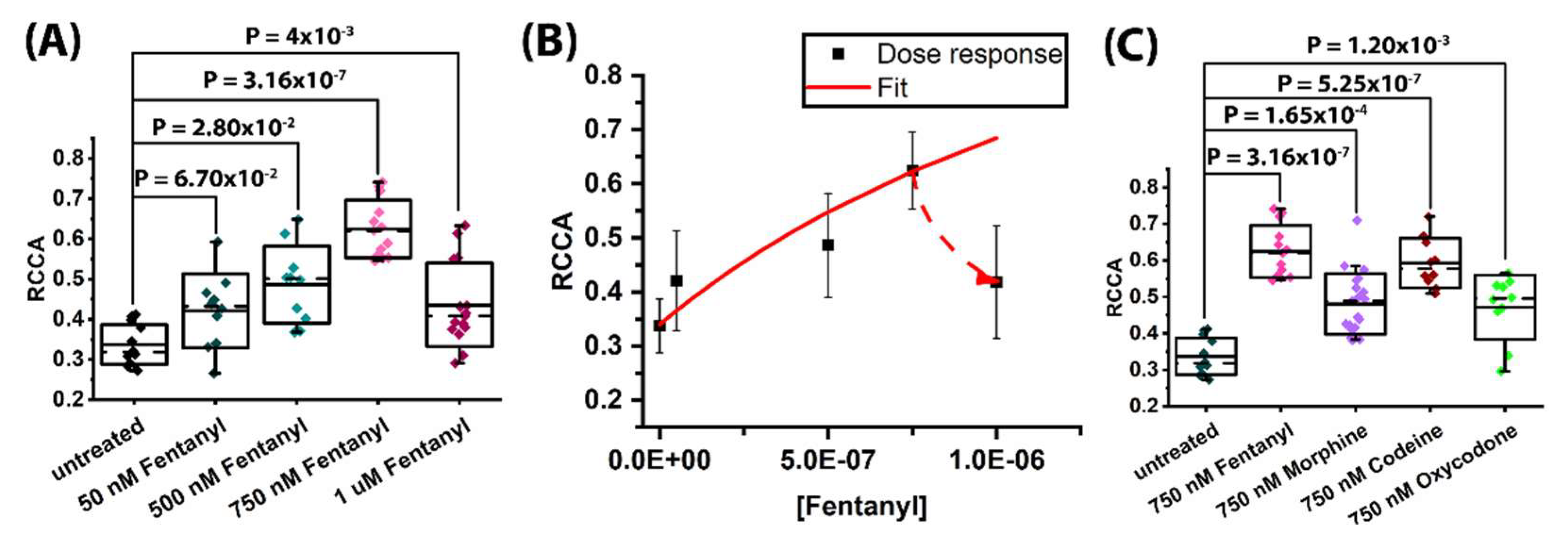
2.1. Non-Peptide Opioids Potentiate MOP and 5-HT1A Heterodimerization to a Different Extent
2.2. Non-Peptide Opioids Increase to a Different Extent the Brightness of eGFP and Tomato
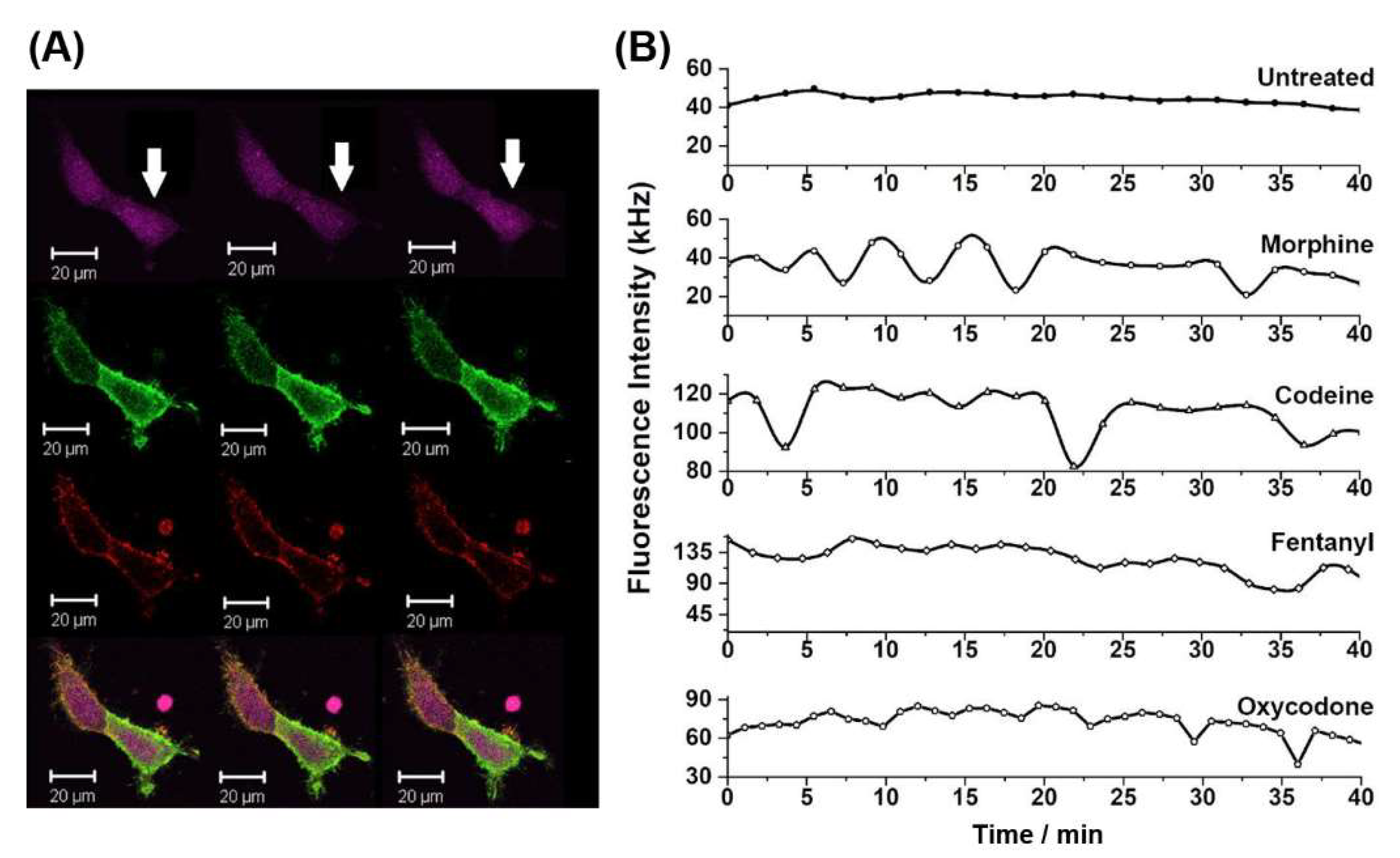
2.3. Non-Peptide Opioids Elicit Different Intracellular Ca2+ Signalling Dynamics
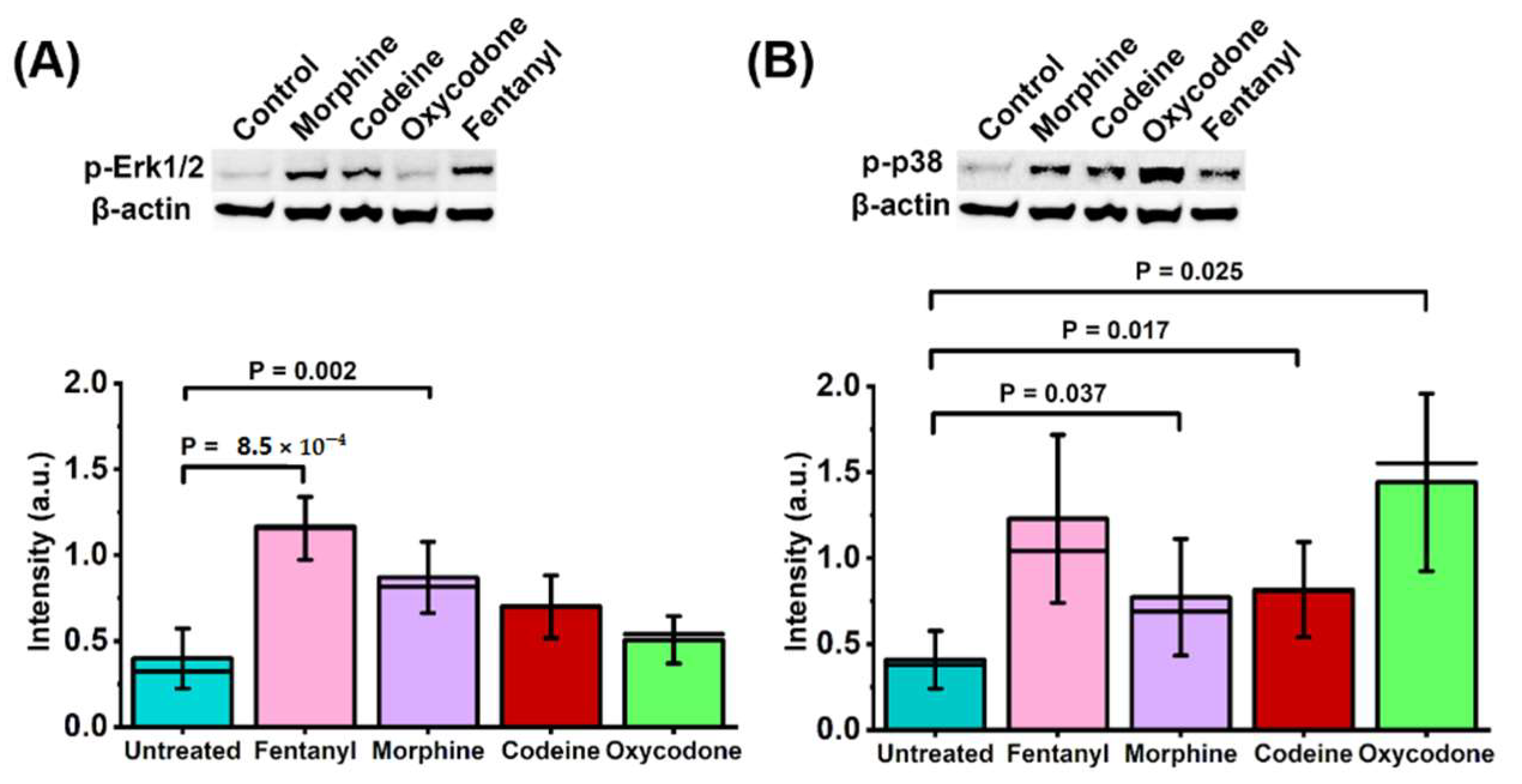
2.4. Non-Peptide Opioids Differ in the Extent to Which They Activate Major Signal Transduction Pathways
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Transfection
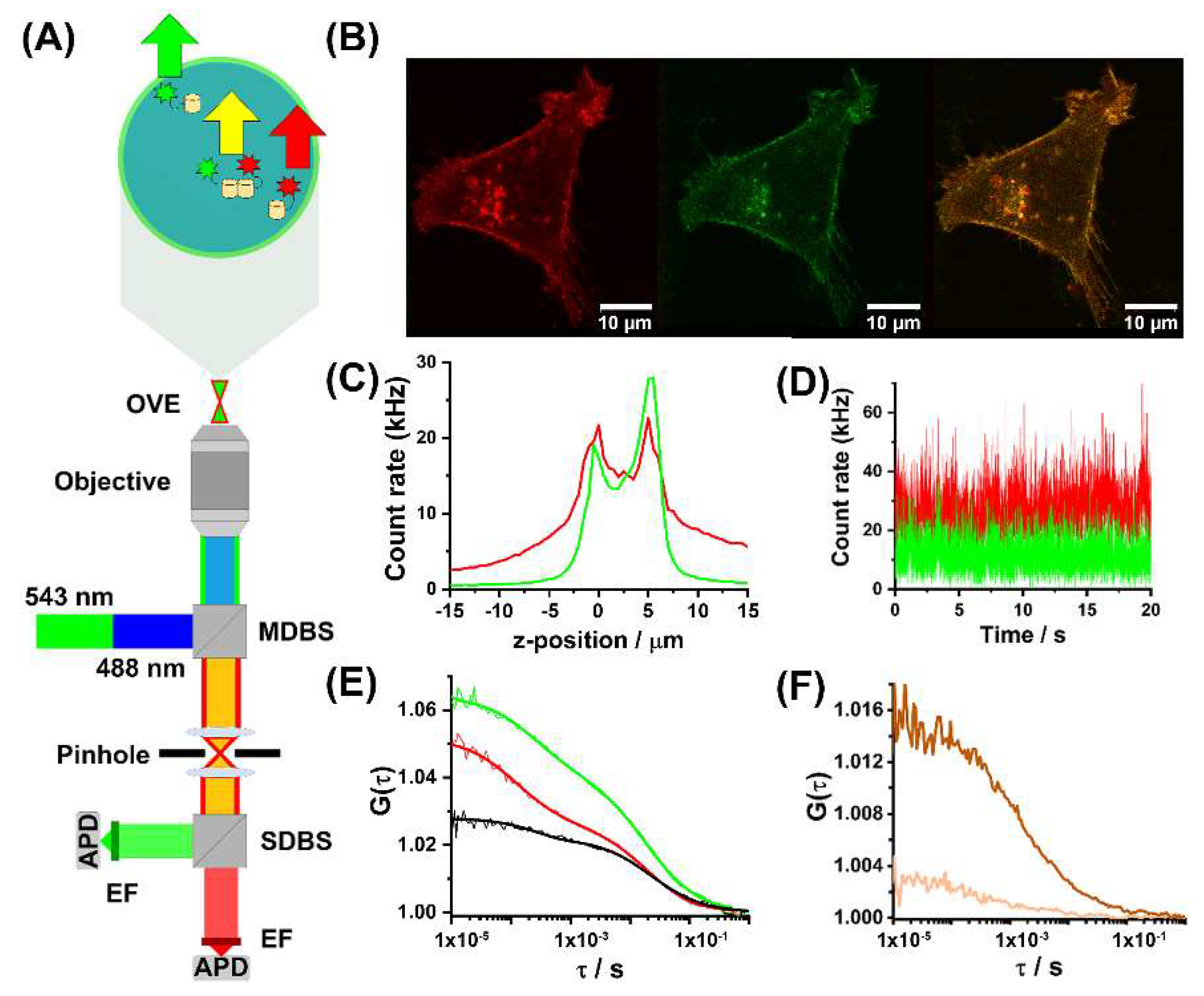
4.2. Confocal Laser Scanning Microscopy (CLSM) Imaging and Fluorescence Correlation and Cross-Correlation Spectroscopy (FCS/FCCS)
4.3. Brief Background on FCS/FCCS
4.4. Opioid Treatment
4.5. Intracellular Ca2+ Imaging
4.6. Western Blotting
4.7. LC-MS/MS Opioid Metabolite Analysis
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Collett, B. The burden of chronic pain. Curr. Med. Res. Opin. 2011, 27, 2065–2066. [Google Scholar] [CrossRef] [PubMed]
- Basbaum, A.I.; Bautista, D.M.; Scherrer, G.; Julius, D. Cellular and molecular mechanisms of pain. Cell 2009, 139, 267–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gosselin, R.D.; Suter, M.R.; Ji, R.R.; Decosterd, I. Glial cells and chronic pain. Neuroscientist 2010, 16, 519–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalso, E.; Edwards, J.E.; Moore, A.R.; McQuay, H.J. Opioids in chronic non-cancer pain: Systematic review of efficacy and safety. Pain 2004, 112, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Ballantyne, J.C. Opioids for the Treatment of Chronic Pain: Mistakes Made, Lessons Learned, and Future Directions. Anesth. Analg. 2017, 125, 1769–1778. [Google Scholar] [CrossRef]
- Haleem, D.J. Serotonin-1A receptor dependent modulation of pain and reward for improving therapy of chronic pain. Pharmacol. Res. 2018, 134, 212–219. [Google Scholar] [CrossRef]
- Xu, X.J.; Colpaert, F.; Wiesenfeld-Hallin, Z. Opioid hyperalgesia and tolerance versus 5-HT1A receptor-mediated inverse tolerance. Trends Pharmacol. Sci. 2003, 24, 634–639. [Google Scholar] [CrossRef]
- Colpaert, F.C.; Deseure, K.; Stinus, L.; Adriaensen, H. High-efficacy 5-hydroxytryptamine 1A receptor activation counteracts opioid hyperallodynia and affective conditioning. J. Pharmacol. Exp. Ther. 2006, 316, 892–899. [Google Scholar] [CrossRef]
- Rojas-Corrales, M.O.; Berrocoso, E.; Micó, J.A. Role of 5-HT1A and 5-HT1B receptors in the antinociceptive effect of tramadol. Eur. J. Pharmacol. 2005, 511, 21–26. [Google Scholar] [CrossRef]
- Mico, J.A.; Berrocoso, E.; Ortega-Alvaro, A.; Gibert-Rahola, J.; Rojas-Corrales, M.O. The role of 5-HT1A receptors in research strategy for extensive pain treatment. Curr. Top. Med. Chem. 2006, 6, 1997–2003. [Google Scholar] [CrossRef]
- Kosek, E.; Jensen, K.B.; Lonsdorf, T.B.; Schalling, M.; Ingvar, M. Genetic variation in the serotonin transporter gene (5-HTTLPR, rs25531) influences the analgesic response to the short acting opioid Remifentanil in humans. Mol. Pain. 2009, 5, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tour, J.; Löfgren, M.; Mannerkorpi, K.; Gerdle, B.; Larsson, A.; Palstam, A.; Bileviciute-Ljungar, I.; Bjersing, J.; Martin, I.; Ernberg, M.; et al. Gene-to-gene interactions regulate endogenous pain modulation in fibromyalgia patients and healthy controls-antagonistic effects between opioid and serotonin-related genes. Pain 2017, 158, 1194–1203. [Google Scholar] [CrossRef] [PubMed]
- Møller, M.; Jakobsen, S.; Gjedde, A. Parametric and regional maps of free serotonin 5HT1A receptor sites in human brain as function of age in healthy humans. Neuropsychopharmacology 2007, 32, 1707–1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrovic, P.; Ingvar, M. Imaging cognitive modulation of pain processing. Pain 2002, 95, 1–5. [Google Scholar] [CrossRef]
- Martikainen, I.K.; Hirvonen, J.; Kajander, J.; Hagelberg, N.; Mansikka, H.; Någren, K.; Hietala, J.; Pertovaara, A. Correlation of human cold pressor pain responses with 5-HT(1A) receptor binding in the brain. Brain Res. 2007, 1172, 21–31. [Google Scholar] [CrossRef]
- Tuominen, L.; Nummenmaa, L.; Keltikangas-Järvinen, L.; Raitakari, O.; Hietala, J. Mapping neurotransmitter networks with PET: An example on serotonin and opioid systems. Hum. Brain Mapp. 2014, 35, 1875–1884. [Google Scholar] [CrossRef]
- Azmitia, E.C.; Gannon, P.J.; Kheck, N.M.; Whitaker-Azmitia, P.M. Cellular localization of the 5-HT1A receptor in primate brain neurons and glial cells. Neuropsychopharmacology 1996, 14, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Grace, P.M.; Maier, S.F.; Watkins, L.R. Opioid-induced central immune signaling: Implications for opioid analgesia. Headache 2015, 55, 475–489. [Google Scholar] [CrossRef] [Green Version]
- Roeckel, L.A.; Le Coz, G.M.; Gavériaux-Ruff, C.; Simonin, F. Opioid-induced hyperalgesia: Cellular and molecular mechanisms. Neuroscience 2016, 338, 160–182. [Google Scholar] [CrossRef]
- Albrecht, D.S.; Forsberg, A.; Sandström, A.; Bergan, C.; Kadetoff, D.; Protsenko, E.; Lampa, J.; Lee, Y.C.; Höglund, C.O.; Catana, C.; et al. Brain glial activation in fibromyalgia—A multi-site positron emission tomography investigation. Brain Behav. Immun. 2019, 75, 72–83. [Google Scholar] [CrossRef]
- Schrepf, A.; Harper, D.E.; Harte, S.E.; Wang, H.; Ichesco, E.; Hampson, J.P.; Zubieta, J.-K.; Clauw, D.J.; Harris, R.E. Endogenous opioidergic dysregulation of pain in fibromyalgia: A PET and fMRI study. Pain 2016, 157, 2217–2225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishimoto, K.; Koyama, S.; Akaike, N. Synergistic mu-opioid and 5-HT1A presynaptic inhibition of GABA release in rat periaqueductal gray neurons. Neuropharmacology 2001, 41, 529–538. [Google Scholar] [CrossRef]
- Jasmin, L.; Wu, M.V.; Ohara, P.T. GABA puts a stop to pain. Curr. Drug Targets CNS Neurol. Disord. 2004, 3, 487–505. [Google Scholar] [CrossRef] [PubMed]
- Cussac, D.; Rauly-Lestienne, I.; Heusler, P.; Finana, F.; Cathala, C.; Bernois, S.; De Vries, L. μ-Opioid and 5-HT1A receptors heterodimerize and show signalling crosstalk via G protein and MAP-kinase pathways. Cell Signal. 2012, 24, 1648–1657. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.G.; Prather, P.L. Chronic exposure to mu-opioid agonists produces constitutive activation of mu-opioid receptors in direct proportion to the efficacy of the agonist used for pretreatment. Mol. Pharmacol. 2001, 60, 53–62. [Google Scholar] [CrossRef]
- Tsai, R.-Y.; Tai, Y.-H.; Tzeng, J.-I.; Lin, S.-L.; Shen, C.-H.; Yang, C.-P.; Hsin, S.-T.; Wang, C.-B.; Wong, C.-S. Ultra-low dose naloxone restores the antinociceptive effect of morphine in pertussis toxin-treated rats and prevents glutamate transporter downregulation by suppressing the p38 mitogen-activated protein kinase signaling pathway. Neuroscience 2009, 159, 1244–1256. [Google Scholar] [CrossRef]
- Radoi, V. Interactions between the opioid and serotonin systems in chronic pain. Quantitative Live Cell Study by Fluorescence Cross-Correlation Spectroscopy (FCCS). Ph.D. Thesis, Karolinska Institutet, Stockholm, Sweden, 2019. [Google Scholar]
- Hulme, E.C.; Trevethick, M.A. Ligand binding assays at equilibrium: Validation and interpretation. Br. J. Pharmacol. 2010, 161, 1219–1237. [Google Scholar] [CrossRef] [Green Version]
- Watabe, M.; Arjunan, S.N.V.; Chew, W.X.; Kaizu, K.; Takahashi, K. Cooperativity transitions driven by higher-order oligomer formations in ligand-induced receptor dimerization. Phys. Rev. E 2019, 100, 062407. [Google Scholar] [CrossRef] [Green Version]
- Heusler, P.; Tardif, S.; Cussac, D. Agonist stimulation at human μ opioid receptors in a [(35)S]GTPγS incorporation assay: Observation of “bell-shaped” concentration-response relationships under conditions of strong receptor G protein coupling. J. Recept. Signal Transduct. Res. 2016, 36, 158–166. [Google Scholar] [CrossRef]
- McIntyre, I.M.; Anderson, D.T. Postmortem Fentanyl Concentrations: A Review. J. Forensic Res. 2012, 3 (Suppl. S1), 2. [Google Scholar] [CrossRef]
- Goldbeter, A. Biochemical Oscillations and Cellular Rhythms: The Molecular Bases of Periodic and Chaotic Behaviour; Cambridge University Press: Cambridge, UK, 1996; pp. 89–160, 349–406. [Google Scholar]
- Macey, T.A.; Lowe, J.D.; Chavkin, C. Mu opioid receptor activation of ERK1/2 is GRK3 and arrestin dependent in striatal neurons. J. Biol. Chem. 2006, 281, 34515–34524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, M.; Walwyn, W.M.; Evans, C.J.; Xie, C.W. p38 MAPK and beta-arrestin 2 mediate functional interactions between endogenous micro-opioid and alpha2A-adrenergic receptors in neurons. J. Biol. Chem. 2009, 284, 6270–6281. [Google Scholar] [CrossRef] [Green Version]
- Kasai, R.S.; Kusumi, A. Single-molecule imaging revealed dynamic GPCR dimerization. Curr. Opin. Cell Biol. 2014, 27, 78–86, Correction in Curr. Opin. Cell Biol. 2014, 27, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, J.; Wright, S.; Rodríguez, D.; Matricon, P.; Lahav, N.; Vromen, A.; Friedler, A.; Strömqvist, J.; Wennmalm, S.; Carlsson, J.; et al. Agonist-induced dimer dissociation as a macromolecular step in G protein-coupled receptor signaling. Nat. Commun. 2017, 8, 226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, S.R.; Bräuner-Osborne, H. Investigating Internalization and Intracellular Trafficking of GPCRs: New Techniques and Real-Time Experimental Approaches. Handb. Exp. Pharmacol. 2018, 245, 41–61. [Google Scholar] [CrossRef]
- Kanaya, N.; Zakhary, D.R.; Murray, P.A.; Damron, D.S. Differential effects of fentanyl and morphine on intracellular Ca2+ transients and contraction in rat ventricular myocytes. Anesthesiology 1998, 89, 1532–1542. [Google Scholar] [CrossRef]
- Fan, R.; Schrott, L.M.; Snelling, S.; Ndi, J.; Arnold, T.; Korneeva, N.L. Chronic oxycodone induces integrated stress response in rat brain. BMC Neurosci. 2015, 16, 58. [Google Scholar] [CrossRef] [Green Version]
- Hutchinson, M.R.; Northcutt, A.L.; Hiranita, T.; Wang, X.; Lewis, S.S.; Thomas, J.; Van Steeg, K.; Kopajtic, T.A.; Loram, L.C.; Sfregola, C.; et al. Opioid activation of toll-like receptor 4 contributes to drug reinforcement. J. Neurosci. 2012, 32, 11187–11200. [Google Scholar] [CrossRef] [Green Version]
- Al-Hasani, R.; Bruchas, M.R. Molecular mechanisms of opioid receptor-dependent signaling and behavior. Anesthesiology 2011, 115, 1363–1381. [Google Scholar] [CrossRef] [Green Version]
- Remillard, D.; Kaye, A.D.; McAnally, H. Oxycodone’s Unparalleled Addictive Potential: Is it Time for a Moratorium? Curr. Pain Headache Rep. 2019, 23, 15. [Google Scholar] [CrossRef]
- Yue, Q.Y.; Hasselström, J.; Svensson, J.O.; Säwe, J. Pharmacokinetics of codeine and its metabolites in Caucasian healthy volunteers: Comparisons between extensive and poor hydroxylators of debrisoquine. Br. J. Clin. Pharmacol. 1991, 31, 635–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortazzo, M.H.; Copenhaver, D.; Fishman, S.M. Major Opioids and Chronic Opioid Therapy. In Practical Management of Pain, 5th ed.; Benzon, H.T., Rathmell, J.P., Wu, C.L., Turk, D.C., Argoff, C.E., Hurley, R.W., Eds.; Elsevier Inc.: Amsterdam, The Netherlands, 2013; pp. 495–507.e493. [Google Scholar] [CrossRef]
- Johnson, J.L.; Rolan, P.E.; Johnson, M.E.; Bobrovskaya, L.; Williams, D.; Johnson, K.F.; Tuke, J.; Hutchinson, M. Codeine-induced hyperalgesia and allodynia: Investigating the role of glial activation. Transl. Psychiatry 2014, 4, e482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mignat, C.; Wille, U.; Ziegler, A. Affinity profiles of morphine, codeine, dihydrocodeine and their glucuronides at opioid receptor subtypes. Life Sci. 1995, 56, 793–799. [Google Scholar] [CrossRef]
- Schmidt, H.; Vormfelde, S.V.; Klinder, K.; Gundert-Remy, U.; Gleiter, C.H.; Skopp, G.; Aderjan, R.; Fuhr, U. Affinities of dihydrocodeine and its metabolites to opioid receptors. Pharmacol. Toxicol. 2002, 91, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Ingelman-Sundberg, M. Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): Clinical consequences, evolutionary aspects and functional diversity. Pharm. J. 2005, 5, 6–13. [Google Scholar] [CrossRef]
- Tzvetkov, M.V.; dos Santos Pereira, J.N.; Meineke, I.; Saadatmand, A.R.; Stingl, J.C.; Brockmöller, J. Morphine is a substrate of the organic cation transporter OCT1 and polymorphisms in OCT1 gene affect morphine pharmacokinetics after codeine administration. Biochem. Pharmacol. 2013, 86, 666–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasai, R.S.; Suzuki, K.G.N.; Prossnitz, E.R.; Koyama-Honda, I.; Nakada, C.; Fujiwara, T.K.; Kusumi, A. Full characterization of GPCR monomer-dimer dynamic equilibrium by single molecule imaging. J. Cell Biol. 2011, 192, 463–480. [Google Scholar] [CrossRef] [Green Version]
- Casadó-Anguera, V.; Moreno, E.; Mallol, J.; Ferré, S.; Canela, E.I.; Cortés, A.; Casadó, V. Reinterpreting anomalous competitive binding experiments within G protein-coupled receptor homodimers using a dimer receptor model. Pharmacol. Res. 2019, 139, 337–347. [Google Scholar] [CrossRef]
- Jorand, R.; Biswas, S.; Wakefield, D.L.; Tobin, S.J.; Golfetto, O.; Hilton, K.; Ko, M.; Ramos, J.W.; Small, A.R.; Chu, P.; et al. Molecular signatures of mu opioid receptor and somatostatin receptor 2 in pancreatic cancer. Mol. Biol. Cell. 2016, 27, 3659–3672. [Google Scholar] [CrossRef] [Green Version]
- Touhara, K.K.; MacKinnon, R. Molecular basis of signaling specificity between GIRK channels and GPCRs. eLife 2018, 7, e42908. [Google Scholar] [CrossRef]
- Volpe, D.A.; Tobin, G.A.M.; Mellon, R.D.; Katki, A.G.; Parker, R.J.; Colatslcy, T.; Kropp, T.J.; Verbois, S.L. Uniform assessment and ranking of opioid Mu receptor binding constants for selected opioid drugs. Regul. Toxicol. Pharmacol. 2011, 59, 385–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rickli, A.; Liakoni, E.; Hoener, M.C.; Liechti, M.E. Opioid-induced inhibition of the human 5-HT and noradrenaline transporters in vitro: Link to clinical reports of serotonin syndrome. Br. J. Pharmacol. 2018, 175, 532–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heiskanen, T.; Langel, K.; Gunnar, T.; Lillsunde, P.; Kalso, E.A. Opioid Concentrations in Oral Fluid and Plasma in Cancer Patients with Pain. J. Pain Symptom Manag. 2015, 50, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Christrup, L.L.; Foster, D.; Popper, L.D.; Troen, T.; Upton, R. Pharmacokinetics, efficacy, and tolerability of fentanyl following intranasal versus intravenous administration in adults undergoing third-molar extraction: A randomized, double-blind, double-dummy, two-way, crossover study. Clin Ther. 2008, 30, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Trescot, A.M. Review of the role of opioids in cancer pain. J. Natl. Compr. Cancer Netw. 2010, 8, 1087–1094. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.; Lawlor, P.; Vigano, A.; Dorgan, M.; Bruera, E. Equianalgesic dose ratios for opioids: A critical review and proposals for long-term dosing. J. Pain Symptom Manag. 2001, 22, 672–687. [Google Scholar] [CrossRef]
- Rennick, A.; Atkinson, T.; Cimino, N.M.; Strassels, S.A.; McPherson, M.L.; Fudin, J. Variability in Opioid Equivalence Calculations. Pain Med. 2016, 17, 892–898. [Google Scholar] [CrossRef] [Green Version]
- Hafner, M.; Niepel, M.; Chung, M.; Sorger, P.K. Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Nat. Methods 2016, 13, 521–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, P.; Smart, T.G. HEK293 cell line: A vehicle for the expression of recombinant proteins. J. Pharmacol. Toxicol. Methods 2005, 51, 187–200. [Google Scholar] [CrossRef]
- Westerink, R.H.; Ewing, A.G. The PC12 cell as model for neurosecretion. Acta Physiol. 2008, 192, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Khan, K.H. Gene expression in Mammalian cells and its applications. Adv. Pharm. Bull. 2013, 3, 257–263. [Google Scholar] [CrossRef]
- Vukojevic, V.; Heidkamp, M.; Ming, Y.; Johansson, B.; Terenius, L.; Rigler, R. Quantitative single-molecule imaging by confocal laser scanning microscopy. Proc. Natl. Acad. Sci. USA 2008, 105, 18176–18181. [Google Scholar] [CrossRef] [Green Version]
- Vukojevic, V.; Papadopoulos, D.K.; Terenius, L.; Gehring, W.J.; Rigler, R. Quantitative study of synthetic Hox transcription factor-DNA interactions in live cells. Proc. Natl. Acad. Sci. USA 2010, 107, 4093–4098. [Google Scholar] [CrossRef] [Green Version]
- Schwille, P. Fluorescence correlation spectroscopy and its potential for intracellular applications. Cell Biochem. Biophys. 2001, 34, 383–408. [Google Scholar] [CrossRef]
- Vukojević, V.; Pramanik, A.; Yakovleva, T.; Rigler, R.; Terenius, L.; Bakalkin, G. Study of molecular events in cells by fluorescence correlation spectroscopy. Cell Mol. Life Sci. 2005, 62, 535–550. [Google Scholar] [CrossRef]
- Kim, S.A.; Heinze, K.G.; Schwille, P. Fluorescence correlation spectroscopy in living cells. Nat. Methods 2007, 4, 963–973. [Google Scholar] [CrossRef]
- Tian, Y.; Martinez, M.M.; Pappas, D. Fluorescence correlation spectroscopy: A review of biochemical and microfluidic applications. Appl. Spectrosc. 2011, 65, 115A–124A. [Google Scholar] [CrossRef] [Green Version]
- Rogacki, M.K.; Golfetto, O.; Tobin, S.J.; Li, T.; Biswas, S.; Jorand, R.; Zhang, H.; Radoi, V.; Ming, Y.; Svenningsson, P.; et al. Dynamic lateral organization of opioid receptors (kappa, muwt and muN40D) in the plasma membrane at the nanoscale level. Traffic 2018, 19, 690–709. [Google Scholar] [CrossRef] [Green Version]
- Schwille, P.; Ries, J. Principles and Applications of Fluorescence Correlation Spectroscopy (Fcs). In Biophotonics: Spectroscopy, Imaging, Sensing, and Manipulation; NATO Security through Science Series B: Physics and Biophysics; Springer: Dordrecht, The Netherlands, 2011; pp. 63–85. [Google Scholar] [CrossRef]
- Bacia, K.; Kim, S.A.; Schwille, P. Fluorescence cross-correlation spectroscopy in living cells. Nat. Methods 2006, 3, 83–89. [Google Scholar] [CrossRef]
- Krieger, J.W.; Singh, A.P.; Bag, N.; Garbe, C.S.; Saunders, T.; Langowski, J.; Wohland, T. Imaging fluorescence (cross-) correlation spectroscopy in live cells and organisms. Nat. Protoc. 2015, 10, 1948–1974. [Google Scholar] [CrossRef]
- Bacia, K.; Petrášek, Z.; Schwille, P. Correcting for spectral cross-talk in dual-color fluorescence cross-correlation spectroscopy. Chemphyschem 2012, 13, 1221–1231. [Google Scholar] [CrossRef] [Green Version]
- Du, Z.; Yu, J.; Li, F.; Deng, L.; Wu, F.; Huang, X.; Bergstrand, J.; Widengren, J.; Dong, C.; Ren, J. In Situ Monitoring of p53 Protein and MDM2 Protein Interaction in Single Living Cells Using Single-Molecule Fluorescence Spectroscopy. Anal. Chem. 2018, 90, 6144–6151. [Google Scholar] [CrossRef]
- Xu, H.; Partilla, J.S.; Wang, X.; Rutherford, J.M.; Tidgewell, K.; Prisinzano, T.E.; Bohn, L.M.; Rothman, R.B. A comparison of noninternalizing (herkinorin) and internalizing (DAMGO) mu-opioid agonists on cellular markers related to opioid tolerance and dependence. Synapse 2007, 61, 166–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burchiel, S.W.; Edwards, B.S.; Kuckuck, F.W.; Lauer, F.T.; Prossnitz, E.; Ransom, J.T.; Sklar, L.A. Analysis of free intracellular calcium by flow cytometry: Multiparameter and pharmacologic applications. Methods 2000, 21, 221–230. [Google Scholar] [CrossRef]
- Tinning, P.W.; Franssen, A.J.P.M.; Hridi, S.U.; Bushell, T.J.; McConnell, G. A 340/380 nm light-emitting diode illuminator for Fura-2 AM ratiometric Ca2+ imaging of live cells with better than 5 nM precision. J. Microsc. 2018, 269, 212–220. [Google Scholar] [CrossRef] [Green Version]
- Murphy, C.M.; Huestis, M.A. LC-ESI-MS/MS analysis for the quantification of morphine, codeine, morphine-3-beta-D-glucuronide, morphine-6-beta-D-glucuronide, and codeine-6-beta-D-glucuronide in human urine. J. Mass Spectrom. 2005, 40, 1412–1416, Correction in J. Mass Spectrom. 2006, 41, 127. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment (750 nM) | Kd (nM) | CPMeGFP (kHz) | CPMTomato (kHz) | ERK1/2 | p38 | CR (kHz) | Ca2+ Dynamics |
|---|---|---|---|---|---|---|---|
| Untreated | 440 ± 70 | 1.1 ± 0.3 | 0.8 ± 0.2 | 40 | Stationary state | ||
| Fentanyl | 80 ± 70 | 1.9 ± 0.7 | 1.1 ± 0.3 | + | + | 40 | Stationary state |
| Morphine | 200 ± 70 | 2.0 ± 0.5 | 1.3 ± 0.7 | + | + | 120 | Small-amplitude oscillations |
| Codeine | 100 ± 70 | 1.9 ± 0.5 | 1.0 ± 0.3 | + | + | 135 | Relaxation oscillations |
| Oxycodone | 200 ± 70 | 1.8 ± 0.7 | 0.9 ± 0.3 | 0 | + | 60 | Relaxation oscillations |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radoi, V.; Jakobsson, G.; Palada, V.; Nikosjkov, A.; Druid, H.; Terenius, L.; Kosek, E.; Vukojević, V. Non-Peptide Opioids Differ in Effects on Mu-Opioid (MOP) and Serotonin 1A (5-HT1A) Receptors Heterodimerization and Cellular Effectors (Ca2+, ERK1/2 and p38) Activation. Molecules 2022, 27, 2350. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27072350
Radoi V, Jakobsson G, Palada V, Nikosjkov A, Druid H, Terenius L, Kosek E, Vukojević V. Non-Peptide Opioids Differ in Effects on Mu-Opioid (MOP) and Serotonin 1A (5-HT1A) Receptors Heterodimerization and Cellular Effectors (Ca2+, ERK1/2 and p38) Activation. Molecules. 2022; 27(7):2350. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27072350
Chicago/Turabian StyleRadoi, Vlad, Gerd Jakobsson, Vinko Palada, Andrej Nikosjkov, Henrik Druid, Lars Terenius, Eva Kosek, and Vladana Vukojević. 2022. "Non-Peptide Opioids Differ in Effects on Mu-Opioid (MOP) and Serotonin 1A (5-HT1A) Receptors Heterodimerization and Cellular Effectors (Ca2+, ERK1/2 and p38) Activation" Molecules 27, no. 7: 2350. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27072350