
Singlet Fission, Polaron Generation and Intersystem Crossing in Hexaphenyl Film

1
School of Sciences, Hangzhou Dianzi University, Hangzhou 310018, China
2
State Key Laboratory of Fine Chemicals, Institute of Artificial Photosynthesis, Dalian University of Technology, Dalian 116024, China
3
School of Materials Science & Engineering, Nanyang Technological University, Singapore 639798, Singapore
4
Center of Artificial Photosynthesis for Solar Fuels, School of Science, Westlake University, Hangzhou 310024, China
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Molecules 2022, 27(16), 5067; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27165067
Submission received: 20 July 2022
/
Revised: 4 August 2022
/
Accepted: 7 August 2022
/
Published: 9 August 2022
(This article belongs to the Special Issue Ultrafast Dynamics in Chemical Processes)
Abstract
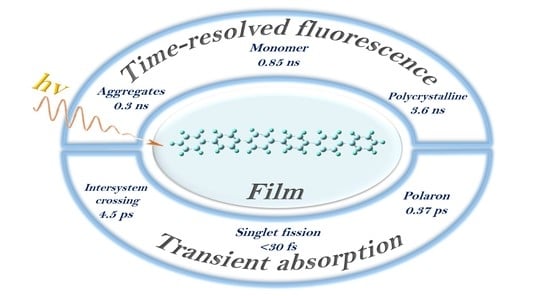
:The ultrafast dynamics of triplet excitons and polarons in hexaphenyl film was investigated by time-resolved fluorescence and femtosecond transient absorption techniques under various excitation photon energies. Two distinct pathways of triplet formation were clearly observed. Long-lived triplet states are populated within 4.5 ps via singlet fission-intersystem crossing, while the short-lived triplet states (1.5 ns) are generated via singlet fission from vibrational electronic states. In the meantime, polarons were formed from hot excitons on a timescale of <30 fs and recombined in ultrafast lifetime (0.37 ps). In addition, the characterization of hexaphenyl film suggests the morphologies of crystal and aggregate to wide applications in organic electronic devices. The present study provides a universally applicable film fabrication in hexaphenyl system towards future singlet fission-based solar cells.
1. Introduction
Organic π-conjugated materials are in the spotlight of interest due to their rich electro-optical properties for wide applications in the field of optoelectronic devices, such as organic light-emitting diodes [1,2,3,4] and solar cells [5]. Among them, short-chain oligomers can be considered as model compounds for unveiling important properties of π electrons in long chain polymers. Para-hexaphenyl, the six phenyls oligomer (Figure 1a), is a promising material, which has been fruitfully applied as efficient blue light emitting diodes [6,7,8,9,10]. It is crucial to explore the fundamental photophysical mechanisms inside this molecule. However, the time-resolved spectroscopic results on triplet formation mechanism are rather rare. The hexaphenyl crystallizes in a monoclinic lattice with the space group P21/a and it arranges in a typical herringbone configuration [11,12,13], resulting in good performance on photoelectric devices.
A unique process, singlet fission (SF), was proposed previously to take place in oligomers. It has aroused an increasing interest over the years because of its potential to enhance the efficiency of organic photovoltaics [14,15] and inorganic silicon solar cells [16,17,18,19]. SF is a spin-allowed process where a singlet excited molecule shares its energy with neighboring molecule in its ground state to produce two neighbor-standing triplet excitons. Generally, SF proceeds in ultrafast time scale of femtosecond to picosecond time [20]. SF was observed in organic single crystals [21,22], polymers [23,24], as well as covalently linked dimers [25,26]. Regarding molecular design for SF, the well-studied candidates are limited to polyacene and its derivatives [27]. Further research on this direction will include design of molecular arrays with new kind of π-conjugated molecules. Hence, it is highly desirable to investigate the para-hexaphenyl short-chain oligomers and reveal photophysical relaxation pathways with involvement of upper excited singlet states.
Related ultrafast spectroscopic studies were previously performed on polycrystalline hexaphenyl films [7,8,28] and on para-hexaphenyl-based organic light-emitting devices [29]. The time-resolved photoluminescence unveils the lowest energy emission from H-type hexaphenyl aggregates [28]. By using the pump-probe technique with 500 fs time resolution, the dominant triplet exciton generation mechanism in hexaphenyl is assigned to nongeminate bimolecular recombination of polarons; fusion of singlet excitons and subsequent fission into correlated triplet pairs was clarified [29].
In the present work, we have prepared hexaphenyl film with the thermal vacuum evaporation method and extended the pump-probe detection range down to 350 nm and increased the time resolution to 20–30 fs. Moreover, we have also applied the fluorescence up-conversion setup with a time resolution of 100 fs. These improvements allow direct detection of the triplet transient at 396 nm, and thus, two pathways of triplet state formation via ISC and SF were clearly identified. By tuning the excitation wavelength, we have observed enhanced efficiency of triplet state T1 formation via SF and ultrafast polaron formation from higher excited states.
2. Materials and Methods
2.1. Preparation of the Hexaphenyl Thin Film
The hexaphenyl thin film was prepared by using vacuum evaporation pump device (QHV-Z350C, Pana Instruments) at temperatures of 373–383 K for 20 min; before being installed in the chamber, the quartz substrates were cleaned by acetone in ultrasonic bath for 10 min and soaked in ethanol for 12 h. After that, the quartz substrates were treated with plasma (FEMTO SR CE, Diener) in an O3-filled environment for 10 min in order to remove organic impurities and contaminants.
2.2. Characterization of the Hexaphenyl Thin Film
XRD data of film were collected using a Rigaku D/Max-2400 X-ray diffractometer in parallel beam geometry employing Cu Kα line focused radiation at 9000 W (45 kV, 200 mA) power and equipped with a position sensitive detector with 10.0 nm radiation entrance slit. Samples were counted on zero background sample holders during the data acquisition. The best counting statistics were achieved by collecting samples using a 0.01° 2θ step scan from 5° to 80° with exposure time of 30 s per step. The structure and morphology of film were investigated by used of field emission scanning electron microscope (FESEM, Nova Nanosem 450). SEM images were measured under 10 kV accelerating voltage and 10 μA current.
2.3. Steady-State Absorption and Fluorescence Spectroscopy
UV-vis absorption and fluorescence spectra were obtained by use of a UV-visible spectrophotometer (Cary 100, Agilent) and spectrofluorometer (Fluorolog-3, Horiba Jobin Yvon), respectively.
2.4. Time-Resolved Fluorescence Spectroscopy
Fluorescence lifetimes ranging from 10 ps to 20 ns were obtained by time-correlated single photon counting (TCSPC) technique (PicoHarp 300, PicoQuant). By use of deconvolution/fit program (FluoFit, PicoQuant), the time detection limit decreased to 10 ps. The second harmonic of a Titanium-sapphire laser (150 fs, 80 MHz) (Mai Tai DeepSee, Spectra-Physics) at 400 nm was used as the excitation source. Femtosecond fluorescence lifetimes were measured by a fluorescence spectrometer (TRFLS, Newport) with 100 fs resolution in combination with a mode-locked Ti-sapphire laser, the same as mentioned in TCSPC. The femtosecond laser system generated light pulses at 800 nm of duration 150 fs at a repetition rate 80 MHz, average power of 2.9 W. The frequency of the laser pulse was doubled with a BBO crystal and served for excitation (pump). The emitted fluorescence was focused into a BBO crystal together with the gate beam (800 nm) to create the up-converted signal at the sum-frequency generation. The overall time resolution of the setup was 100 fs. Fluorescent images were acquired with confocal microscopy Olympus FV1000.
2.5. Transient Absorption Spectroscopy
The transient absorption (TA) spectra were measured by use of a home-made pump-probe setup, described in detail previously [26,30]. The output of a mode-locked Ti-sapphire amplified laser system (Spitfire Ace, Spectra-Physics), with a wavelength of 800 nm, pulse-width of 35 fs, repetition rate of 1 kHz and average power of 4 W, was split into two beams (10:1). Pump beam in the range of 240–2400 nm is acquired by use of an Optical Parametric Amplifier (TOPAS, Light Conversion). The pump pulse duration was 42 fs (measured from the risetime of TA kinetics for Rhodamine 6G). Deconvolution fit process allows obtaining three times higher resolution than the pulse width of the laser, i.e., 20–30 fs [31,32,33]. Part of 800 nm laser pulse was focused in a 3 mm thick, rotated CaF2 plate to produce a white light continuum (WLC), which was used as a probe beam ranging from 350 to 850 nm. WLC propagated through the sample and directed into the diode array attached to spectrograph (Jobin Yvon CP140) with detection range 250–850 nm (Entwicklungsbuero Stresing, Germany).
The entire setup was controlled by a PC with the help of LabView software (National Instruments). All measurements were performed at room temperature under aerated conditions.
The experimental data were fitted to a multiexponential decay function convoluted with the instrument response function B (t − t0) centered at t0:
where ΔA(t) is the difference absorption at time t, ΔAi is the amplitude of the component with lifetime τi and ΔA0 is the offset due to long-living species. The instrument response function was modeled by a Gaussian with a variable Full Width Half Maximum (FWHM).
2.6. Global Analysis
Global lifetime analysis of time-resolved experimental results was performed by Glotaran software [34]. The Decay Associated Spectra (DAS) allow separating several overlapping spectra in parallel model, whereas the Evolution Associated Spectra (EAS) shows the global fit results in a sequential model. The Species Associated Spectra (SAS) use to separate the process which is a combination of the parallel and sequential model and involves branching, back-reactions or multiple compartment excitation.
3. Results and Discussion
3.1. Steady-State Absorption and Fluorescence Spectra of Hexaphenyl Film
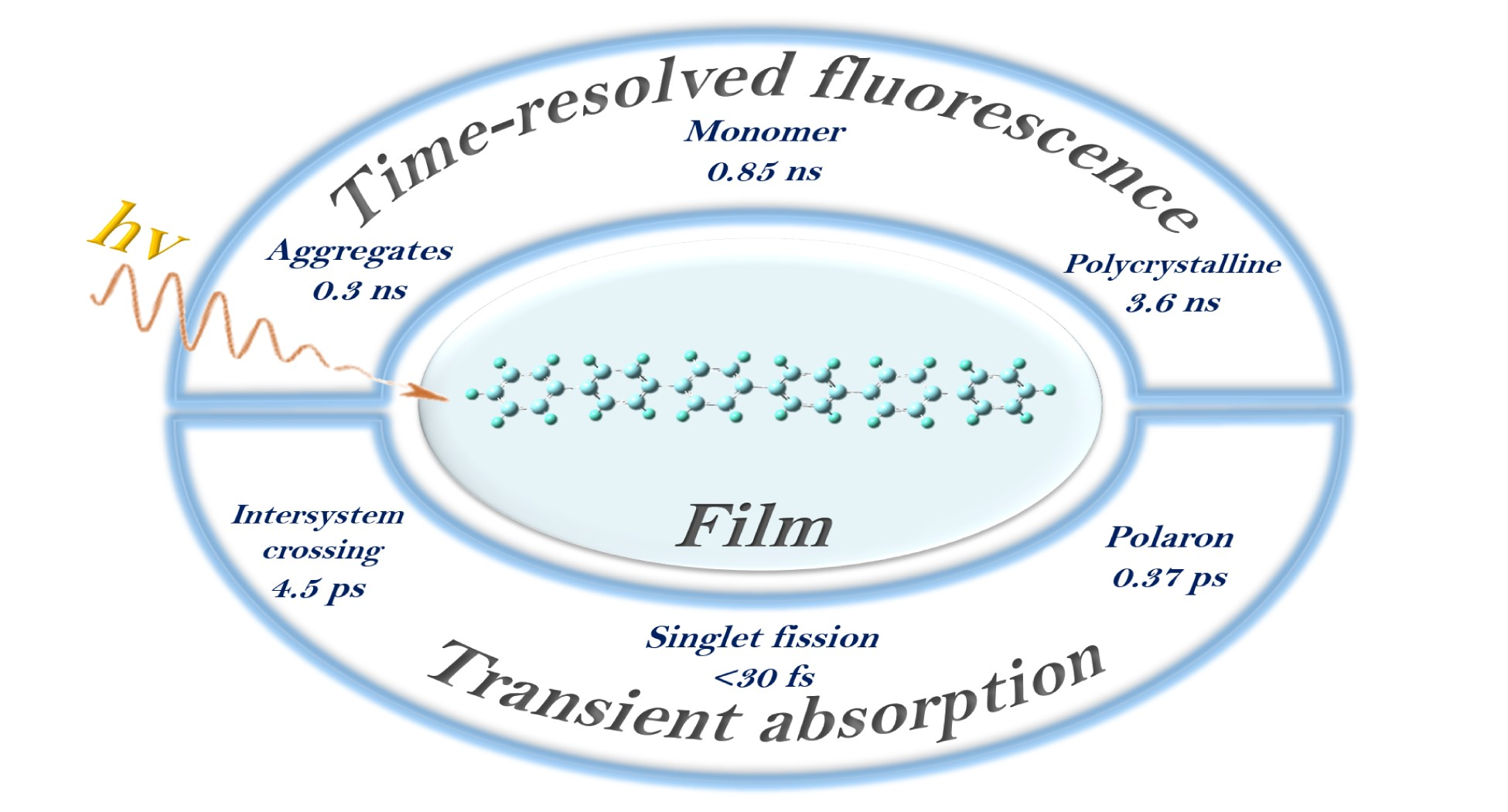
Steady-state absorption, fluorescence and fluorescence excitation spectra of hexaphenyl film were measured, presented in Figure 1b. The film absorption spectrum (Figure 1b red curve) shows two broad bands between 200 and 480 nm with the maximum at 285 and 220 nm. Compared to hexaphenyl solution (Figure S1), the absorption spectra of film are broad and unstructured.
The spectroscopic feature of film ascribes to its conformational disorder (inhomogeneous broadening), i.e., the variety of oligomer geometrical arrangements due to the torsional degree of freedom of adjacent rings. Inhomogeneous broadening of the optical absorption in conjugated oligomers is attributed to the conjugation length distribution [35]. The fluorescence spectrum exhibits a broad band with three maxima at 402, 422 and 449 nm. These vibronic structures are ascribed to a strong electron-vibronic coupling that favors the formation of polaron-excitons during the optical excitation of the molecules, in agreement with previous studies on luminescent polymers and oligomers [36,37].
Surprisingly, the fluorescence excitation spectra with various probe wavelengths at λ = 420 and 550 nm are entirely different (Figure 1b). At 550 nm, the excitation spectra fit very well with absorption. Understandably, it should follow Kasha–Vavilov’s rule [38,39]. However, at 420 nm there is a strong deviation between 280 and 380 nm, indicating that fluorescence emission at 420 and 550 nm is originating from different species of monomer and aggregate, respectively. Another possibility is that the fluorescence is affected by reabsorption [40,41]. In order to clarify the origin of the emission species, the photoluminescence spectra of hexaphenyl in cyclohexane and single crystal were recorded (Figure S2). To follow the transition assignments, Gaussian multipeak analysis was performed for both film and crystal (Figure S3), and the resulting maxima were recorded in Table S1. In the film, constant energy spacing is 0.17 eV (as obtained from the fluorescence spectrum), corresponding to the stretching vibration of the carbon backbone [42]. The crystal emission shows three maxima of 2.60, 2.77 and 2.89 eV, which exist in the film as well. As known, the hexaphenyl crystal is slip-stacked and aligned in a face-to-tail herringbone arrangement (J-aggregations). Similar to crystal, J-aggregated morphology of the film contributes to the emission between 425 and 600 nm. Furthermore, hexaphenyl monomer in solution shows one broad band at shorter wavelength 325–450 nm, which is not observed in hexaphenyl film.
3.2. Various Emission Species in Hexaphenyl Film
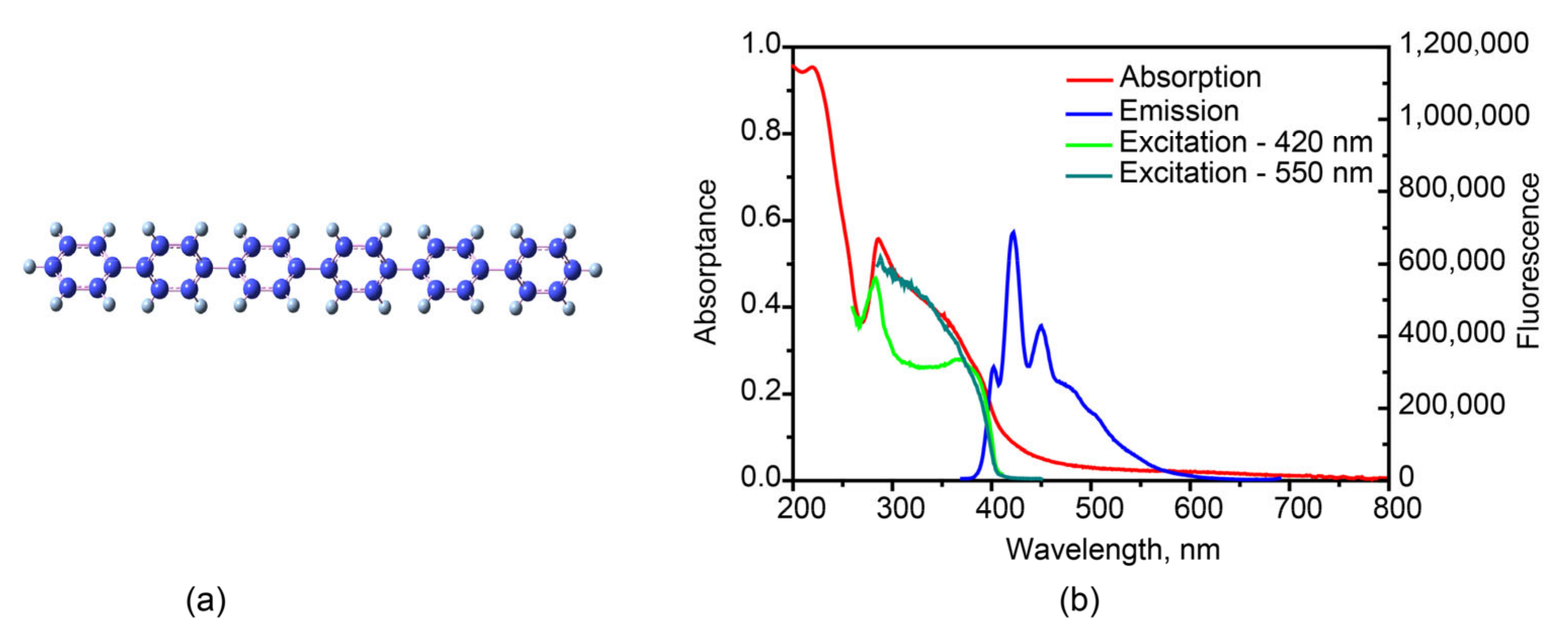
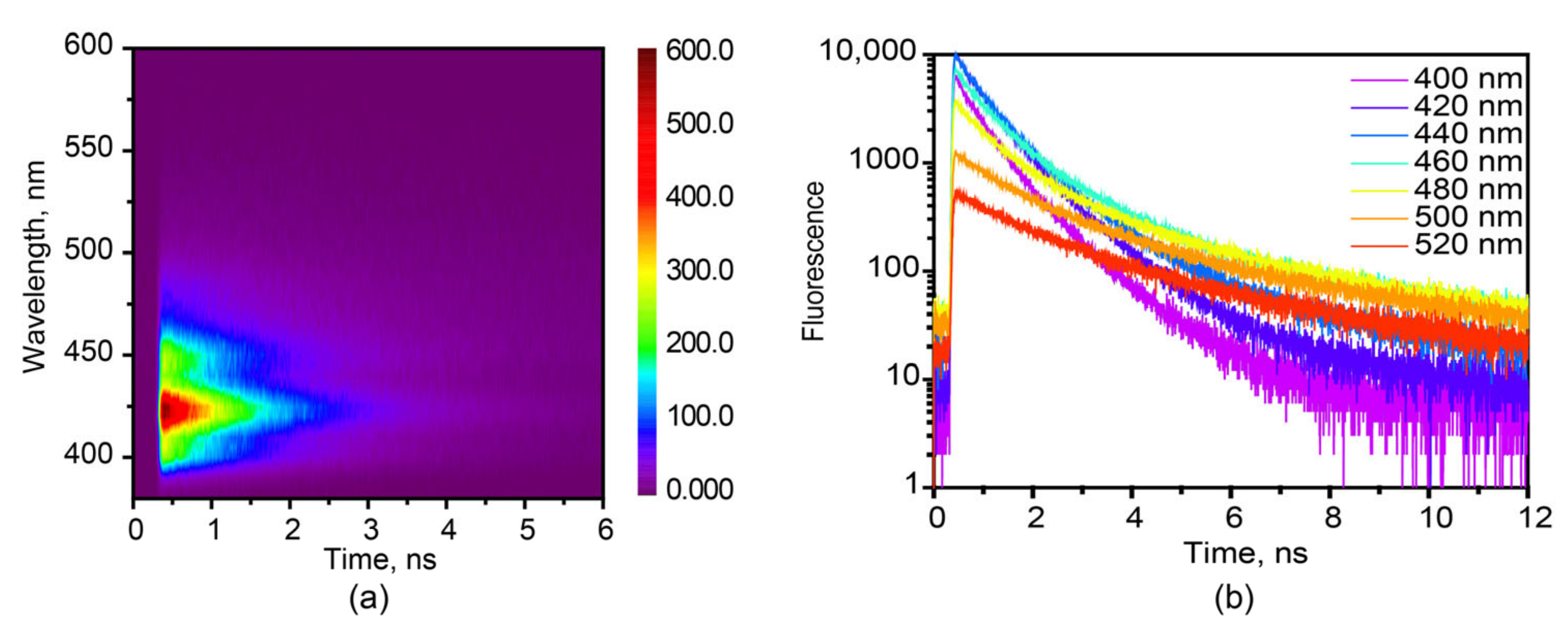
The fluorescence map and kinetics of hexaphenyl film in 380–600 nm are recorded in Figure 2 at λexc = 360 nm, by use of TCSPC technique. The fluorescence lifetime of hexaphenyl in hexane is presented in Figure S4a with single-exponential decay τ = 0.77 ns. The fitting results of film fluorescence decay kinetics are shown in the Table S2: τ1 = 0.24–0.39 ns (A1 = 0.42–0.67), τ2 = 0.64–1.05 ns (A2 = 0.30–0.43) and τ3 = 1.6–3.9 ns (A3 = 0.05–0.18). In order to properly assign the origin of each time component, the map data were globally fit, resulting in τ1 = 0.30, τ2 = 0.85 and τ3 = 3.6 ns. Decay-associated spectra (DAS) are shown in Figure S5.
The shorter lifetimes τ1 = 0.3 and τ1 = 0.85 ns are due to the heterogeneity in organic film. They shared similar DAS, and therefore, we ascribe to the aggregate state in slightly different environments in film. These two times are in line with reported fluorescence lifetime τ = 279 ps from aggregated hexaphenyl film [28]. The shorter lifetimes τ1 = 0.3 ns and τ2 = 0.85 ns are due to the heterogeneity of organic film. They shared similar DAS, and therefore, we attribute them to the aggregated states in slightly different environment.
The longest lifetime, τ3 = 3.6 ns, we assign to the fluorescence from crystalline morphology in film. It agrees well with the fluorescence lifetime of 2.6 ns obtained for hexaphenyl single crystal (Figure S4b). The XRD data of hexaphenyl film reveal the sharp peak in the same position of crystal (Figure S6), in agreement with previous publications [13,43], suggesting the existence of crystalline morphology in film. Moreover, fluorescence confocal microscopy was performed (Figure S7). The resulting fluorescence image shows several types of fluorescing ingredients on the surface with lifetimes ranging from 0.13 to 1.9 ns, indicating the heterogeneity of the film. Besides, the SEM image (Figure S8) exhibits clearly both aggregated and crystalline morphologies. To sum up, the obtained fluorescence lifetimes depend on probe wavelength and are explained by the inhomogeneous broadening. This heterogeneous phenomenon can be attributed to the existence of different lattice locations where the molecules experience various local electric and magnetic fields.
Furthermore, the up-conversion technique with higher time-resolution was applied in order to resolve the ultrafast fluorescence in hexaphenyl film at λexc = 400 nm (Figure S9). It gives two decays: τ1 = 22–35 ps (A1 = 0.22–0.25) and τ2 = 220–300 ps (A2 = 0.69–0.74) (Table S3). They accord to the aggregates’ lifetime [28].
3.3. Transient Absorption Spectra of Hexaphenyl Film
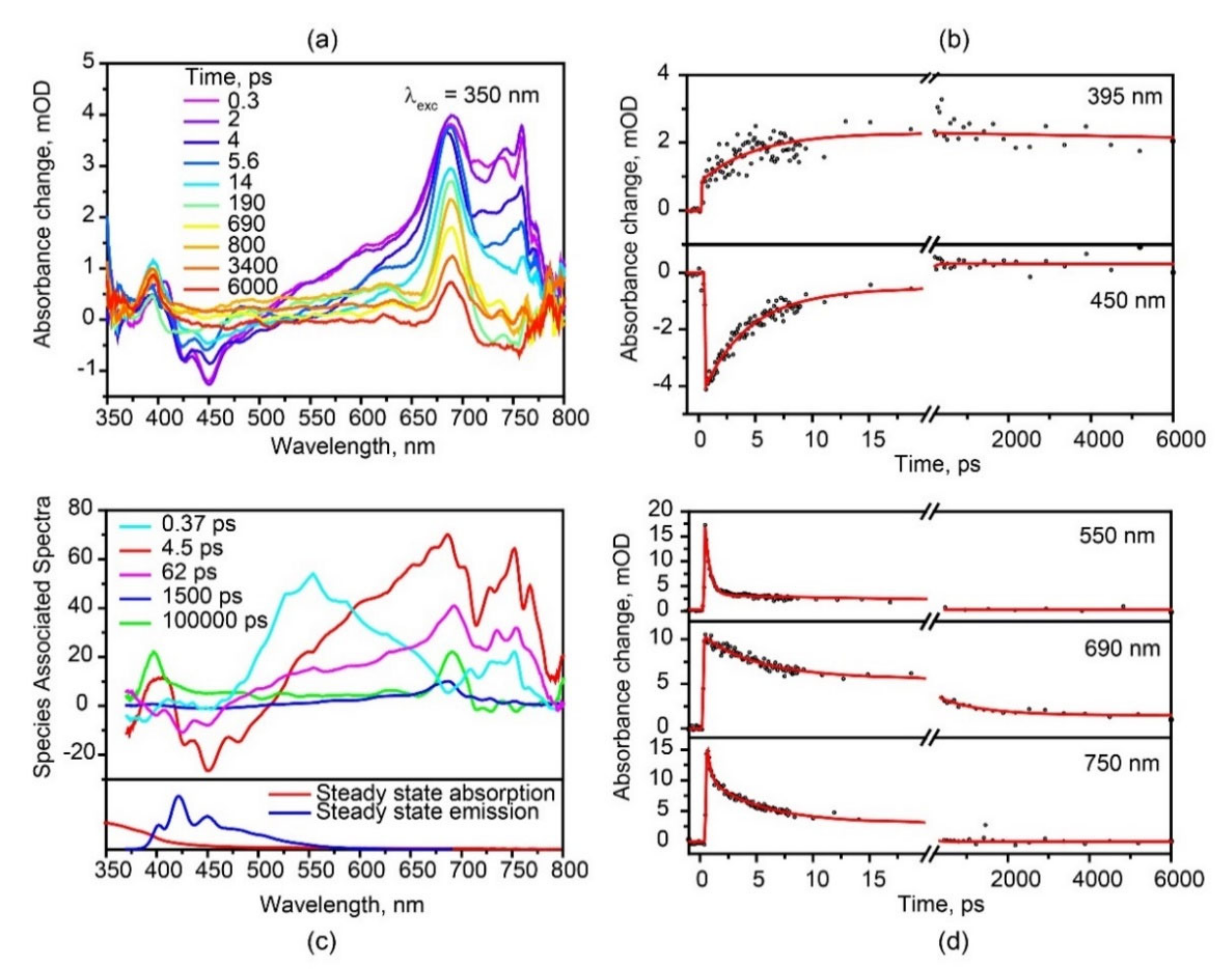
Femtosecond transient absorption (TA) spectra of hexaphenyl film at λexc = 350 nm are shown in Figure 3a. The TA kinetics at different probe wavelengths are shown in Figure 3b,d; fit data at λexc = 350 nm are summarized in Table 1. The TA spectra display three excited state absorption (ESA) bands at 397, 687 and 752 nm and a broad unstructured band between 510 and 650 nm. The negative band at 410–490 nm is a mirror image of the steady-state fluorescence spectrum. Therefore, this band is assigned to stimulated emission (SE), which exhibits the two time constants of 4.5 and 89 ps, in agreement with the above fluorescence data. Note that SE is distorted by positive transients at longer delay times. It should be mentioned that we did not observe a ground state bleaching (GSB) signal because it overlaps with the strong ESA band at 397 nm.
The positive ESA at 752 nm is due to singlet-singlet absorption, in line with the TA data of [28]. This band decays tri-exponentially with 0.3, 4.3 and 84 ps; the latter two lifetimes are similar to SE decay (4.5 and 89 ps). The ultrafast time of 0.3 ps we ascribe to polaron formation [29]. When the lattice phonons are strongly coupled to the charge carriers in the molecular crystal, it results in the formation of localized polaron [44]. Polarons of several hundreds of femtoseconds with and without an applied external field were previously observed in hexaphenyl film by Zenz et al. [29].
3.4. Two Pathways of Triplet Formation
Furthermore, in our TA spectra, there are two longer lived ESA species at 397 and 687 nm. The 687 nm transient band we assign to triplet-triplet absorption, in agreement with previous publications of hexaphenyl [6,28,29] and oligothiophene films [45,46]. In our film, this triplet state forms instantaneously (<30 fs, i.e., within time resolution) and decays with two time constants: 1 ns and >>100 ns. We conclude that ultrafast formation of triplet excitons is due to singlet fission in hexaphenyl aggregates. With 350 nm (3.54 eV) excitation, we are populating the higher vibrational levels of S1. Thus, direct SF proceeds from the upper vibrational levels of the S1 within <30 fs. Note that usually the triplet states live much longer, i.e., micro- and milliseconds. Our triplets are relatively short-lived due to efficient triplet-triplet annihilation between nearby triplet excitons formed via SF. The 687 nm TA band is overlapped with strong singlet-singlet and polaron transient spectra; therefore, global analysis is made in order to separate the time constants. Furthermore, in Figure 3a, the “clean” ESA band at 397 nm has a long-lived component (>100 ns); we assign this long-lived transient to triplet-triplet absorption as well. This triplet band rises with two time constants: ultrafast (<30 fs) and 4.5 ps, which is indicative of two pathways of triplet formation, i.e., via SF and intersystem crossing (ISC), respectively (see discussion below)
In order to clarify the full photophysical pathways, the fs-TA data at λexc = 350 nm were analyzed by Glotaran software. We have applied the target model where two pathways of triplet formation are involved, i.e., one from the lowest singlet state and another from upper vibrational states of S1. It includes also the processes of polaron formation and energy transfer from the hot S1 state to the aggregate S1 state (Figure S10). Resulting Species Associated Spectra (SAS), with the five components of 0.37, 4.5, 62 and 1500 ps and >100 ns, are presented in Figure 3c. These time constants are assigned to polaron, two aggregates S1, short-lived states and long-lived triplet states, respectively. The broad spectrum of ultrashort-lived polaron is correlated with the previous publication [29]. The SAS of monomer and aggregate singlet reveal negative SE transients. They both correspond to various bands of the steady-state fluorescence spectrum. This is indicative of presence of different emissive species.
Obviously, triplet transient(s) should be the same independent on how they are formed, whether directly via ISC or via SF. Global analysis clearly reveals that the spectra of short-lived and long-lived triplets display the same maxima, both at 397 and 687 nm, indicating the same origin of these bands. Thus, triplets formed directly via ISC or via SF show the same TA. However, their formation and decay kinetics are quite different. In our case, triplet states formed via ISC have a 4.5 ps rise time, which corresponds to the singlet decay τ = 4.3 ps; the lifetime of this [single] triplet state is long (>100 ns). In contrary, triplet states via SF are formed instantaneously (rise time <30 fs); this pair of triplet states has a short lifetime, τ = 1.5 ns (because of efficient triplet-triplet annihilation).
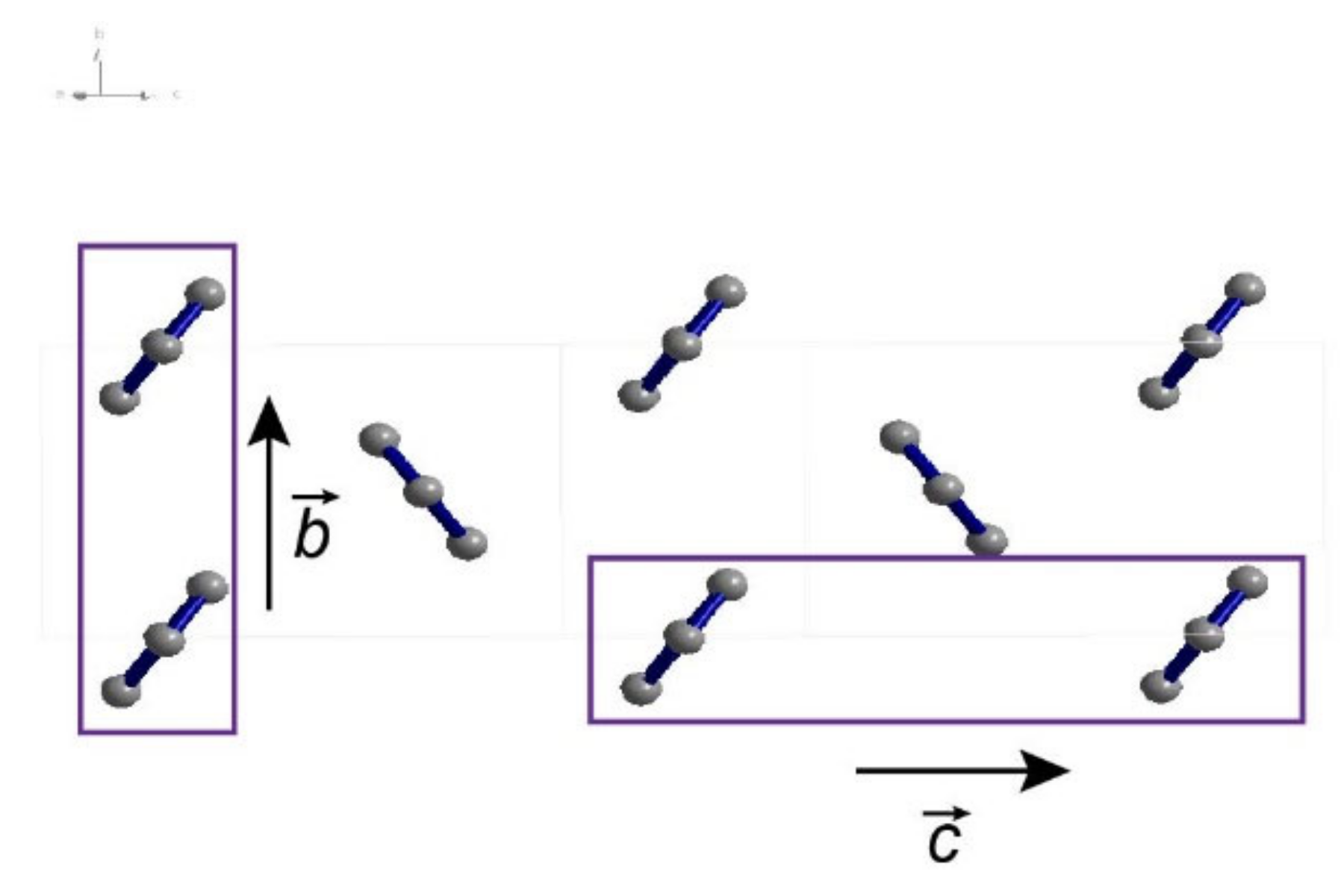
First-principles TD-DFT calculations based on b31yp/6-31g(d) were performed and the resulting the energy levels of the singlet and triplet states were obtained at the optimized excited state geometry. The first excited singlet state is located at 3.00 eV (Figure S11), in agreement with previous reports [47]. The lowest triplet state T1 energy optimized by the same method is 1.04 eV (Figure S12). Thus, it possesses the relationship 2E(T1) < E(S1) required for singlet fission. The schematic stacking patterns of hexaphenyl single crystal along the b- and c-axes is shown in Figure 4. The hexaphenyl molecules are slip-stacked and aligned in a face-to-tail herringbone arrangement. In organic semiconductors, the packing configuration of molecules influences strongly the SF efficiency: slip-stacking favors delocalization of excitation and facilitate exciton fission [48]. Hence, in hexaphenyl film, triplets can be generated via SF in such optimal packings.
In previous publications, the Wasielewski group [49] has reported in the solution of anthracene derivatives, spin-orbit-induced intersystem crossing (SO-ISC) contributes to 53% of triplet formation and SF attributes to the remaining 47%. In this system, SO-ISC originates from individual molecule and SF results from aggregates [49]. In crystalline tetracene [50], the switching between singlet fission and intersystem crossing was manipulated by suppressing intermolecular coupling and lowering the temperature. Moreover, they proposed that the spin dephasing time of triplet pair generated by SF is 40 ns and the lifetime of triplets formed in isolated molecule via ISC is 100 times longer [50]. These observations are in line with our short-lived triplet states generated via SF and long-lived triplets formed via ISC. In the case of tetrachlorinated phenazinothiadiazole [51], the formation mechanism of triplet state depends on concentration. At higher concentrations, TA spectra show the occurrence of a second triplet forming process besides ISC, which is SF, while at lower concentrations, there is only ISC [51]. In pentacene derivatives [52], the introduction of a radical substituent greatly accelerated intersystem crossing and notably enhanced singlet fission [52]. Moreover, in nonconjugated covalent pentacene dimer SF dominates in a dimer, while ISC occurs in its remaining monomeric moiety with much lower efficiency [53]. Note that the triplet states of the abovementioned molecules are located very far away from the singlet state (E(S1) ≈ 2E(T1)); still, both ISC and SF may happen under this condition. Therefore, we believe that both ISC and SF exist in hexaphenyl film.
We explained the mechanism of ISC in hexaphenyl film in the term of singlet fission intersystem crossing (SF-ISC), which was proposed previously for anthanthrene derivatives [49]. The triplet states were populated efficiently with quantum yield of 30% despite the large S1-T1 energy gap (>1 eV) and the lack of carbonyl groups or heavy atoms. This mechanism was based on previous studies in both polyacenes and terrylenediiimides [53,54,55,56,57]: singlet fission can proceed with formation of an initial spin-correlated triplet pair state 1(T1T1) that can intersystem cross to a quintet spin state 5(T1T1), which further separated to form two T1 states. The quintet state is formed with its ms = 0 sublevel greatly over-populated, so that this initial non-Boltzmann spin population is carried over to the ms = 0 sublevel of the T1 states. This population of T1 is labeled as SF-ISC. They attribute the appearance of the SF-ISC triplet to a change in the electronic coupling among the aggregated molecules. Similarly, in the case of hexaphenyl film, we identified the heterogeneity of the film from various herringbone and parallel orientations of hexaphenyl due to a change in the dipolar coupling. Therefore, the SF channel in crystalline hexaphenyls result in the population of 5(T1T1). Another channel of triplet formation will involve SF-ISC mechanism with lifetime of 4.5 ps, which corresponds to the singlet state lifetime.
As already mentioned, our film contains aggregated and crystalline morphologies; ISC proceeds in aggregated hexaphenyl and resulting triplets are long-lived (>100 ns). While SF takes place in this film, the generated nearby triplets live much shorter (1.5 ns) due to efficient triplet-triplet annihilation.
Microscopically, both SF and ISC are electronically nonadiabatic processes. Since SF is spin allowed and ISC is spin forbidden, the nonadiabatic coupling constant (and therefore, transfer rate) is usually much higher for SF than for ISC. Direct SF, notably if it is mediated by a conical intersection as for rubrene crystal [58], can occur on a sub 100 fs timescale, e.g., theoretical simulations for pentacene [59] and rubrene [60]. As for ISC, it is well known that it can become faster in molecular crystals/films, due to favorable geometric constraints, which may lead to (i) enhancement of through space/bond couplings and (ii) alignment of electronic and vibrational levels causing vibration-mediated coherent effects [61,62]. Therefore, triplet formation will occur both via ISC and SF. Further concerted experimental and theoretical efforts are necessary to clarify microscopic origin and pathways of the triplet formation in hexaphenyl films. For pentacene, for example, such a study exists [53].
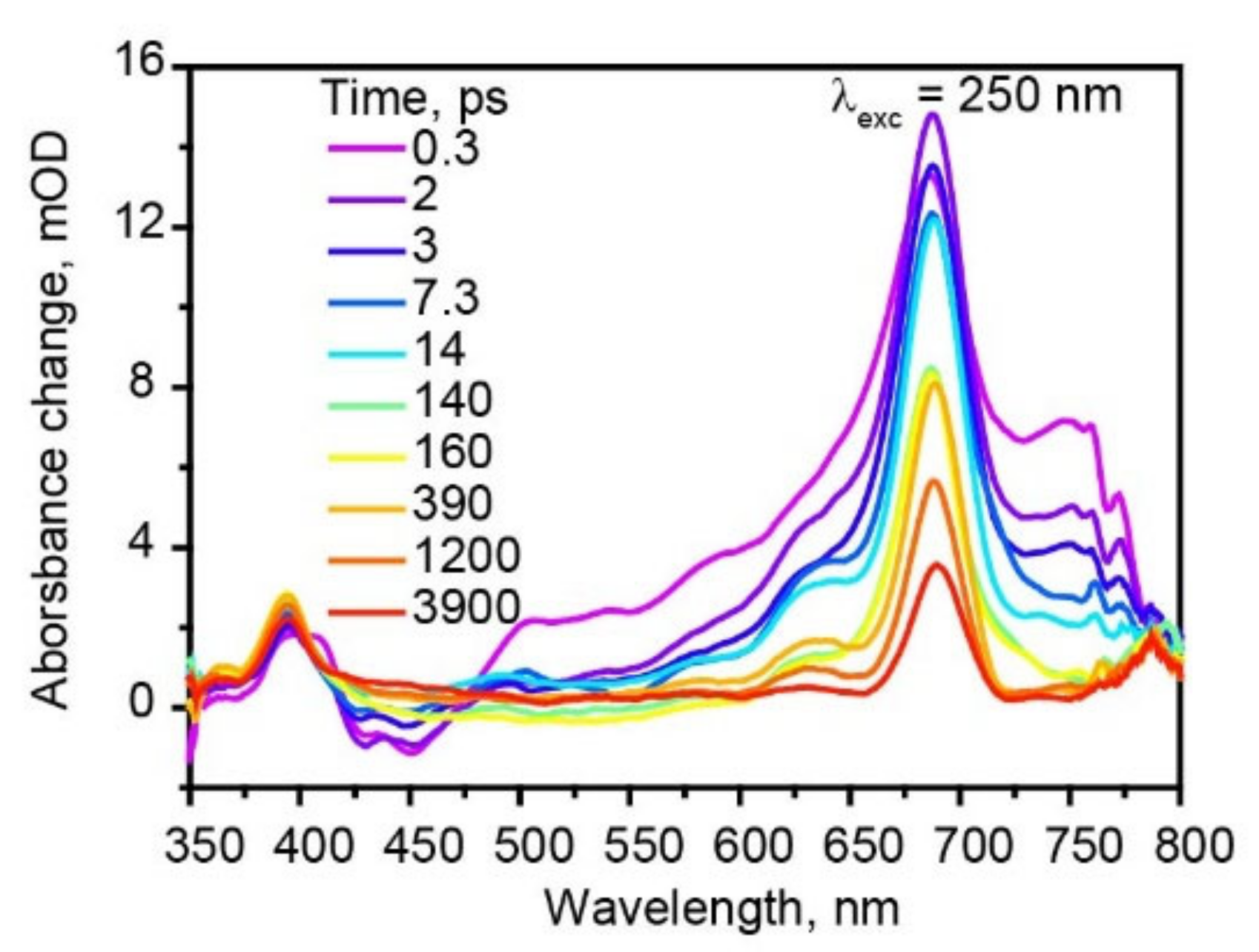
Furthermore, we have excited hexaphenyl film with 250 nm (Figure 5), thus populating higher-lying electronic states. The TA kinetics at different probe wavelengths are shown in Figure S13; fit data at λexc = 250 nm are summarized in Table S4. Two triplet state absorption bands are more pronounced compared with 350 nm excitation. From the comparison of the kinetics and the amplitudes of two triplet absorption bands at 398 and 689 nm, we found that at λexc = 350 nm 61% triplet excitons are formed via ISC and 26% via SF. However, at λexc = 250 nm, 47% triplet excitons are formed via ISC, while 39% via SF. Thus, larger excitation photon energies lead to more efficient SF.
4. Conclusions
We have studied the excited state dynamics of hexaphenyl film with a femtosecond pump-probe and fluorescence spectroscopy. In this heterogeneous film we found different emission species: aggregated and crystalline parts. From transient absorption, an ultrafast (30 fs) singlet fission directly from higher vibrational levels was observed. Another route of triplet state formation is intersystem crossing from the lowest singlet state within 4.5 ps. Moreover, an ultrafast polaron generation was also detected at different excitation conditions. DFT calculations support our experimental data and conclusions.
Our film with coexistence of aggregated and crystalline morphologies could exhibit efficient singlet fission. The present results give a new insight into the excited state dynamics in hexaphenyl film, further contributing to the understanding of singlet fission in organic crystals and films. Moreover, the simple-fabricated commercially available hexaphenyl film is highly promising material for organic photovoltaic applications.
Supplementary Materials
The following supporting information can be downloaded at: https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/molecules27165067/s1, Figure S1: Steady state absorption and fluorescence spectra of hexaphenyl in hexane (λexc = 300 nm); Figure S2: Fluorescence of hexaphenyl solution (λexc = 300 nm), film (λexc = 350 nm) and single crystal (λexc = 350 nm); Figure S3: Gaussian fit for steady state fluorescence spectra of (a) hexaphenyl film and (b) single crystal; Figure S4: Fluorescence decay kinetics of the (a) hexaphenyl solution and (b) single crystal λexc = 267 nm at various probe wavelengths; Figure S5: Global fit spectra of TCSPC results for hexaphenyl film at λexc = 360 nm; Figure S6: X-ray diffraction (XRD) patterns of hexaphenyl film and single crystal presented on the same scale; Figure S7: Fluorescence lifetime imaging acquired with confocal microscopy. Range size (a): 1 × 1 μm, (b): 10 × 10 μm; Figure S8: Scanning electron microscope (SEM) of hexaphenyl film; Figure S9: Up-conversion fluorescence decay kinetics of hexaphenyl film at λexc = 400 nm; Figure S10: Global fit target model (a) and kinetics (b) of Species Associated Spectra; Figure S11: TD-DFT/(b3lyp/6-31g(d)) calculation of excited singlet state energies of hexaphenyl isolated molecule at S1 geometry; Figure S12: TD-DFT/(b3lyp/6-31g(d)) calculation of excited triplet state energies of hexaphenyl isolated molecule at T1 geometry; Figure S13: Decay kinetics of the hexaphenyl film λexc = 250 nm at different wavelengths; Table S1: Gaussian multipeak fit maxima of the steady state fluorescence spectra of hexaphenyl film and single crystal; Table S2: Fluorescence decay kinetics of hexaphenyl film, solution and single crystal at various emission wavelengths (TCSPC data); Table S3: Fluorescence decay kinetics of hexaphenyl film at various emission wavelengths (up-conversion data). τ3 is fixed to respective TCSPC data; Table S4: Lifetimes obtained from fit/deconvolution of TA spectra for the hexaphenyl film at λexc = 250 nm; f = fixed.
Author Contributions
Conceptualization, G.G.G.; methodology, W.N., T.L. and G.G.G.; formal analysis, W.N. and T.L.; data curation, T.L.; writing—original draft preparation, W.N., T.L. and G.G.G.; writing—review and editing, G.G.G., C.K. and L.S.; supervision, G.G.G. and L.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by a DUT startup grant and the National Natural Science Foundation of China (Grant No. 22088102).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are not available from the authors.
References
- Burroughes, J.H.; Bradley, D.D.; Brown, A.R.; Marks, R.N.; Mackay, K.; Friend, R.H.; Holmes, A.B. Light-Emitting Diodes Based on Conjugated Polymers. Nature 1990, 347, 539–541. [Google Scholar] [CrossRef]
- Graupner, W.; Grem, G.; Meghdadi, F.; Paar, C.; Leising, G.; Scherf, U.; Mullen, K.; Fischer, W.; Stelzer, F. Electroluminescence with Conjugated Polymers and Oligomers. Mol. Cryst. Liq. Cryst. 1994, 256, 549–554. [Google Scholar]
- Era, M.; Tsutsui, T.; Saito, S. Polarized Electroluminescence from Oriented P-Sexiphenyl Vacuum-Deposited Film. Appl. Phys. Lett. 1995, 67, 2436–2438. [Google Scholar] [CrossRef]
- Leising, G.; Tasch, S.; Brandstatter, C.; Meghdadi, F.; Froyer, G.; Athouel, L. Red–Green–Blue Light Emission from A Thin Film Electroluminescence Device Based on Parahexaphenyl. Adv. Mater. 1997, 9, 33–36. [Google Scholar]
- Sariciftci, N.S.; Smilowitz, L.; Heeger, A.J.; Wudl, F. Photoinduced Electron Transfer from A Conducting Polymer to Buckminsterfullerene. Science 1992, 258, 1474–1476. [Google Scholar] [CrossRef]
- Leising, G.; Ekstroem, O.; Graupner, W.; Meghdadi, F.; Moser, M.; Kranzelbinder, G.; Jost, T.; Tasch, S.; Winker, B.; Athouel, L.; et al. Multicolor Electroluminescence and Stimulated Emission of Conjugated Polymers and Oligomers. Proc. SPIE—Int. Soc. Opt. Eng. 1996, 2852, 189–200. [Google Scholar]
- Piaggi, A.; Lanzani, G.; Bongiovanni, G.; Mura, A.; Graupner, W.; Meghdadi, F.; Leising, G.; Nisoli, M. Emission Properties of Para-Hexaphenyl Polycrystalline Films. Phys. Rev. B 1997, 56, 10133. [Google Scholar] [CrossRef] [Green Version]
- Piaggi, A.; Lanzani, G.; Bongiovanni, G.; Loi, M.A.; Mura, A.; Graupner, W.; Meghdadi, F.; Leising, G. Optical Properties of Polycrystalline Films. Opt. Mater. 1998, 9, 489–493. [Google Scholar]
- Frank, P.; Hlawacek, G.; Lengyel, O.; Satka, A.; Teichert, C.; Resel, R.; Winkler, A. Influence of Surface Temperature and Surface Modifications on The Initial Layer Growth of Para-Hexaphenyl on Mica (001). Surf. Sci. 2007, 601, 2152–2160. [Google Scholar] [CrossRef]
- Leising, G.; Tasch, S.; Meghdadi, F.; Athouel, L.; Froyer, G.; Scherf, U. Blue Electroluminescence with Ladder-Type Poly(Para-Phenylene) and Para-Hexaphenyl. Synth. Met. 1996, 81, 185–189. [Google Scholar] [CrossRef]
- Resel, R.; Koch, N.; Meghdadi, F.; Leising, G.; Athouel, L.; Froyer, G.; Hofer, F.A. Polymorph Crystal Structure of Hexaphenyl Observed in Thin Films. Cryst. Res. Technol. 2001, 36, 47–54. [Google Scholar]
- Baker, K.N.; Fratini, A.V.; Resch, T.; Knachel, H.C.; Adams, W.W.; Socci, E.P.; Farmer, B.L. Crystal Structures, Phase Transitions and Energy Calculations of Poly (P-Phenylene) Oligomers. Polymer 1993, 34, 1571–1587. [Google Scholar] [CrossRef]
- Athouel, L.; Froyer, G.; Riou, M.T.; Schott, M. Structural Studies of Parasexiphenyl Thin Films: Importance of The Deposition Parameters. Thin Solid Films 1996, 274, 35–45. [Google Scholar] [CrossRef]
- Congreve, D.N.; Lee, J.; Thompson, N.J.; Hontz, E.; Yost, S.R.; Reusswig, P.D.; Bahlke, M.E.; Reineke, S.; Voorhis, T.; Baldo, M.A. External Quantum Efficiency Above 100% in A Singlet-Exciton-Fission–Based Organic Photovoltaic Cell. Science 2013, 340, 334–337. [Google Scholar] [CrossRef]
- Qiao, X.; Ma, D. Nonlinear Optoelectronic Processes in Organic Optoelectronic Devices: Triplet-Triplet Annihilation and Singlet Fission. Mater. Sci. Eng. R 2020, 139, 100519. [Google Scholar]
- Rao, A.; Friend, R.H. Harnessing Singlet Exciton Fission to Break the Shockley-Queisser Limit. Nat. Rev. Mater. 2017, 2, 17063. [Google Scholar] [CrossRef]
- Futscher, M.H.; Rao, A.; Ehrler, B. The Potential of Singlet Fission Photon Multipliers as an Alternative to Silicon-Based Tandem Solar Cells. ACS Energy Lett. 2018, 3, 2587–2592. [Google Scholar] [CrossRef] [Green Version]
- MacQueen, R.W.; Liebhaber, M.; Niederhausen, J.; Mews, M.; Gersmann, C.; Jäckle, S.; Jäger, K.; Tayebjee, M.J.Y.; Schmidt, T.W.; Rech, B.; et al. Crystalline Silicon Solar Cells with Tetracene Interlayers: The Path to Silicon-Singlet Fission Heterojunction Devices. Mater. Horiz. 2018, 5, 1065–1075. [Google Scholar] [CrossRef] [Green Version]
- Casillas, R.; Papadopoulos, I.; Ullrich, T.; Thiel, D.; Kunzmann, A.; Guldi, D.M. Molecular Insights and Concepts to Engineer Singlet Fission Energy Conversion Devices. Energy Environ. Sci. 2020, 13, 2741–2804. [Google Scholar] [CrossRef]
- Smith, M.B.; Michl, J. Recent Advances in Singlet Fission. Annu. Rev. Phys. Chem. 2013, 64, 361–386. [Google Scholar] [CrossRef]
- Wilson, M.W.B.; Rao, A.; Clark, J.; Kumar, R.S.S.; Brida, D.; Cerullo, G.; Friend, R.H. Ultrafast Dynamics of Exciton Fission in Polycrystalline Pentacene. J. Am. Chem. Soc. 2011, 133, 11830–11833. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zhang, K.; Kloc, C.; Sun, H.; Michel-Beyerle, M.E.; Gurzadyan, G.G. Singlet Fission in Rubrene Single Crystal: Direct Observation by Femtosecond Pump–Probe Spectroscopy. Phys. Chem. Chem. Phys. 2012, 14, 8307–8312. [Google Scholar] [CrossRef] [PubMed]
- Pun, A.B.; Sanders, S.N.; Kumarasamy, E.; Sfeir, M.Y.; Congreve, D.N.; Campos, L.M. Triplet Harvesting from Intramolecular Singlet Fission in Polytetracene. Adv. Mater. 2017, 29, 1701416. [Google Scholar] [CrossRef] [PubMed]
- Grieco, C.; Doucette, G.S.; Munson, K.T.; Swartzfager, J.R.; Munro, J.M.; Anthony, J.E.; Dabo, I.; Asbury, J.B. Vibrational Probe of the Origin of Singlet Exciton Fission in TIPS-Pentacene Solutions. J. Chem. Phys. 2019, 151, 154701. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Bae, Y.J.; Mauck, C.M.; Mandal, A.; Young, R.M.; Wasielewski, M.R. Singlet Fission in Covalent Terrylenediimide Dimers: Probing the Nature of the Multiexciton State Using Femtosecond Mid-Infrared Spectroscopy. J. Am. Chem. Soc. 2018, 140, 9184–9192. [Google Scholar] [CrossRef]
- Ni, W.; Gurzadyan, G.G.; Zhao, J.; Che, Y.; Li, X.; Sun, L. Singlet Fission from Upper Excited Electronic States of Cofacial Perylene Dimer. J. Phys. Chem. Lett. 2019, 10, 2428–2433. [Google Scholar] [CrossRef]
- Smith, M.B.; Michl, J. Singlet Fission. Chem. Rev. 2010, 110, 6891–6936. [Google Scholar] [CrossRef]
- Graupner, W.; Meghdadi, F.; Leising, G.; Lanzani, G.; Nisoli, M.; De Silvestri, S.; Fischer, W.; Stelzer, F. Photoexcitations in Para-Hexaphenyl. Phys. Rev. B 1997, 56, 10128. [Google Scholar] [CrossRef]
- Zenz, C.; Cerullo, G.; Lanzani, G.; Graupner, W.; Meghdadi, F.; Leising, G.; De Silvestri, S. Ultrafast Photogeneration Mechanisms of Triplet States in Para-Hexaphenyl. Phys. Rev. B 1999, 59, 14336. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Gong, C.; Gurzadyan, G.G.; Gelin, M.F.; Liu, J.; Sun, L. Ultrafast Relaxation Dynamics in Zinc Tetraphenylporphyrin Surface-Mounted Metal Organic Framework. J. Phys. Chem. C 2017, 122, 50–61. [Google Scholar] [CrossRef]
- Ullrich, S.; Schultz, T.; Zgierski, M.Z.; Stolow, A. Electronic Relaxation Dynamics in DNA and RNA Bases Studied by Time-Resolved Photoelectron Spectroscopy. Phys. Chem. Chem. Phys. 2004, 6, 2796–2801. [Google Scholar] [CrossRef]
- Shang, J.; Yan, S.; Cong, C.; Tan, H.; Yu, T.; Gurzadyan, G.G. Probing Near Dirac Point Electron-Phonon Interaction in Graphene. Opt. Mater. Express 2012, 2, 1713–1722. [Google Scholar] [CrossRef]
- Shang, J.; Yu, T.; Lin, J.; Gurzadyan, G.G. Ultrafast Electron-Optical Phonon Scattering and Quasiparticle Lifetime in CVD-Grown Graphene. ACS Nano 2011, 5, 3278–3283. [Google Scholar] [CrossRef]
- Snellenburg, J.J.; Laptenok, S.P.; Seger, R.; Mullen, K.M.; Van Stokkum, I.H.M. Glotaran: A Java-Based Graphical User Interface for the R Package TIMP. J. Stat. Softw. 2012, 49, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Kersting, R.; Lemmer, U.; Mahrt, R.F.; Leo, K.; Kurz, H.; Bässler, H.; Göbel, E.O. Femtosecond Energy Relaxation in π-Conjugated Polymers. Phys. Rev. Lett. 1993, 70, 3820–3823. [Google Scholar] [CrossRef] [PubMed]
- Faulques, E.; Wéry, J.; Lefrant, S.; Ivanov, V.G.; Jonusauskas, G. Transient Photoluminescence of Para-Hexaphenyl Layers. Phys. Rev. B 2002, 65, 212202. [Google Scholar] [CrossRef]
- Rzepka, E.; Jin, C.Q.; Lefrant, S.; Pelous, Y.; Froyer, G.; Siove, A. Luminescence Studies in Polyparaphenylene Samples. Synth. Met. 1989, 29, 23–28. [Google Scholar] [CrossRef]
- Kasha, M. Characterization of Electronic Transitions in Complex Molecules. Discuss. Faraday Soc. 1950, 9, 14–19. [Google Scholar] [CrossRef]
- Hestand, N.J.; Spano, F.C. Molecular Aggregate Photophysics Beyond the Kasha Model: Novel Design Principles for Organic Materials. Acc. Chem. Res. 2017, 50, 341–350. [Google Scholar] [CrossRef]
- Nakanishi, T.; Mizuno, K.I.; Matsui, A.; Nishimura, H. Reabsorption of Excitonic Luminescence in Pyrene Crystals. J. Lumin. 1991, 47, 303–308. [Google Scholar] [CrossRef]
- Kobayashi, T.; Nagakura, S. Reabsorption and High Density Excitation Effects on The Time-Resolved Fluorescence Spectra of Anthracene Crystal. Mol. Cryst. Liq. Cryst. 1974, 26, 33–43. [Google Scholar] [CrossRef]
- Louarn, G.; Athouël, L.; Froyer, G.; Buisson, J.P.; Lefrant, S. Optical Characterization of Parasexiphenyl: A Model Compound of Polyparaphenylene. Synth. Met. 1993, 57, 4762–4767. [Google Scholar] [CrossRef]
- Resel, R.; Koch, N.; Meghdadi, F.; Leising, G.; Unzog, W.; Reichmann, K. Growth and Preferred Crystallographic Orientation of Hexaphenyl thin Films. Thin Solid Films 1997, 305, 232–242. [Google Scholar] [CrossRef]
- Pope, M.; Swenberg, C.E. Electronic Processes in Organic Crystals and Polymers; Oxford University Press: New York, NY, USA, 1999. [Google Scholar]
- Grebner, D.; Helbig, M.; Rentsch, S. Size-Dependent Properties of Oligothiophenes by Picosecond Time-Resolved Spectroscopy. J. Phys. Chem. 1995, 99, 16991–16998. [Google Scholar] [CrossRef]
- Janssen, R.A.J.; Smilowitz, L.; Sariciftci, N.S.; Moses, D. Triplet-State Photoexcitations of Oligothiophene Films and Solutions. J. Chem. Phys. 1994, 101, 1787–1798. [Google Scholar] [CrossRef]
- Tavares, L.; Cadelano, M.; Quochi, F.; Simbrunner, C.; Schwabegger, G.; Saba, M.; Mura, A.; Bongiovanni, G.; Silva Fiho, D.A.; Cunha, W.F.; et al. Efficient Exciton Diffusion and Resonance-Energy Transfer in Multilayered Organic Epitaxial Nanofibers. J. Phys. Chem. C 2015, 119, 15689–15697. [Google Scholar] [CrossRef] [PubMed]
- Kolata, K.; Breuer, T.; Witte, G.; Chatterjee, S. Molecular Packing Determines Singlet Exciton Fission in Organic Semiconductors. ACS Nano 2014, 8, 7377–7383. [Google Scholar] [CrossRef]
- Jue Bae, Y.; Krzyaniak, M.D.; Majewski, M.B.; Desroches, M.; Morin, J.F.; Wu, Y.L.; Wasielewski, M.R. Competition Between Singlet Fission and Spin-Orbit-Induced Intersystem Crossing in Anthanthrene and Anthanthrone Derivatives. ChemPlusChem 2019, 84, 1432–1438. [Google Scholar] [CrossRef]
- Bayliss, S.L.; Kraffert, F.; Wang, R.; Zhang, C.; Bittl, R.; Behrends, J. Tuning Spin Dynamics in Crystalline Tetracene. J. Phys. Chem. Lett. 2019, 10, 1908–1913. [Google Scholar] [CrossRef]
- Wollscheid, N.; Alagna, N.; Lustres, J.L.P.; Buckup, T.; Hahn, S.; Bunz, U.H.; Motzkus, M. Unveiling the Concentration Dependent Direct Triplet Formation Via Singlet Fission in A Tetracene Derivative. EPJ Web Conf. 2019, 205, 09031. [Google Scholar] [CrossRef]
- Ito, A.; Shimizu, A.; Kishida, N.; Kawanaka, Y.; Kosumi, D.; Hashimoto, H.; Teki, Y. Excited-State Dynamics of Pentacene Derivatives with Stable Radical Substituents. Angew. Chem. Int. Ed. 2014, 126, 6833–6837. [Google Scholar] [CrossRef]
- Basel, B.S.; Zirzlmeier, J.; Hetzer, C.; Phelan, B.T.; Krzyaniak, M.D.; Reddy, S.R.; Coto, P.B.; Horwitz, N.E.; Young, R.M.; White, F.J.; et al. Unified Model for Singlet Fission Within A Non-Conjugated Covalent Pentacene Dimer. Nat. Commun. 2017, 8, 15171. [Google Scholar] [CrossRef] [PubMed]
- Weiss, L.R.; Bayliss, S.L.; Kraffert, F.; Thorley, K.J.; Anthony, J.E.; Bittl, R.; Friend, R.H.; Rao, A.; Greenham, N.C.; Behrends, J. Strongly Exchange-Coupled Triplet Pairs in an Organic Semiconductor. Nat. Phys. 2017, 13, 176–181. [Google Scholar] [CrossRef]
- Bayliss, S.L.; Weiss, L.R.; Rao, A.; Friend, R.H.; Chepelianskii, A.D.; Greenham, N.C. Spin Signatures of Exchange-Coupled Triplet Pairs Formed by Singlet Fission. Phys. Rev. B 2016, 94, 045204. [Google Scholar] [CrossRef] [Green Version]
- Tayebjee, M.J.Y.; Sanders, S.N.; Kumarasamy, E.; Campos, L.M.; Sfeir, M.Y.; McCamey, D.R. Quintet Multiexciton Dynamics in Singlet Fission. Nat. Phys. 2017, 13, 182–188. [Google Scholar] [CrossRef]
- Basel, B.S.; Young, R.M.; Krzyaniak, M.D.; Papadopoulos, I.; Hetzer, C.; Gao, Y.; La Porte, N.T.; Phelan, B.T.; Clark, T.; Tykwinski, R.R.; et al. Influence of the Heavy-Atom Effect on Singlet Fission: A Study of Platinum-Bridged Pentacene Dimers. Chem. Sci. 2019, 10, 11130–11140. [Google Scholar] [CrossRef] [Green Version]
- Miyata, K.; Kurashige, Y.; Watanabe, K.; Sugimoto, T.; Takahashi, S.; Tanaka, S.; Takeya, J.; Yanai, T.; Matsumoto, Y. Coherent Singlet Fission Activated by Symmetry Breaking. Nat. Chem. 2017, 9, 983–989. [Google Scholar] [CrossRef]
- Reddy, S.R.; Coto, P.B.; Thoss, M. Quantum Dynamical Simulation of Intramolecular Singlet Fission in Covalently Coupled Pentacene Dimers. J. Chem. Phys. 2019, 151, 044307. [Google Scholar] [CrossRef]
- Sun, K.; Xu, Q.; Chen, L.; Gelin, M.F.; Zhao, Y. Temperature Effects on Singlet Fission Dynamics Mediated by A Conical Intersection. J. Chem. Phys. 2020, 153, 194106. [Google Scholar] [CrossRef]
- Serevicius, T.; Komskis, R.; Adomėnas, P.; Adomėnienė, O.; Kreiza, G.; Jankauskas, V.; Kazlauskas, K.; Miasojedovas, A.; Jankus, V.; Monkman, A.P.; et al. Triplet-Triplet Annihilation in 9, 10-Diphenylanthracene Derivatives: The Role of Intersystem Crossing and Exciton Diffusion. J. Phys. Chem. C 2017, 121, 8515–8524. [Google Scholar] [CrossRef]
- Ford, T.A.; Avilov, I.; Beljonne, D.; Greenham, N.C. Enhanced Triplet Exciton Generation in Polyfluorene Blends. Phys. Rev. B 2005, 71, 125212. [Google Scholar] [CrossRef]
Figure 1.
(a) Structure of hexaphenyl. (b) Steady-state absorption, fluorescence and excitation spectra of hexaphenyl film (λexc = 350 nm, λem = 420 and 550 nm).
Figure 1.
(a) Structure of hexaphenyl. (b) Steady-state absorption, fluorescence and excitation spectra of hexaphenyl film (λexc = 350 nm, λem = 420 and 550 nm).

Figure 2.
(a) Fluorescence TCSPC map of the hexaphenyl film, λexc = 360 nm. (b) Fluorescence kinetics of hexaphenyl film at various probe wavelengths, λexc = 360 nm.
Figure 2.
(a) Fluorescence TCSPC map of the hexaphenyl film, λexc = 360 nm. (b) Fluorescence kinetics of hexaphenyl film at various probe wavelengths, λexc = 360 nm.

Figure 3.
Transient absorption spectra (a) and Species Associated Spectra by global fit (c) of hexaphenyl film; decay kinetics (b,d) at various probe wavelengths. λexc = 350 nm.
Figure 3.
Transient absorption spectra (a) and Species Associated Spectra by global fit (c) of hexaphenyl film; decay kinetics (b,d) at various probe wavelengths. λexc = 350 nm.

Figure 4.
Correlation of packing scheme along different crystal axes. Schematics of hexaphenyl stacking patterns along the b- and c-axes.
Figure 4.
Correlation of packing scheme along different crystal axes. Schematics of hexaphenyl stacking patterns along the b- and c-axes.

Figure 5.
Femtosecond transient absorption spectra of hexaphenyl film at λexc = 250 nm.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Lifetimes obtained from Fit/Deconvolution of TA dynamics for the hexaphenyl film at λexc = 350 nm; f = fixed.
Table 1.
Lifetimes obtained from Fit/Deconvolution of TA dynamics for the hexaphenyl film at λexc = 350 nm; f = fixed.
| λexc, nm | λprobe, nm | τ1, ps | A1 | τ2, ps | A2 | τ3, ps | A3 | τ4, ps | A4 |
|---|---|---|---|---|---|---|---|---|---|
| 350 | 395 | 4.5 | −0.61 | 100,000 f | 1 | ||||
| 450 | 4.5 | −0.75 | 89 | −0.25 | 100,000 f | 1 | |||
| 550 | 0.45 | 0.85 | 62 | 0.15 | |||||
| 690 | 4.6 | 0.41 | 65 | 0.18 | 1000 | 0.26 | 100,000 f | 0.15 | |
| 750 | 0.30 | 0.43 | 4.3 | 0.37 | 84 | 0.20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ni, W.; Li, T.; Kloc, C.; Sun, L.; Gurzadyan, G.G. Singlet Fission, Polaron Generation and Intersystem Crossing in Hexaphenyl Film. Molecules 2022, 27, 5067. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27165067
AMA Style
Ni W, Li T, Kloc C, Sun L, Gurzadyan GG. Singlet Fission, Polaron Generation and Intersystem Crossing in Hexaphenyl Film. Molecules. 2022; 27(16):5067. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27165067
Chicago/Turabian StyleNi, Wenjun, Tianjiao Li, Christian Kloc, Licheng Sun, and Gagik G. Gurzadyan. 2022. "Singlet Fission, Polaron Generation and Intersystem Crossing in Hexaphenyl Film" Molecules 27, no. 16: 5067. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27165067