
Biosynthesis of DNA-Alkylating Antitumor Natural Products


1
State Key Laboratory of Bioorganic and Natural Products Chemistry, Center for Excellence in Molecular Synthesis, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China
2
School of Chemistry and Materials Science, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, 1 Sub-lane Xiangshan, Hangzhou 310024, China
*
Author to whom correspondence should be addressed.
Molecules 2022, 27(19), 6387; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27196387
Submission received: 31 August 2022
/
Revised: 23 September 2022
/
Accepted: 23 September 2022
/
Published: 27 September 2022
(This article belongs to the Special Issue Enzymes, Pathways and Intermediates of Bacterial Metabolism)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
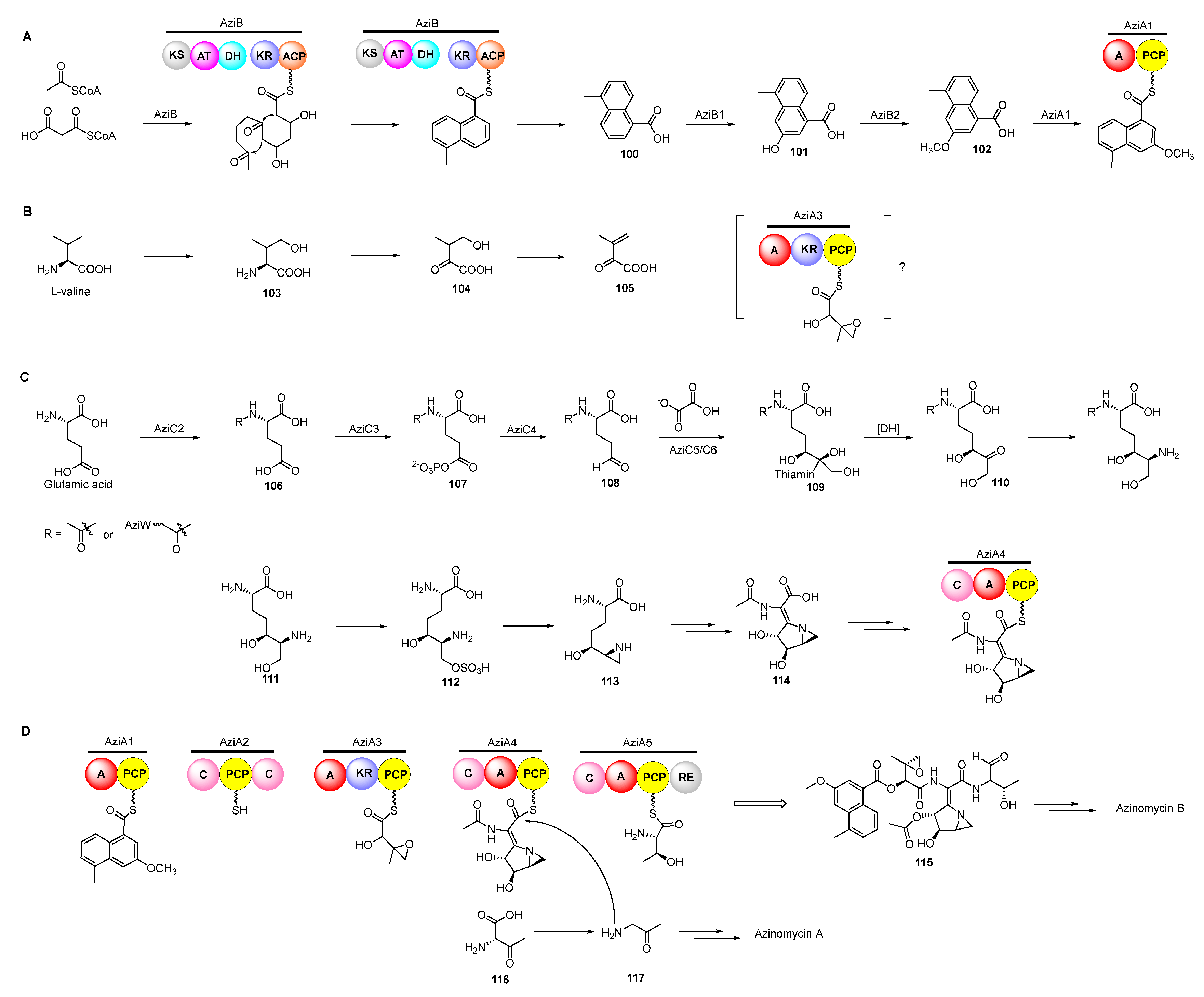{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:DNA-alkylating natural products play an important role in drug development due to their significant antitumor activities. They usually show high affinity with DNA through different mechanisms with the aid of their unique scaffold and highly active functional groups. Therefore, the biosynthesis of these natural products has been extensively studied, especially the construction of their pharmacophores. Meanwhile, their producing strains have evolved corresponding self-resistance strategies to protect themselves. To further promote the functional characterization of their biosynthetic pathways and lay the foundation for the discovery and rational design of DNA alkylating agents, we summarize herein the progress of research into DNA-alkylating antitumor natural products, including their biosynthesis, modes of action, and auto-resistance mechanisms.
1. Introduction
Natural products (NPs) are an important source of pharmaceuticals due to their diverse bioactivities [1]. Since DNA is essential for living organisms, DNA-targeting NPs, which usually function as carcinogenesis or cancer treatment, constitute an indispensable family of bioactive NPs [2,3]. Although the genotoxic metabolite colibactin, produced by human gut bacteria, is shown to cause colorectal cancer by alkylating DNA to generate DNA mutation [4,5,6], some DNA-targeting NPs are applied in chemotherapy. They can interact with specific DNA duplex structures and cause DNA damage via different modes of action [7]. One of the mechanisms is the cleavage of DNA through inducing the production of radical DNA by redox reactions or nucleophilic addition. Broad anti-cancer antibiotic bleomycin (BLM) can be transformed to HOO-Fe(III)-BLM in the presence of Fe/O2 to damage DNA [8]. The enediyne-containing NPs dynemicin A and calicheamicin can generate biradical intermediates to cleave DNA activated by reducing the quinone moiety and the attack of a thiol, respectively [9,10,11,12]. Some chemicals, such as streptozotocin, conduct the methylation of DNA [13,14]. Additionally, another family of DNA-targeting antitumor agents can alkylate DNA in situ with covalent bonds. They can directly react with DNA using highly active functional groups such as epoxide, cyclopropane, and aziridine to form bulky DNA adducts [15,16]. Furthermore, as a result of the potent cytotoxicity of DNA-alkylating NPs, it is preferable for their producers to possess resistant genes located in biosynthetic gene clusters (BGCs) to protect themselves. BGCs-associated self-resistance is mainly achieved through excision of the abnormal base, degradation of active functional groups, and the binding or transport of toxins [17].
The biosynthesis and resistance of radical-based DNA damage agents, including BLMs and enediynes, have already been well reviewed [18]; herein, we mainly discuss DNA-alkylating (except DNA-methylation) antitumor NPs, including their modes of action, BGC-associated self-resistance, and biosynthetic pathways, especially the construction of their highly active groups as a warhead.
2. Spirocyclopropane-Containing Cyclohexadienone Natural Products
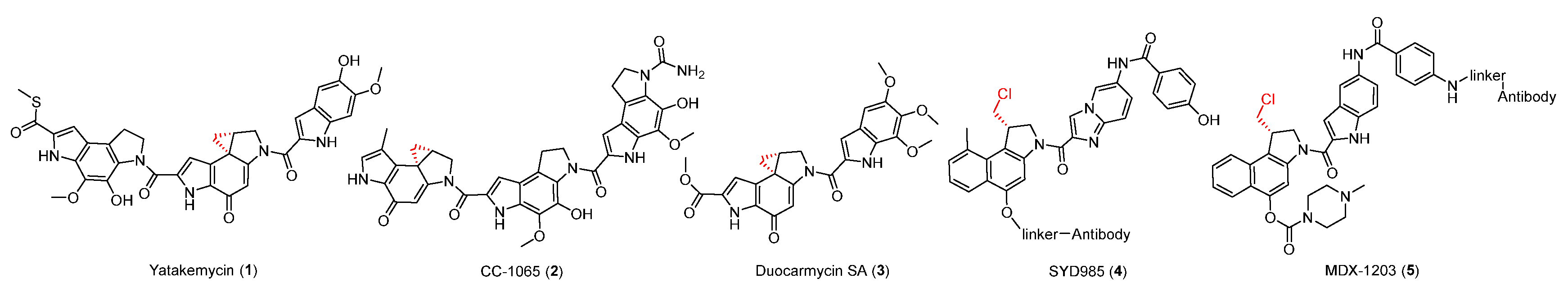
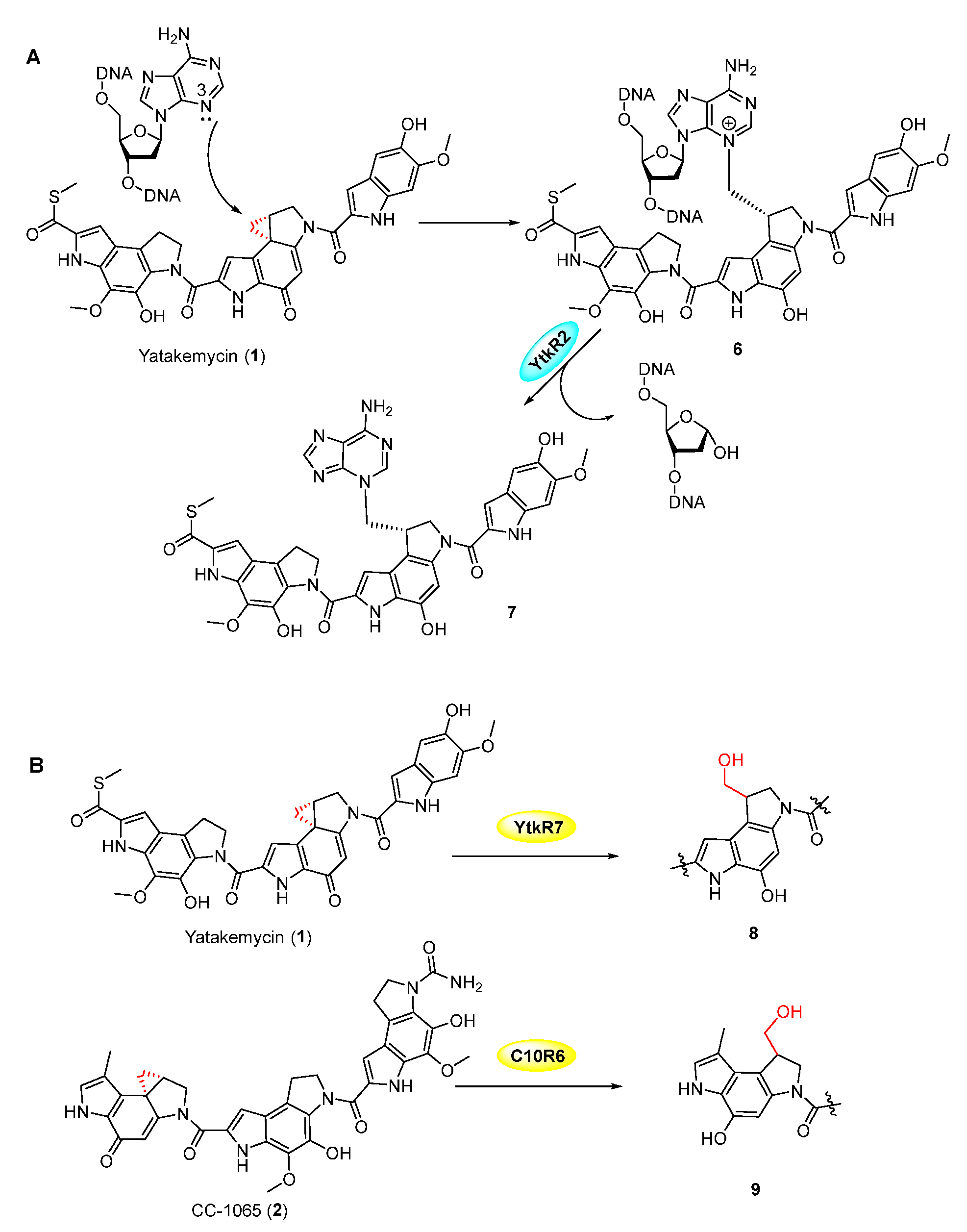
The spirocyclopropylcyclohexadienone family, including yatakemycin (YTM, 1), CC-1065 (2), and duocarmycin SA (3), all contain a highly active cyclopropane moiety and exhibit potent antitumor activities (Figure 1) [19,20,21,22]. Duocarmycin-based antibody-drug conjugates (ADC, SYD985 (4), and MDX-1203 (5)) have entered clinical trials for the treatment of specific cancers as prodrugs (Figure 1) [23,24]. They can selectively bind AT-rich regions in the DNA minor groove by non-covalent interaction, then form a covalent bond with DNA in which the cyclopropanol group is attacked by the N-3 of adenine (Figure 2A) [25]. The YTM-producer was first identified as protecting itself with DNA glycosylases YtkR2 through the base-excision repair mechanism (Figure 2A). The homologous enzyme C10R5 exhibited a similar function in the CC-1065-producing strain. [26] Because the cyclopropane warhead exhibits strong potency, additional self-protection of their hosts can also be achieved by the cleavage of this moiety. A GyrI-like protein was verified to hydrolyze the cyclopropane moiety in YTM and CC-1065 to facilitate detoxification (Figure 2B) [27,28,29].
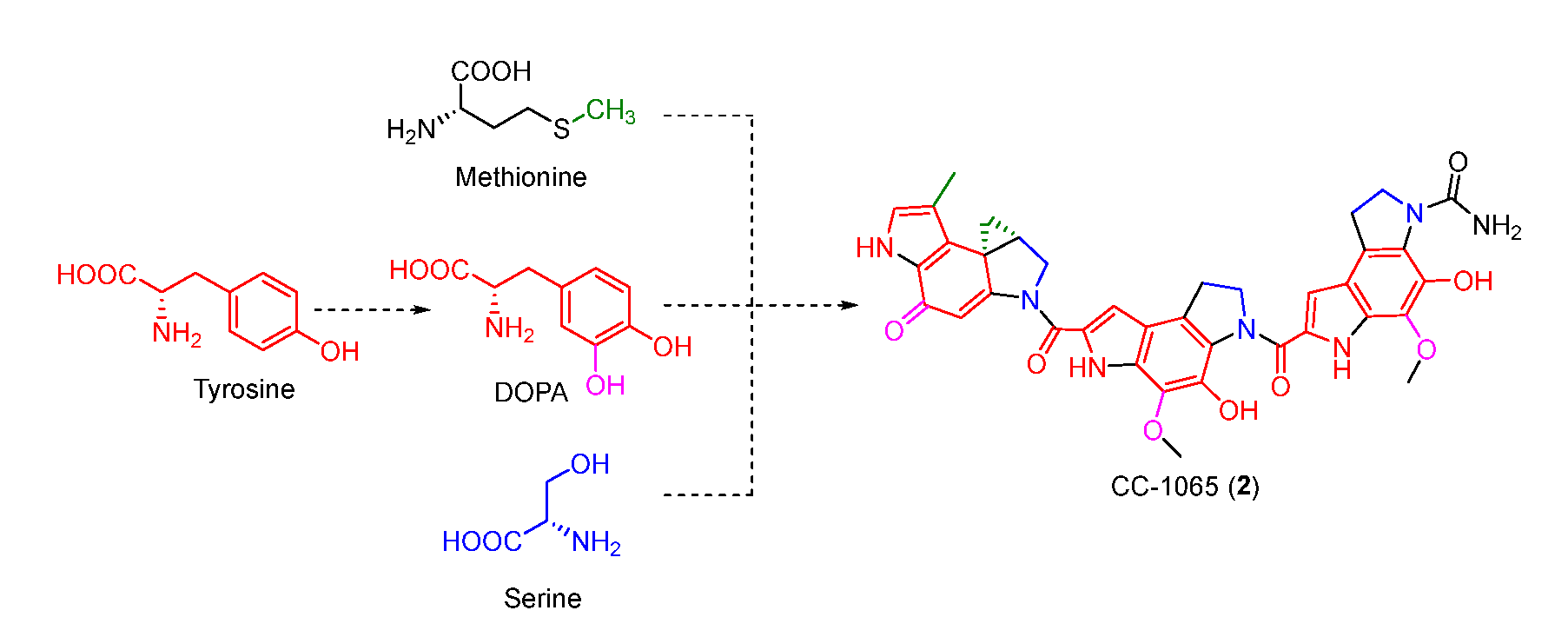
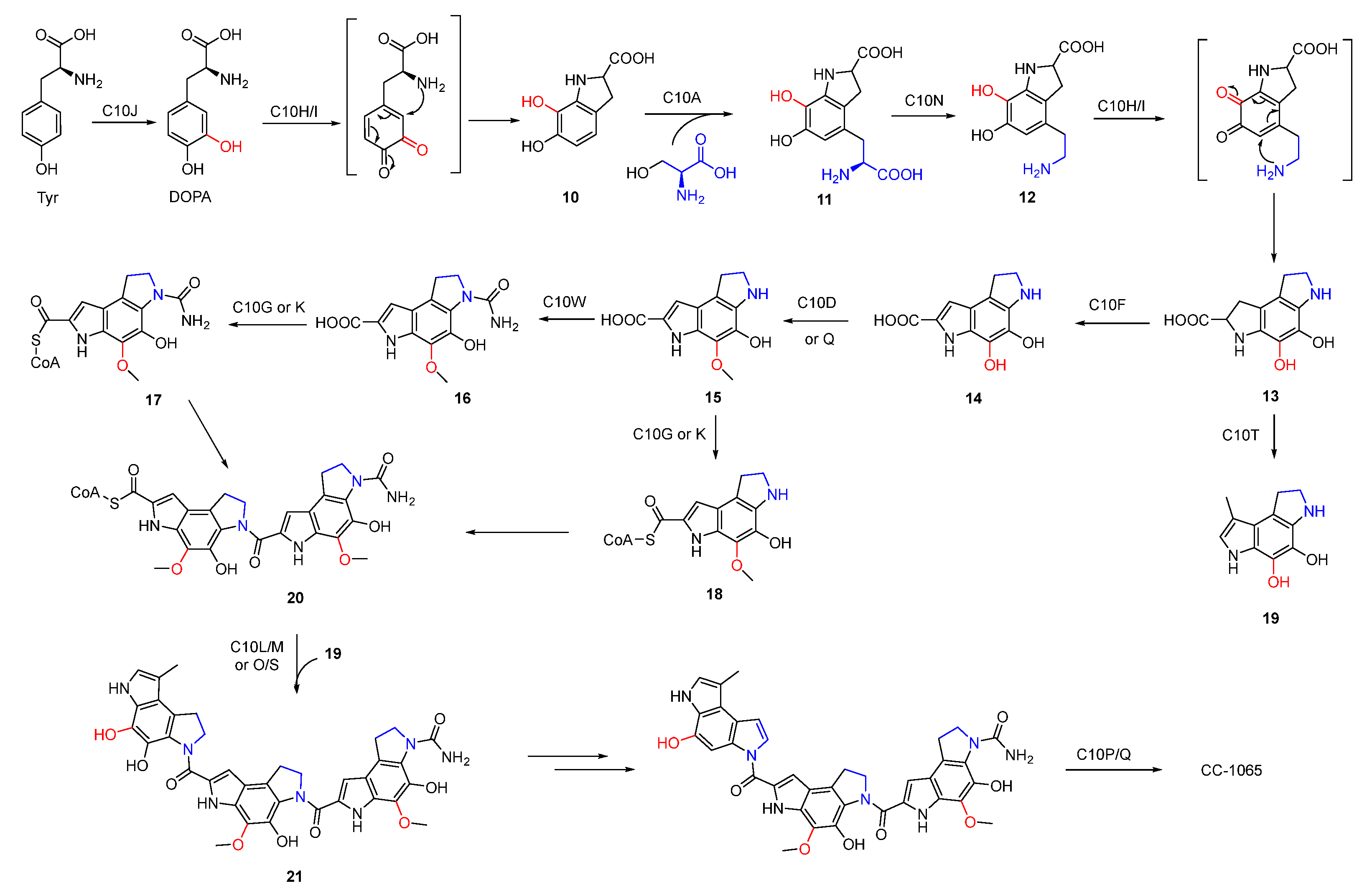
The benzodipyrrole scaffold in CC-1065 was derived from serine, methionine, and tyrosine-derived DOPA, and was revealed by isotopic feeding experiments (Figure 3) [30,31]. Wu et al. proposed the possible biosynthetic pathway of CC-1065. Tyrosine was first oxidized to DOPA which underwent intramolecular cyclization to afford 10. Next, 11 produced by the combination of serine and 10 was decarboxylated and cyclized to yield 13 which was further modified to form three different types of building blocks (17, 18, and 19). The assembly of these building blocks generated the final core structure 21 (Figure 4).
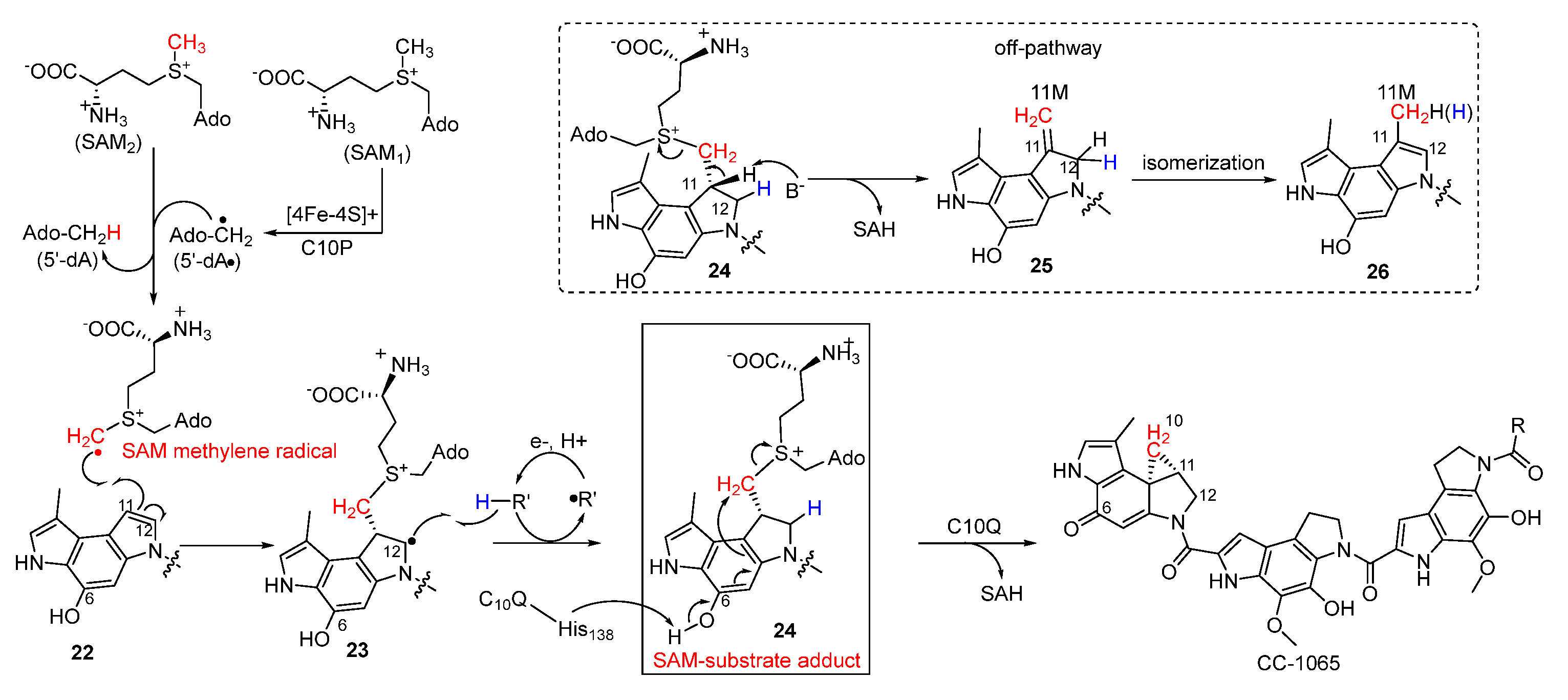
Strategies for the incorporation of cyclopropane have long fascinated chemists, since it is an important synthetic building block and a common pharmacophoric group. The chemical synthesis of cyclopropane in this family of NPs was mainly achieved by nucleophilic cyclopropanation [32]. In the biosynthesis of CC-1065, Jin et al. reported that a two-component cyclopropanase system consisting of a HemN-like radical S-adenosylmethionine (SAM) enzyme C10P and a methyltransferase C10Q was responsible for generating the essential cyclopropane moiety involving a unique enzymatic mechanism (Figure 5) [33,34]. To explain in detail, the highly active SAM methylene radical attacks the C-11 position of 22 to generate the radical intermediate 23, which subsequently abstracts hydrogen to yield the SAM-substrate adduct 24. Following this, the deprotonation of the phenolic hydroxyl group in virtue of His-138 residue in C10Q induced SN2 cyclopropanation to produce CC-1065 with S-adenosylhomocysteine (SAH) as the leaving group. Additionally, 24 could also be converted to 25 by non-enzymatic reaction with the release of SAH, and the following isomerization produces the methylated compound 26.
3. DNA-Alkylating Natural Products with Heterocyclic Propane as Pharmacophore
3.1. Pluramycins
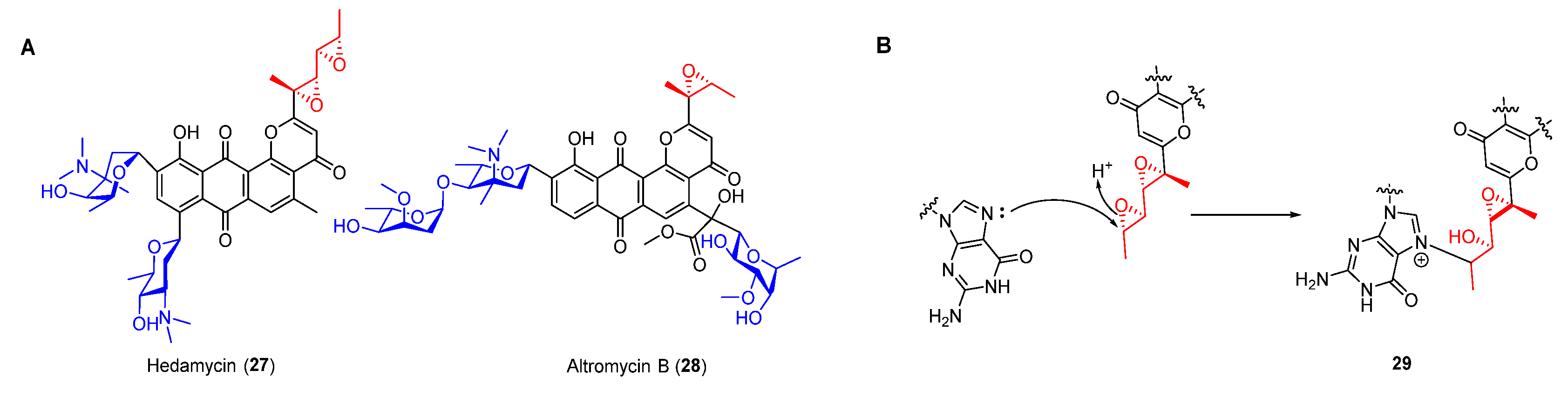
As an important family of NPs, type II polyketides display various structurally diverse biological activities [35,36]. Anthracycline compounds such as daunomycin and nogalamycin exhibit antitumor activities by intercalating into grooves of DNA, while most of these compounds are unable to form a covalent bond with duplex DNA [37,38]. Nevertheless, pluramycin antibiotics including hedamycin (27) and altromycin B (28) (Figure 6A), which usually contain an epoxide moiety, can intercalate and alkylate DNA simultaneously. Similar to daunomycin, their anthraquinone ring was characterized as intercalating into DNA and binding saccharides in the minor or major groove, thereby contributing to the stabilization of the drug–DNA complex [39,40,41]. Furthermore, their epoxides could be opened via nucleophilic attack of N-7 of guanine, resulting in the formation of an adduct by covalent bond (Figure 6B).
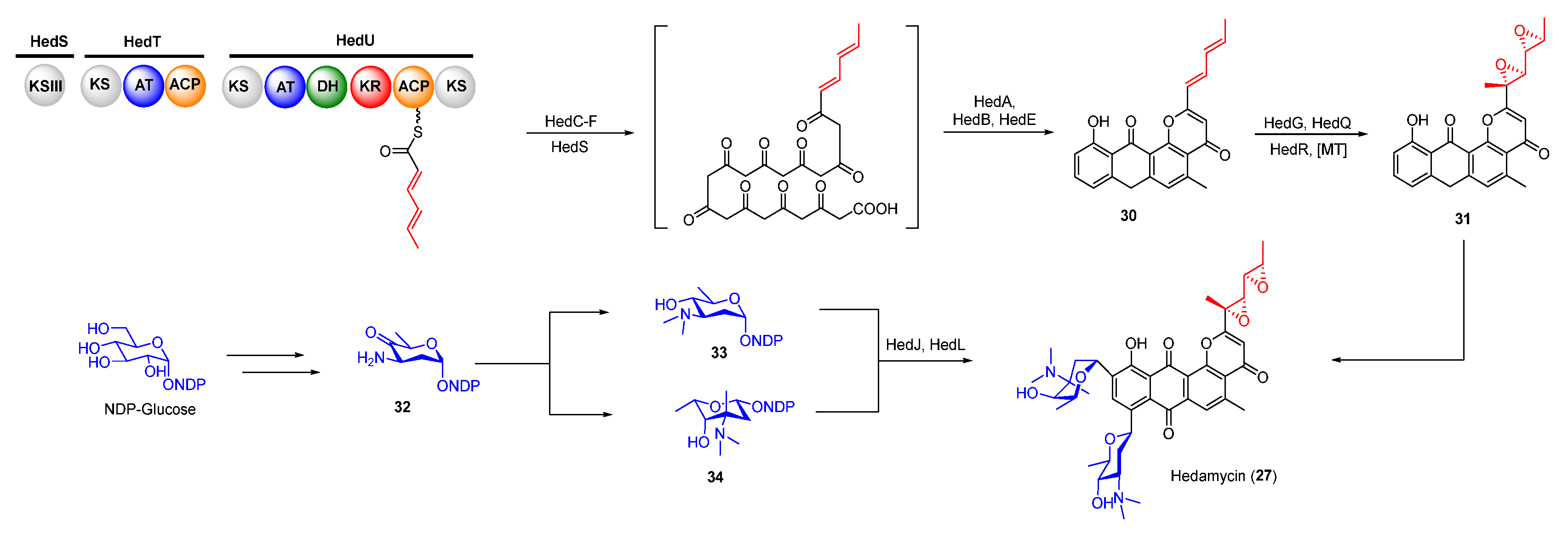
Biosynthetically, the epoxides in hedamycin were formed on its non-acetyl starter unit generated by two separate type I polyketide synthases (PKSs, HedT, and HedU). HedU was proposed to catalyze two rounds of chain elongation employing the acetyl starter unit provided by HedT (Figure 7) [42,43,44]. The obtained unsaturated 2,4-hexadienyl unit was then transferred to the downstream type II PKS to produce the aromatic precursor. The following oxidation of the C2-alkyl side in intermediate 30 afforded the epoxide intermediate 31 which was further modified by methyltransferase and two C-glycosyltransferases to yield hedamycin (Figure 7).
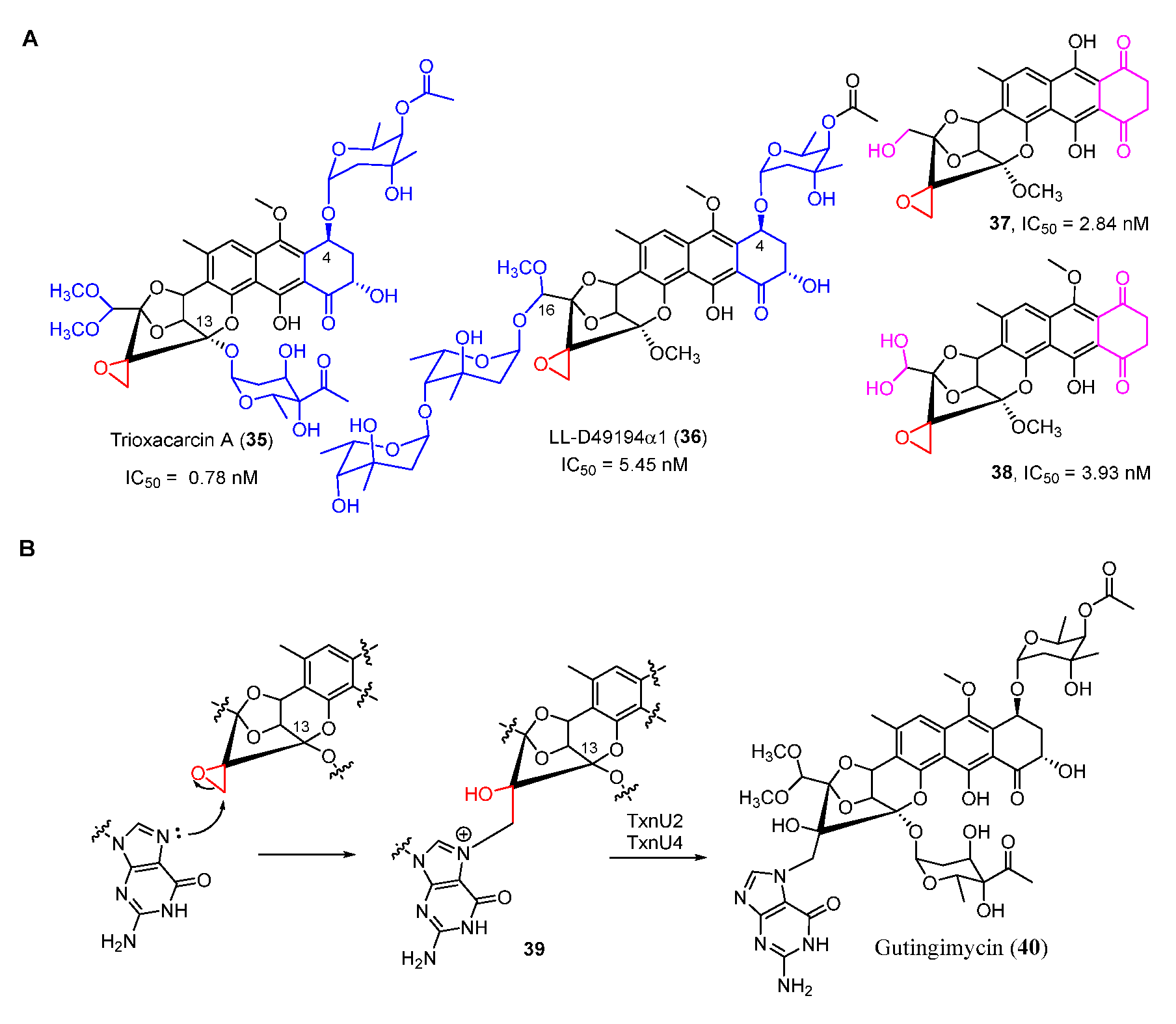
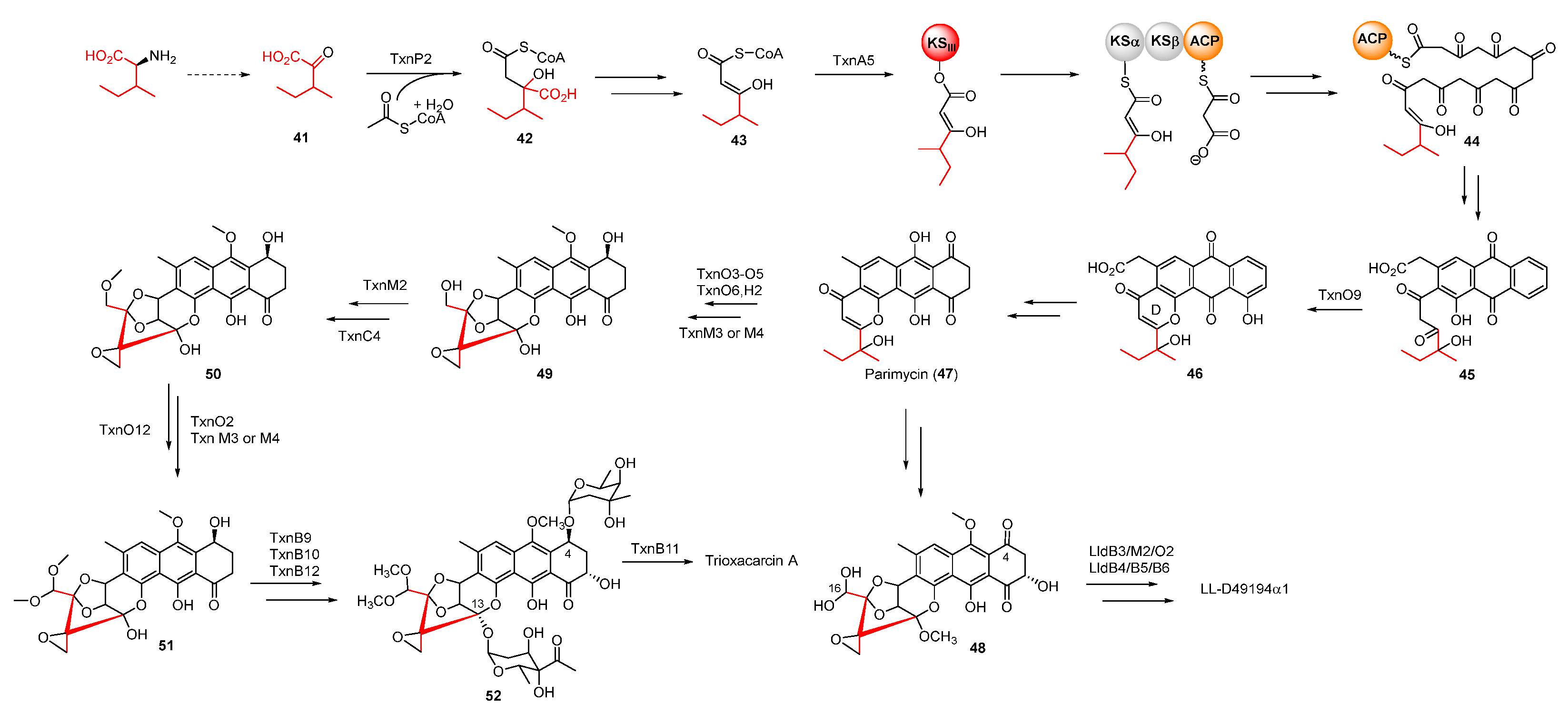
Trioxacarcins (TXNs), firstly isolated from Streptomyces bottropensis NRRL 12051 in 1981, exhibit extraordinary antibacterial, antimalarial, and antitumor activities [45]. Among the TXNs, TXN A (35) showed the most potent antitumor activities with sub-nanomolar IC50 values against various cancer cell lines (Figure 8A). The unique fused spiro-epoxide of TXN A is essential for its bioactivities because the epoxide can react with N-7 of guanine to form a DNA–TXN A complex (Figure 8B). The crystal structure of this complex revealed that glycosyl groups at C-4 and C-13 were docked with the minor and major groove, respectively [46]. It also displayed an unexpected flipping out of the base at the intercalation site, which might be important for DNA–protein interaction. Recently, the study of the TXN analog LL-D49194α1 (36) showed that the deglycosylated compounds (37 and 38) exhibited more potent anticancer activities than 36, possibly suggesting a new mode of interaction with DNA (Figure 8A) [47]. Furthermore, the DNA–TXN complex (39) could be cleaved to yield gutingimycin (40) involving a self-resistance mechanism of an excising base (Figure 8B) [48,49]. Recently, four DNA glycosylases, TxnU2, TxnU4, LldU1, and LldU5, were reported to be responsible for excising the intercalated guanine adducts [50].
Based on the work of Zhang et al., TXNs were also biosynthesized from anthraquinone-intermediate parimycin (47), similarly to the precursor of hedamycin, as revealed by gene-deletion experiments [45]. According to isotope-labelled precursor feeding experiments, the unusual starter unit 2-methylbutyryl of TXNs was derived from L-isoleucine through transamination. After a series of modifications, including condensation with acetyl-CoA and decarboxylation, this starter unit was incorporated into the polyketide chain in virtue of KSIII (Figure 9). The formation and subsequent cyclization of the polyketide chain provided intermediate 46, whose pyrone ring was formed by a CalC-like protein, TxnO9 [51]. The decarboxylated intermediate 47 underwent complex tailoring steps to afford intermediate 51 with a unique spiro-epoxide structure, but the specific enzymatic process and mechanism remained uncharacterized. Following that, the methylation of 51 at C-4 and C-13 yielded 52, whose C4-sugar was finally acetylated by the membrane-bound O-acetyltransferase TxnB11 to form 35 (Figure 9) [52]. Unlike TXNs, the C-16 and C-4 of 45 were glycosylated and methylated to produce LL-D49194α1.
3.2. Mitomycins
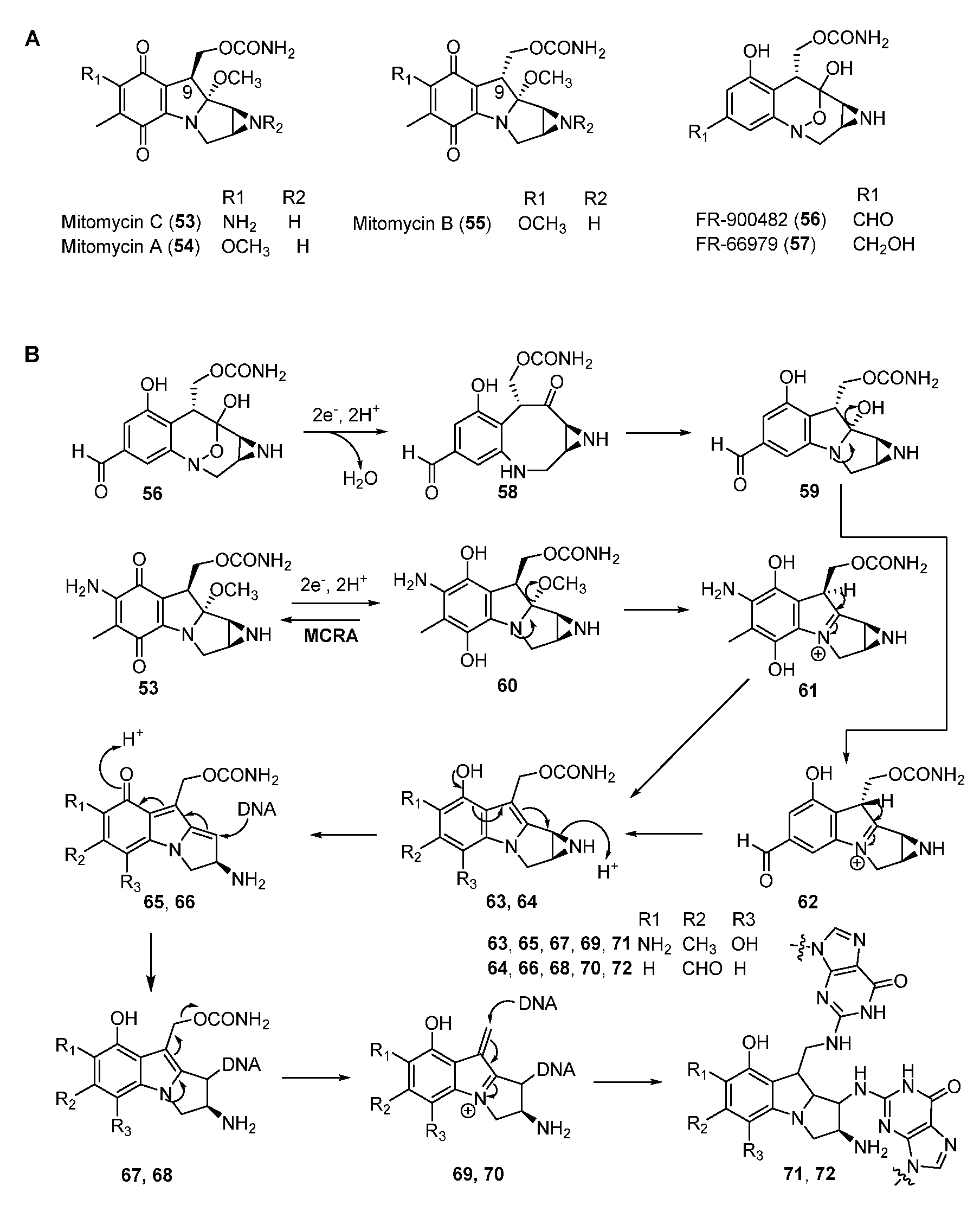
Mitomycins (MMs, such as MMA, B, and C) are antitumor NPs discovered in Streptomyces. They all contain the quinone backbone and a unique azabicycle moiety (Figure 10A) [18,53]. Among these compounds, MMC has been used as a chemotherapeutic agent in the clinic for more than five decades. MMC can form inter-strand and intra-strand cross-linking with DNA at the selective sequence (5’-CG-3’) and resides in the minor groove [54]. Other compounds of the mitomycin family, such as FR900482 and FR66979, also showed potent DNA cross-linking activity as well as bioactivities against cancer cell lines (Figure 10A). FR900482 was superior to MMC in both efficacy and safety [55].
The mode of action of MMs is well studied. Firstly, a reductive pathway is required to activate the quinone moiety of MMC by either enzymatic or chemical means to form the hydroquinone intermediate 60 [18,53]. Subsequent elimination of methanol in 60 affords 61 which undergoes tautomerization and the ring-open reaction of aziridine ring to yield 65 (Figure 10B). The N-2 of guanine attacks the C-1 position to generate the DNA–compound complex, then the departure of carbamate produces the iminium intermediate 69, which is attached by the second guanine of DNA in the same way to form 71. Furthermore, the first reductive activation could be inhibited by a FAD-dependent oxidoreductase MCRA (encoded by mcrA) which enables the oxidization of the hydroquinone form to the quinone form to confer self-resistance [56,57,58,59]. Although the alkylating mechanism of FR900482 is similar to that of MMC, it is activated by cleaving the N-O bond to form 59 (Figure 10B).
Since these compounds possess excellent bioactivities and the common pharmacophoric group azabicycle, their synthesis has attracted extensive attention. In chemical synthesis, the azabicycle moiety of MMs is installed from benzazocane intermediates via intramolecular substitution [60], but their biosynthetic pathways are still not well elucidated. According to isotopic precursors feeding experiments conducted by Hornemann et al., the origins of the O-methyl group and the carbamate were methionine and L-citrulline, respectively, while the mitosane core was derived from 3-amino-5-hydroxybenzoic acid (AHBA, 81) and glucosamine [61,62]. The precursor AHBA was formed via the amino-shikimate pathway related to rifamycin and kanosamine biosynthesis [63,64]. After the formation of AHBA, it was firstly activated by acyl AMP-ligase MitE and was then loaded onto acyl carrier protein (ACP) MmcB (Figure 11). The glycosyltransferase MitB was verified to catalyze the glycosylation of AHBA-MmcB with UDP-GlcNAc [65,66,67]. Recently, Wang et al. traced all the ACP-channelled MM intermediates indicating that AHBA-MmcB-GlcNAc intermediate 85 should undergo the deacetylation by MitC to form 86 which was further transformed to 88 by MitF and MitD [68]. The epoxide intermediate might be cyclized to provide benzazocine 92. 92 then underwent oxidation and several uncovered modifications to generate hydroxyquinone intermediate 95 which was methylated to afford MMA, the direct precursor of MMC [69]. Sherman and co-workers also identified a methyltransferase MitM which methylated the nitrogen of aziridine in MMA rather than MMC to yield MMF (96) [70]. In addition, the epoxide of 88 could be opened by the nucleophilic attack to afford 89 which was the precursor of the MMs with α-C9. Moreover, the oxidation of the aniline amine in 89 facilitated forming the core structure of FR900482.
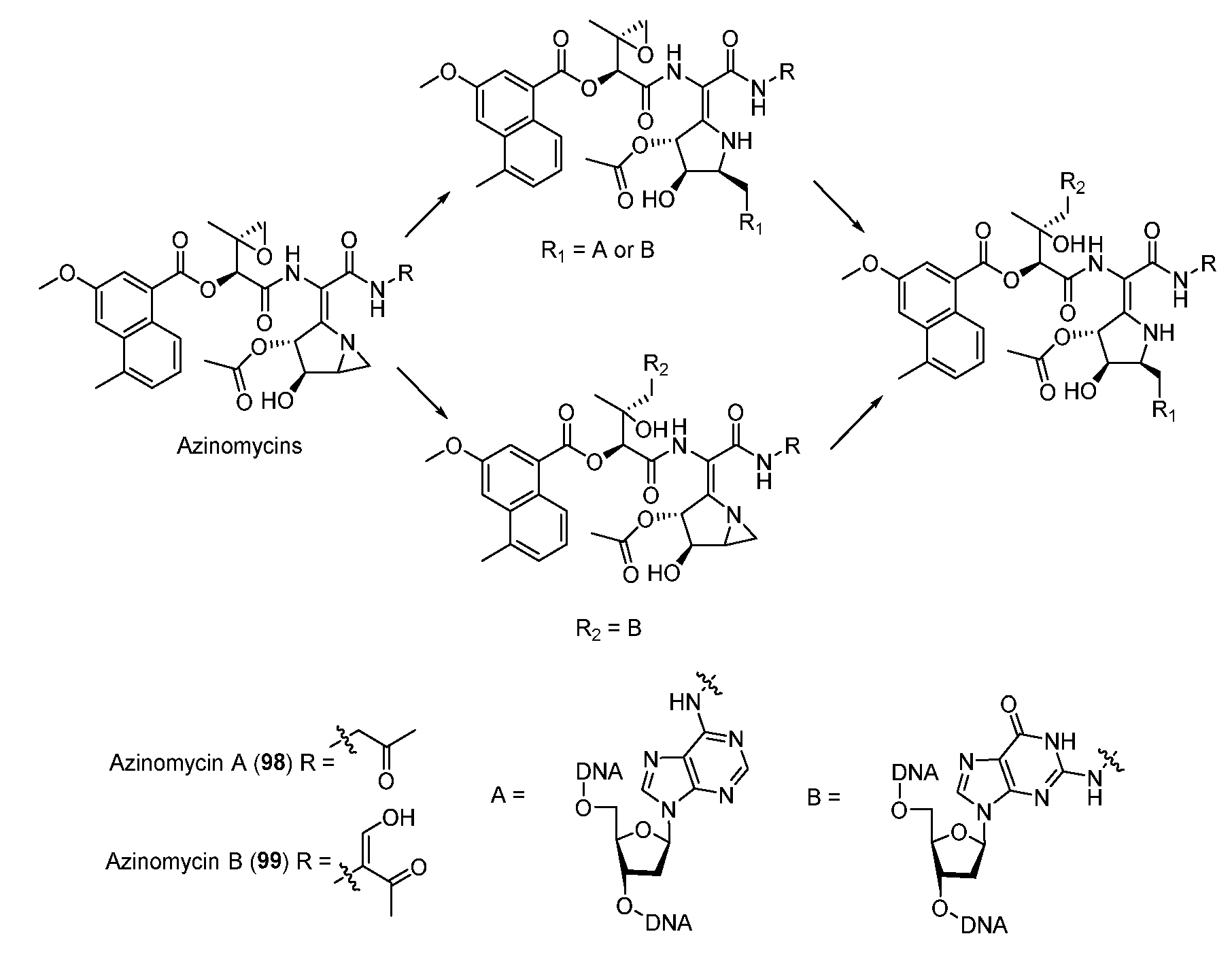
3.3. Azinomycins
The antitumor antibiotics azinomycin A (98) and B (99) contain naphthoic acid (NPA) moiety, epoxide, and azabicyclohexane ring which all contribute to alkylating DNA (Figure 12) [71]. The electrophilic epoxide and aziridine can both be attacked by N-7 of guanine and the latter can even be opened by N-7 of adenine, leading to the formation of interstrand DNA cross-links (Figure 12) [72,73]. NPA moiety also plays an important role in the DNA alkylating activity by virtue of non-covalent interactions [74]. In 2011, the aminoglycoside transferase AziR was identified to mediate the self-resistance of azinomycin and reduce the DNA damage via binding azinomycin. Recently, a novel DNA glycosylase Orf1 and an endonuclease AziN were reported to repair the DNA damage to achieve self-protection [75,76,77,78].
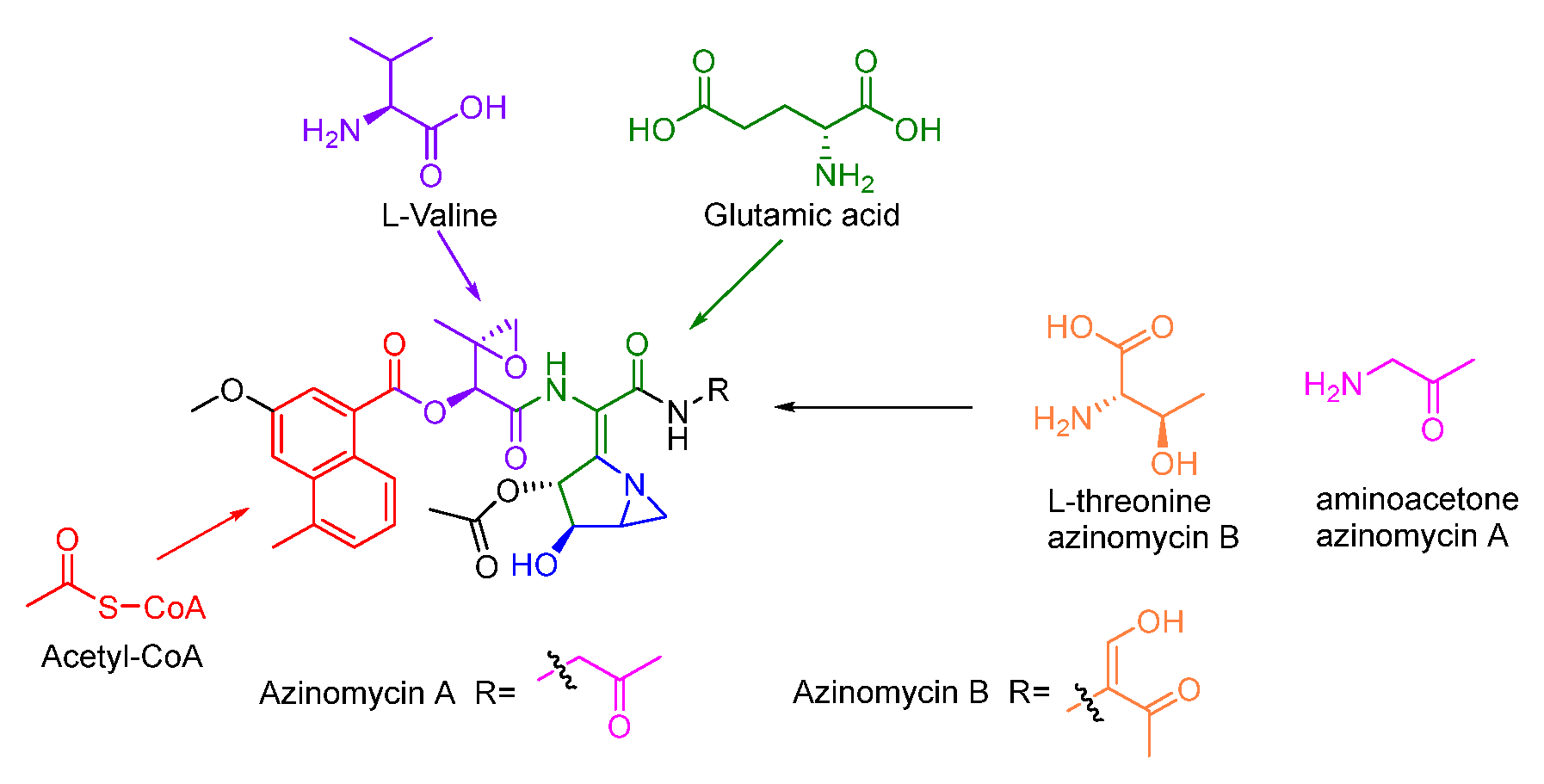
Previous isotope-labelled precursor feeding experiments revealed that the epoxy moiety, the azabicyclic fragment, and the terminal part in azinomycin B were derived from acetyl-CoA, valine, glutamic, and threonine, respectively (Figure 13) [79,80]. The feeding experiments with isotopically labelled substrates showed that 3-methyl-2-oxobutenoate (105) was incorporated into the azinomycin epoxide as the penultimate precursor (Figure 14A). The formation of 105 was achieved by oxidation, transamination as well as dehydration beginning with L-valine. Even so, the exact timing of forming epoxy amide remains unclear up to now [81].
In the biosynthesis of this class of non-ribosomal peptide-polyketide hybrid compounds, iterative type I PKS AziB catalysed the formation of 5-methyl-NPA (100) which was further transformed to 3-methoxy-5-methyl-NPA (102) by a P450 hydroxylase AziB1 and the O-methyltransferase AziB2 (Figure 14B) [82]. The first building block 102 was activated by the distinct adenylation (A) domain of the di-domain non-ribosomal peptide synthetase (NRPS) AziA1 to initiate the backbone formation of azinomycins [83,84].
The azabicycle moiety was constructed from 3,4-epoxypiperidine derivatives via spontaneously intramolecular substitution in chemical synthesis [85]. Watanabe and co-workers unraveled the biosynthetic pathway of the azabicyclic fragment in azinomycin, wherein the glutamic acid was initially acetylated at the amino group by N-acetyltransferase AziC2 to form N-acetyl glutamate 106. The N-acetyl glutamate kinase AziC3 subsequently phosphorylated the carboxyl to afford the N-acetyl-glutamyl 5-phosphate (107) which was subsequently reduced to N-acetyl-glutamate-5-semialdehyde (108) by an N-acetyl-γ-glutamate phosphate reductase AziC4 (Figure 14C), and the key two-carbon extension on aldehyde intermediate catalyzed by the transketolase AziC5/C6 afforded 110 which was further converted to the acetylated nonproteinogenic amino acid diamino-dihydroxy-heptanoic acid (DADH, 111) by an aminotransferase (AziC1 or AziC7) [86,87,88]. Recently, Kurosawa et al. demonstrated that 111 could be further sulphated to 112 and the sulfate group in 112 was finally attacked by the ortho amino group to form the aziridine ring intermediate 113 which may be subsequently acetylated and cyclized to form the azabicyclic fragment 114 (Figure 14C) [89]. Additionally, glutamic acid might be firstly activated by the amino-group carrier protein (AmCP) and was further modified to produce DADH which was then introduced into the azabicyclic structure according to a recent study about the biosynthesis of vazabitide A [90]. Moreover, the enol in the final building block of azinomycin B was generated by the oxidation of L-threonine, while the decarboxylation of the intermediate 116 afforded the aminoacetone 117 in azinomycin A [91].
4. DNA-Alkylating Natural Products with Imine as Warheads
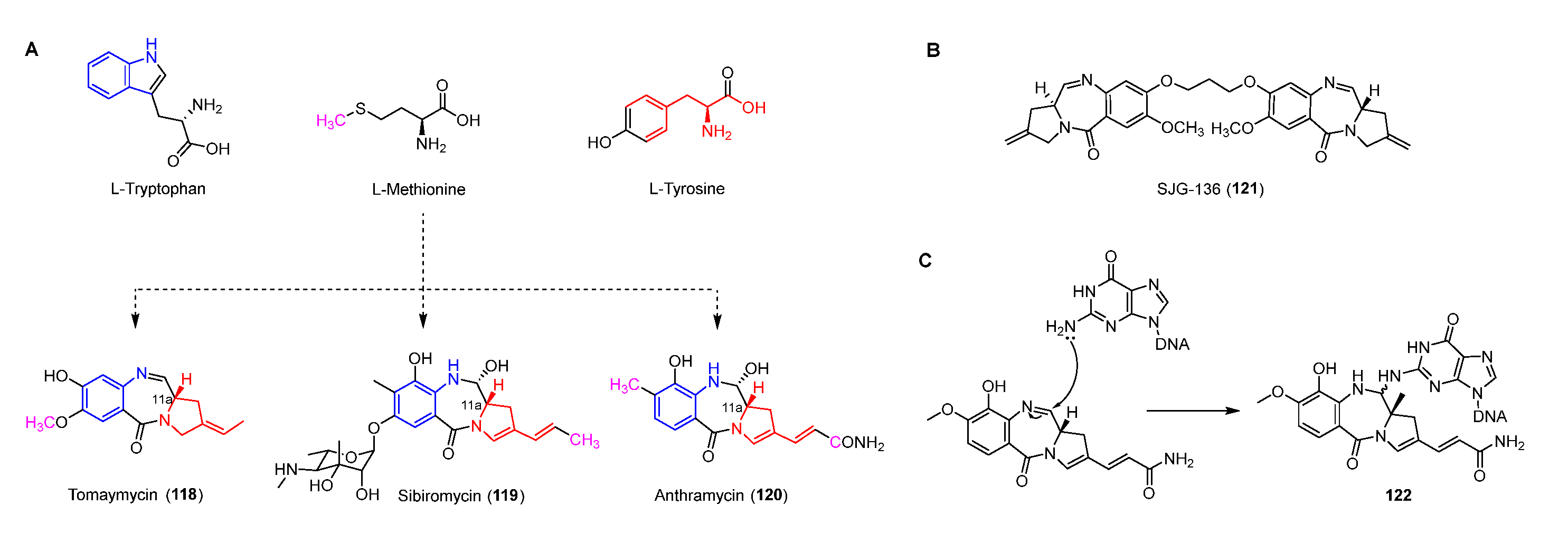
4.1. Pyrrolobenzodiazepines
Antitumor antibiotics pyrrolobenzodiazepines (PDBs), including anthramycin, sibiromycin, and tomaymycin, all contain three parts: anthranilate, diazepine, and hydropyrrole (Figure 15A) [92,93]. The imine in the diazepine can be attacked by N-2 of guanine to form a stable covalent bond resulting in inhibiting DNA synthesis (Figure 15C) [94]. Moreover, the crystal structure of the anthramycin-DNA complex indicates that the S-configuration of C-11a make it suitable for docking in the minor groove of DNA [95,96]. In addition, a PDB dimer, SJG-136 (121), which has completed the phase II clinical trial for treating leukemia and ovarian cancer, can form DNA inter-strand and intra-strand cross-linking of DNA (Figure 15B) [97,98].
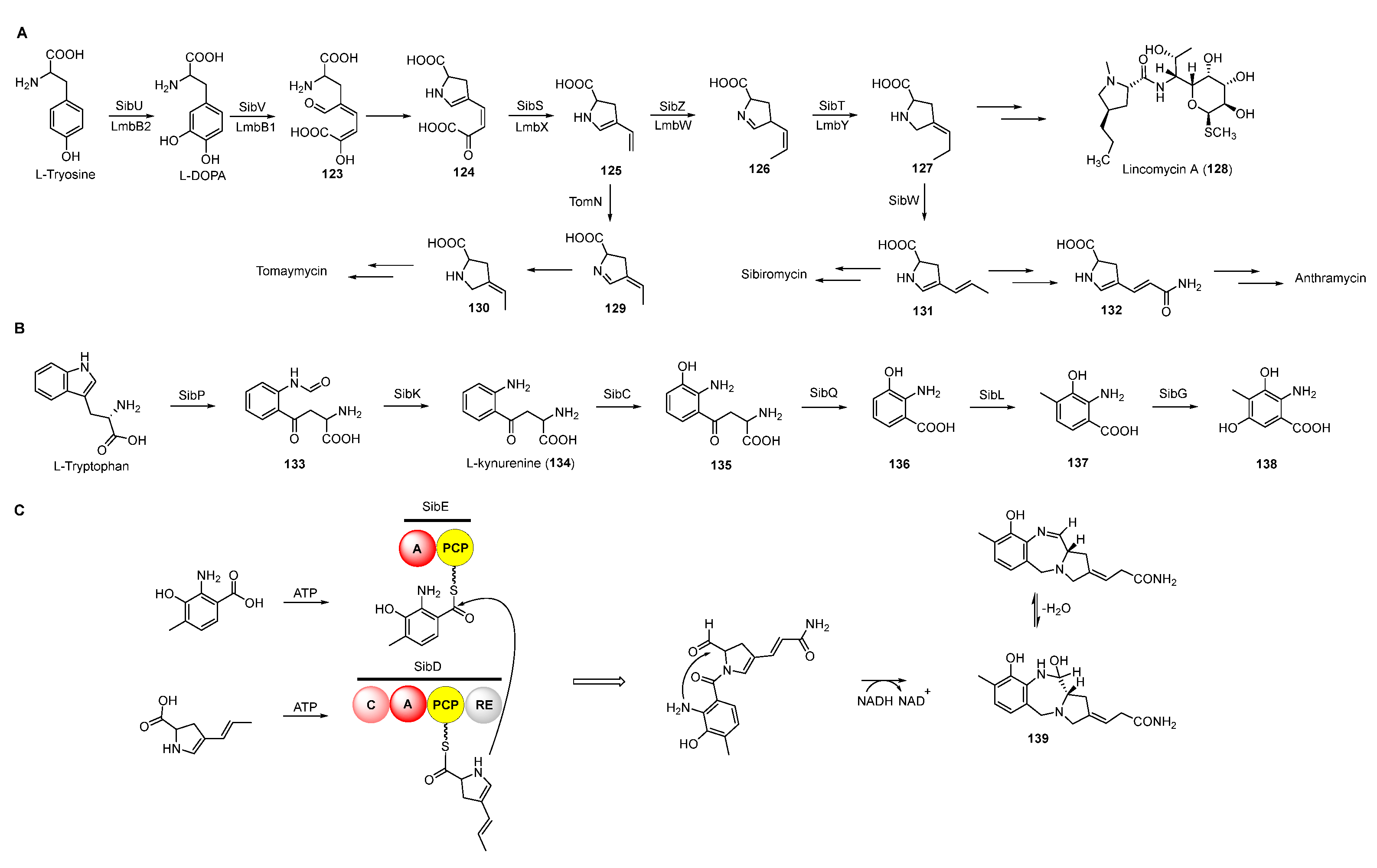
Based on previous feeding experiments, L-methionine, L-tyrosine, and L-tryptophan were supposed to be the biosynthetic precursors of pyrrolobenzodiazepines (Figure 15A) [99]. Like lincomycin biosynthesis, L-dopa from L-tyrosine is cleaved to yield semialdehyde intermediate 123, followed by intramolecular cyclization to form dihydropyrrole intermediate 124 (Figure 16A) [100]. The decarboxylation of intermediate 124 generates 125, which is further converted into a variety of dihydropyrrole precursors (130-132) to be introduced into PDB biosynthesis [101,102,103]. For the biosynthesis of hydroxyanthranilic acid intermediates, L-tryptophan was firstly degraded to L-kynurenine (134), the biosynthetic precursor of important NPs including actinomycin, quinolobactin, and daptomycin [104,105,106]. Following this, 134 goes through three continuous tailoring steps mediated by monooxygenase, kynurenine hydrolase, and methyltransferase, respectively, to generate 3-hydroxyl-4-methyl-anthranilic acid (137), which is subsequently introduced into the anthramycin or oxidized to be the precursor (138) of sibiromycin (Figure 16B) [107]. Two NRPSs containing two (A-PCP) and four domains (C-A-PCP-RE), respectively, are responsible for the formation of the amide bond between two building blocks, and the release and intermolecular cyclization of the chain to afford 139, which is dehydrated to form the final compounds with imine moieties (Figure 16C) [108].
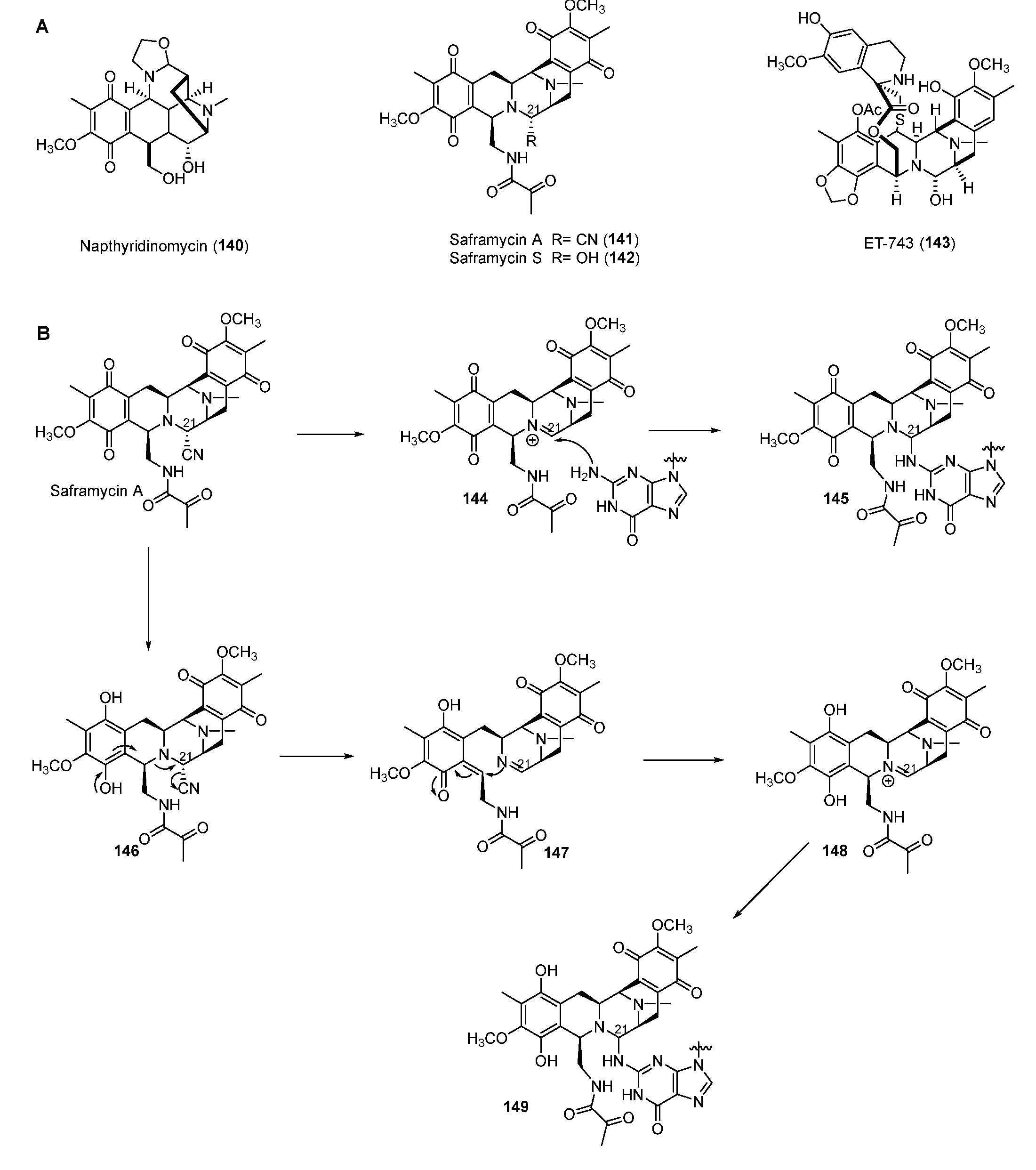
4.2. Tetrahydroisoquinolines
Tetrahydroisoquinoline NPs are mainly classified into three subfamilies composed of the saframycin (SFM) family including SFM A (141) and ET-743 (143) and the naphthyridinomycin (NDM) family, including NDM A (140), as well as the quinocarcin family compounds (Figure 17A) [109]. Most SFMs exhibit inhibitory activity against cancer cell lines by alkylating DNA. ET-743, produced by the bacterial symbiont Candidatus Endoecteinascidia frumentensis, displayed the most potent antitumor activities and has been used clinically to treat ovarian neoplasms and sarcomas [110]. Two mechanisms of SFM A for alkylating DNA were reported. One way was the formation of iminium intermediate 148 through reduction of the quinone moiety (Figure 17B) [111]. In the other way, the ortho-position nitrogen could directly promote the departure of the functional group in C-21 to yield the iminium intermediate 144. Both 144 and 148 could be attacked by nucleophilic N-2 residue of guanine in GC-rich regions of DNA to form the DNA–drug complex (Figure 17B) [112]. Furthermore, a FAD-binding oxidoreductase NapU encoded in BGC of NDM was reported to activate and inactivate the matured prodrug by extracellular oxidation conferring self-protection [113]. Recently, a short-chain dehydrogenase NapW mediated the reduction of the hemiaminal pharmacophore, implicating another level of the self-resistance mechanism of the tetrahydroisoquinoline family [114].
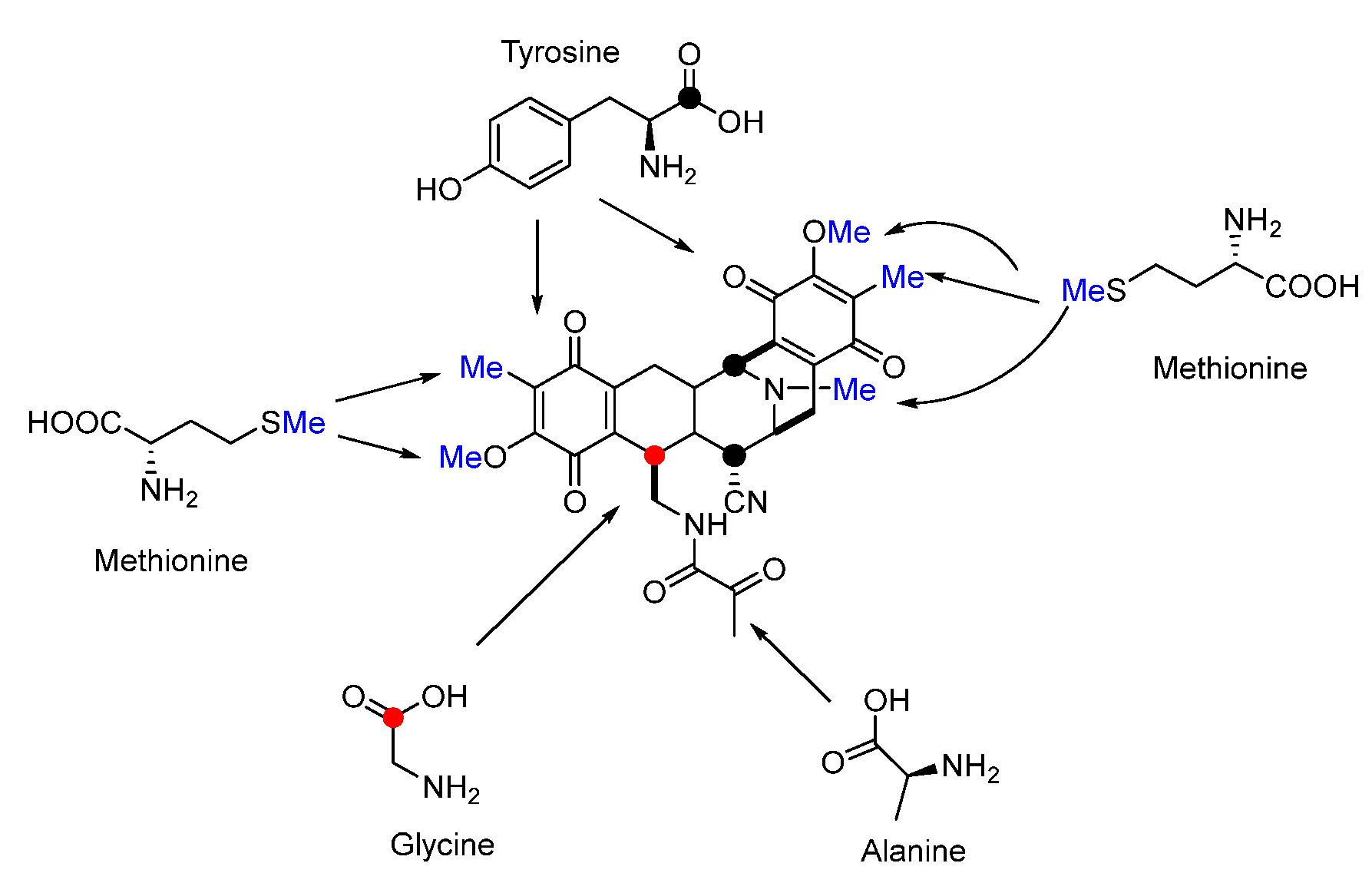
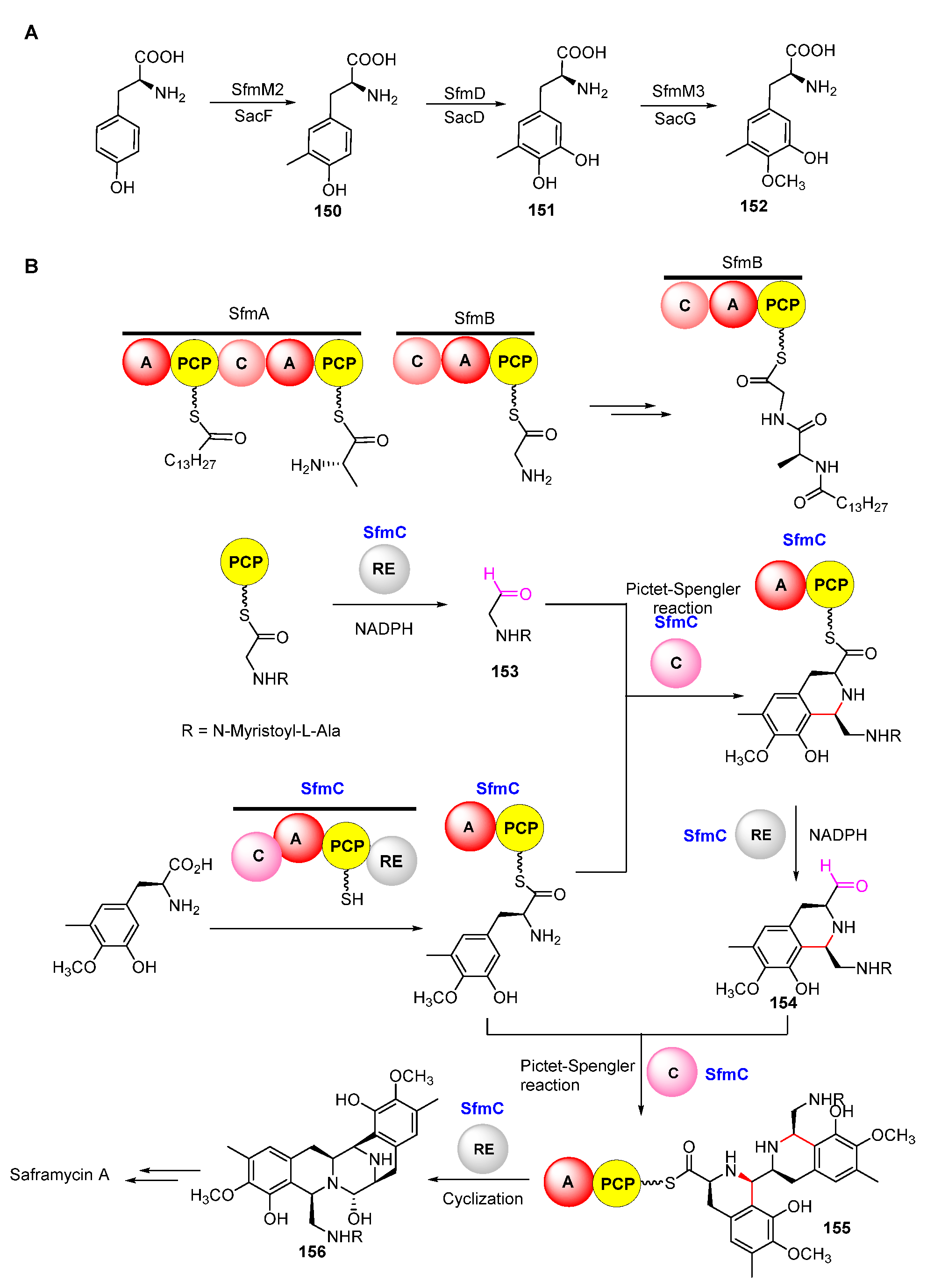
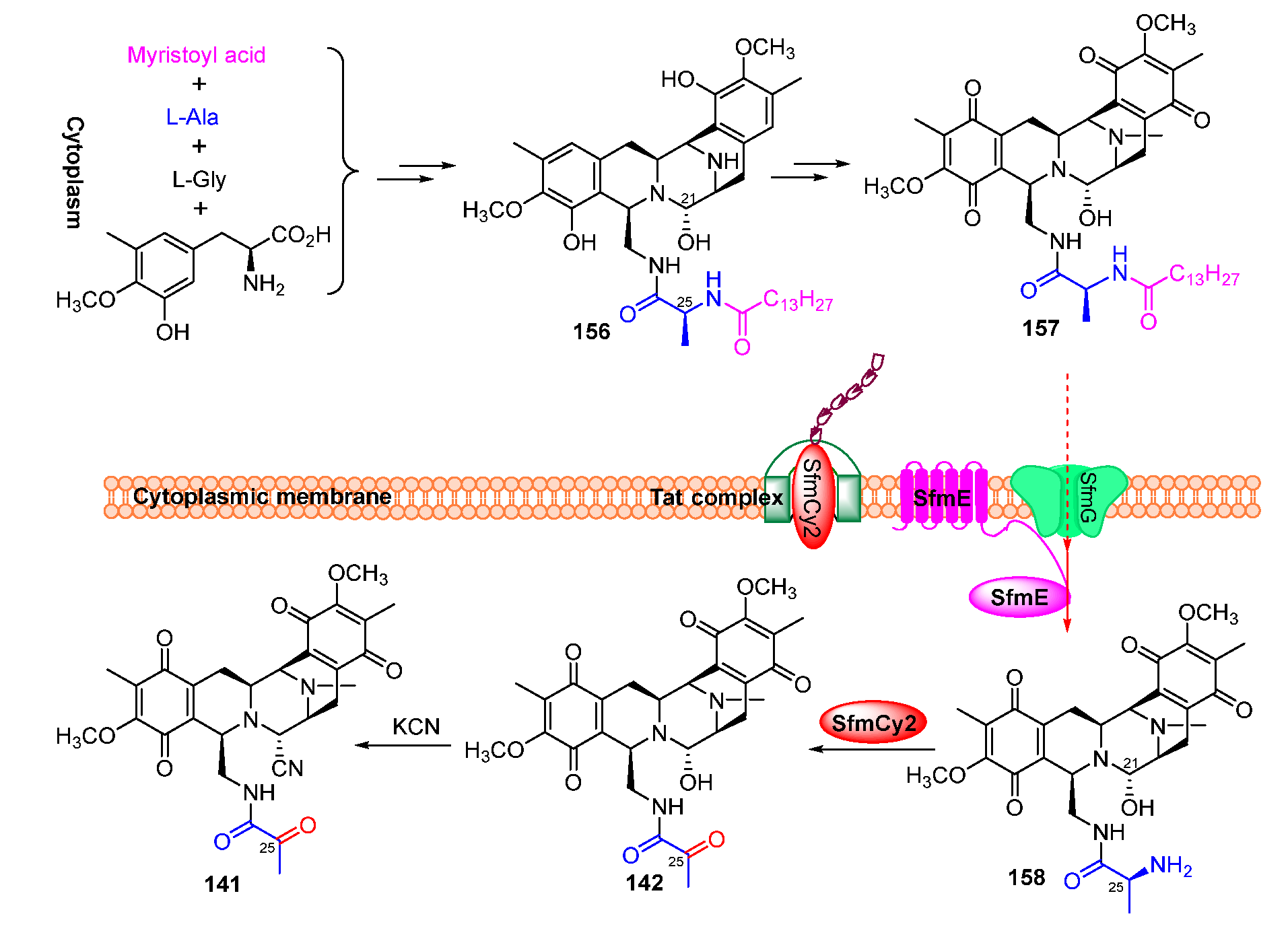
Isotope-labeled precursor feeding experiments have revealed that the skeleton structure of SFMs is derived from tyrosine, alanine, glycine, and methionine (Figure 18) [115,116]. L-tyrosine undergoes C-methylation, oxidation, and O-methylation to afford the precursor 3-hydroxy-5-methyl-O-methyltyrosine (152) (Figure 19A) [117,118]. In 2010, Oikawa and co-workers reconstituted the formation of the core structure in vitro, revealing that two Pictet–Spengler (PS) reactions were involved in this process. Specifically, 152 was firstly activated and uploaded onto PCP of NRPS SfmC, assembling with intermediate 153 produced by NRPSs SfmA and SfmB via the first PS reaction. Following this, 154 formed by the subsequent reduction underwent a second PS reaction and reduction to generate 155 (Figure 19B) [119,120]. The following reduction and intramolecular cyclization of 155 yielded 156 which was then oxidized and methylated to generate 157. Subsequently, 157 was transported outside the cell in company with its fatty acid chain and was removed by the membrane-anchored protein SfmE to produce 158. Finally, SfmCy2 catalyzed the extracellular deamination of 158 to form 142, indicating a prodrug maturation process (Figure 20) [121].
5. Others
5.1. Leinamycin
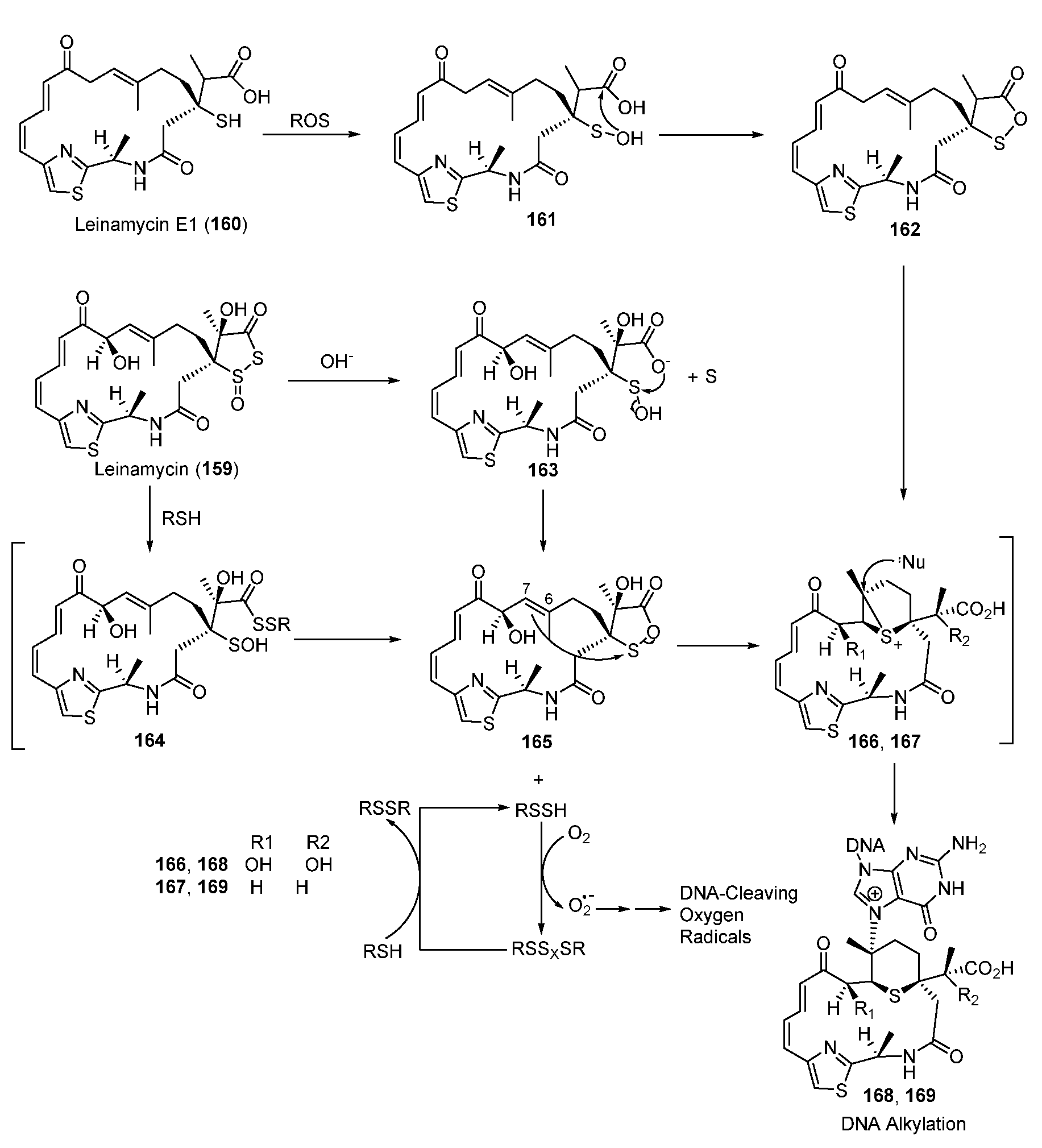
Antitumor agent leinamycin (LNM, 159) is a hybrid peptide-polyketide NP and contains a unique 1,3-dioxo-1,2-dithiolane moiety, which is essential for its anticancer activity (Figure 21). The alkylation of DNA involves a rearrangement reaction in which LNM is initially activated via the attack of thiol to form the sulfenic acid intermediate 165, which also enabled, triggering oxidative DNA cleavage via generating unstable hydrodisulfide intermediate (RSSH) (Figure 21) [122,123,124,125]. The oxathiolane intermediate 165 could be produced by breaking the S-S bond of LNM via thiol attack or hydrolysis. The C6-C7 alkene of 165 then attacks the electrophilic sulfur of the oxathiolanone group to generate an episulfonium ion intermediate, 166, following an intermolecular nucleophilic attack with the N7 of guanine residues in duplex DNA to yield the DNA–drug adduct 168. Unlike the reduction-mediated alkylation of DNA by LNM, LNM E1(160) could be transformed to its episulfonium ion intermediate 167 through an oxidative reaction catalyzed by reactive oxygen species (Figure 21) [126].
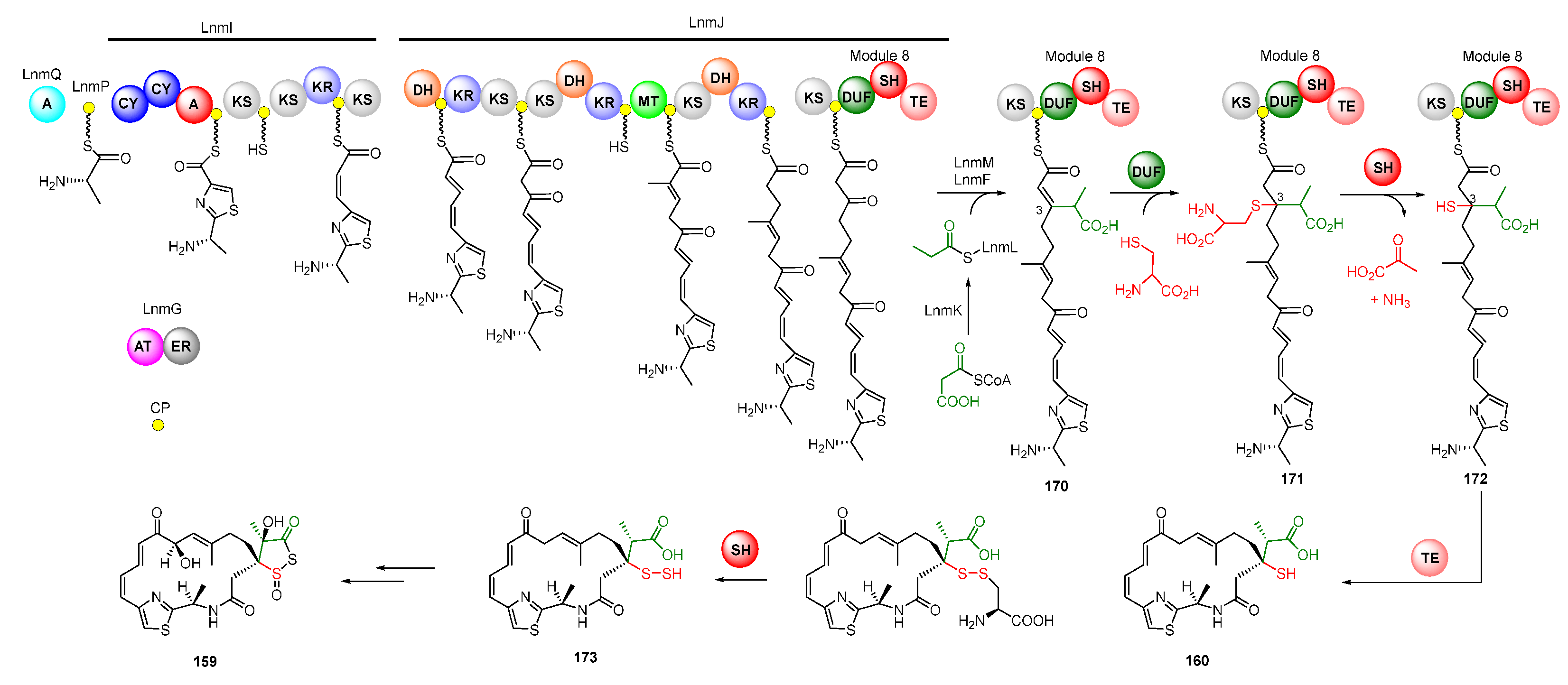
The skeleton of LNM was biosynthesized by hybrid NRPS-PKS assembly lines [127]. The LnmQ, LnmP, and NRPS module of LnmI are responsible for the unique thiazole-containing starter unit. The polyketide chain of LNM is elongated by two AT-less type I PKSs, LnmI and LnmJ, as well as a trans-AT enzyme LnmG [128,129,130,131,132]. A β-branched C3 unit derived from methylmalonyl-CoA was then installed by a set of proteins including a free-standing ACP (LnmL), a bifunctional AT-decarboxylase (LnmK), an HMGS homolog (LnmM), and an ECH homolog (LnmF) (Figure 22) [133,134]. Module 8 in LnmJ, containing a domain of unknown function domain (DUF), added an L-cysteine into C-3 of intermediate 170 to produce intermediate 171. The PLP-dependent cysteine lyase domain (SH) can catalyze the cleavage of the C-S bond to yield 172, which is cyclized to release the chain to generate LNM E1 (Figure 22) [135]. Recently, Meng et al. demonstrated that LnmJ-SH domain directly installed a -SSH group into the LNM polyketide scaffold via cleavage of the C-S bond linking the thiocysteine to form LNM E (173), which might be further transferred to LNM by a set of uncharacterized enzymes [136,137].
5.2. Gilvocarcins
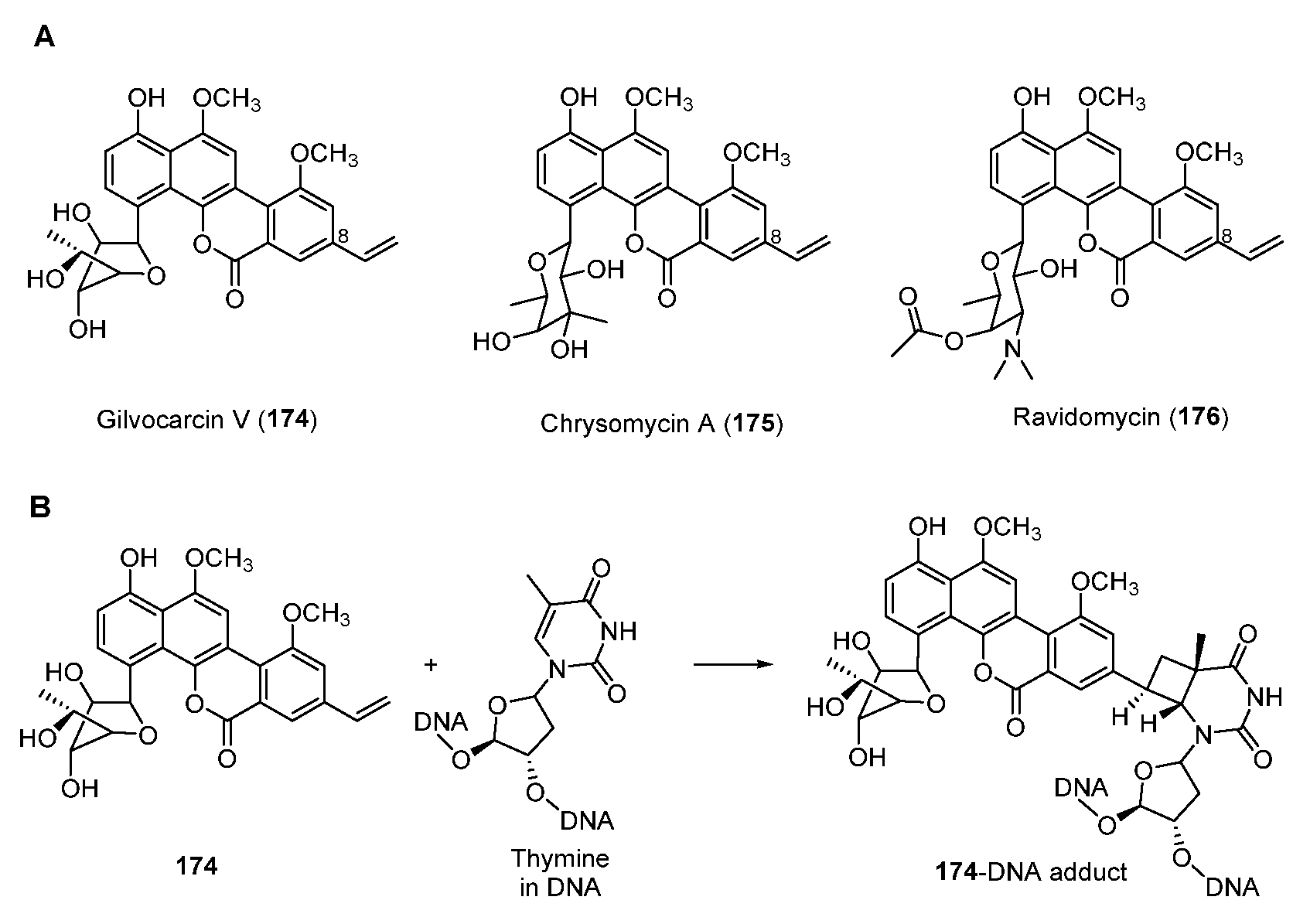
Antitumor antibiotic gilvocarcins are a subfamily of C-glycoside aromatic polyketides and are derived from the typical angucycline scaffold [138]. Part of gilvocarcin-type natural products including gilvocarcin V (174), chrysomycin A (175), and ravidomycin (176) (Figure 23A) possess the vinyl substituent at C-8 which mediates a photo-activated [2 + 2] photocycloaddition with the thymidyl residue on DNA (Figure 23B) [139,140].
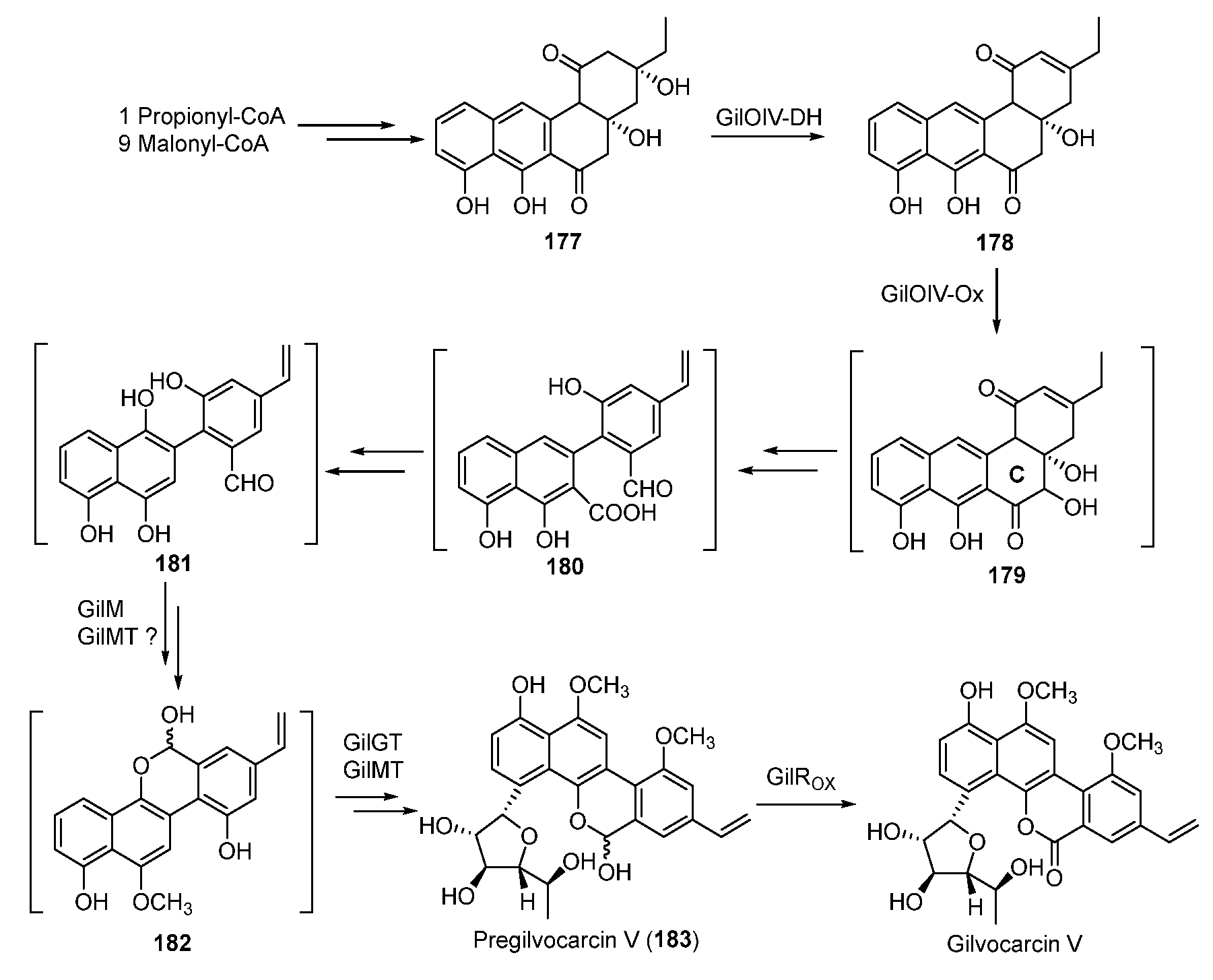
During the biosynthesis of 174, the type II PKS and cyclases afford the angucycline precursor 177 starting from the starter unit propionyl-CoA [141,142]. Subsequent dehydration of 177 generates 178, whose C-ring is oxidatively rearranged to yield 182 through possible intermediates 180 and 181. Following this, 182 is glycosylated to form pregilvocarcin V, which is finally oxidized to gilvocarcin V (Figure 24) [143,144,145,146].
6. Perspective
Since DNA-alkylating NPs exhibit potent antitumor activity, their biosynthesis has received extensive attention. Obviously, the highly active functional groups including epoxide, cyclopropane, aziridine, and imine in their chemical structures play an important role in alkylating DNA to form the DNA–drug adduct. Elucidation of their biosynthetic pathways not only facilitates the discovery of new NPs with these biological active groups by genome mining but also is valuable for engineering NPs and drug design [147]. The unprecedented enzymology involved in their biosynthetic pathways can exert a positive influence on the development of biocatalysts as well. As a result of their strong DNA-alkylating activities, their producers have to confer self-resistance strategies to avoid damaging themselves, mainly through the cleavage of the DNA–drug complex or modification of the functional groups. Therefore, resistant gene-guided genome mining also contributes to discovering new DNA-alkylating antibiotics [148]. In addition, other moieties which contribute to affinity and reactivity with DNA are also indispensable for their ability to alkylate DNA. The modification of these moieties may enhance their activity or sensitivity and facilitate linking the NPs to antibodies via chemical synthesis.
Author Contributions
Conceptualization, Q.-Y.N., Y.H. and G.-L.T.; writing—original draft preparation, Q.-Y.N. and Y.H.; writing—review and editing, Q.-Y.N., Y.H., X.-F.H. and G.-L.T.; supervision, G.-L.T.; All authors have read and agreed to the published version of the manuscript.
Funding
National Natural Science Foundation of China (31930002 and 21621002) and the Youth Innovation Promotion Association Chinese Academy of Sciences (2019257).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed]
- Rajski, S.R.; Williams, R.M. DNA Cross-Linking Agents as Antitumor Drugs. Chem. Rev. 1998, 98, 2723–2796. [Google Scholar] [CrossRef] [PubMed]
- Klaunig, J.E.; Kamendulis, L.M. Carcinogenicity. In Comprehensive Toxicology; Elsevier: Amsterdam, The Netherlands, 2010; pp. 117–138. ISBN 978-0-08-046884-6. [Google Scholar]
- Nougayrède, J.-P.; Homburg, S.; Taieb, F.; Boury, M.; Brzuszkiewicz, E.; Gottschalk, G.; Buchrieser, C.; Hacker, J.; Dobrindt, U.; Oswald, E. Escherichia Coli Induces DNA Double-Strand Breaks in Eukaryotic Cells. Science 2006, 313, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Kim, C.S.; Healy, A.R.; Wernke, K.M.; Wang, Z.; Frischling, M.C.; Shine, E.E.; Wang, W.; Herzon, S.B.; Crawford, J.M. Structure Elucidation of Colibactin and Its DNA Cross-Links. Science 2019, 365, eaax2685. [Google Scholar] [CrossRef]
- Wilson, M.R.; Jiang, Y.; Villalta, P.W.; Stornetta, A.; Boudreau, P.D.; Carrá, A.; Brennan, C.A.; Chun, E.; Ngo, L.; Samson, L.D.; et al. The Human Gut Bacterial Genotoxin Colibactin Alkylates DNA. Science 2019, 363, eaar7785. [Google Scholar] [CrossRef]
- Wolkenberg, S.E.; Boger, D.L. Mechanisms of in Situ Activation for DNA-Targeting Antitumor Agents. Chem. Rev. 2002, 102, 2477–2496. [Google Scholar] [CrossRef]
- Barr, J.R.; Van Atta, R.B.; Natrajan, A.; Hecht, S.M.; Van der Marel, G.A.; Van Boom, J.H. Iron(II) Bleomycin-Mediated Reduction of Oxygen to Water: An Oxygen-17 NMR Study. J. Am. Chem. Soc. 1990, 112, 4058–4060. [Google Scholar] [CrossRef]
- Konishi, M.; Ohkuma, H.; Tsuno, T.; Oki, T.; VanDuyne, G.D.; Clardy, J. Crystal and Molecular Structure of Dynemicin A: A Novel 1,5-Diyn-3-Ene Antitumor Antibiotic. J. Am. Chem. Soc. 1990, 112, 3715–3716. [Google Scholar] [CrossRef]
- Snyder, J.P.; Tipsword, G.E. Proposal for Blending Classical and Biradical Mechanisms in Antitumor Antibiotics: Dynemicin A. J. Am. Chem. Soc. 1990, 112, 4040–4042. [Google Scholar] [CrossRef]
- Ellestad, G.A.; Hamann, P.R.; Zein, N.; Morton, G.O.; Siegel, M.M.; Pastel, M.; Borders, D.B.; McGahren, W.J. Reactions of the Trisulfide Moiety in Calicheamicin. Tetrahedron Lett. 1989, 30, 3033–3036. [Google Scholar] [CrossRef]
- Myers, A.G.; Cohen, S.B.; Kwon, B.M. A Study of the Reaction of Calicheamicin.Gamma.1 with Glutathione in the Presence of Double-Stranded DNA. J. Am. Chem. Soc. 1994, 116, 1255–1271. [Google Scholar] [CrossRef]
- Ng, T.L.; Rohac, R.; Mitchell, A.J.; Boal, A.K.; Balskus, E.P. An N-Nitrosating Metalloenzyme Constructs the Pharmacophore of Streptozotocin. Nature 2019, 566, 94–99. [Google Scholar] [CrossRef]
- McBride, M.J.; Sil, D.; Ng, T.L.; Crooke, A.M.; Kenney, G.E.; Tysoe, C.R.; Zhang, B.; Balskus, E.P.; Boal, A.K.; Krebs, C.; et al. A Peroxodiiron(III/III) Intermediate Mediating Both N -Hydroxylation Steps in Biosynthesis of the N -Nitrosourea Pharmacophore of Streptozotocin by the Multi-Domain Metalloenzyme SznF. J. Am. Chem. Soc. 2020, 142, 11818–11828. [Google Scholar] [CrossRef]
- Veglia, F.; Matullo, G.; Vineis, P. Bulky DNA Adducts and Risk of Cancer: A Meta-Analysis. Cancer Epidemiol. Biomark. Prev. 2003, 12, 157–160. [Google Scholar]
- Motwani, H.V. DNA as an in Vitro Trapping Agent for Detection of Bulky Genotoxic Metabolites. J. Chromatogr. B 2020, 1152, 122276. [Google Scholar] [CrossRef]
- O’Neill, E.C.; Schorn, M.; Larson, C.B.; Millán-Aguiñaga, N. Targeted Antibiotic Discovery through Biosynthesis-Associated Resistance Determinants: Target Directed Genome Mining. Crit. Rev. Microbiol. 2019, 45, 255–277. [Google Scholar] [CrossRef]
- Galm, U.; Hager, M.H.; Van Lanen, S.G.; Ju, J.; Thorson, J.S.; Shen, B. Antitumor Antibiotics: Bleomycin, Enediynes, and Mitomycin. Chem. Rev. 2005, 105, 739–758. [Google Scholar] [CrossRef]
- Igarashi, Y.; Futamata, K.; Fujita, T.; Sekine, A.; Senda, H.; Naoki, H.; Furumai, T. Yatakemycin, a Novel Antifungal Antibiotic Produced by Streptomyces Sp. TP-A0356. J. Antibiot. 2003, 56, 107–113. [Google Scholar] [CrossRef]
- Hanka, L.J.; Dietz, A.; Gerpheide, S.A.; Kuentzel, S.L.; Martin, D.G. CC-1065 (NSC-298223), a New Antitumor Antibiotic. Production, in Vitro Biological Activity, Microbiological Assays and Taxonomy of the Producing Microorganism. J. Antibiot. 1978, 31, 1211–1217. [Google Scholar] [CrossRef]
- Ichimura, M.; Ogawa, T.; Takahashi, K.-I.; Kobayashi, E.; Kawamoto, I.; Yasuzawa, T.; Takahashi, I.; Nakano, H. Duocarmycin SA, a New Antitumor Antibiotic from Streptomyces Sp. J. Antibiot. 1990, 43, 1037–1038. [Google Scholar] [CrossRef]
- Martin, D.G.; Chidester, C.G.; Duchamp, D.J.; Mizsak, S.A. Structure of CC-1065 (NSC-298223), a New Antitumor Antibiotic. J. Antibiot. 1980, 33, 902–903. [Google Scholar] [CrossRef] [Green Version]
- Menderes, G.; Bonazzoli, E.; Bellone, S.; Black, J.; Predolini, F.; Pettinella, F.; Masserdotti, A.; Zammataro, L.; Altwerger, G.; Buza, N.; et al. SYD985, a Novel Duocarmycin-Based HER2-Targeting Antibody–Drug Conjugate, Shows Antitumor Activity in Uterine and Ovarian Carcinosarcoma with HER2/Neu Expression. Clin. Cancer Res. 2017, 23, 5836–5845. [Google Scholar] [CrossRef]
- Gerber, H.-P.; Koehn, F.E.; Abraham, R.T. The Antibody-Drug Conjugate: An Enabling Modality for Natural Product-Based Cancer Therapeutics. Nat. Prod. Rep. 2013, 30, 625. [Google Scholar] [CrossRef]
- Boger, D.L.; Garbaccio, R.M. Shape-Dependent Catalysis: Insights into the Source of Catalysis for the CC-1065 and Duocarmycin DNA Alkylation Reaction. Acc. Chem. Res. 1999, 32, 1043–1052. [Google Scholar] [CrossRef]
- Mullins, E.A.; Dorival, J.; Tang, G.-L.; Boger, D.L.; Eichman, B.F. Structural Evolution of a DNA Repair Self-Resistance Mechanism Targeting Genotoxic Secondary Metabolites. Nat. Commun. 2021, 12, 6942. [Google Scholar] [CrossRef]
- Xu, H.; Huang, W.; He, Q.-L.; Zhao, Z.-X.; Zhang, F.; Wang, R.; Kang, J.; Tang, G.-L. Self-Resistance to an Antitumor Antibiotic: A DNA Glycosylase Triggers the Base-Excision Repair System in Yatakemycin Biosynthesis. Angew. Chem. Int. Ed. 2012, 51, 10532–10536. [Google Scholar] [CrossRef]
- Mullins, E.A.; Shi, R.; Eichman, B.F. Toxicity and Repair of DNA Adducts Produced by the Natural Product Yatakemycin. Nat. Chem. Biol. 2017, 13, 1002–1008. [Google Scholar] [CrossRef]
- Yuan, H.; Zhang, J.; Cai, Y.; Wu, S.; Yang, K.; Chan, H.C.S.; Huang, W.; Jin, W.-B.; Li, Y.; Yin, Y.; et al. GyrI-like Proteins Catalyze Cyclopropanoid Hydrolysis to Confer Cellular Protection. Nat. Commun. 2017, 8, 1485. [Google Scholar] [CrossRef]
- Hurley, L.H.; Rokem, J.S. Biosynthesis of the Antitumor Antibiotic CC-1065 by Streptomyces zelensis. J. Antibiot. 1983, 36, 383–390. [Google Scholar] [CrossRef]
- Wu, S.; Jian, X.-H.; Yuan, H.; Jin, W.-B.; Yin, Y.; Wang, L.-Y.; Zhao, J.; Tang, G.-L. Unified Biosynthetic Origin of the Benzodipyrrole Subunits in CC-1065. ACS Chem. Biol. 2017, 12, 1603–1610. [Google Scholar] [CrossRef]
- Tichenor, M.S.; Boger, D.L. Yatakemycin: Total Synthesis, DNA Alkylation, and Biological Properties. Nat. Prod. Rep. 2008, 25, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.-B.; Wu, S.; Xu, Y.-F.; Yuan, H.; Tang, G.-L. Recent Advances in HemN-like Radical S-Adenosyl-L-Methionine Enzyme-Catalyzed Reactions. Nat. Prod. Rep. 2020, 37, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.-B.; Wu, S.; Jian, X.-H.; Yuan, H.; Tang, G.-L. A Radical S-Adenosyl-L-Methionine Enzyme and a Methyltransferase Catalyze Cyclopropane Formation in Natural Product Biosynthesis. Nat. Commun. 2018, 9, 2771. [Google Scholar] [CrossRef] [PubMed]
- Hertweck, C.; Luzhetskyy, A.; Rebets, Y.; Bechthold, A. Type II Polyketide Synthases: Gaining a Deeper Insight into Enzymatic Teamwork. Nat. Prod. Rep. 2007, 24, 162–190. [Google Scholar] [CrossRef]
- Das, A.; Khosla, C. Biosynthesis of Aromatic Polyketides in Bacteria. Acc. Chem. Res. 2009, 42, 631–639. [Google Scholar] [CrossRef]
- Wang, A.H.J.; Ughetto, G.; Quigley, G.J.; Rich, A. Interactions between an Anthracycline Antibiotic and DNA: Molecular Structure of Daunomycin Complexed to d(CpGpTpApCpG) at 1.2-.ANG. Resolution. Biochemistry 1987, 26, 1152–1163. [Google Scholar] [CrossRef]
- Williams, L.D.; Egli, M.; Qi, G.; Bash, P.; van der Marel, G.A.; van Boom, J.H.; Rich, A.; Frederick, C.A. Structure of Nogalamycin Bound to a DNA Hexamer. Proc. Natl. Acad. Sci. USA 1990, 87, 2225–2229. [Google Scholar] [CrossRef]
- Hansen, M.; Yun, S.; Hurley, L. Hedamycin Intercalates the DNA Helix and, through Carbohydrate-Mediated Recognition in the Minor Groove, Directs N7-Alkylation of Guanine in the Major Groove in a Sequence-Specific Manner. Chem. Biol. 1995, 2, 229–240. [Google Scholar] [CrossRef]
- Sun, D.; Hansen, M.; Clement, J.J.; Hurley, L.H. Structure of the Altromycin B (N7-Guanine)-DNA Adduct. A Proposed Prototypic DNA Adduct Structure for the Pluramycin Antitumor Antibiotics. Biochemistry 1993, 32, 8068–8074. [Google Scholar] [CrossRef]
- Pavlopoulos, S.; Bicknell, W.; Wickham, G.; Craik, D.J. Characterization of the Sequential Non-Covalent and Covalent Interactions of the Antitumour Antibiotic Hedamycin with Double Stranded DNA by NMR Spectroscopy. J. Mol. Recognit. 1999, 12, 346–354. [Google Scholar] [CrossRef]
- Bililign, T.; Griffith, B.R.; Thorson, J.S. Structure, Activity, Synthesis and Biosynthesis of Aryl-C-Glycosides. Nat. Prod. Rep. 2005, 22, 742. [Google Scholar] [CrossRef]
- Das, A.; Khosla, C. In Vivo and In Vitro Analysis of the Hedamycin Polyketide Synthase. Chem. Biol. 2009, 16, 1197–1207. [Google Scholar] [CrossRef]
- Bililign, T.; Hyun, C.-G.; Williams, J.S.; Czisny, A.M.; Thorson, J.S. The Hedamycin Locus Implicates a Novel Aromatic PKS Priming Mechanism. Chem. Biol. 2004, 11, 959–969. [Google Scholar] [CrossRef]
- Zhang, M.; Hou, X.-F.; Qi, L.-H.; Yin, Y.; Li, Q.; Pan, H.-X.; Chen, X.-Y.; Tang, G.-L. Biosynthesis of Trioxacarcin Revealing a Different Starter Unit and Complex Tailoring Steps for Type II Polyketide Synthase. Chem. Sci. 2015, 6, 3440–3447. [Google Scholar] [CrossRef]
- Pfoh, R.; Laatsch, H.; Sheldrick, G.M. Crystal Structure of Trioxacarcin A Covalently Bound to DNA. Nucleic Acids Res. 2008, 36, 3508–3514. [Google Scholar] [CrossRef]
- Dong, L.; Shen, Y.; Hou, X.-F.; Li, W.-J.; Tang, G.-L. Discovery of Druggability-Improved Analogues by Investigation of the LL-D49194α1 Biosynthetic Pathway. Org. Lett. 2019, 21, 2322–2325. [Google Scholar] [CrossRef]
- Pröpper, K.; Dittrich, B.; Smaltz, D.J.; Magauer, T.; Myers, A.G. Crystalline Guanine Adducts of Natural and Synthetic Trioxacarcins Suggest a Common Biological Mechanism and Reveal a Basis for the Instability of Trioxacarcin A. Bioorg. Med. Chem. Lett. 2014, 24, 4410–4413. [Google Scholar] [CrossRef]
- Fitzner, A.; Frauendorf, H.; Laatsch, H.; Diederichsen, U. Formation of Gutingimycin: Analytical Investigation of Trioxacarcin A-Mediated Alkylation of DsDNA. Anal. Bioanal. Chem. 2008, 390, 1139–1147. [Google Scholar] [CrossRef]
- Chen, X.; Bradley, N.P.; Lu, W.; Wahl, K.L.; Zhang, M.; Yuan, H.; Hou, X.-F.; Eichman, B.F.; Tang, G.-L. Base Excision Repair System Targeting DNA Adducts of Trioxacarcin/LL-D49194 Antibiotics for Self-Resistance. Nucleic Acids Res. 2022, 50, 2417–2430. [Google Scholar] [CrossRef]
- Hou, X.-F.; Song, Y.-J.; Zhang, M.; Lan, W.; Meng, S.; Wang, C.; Pan, H.-X.; Cao, C.; Tang, G.-L. Enzymology of Anthraquinone-γ-Pyrone Ring Formation in Complex Aromatic Polyketide Biosynthesis. Angew. Chem. Int. Ed. 2018, 57, 13475–13479. [Google Scholar] [CrossRef]
- Yin, Y.; Shen, Y.; Meng, S.; Zhang, M.; Pan, H.; Tang, G. Characterization of a Membrane-Bound O-acetyltransferase Involved in Trioxacarcin Biosynthesis Offers Insights into Its Catalytic Mechanism. Chin. J. Chem. 2020, 38, 1607–1611. [Google Scholar] [CrossRef]
- Bass, P.D.; Gubler, D.A.; Judd, T.C.; Williams, R.M. Mitomycinoid Alkaloids: Mechanism of Action, Biosynthesis, Total Syntheses, and Synthetic Approaches. Chem. Rev. 2013, 113, 6816–6863. [Google Scholar] [CrossRef]
- Suresh Kumar, G.; Lipman, R.; Cummings, J.; Tomasz, M. Mitomycin C−DNA Adducts Generated by DT-Diaphorase. Revised Mechanism of the Enzymatic Reductive Activation of Mitomycin C. Biochemistry 1997, 36, 14128–14136. [Google Scholar] [CrossRef]
- Hirai, O.; Shimomura, K.; Mizota, T.; Matsumoto, S.; Mori, J.; Kikuchi, H. A New Antitumor Antibiotic, FR-900482. IV. Hematological Toxicity in Mice. J. Antibiot. 1987, 40, 607–611. [Google Scholar] [CrossRef]
- August, P.R.; Flickinger, M.C.; Sherman, D.H. Cloning and Analysis of a Locus (Mcr) Involved in Mitomycin C Resistance in Streptomyces lavendulae. J. Bacteriol. 1994, 176, 4448–4454. [Google Scholar] [CrossRef]
- Sheldon, P.J.; Johnson, D.A.; August, P.R.; Liu, H.W.; Sherman, D.H. Characterization of a Mitomycin-Binding Drug Resistance Mechanism from the Producing Organism, Streptomyces lavendulae. J. Bacteriol. 1997, 179, 1796–1804. [Google Scholar] [CrossRef]
- Johnson, D.A.; August, P.R.; Shackleton, C.; Liu, H.; Sherman, D.H. Microbial Resistance to Mitomycins Involves a Redox Relay Mechanism. J. Am. Chem. Soc. 1997, 119, 2576–2577. [Google Scholar] [CrossRef]
- He, M.; Sheldon, P.J.; Sherman, D.H. Characterization of a Quinone Reductase Activity for the Mitomycin C Binding Protein (MRD): Functional Switching from a Drug-Activating Enzyme to a Drug-Binding Protein. Proc. Natl. Acad. Sci. USA 2001, 98, 926–931. [Google Scholar] [CrossRef]
- Namiki, H.; Chamberland, S.; Gubler, D.A.; Williams, R.M. Synthetic and Biosynthetic Studies on FR900482 and Mitomycin C: An Efficient and Stereoselective Hydroxymethylation of an Advanced Benzazocane Intermediate. Org. Lett. 2007, 9, 5341–5344. [Google Scholar] [CrossRef]
- Hornemann, U.; Kehrer, J.P.; Nunez, C.S.; Ranieri, R.L. D-Glucosamine and L-Citrulline, Precursors in Mitomycin Biosynthesis by Streptomyces verticillatus. J. Am. Chem. Soc. 1974, 96, 320–322. [Google Scholar] [CrossRef]
- Mao, Y.; Varoglu, M.; Sherman, D.H. Molecular Characterization and Analysis of the Biosynthetic Gene Cluster for the Antitumor Antibiotic Mitomycin C from Streptomyces lavendulae NRRL 2564. Chem. Biol. 1999, 6, 251–263. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.-G.; Yu, T.-W.; Fryhle, C.B.; Handa, S.; Floss, H.G. 3-Amino-5-Hydroxybenzoic Acid Synthase, the Terminal Enzyme in the Formation of the Precursor of MC7N Units in Rifamycin and Related Antibiotics. J. Biol. Chem. 1998, 273, 6030–6040. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Frost, J.W. Kanosamine Biosynthesis: A Likely Source of the Aminoshikimate Pathway’s Nitrogen Atom. J. Am. Chem. Soc. 2002, 124, 10642–10643. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.P.; Yokoyama, K. Characterization of Acyl Carrier Protein-Dependent Glycosyltransferase in Mitomycin C Biosynthesis. Biochemistry 2019, 58, 2804–2808. [Google Scholar] [CrossRef]
- Thibodeaux, C.J.; Chang, W.; Liu, H. Enzymatic Chemistry of Cyclopropane, Epoxide, and Aziridine Biosynthesis. Chem. Rev. 2012, 112, 1681–1709. [Google Scholar] [CrossRef]
- Ogasawara, Y.; Nakagawa, Y.; Maruyama, C.; Hamano, Y.; Dairi, T. In Vitro Characterization of MitE and MitB: Formation of N-Acetylglucosaminyl-3-Amino-5-Hydroxybenzoyl-MmcB as a Key Intermediate in the Biosynthesis of Antitumor Antibiotic Mitomycins. Bioorg. Med. Chem. Lett. 2019, 29, 2076–2078. [Google Scholar] [CrossRef]
- Wang, S.; Cheng, Y.; Wang, X.; Yang, Q.; Liu, W. Tracing of Acyl Carrier Protein-Channeled Mitomycin Intermediates in Streptomyces caespitosus Facilitates Characterization of the Biosynthetic Steps for AHBA–GlcN Formation and Processing. J. Am. Chem. Soc. 2022, 144, 14945–14956. [Google Scholar] [CrossRef]
- Grüschow, S.; Chang, L.-C.; Mao, Y.; Sherman, D.H. Hydroxyquinone O-Methylation in Mitomycin Biosynthesis. J. Am. Chem. Soc. 2007, 129, 6470–6476. [Google Scholar] [CrossRef]
- Varoglu, M.; Mao, Y.; Sherman, D.H. Mapping the Mitomycin Biosynthetic Pathway by Functional Analysis of the MitM Aziridine N -Methyltransferase. J. Am. Chem. Soc. 2001, 123, 6712–6713. [Google Scholar] [CrossRef]
- Foulke-Abel, J.; Agbo, H.; Zhang, H.; Mori, S.; Watanabe, C.M.H. Mode of Action and Biosynthesis of the Azabicycle-Containing Natural Products Azinomycin and Ficellomycin. Nat. Prod. Rep. 2011, 28, 693–704. [Google Scholar] [CrossRef]
- Zang, H.; Gates, K.S. DNA Binding and Alkylation by the “Left Half” of Azinomycin B. Biochemistry 2000, 39, 14968–14975. [Google Scholar] [CrossRef]
- Alcaro, S.; Ortuso, F.; Coleman, R.S. Molecular Modeling of DNA Cross-Linking Analogues Based on the Azinomycin Scaffold. J. Chem. Inf. Model. 2005, 45, 602–609. [Google Scholar] [CrossRef]
- Coleman, R.S.; Perez, R.J.; Burk, C.H.; Navarro, A. Studies on the Mechanism of Action of Azinomycin B: Definition of Regioselectivity and Sequence Selectivity of DNA Cross-Link Formation and Clarification of the Role of the Naphthoate. J. Am. Chem. Soc. 2002, 124, 13008–13017. [Google Scholar] [CrossRef]
- Foulke-Abel, J.; Kelly, G.T.; Zhang, H.; Watanabe, C.M.H. Characterization of AziR, a Resistance Protein of the DNA Cross-Linking Agent Azinomycin B. Mol. BioSyst. 2011, 7, 2563. [Google Scholar] [CrossRef]
- Wang, S.; Liu, K.; Xiao, L.; Yang, L.; Li, H.; Zhang, F.; Lei, L.; Li, S.; Feng, X.; Li, A.; et al. Characterization of a Novel DNA Glycosylase from S. sahachiroi Involved in the Reduction and Repair of Azinomycin B Induced DNA Damage. Nucleic Acids Res. 2016, 44, 187–197. [Google Scholar] [CrossRef]
- Chen, X.; Sun, Y.; Wang, S.; Ying, K.; Xiao, L.; Liu, K.; Zuo, X.; He, J. Identification of a Novel Structure-Specific Endonuclease AziN That Contributes to the Repair of Azinomycin B-Mediated DNA Interstrand Crosslinks. Nucleic Acids Res. 2020, 48, 709–718. [Google Scholar] [CrossRef]
- Mullins, E.A.; Warren, G.M.; Bradley, N.P.; Eichman, B.F. Structure of a DNA Glycosylase That Unhooks Interstrand Cross-Links. Proc. Natl. Acad. Sci. USA 2017, 114, 4400–4405. [Google Scholar] [CrossRef]
- Kelly, G.T.; Sharma, V.; Watanabe, C.M.H. An Improved Method for Culturing Streptomyces sahachiroi: Biosynthetic Origin of the Enol Fragment of Azinomycin B. Bioorg. Chem. 2008, 36, 4–15. [Google Scholar] [CrossRef]
- Kelly, G.T.; Washburn, L.A.; Watanabe, C.M.H. The Fate of Molecular Oxygen in Azinomycin Biosynthesis. J. Org. Chem. 2019, 84, 2991–2996. [Google Scholar] [CrossRef]
- Sharma, V.; Kelly, G.T.; Watanabe, C.M.H. Exploration of the Molecular Origin of the Azinomycin Epoxide: Timing of the Biosynthesis Revealed. Org. Lett. 2008, 10, 4815–4818. [Google Scholar] [CrossRef]
- Zhao, Q.; He, Q.; Ding, W.; Tang, M.; Kang, Q.; Yu, Y.; Deng, W.; Zhang, Q.; Fang, J.; Tang, G.; et al. Characterization of the Azinomycin B Biosynthetic Gene Cluster Revealing a Different Iterative Type I Polyketide Synthase for Naphthoate Biosynthesis. Chem. Biol. 2008, 15, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Mori, S.; Simkhada, D.; Zhang, H.; Erb, M.S.; Zhang, Y.; Williams, H.; Fedoseyenko, D.; Russell, W.K.; Kim, D.; Fleer, N.; et al. Polyketide Ring Expansion Mediated by a Thioesterase, Chain Elongation and Cyclization Domain, in Azinomycin Biosynthesis: Characterization of AziB and AziG. Biochemistry 2016, 55, 704–714. [Google Scholar] [CrossRef]
- Ding, W.; Deng, W.; Tang, M.; Zhang, Q.; Tang, G.; Bi, Y.; Liu, W. Biosynthesis of 3-Methoxy-5-Methyl Naphthoic Acid and Its Incorporation into the Antitumor Antibiotic Azinomycin B. Mol. BioSyst. 2010, 6, 1071. [Google Scholar] [CrossRef]
- Miyashita, K.; Park, M.; Adachi, S.; Seki, S.; Obika, S.; Imanishi, T. A 3,4-Epoxypiperidine Structure as a Novel and Simple DNA-Cleavage Unit. Bioorg. Med. Chem. Lett. 2002, 12, 1075–1077. [Google Scholar] [CrossRef]
- Nepal, K.K.; Lee, R.P.; Rezenom, Y.H.; Watanabe, C.M.H. Probing the Role of N -Acetyl-Glutamyl 5-Phosphate, an Acyl Phosphate, in the Construction of the Azabicycle Moiety of the Azinomycins. Biochemistry 2015, 54, 4415–4418. [Google Scholar] [CrossRef]
- Mori, S.; Nepal, K.K.; Kelly, G.T.; Sharma, V.; Simkhada, D.; Gowda, V.; Delgado, D.; Watanabe, C.M.H. Priming of Azabicycle Biosynthesis in the Azinomycin Class of Antitumor Agents. Biochemistry 2017, 56, 805–808. [Google Scholar] [CrossRef]
- Washburn, L.A.; Foley, B.; Martinez, F.; Lee, R.P.; Pryor, K.; Rimes, E.; Watanabe, C.M.H. Transketolase Activity in the Formation of the Azinomycin Azabicycle Moiety. Biochemistry 2019, 58, 5255–5258. [Google Scholar] [CrossRef]
- Kurosawa, S.; Hasebe, F.; Okamura, H.; Yoshida, A.; Matsuda, K.; Sone, Y.; Tomita, T.; Shinada, T.; Takikawa, H.; Kuzuyama, T.; et al. Molecular Basis for Enzymatic Aziridine Formation via Sulfate Elimination. J. Am. Chem. Soc. 2022, 144, 16164–16170. [Google Scholar] [CrossRef] [PubMed]
- Hasebe, F.; Matsuda, K.; Shiraishi, T.; Futamura, Y.; Nakano, T.; Tomita, T.; Ishigami, K.; Taka, H.; Mineki, R.; Fujimura, T.; et al. Amino-Group Carrier-Protein-Mediated Secondary Metabolite Biosynthesis in Streptomyces. Nat. Chem. Biol. 2016, 12, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Kelly, G.T.; Foulke-Abel, J.; Watanabe, C.M.H. Aminoacetone as the Penultimate Precursor to the Antitumor Agent Azinomycin A. Org. Lett. 2009, 11, 4006–4009. [Google Scholar] [CrossRef] [PubMed]
- Antonow, D.; Thurston, D.E. Synthesis of DNA-Interactive Pyrrolo [2,1-c][1,4]Benzodiazepines (PBDs). Chem. Rev. 2011, 111, 2815–2864. [Google Scholar] [CrossRef]
- Gerratana, B. Biosynthesis, Synthesis, and Biological Activities of Pyrrolobenzodiazepines: Activities of Pyrrolobenzodiazepines. Med. Res. Rev. 2012, 32, 254–293. [Google Scholar] [CrossRef]
- Barkley, M.D.; Cheatham, S.; Thurston, D.E.; Hurley, L.H. Pyrrolo[1,4]Benzodiazepine Antitumor Antibiotics: Evidence for Two Forms of Tomaymycin Bound to DNA. Biochemistry 1986, 25, 3021–3031. [Google Scholar] [CrossRef]
- Kopka, M.L.; Goodsell, D.S.; Baikalov, I.; Grzeskowiak, K.; Cascio, D.; Dickerson, R.E. Crystal Structure of a Covalent DNA-Drug Adduct: Anthramycin Bound to C-C-A-A-C-G-T-T-G-G and a Molecular Explanation of Specificity. Biochemistry 1994, 33, 13593–13610. [Google Scholar] [CrossRef]
- Arora, S.K. Structure of Tomaymycin, a DNA Binding Antitumor Antibiotic. J. Antibiot. 1981, 34, 462–464. [Google Scholar] [CrossRef]
- Campbell, A.D.; Tomasi, S.; Tiberghien, A.C.; Parker, J.S. An Isomerization Approach to Tesirine and Pyrrolobenzodiazepines. Org. Process Res. Dev. 2019, 23, 2543–2548. [Google Scholar] [CrossRef]
- Mantaj, J.; Jackson, P.J.M.; Rahman, K.M.; Thurston, D.E. From Anthramycin to Pyrrolobenzodiazepine (PBD)-Containing Antibody-Drug Conjugates (ADCs). Angew. Chem. Int. Ed. 2017, 56, 462–488. [Google Scholar] [CrossRef]
- Li, W.; Khullar, A.; Chou, S.; Sacramo, A.; Gerratana, B. Biosynthesis of Sibiromycin, a Potent Antitumor Antibiotic. Appl. Environ. Microbiol. 2009, 75, 2869–2878. [Google Scholar] [CrossRef]
- Kamenik, Z.; Gazak, R.; Kadlcik, S.; Steiningerova, L.; Rynd, V.; Janata, J. C-C Bond Cleavage in Biosynthesis of 4-Alkyl-L-Proline Precursors of Lincomycin and Anthramycin Cannot Precede C-Methylation. Nat. Commun. 2018, 9, 3167. [Google Scholar] [CrossRef]
- Hurley, L.H. Elucidation and Formulation of Novel Biosynthetic Pathways Leading to the Pyrrolo[1,4]Benzodiazepine Antibiotics Anthramycin, Tomaymycin, and Sibiromycin. Acc. Chem. Res. 1980, 13, 263–269. [Google Scholar] [CrossRef]
- Li, W.; Chou, S.; Khullar, A.; Gerratana, B. Cloning and Characterization of the Biosynthetic Gene Cluster for Tomaymycin, an SJG-136 Monomeric Analog. Appl. Environ. Microbiol. 2009, 75, 2958–2963. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.; Li, W.; Gerratana, B.; Rokita, S.E. Identification of the Dioxygenase-Generated Intermediate Formed during Biosynthesis of the Dihydropyrrole Moiety Common to Anthramycin and Sibiromycin. Bioorg. Med. Chem. 2015, 23, 449–454. [Google Scholar] [CrossRef]
- Keller, U. Acyl Pentapeptide Lactone Synthesis in Actinomycin-Producing Streptomycetes by Feeding with Structural Analogs of 4-Methyl-3-Hydroxyanthranilic Acid. J. Biol. Chem. 1984, 259, 8226–8231. [Google Scholar] [CrossRef]
- Matthijs, S.; Baysse, C.; Koedam, N.; Tehrani, K.A.; Verheyden, L.; Budzikiewicz, H.; Schäfer, M.; Hoorelbeke, B.; Meyer, J.-M.; De Greve, H.; et al. The Pseudomonas Siderophore Quinolobactin Is Synthesized from Xanthurenic Acid, an Intermediate of the Kynurenine Pathway: Quinolobactin Siderophore from P. Fluorescens. Mol. Microbiol. 2004, 52, 371–384. [Google Scholar] [CrossRef]
- Nguyen, K.T.; Kau, D.; Gu, J.-Q.; Brian, P.; Wrigley, S.K.; Baltz, R.H.; Miao, V. A Glutamic Acid 3-Methyltransferase Encoded by an Accessory Gene Locus Important for Daptomycin Biosynthesis in Streptomyces roseosporus. Mol. Microbiol. 2006, 61, 1294–1307. [Google Scholar] [CrossRef]
- Hu, Y.; Phelan, V.; Ntai, I.; Farnet, C.M.; Zazopoulos, E.; Bachmann, B.O. Benzodiazepine Biosynthesis in Streptomyces refuineus. Chem. Biol. 2007, 14, 691–701. [Google Scholar] [CrossRef]
- Giessen, T.W.; Kraas, F.I.; Marahiel, M.A. A Four-Enzyme Pathway for 3,5-Dihydroxy-4-Methylanthranilic Acid Formation and Incorporation into the Antitumor Antibiotic Sibiromycin. Biochemistry 2011, 50, 5680–5692. [Google Scholar] [CrossRef]
- Scott, J.D.; Williams, R.M. Chemistry and Biology of the Tetrahydroisoquinoline Antitumor Antibiotics. Chem. Rev. 2002, 102, 1669–1730. [Google Scholar] [CrossRef]
- Le, V.H.; Inai, M.; Williams, R.M.; Kan, T. Ecteinascidins. A Review of the Chemistry, Biology and Clinical Utility of Potent Tetrahydroisoquinoline Antitumor Antibiotics. Nat. Prod. Rep. 2015, 32, 328–347. [Google Scholar] [CrossRef]
- Lown, J.W.; Joshua, A.V.; Lee, J.S. Molecular Mechanisms of Binding and Single-Strand Scission of DNA by the Antitumor Antibiotics Saframycins A and C. Biochemistry 1982, 21, 419–428. [Google Scholar] [CrossRef]
- Hill, G.C.; Remers, W.A. Computer Simulation of the Binding of Saframycin A to d(GATGCATC)2. J. Med. Chem. 1991, 34, 1990–1998. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wen, W.-H.; Pu, J.-Y.; Tang, M.-C.; Zhang, L.; Peng, C.; Xu, Y.; Tang, G.-L. Extracellularly Oxidative Activation and Inactivation of Matured Prodrug for Cryptic Self-Resistance in Naphthyridinomycin Biosynthesis. Proc. Natl. Acad. Sci. USA 2018, 115, 11232–11237. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.-H.; Zhang, Y.; Zhang, Y.-Y.; Yu, Q.; Jiang, C.-C.; Tang, M.-C.; Pu, J.-Y.; Wu, L.; Zhao, Y.-L.; Shi, T.; et al. Reductive Inactivation of the Hemiaminal Pharmacophore for Resistance against Tetrahydroisoquinoline Antibiotics. Nat. Commun. 2021, 12, 7085. [Google Scholar] [CrossRef] [PubMed]
- Mikami, Y.; Takahashi, K.; Yazawa, K.; Arai, T.; Namikoshi, M.; Iwasaki, S.; Okuda, S. Biosynthetic Studies on Saframycin A, a Quinone Antitumor Antibiotic Produced by Streptomyces lavendulae. J. Biol. Chem. 1985, 260, 344–348. [Google Scholar] [CrossRef]
- Arai, T.; Yazawa, K.; Takahashi, K.; Maeda, A.; Mikami, Y. Directed Biosynthesis of New Saframycin Derivatives with Resting Cells of Streptomyces lavendulae. Antimicrob. Agents Chemother. 1985, 28, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.-C.; Fu, C.-Y.; Tang, G.-L. Characterization of SfmD as a Heme Peroxidase That Catalyzes the Regioselective Hydroxylation of 3-Methyltyrosine to 3-Hydroxy-5-Methyltyrosine in Saframycin A Biosynthesis. J. Biol. Chem. 2012, 287, 5112–5121. [Google Scholar] [CrossRef]
- Fu, C.-Y.; Tang, M.-C.; Peng, C.; Li, L.; He, Y.-L.; Liu, W.; Tang, G.-L. Biosynthesis of 3-Hydroxy-5-Methyl-o-Methyltyrosine in the Saframycin/ Safracin Biosynthetic Pathway. J. Microbiol. Biotechnol. 2009, 19, 439–446. [Google Scholar] [CrossRef]
- Koketsu, K.; Watanabe, K.; Suda, H.; Oguri, H.; Oikawa, H. Reconstruction of the Saframycin Core Scaffold Defines Dual Pictet-Spengler Mechanisms. Nat. Chem. Biol. 2010, 6, 408–410. [Google Scholar] [CrossRef]
- Stöckigt, J.; Antonchick, A.P.; Wu, F.; Waldmann, H. The Pictet-Spengler Reaction in Nature and in Organic Chemistry. Angew. Chem. Int. Ed. 2011, 50, 8538–8564. [Google Scholar] [CrossRef]
- Song, L.-Q.; Zhang, Y.-Y.; Pu, J.-Y.; Tang, M.-C.; Peng, C.; Tang, G.-L. Catalysis of Extracellular Deamination by a FAD-Linked Oxidoreductase after Prodrug Maturation in the Biosynthesis of Saframycin A. Angew. Chem. Int. Ed. 2017, 129, 9244–9248. [Google Scholar] [CrossRef]
- Gates, K.S. Mechanisms of DNA Damage by Leinamycin. Chem. Res. Toxicol. 2000, 13, 953–956. [Google Scholar] [CrossRef]
- Fekry, M.I.; Szekely, J.; Dutta, S.; Breydo, L.; Zang, H.; Gates, K.S. Noncovalent DNA Binding Drives DNA Alkylation by Leinamycin: Evidence That the Z, E -5-(Thiazol-4-Yl)-Penta-2,4-Dienone Moiety of the Natural Product Serves as an Atypical DNA Intercalator. J. Am. Chem. Soc. 2011, 133, 17641–17651. [Google Scholar] [CrossRef]
- Asai, A.; Hara, M.; Kakita, S.; Kanda, Y.; Yoshida, M.; Saito, H.; Saitoh, Y. Thiol-Mediated DNA Alkylation by the Novel Antitumor Antibiotic Leinamycin. J. Am. Chem. Soc. 1996, 118, 6802–6803. [Google Scholar] [CrossRef]
- Breydo, L.; Zang, H.; Mitra, K.; Gates, K.S. Thiol-Independent DNA Alkylation by Leinamycin. J. Am. Chem. Soc. 2001, 123, 2060–2061. [Google Scholar] [CrossRef]
- Huang, S.-X.; Yun, B.-S.; Ma, M.; Basu, H.S.; Church, D.R.; Ingenhorst, G.; Huang, Y.; Yang, D.; Lohman, J.R.; Tang, G.-L.; et al. Leinamycin E1 Acting as an Anticancer Prodrug Activated by Reactive Oxygen Species. Proc. Natl. Acad. Sci. USA 2015, 112, 8278–8283. [Google Scholar] [CrossRef]
- Tang, G.-L.; Cheng, Y.-Q.; Shen, B. Chain Initiation in the Leinamycin-Producing Hybrid Nonribosomal Peptide/Polyketide Synthetase from Streptomyces Atroolivaceus S-140. Discrete, Monofunctional Adenylation Enzyme and Peptidyl Carrier Protein That Directly Load D-Alanine. J. Biol. Chem. 2007, 282, 20273–20282. [Google Scholar] [CrossRef]
- Cheng, Y.-Q.; Tang, G.-L.; Shen, B. Type I Polyketide Synthase Requiring a Discrete Acyltransferase for Polyketide Biosynthesis. Proc. Natl. Acad. Sci. USA 2003, 100, 3149–3154. [Google Scholar] [CrossRef]
- Helfrich, E.J.N.; Piel, J. Biosynthesis of Polyketides by Trans-AT Polyketide Synthases. Nat. Prod. Rep. 2016, 33, 231–316. [Google Scholar] [CrossRef]
- Tang, G.-L.; Cheng, Y.-Q.; Shen, B. Polyketide Chain Skipping Mechanism in the Biosynthesis of the Hybrid Nonribosomal Peptide−Polyketide Antitumor Antibiotic Leinamycin in Streptomyces atroolivaceus S-140. J. Nat. Prod. 2006, 69, 387–393. [Google Scholar] [CrossRef]
- Huang, Y.; Tang, G.-L.; Pan, G.; Chang, C.-Y.; Shen, B. Characterization of the Ketosynthase and Acyl Carrier Protein Domains at the LnmI Nonribosomal Peptide Synthetase–Polyketide Synthase Interface for Leinamycin Biosynthesis. Org. Lett. 2016, 18, 4288–4291. [Google Scholar] [CrossRef]
- Cheng, Y.-Q.; Tang, G.-L.; Shen, B. Identification and Localization of the Gene Cluster Encoding Biosynthesis of the Antitumor Macrolactam Leinamycin in Streptomyces atroolivaceus S-140. J. Bacteriol. 2002, 184, 7013–7024. [Google Scholar] [CrossRef] [Green Version]
- Dunbar, K.L.; Scharf, D.H.; Litomska, A.; Hertweck, C. Enzymatic Carbon–Sulfur Bond Formation in Natural Product Biosynthesis. Chem. Rev. 2017, 117, 5521–5577. [Google Scholar] [CrossRef]
- Ma, M.; Lohman, J.R.; Liu, T.; Shen, B. C-S Bond Cleavage by a Polyketide Synthase Domain. Proc. Natl. Acad. Sci. USA 2015, 112, 10359–10364. [Google Scholar] [CrossRef]
- Huang, Y.; Huang, S.-X.; Ju, J.; Tang, G.; Liu, T.; Shen, B. Characterization of the LnmKLM Genes Unveiling Key Intermediates for β-Alkylation in Leinamycin Biosynthesis. Org. Lett. 2011, 13, 498–501. [Google Scholar] [CrossRef]
- Meng, S.; Steele, A.D.; Yan, W.; Pan, G.; Kalkreuter, E.; Liu, Y.-C.; Xu, Z.; Shen, B. Thiocysteine Lyases as Polyketide Synthase Domains Installing Hydropersulfide into Natural Products and a Hydropersulfide Methyltransferase. Nat. Commun. 2021, 12, 5672. [Google Scholar] [CrossRef]
- Kwong, T.; Ma, M.; Pan, G.; Hindra; Yang, D.; Yang, C.; Lohman, J.R.; Rudolf, J.D.; Cleveland, J.L.; Shen, B. P450-Catalyzed Tailoring Steps in Leinamycin Biosynthesis Featuring Regio- and Stereoselective Hydroxylations and Substrate Promiscuities. Biochemistry 2018, 57, 5005–5013. [Google Scholar] [CrossRef]
- Kharel, M.K.; Pahari, P.; Shepherd, M.D.; Tibrewal, N.; Nybo, S.E.; Shaaban, K.A.; Rohr, J. Angucyclines: Biosynthesis, Mode-of-Action, New Natural Products, and Synthesis. Nat. Prod. Rep. 2012, 29, 264–325. [Google Scholar] [CrossRef]
- Chen, J.-M.; Shepherd, M.D.; Horn, J.; Leggas, M.; Rohr, J. Enzymatic Methylation and Structure-Activity-Relationship Studies on Polycarcin V, a Gilvocarcin-Type Antitumor Agent. ChemBioChem 2014, 15, 2729–2735. [Google Scholar] [CrossRef]
- McGee, L.R.; Misra, R. Gilvocarcin Photobiology. Isolation and Characterization of the DNA Photoadduct. J. Am. Chem. Soc. 1990, 112, 2386–2389. [Google Scholar] [CrossRef]
- Ueberschaar, N.; Xu, Z.; Scherlach, K.; Metsä-Ketelä, M.; Bretschneider, T.; Dahse, H.-M.; Görls, H.; Hertweck, C. Synthetic Remodeling of the Chartreusin Pathway to Tune Antiproliferative and Antibacterial Activities. J. Am. Chem. Soc. 2013, 135, 17408–17416. [Google Scholar] [CrossRef]
- Fischer, C.; Lipata, F.; Rohr, J. The Complete Gene Cluster of the Antitumor Agent Gilvocarcin V and Its Implication for the Biosynthesis of the Gilvocarcins. J. Am. Chem. Soc. 2003, 125, 7818–7819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Fischer, C.; Beninga, C.; Rohr, J. Oxidative Rearrangement Processes in the Biosynthesis of Gilvocarcin V. J. Am. Chem. Soc. 2004, 126, 12262–12263. [Google Scholar] [CrossRef] [PubMed]
- Kharel, M.K.; Zhu, L.; Liu, T.; Rohr, J. Multi-Oxygenase Complexes of the Gilvocarcin and Jadomycin Biosyntheses. J. Am. Chem. Soc. 2007, 129, 3780–3781. [Google Scholar] [CrossRef] [PubMed]
- Kharel, M.K.; Nybo, S.E.; Shepherd, M.D.; Rohr, J. Cloning and Characterization of the Ravidomycin and Chrysomycin Biosynthetic Gene Clusters. ChemBioChem 2010, 11, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Noinaj, N.; Bosserman, M.A.; Schickli, M.A.; Piszczek, G.; Kharel, M.K.; Pahari, P.; Buchanan, S.K.; Rohr, J. The Crystal Structure and Mechanism of an Unusual Oxidoreductase, GilR, Involved in Gilvocarcin V Biosynthesis. J. Biol. Chem. 2011, 286, 23533–23543. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.-Y.; Ohashi, M.; Tang, Y. Deciphering Chemical Logic of Fungal Natural Product Biosynthesis Through Heterologous Expression and Genome Mining. Nat. Prod. Rep. 2022. [Google Scholar] [CrossRef] [PubMed]
- Bradley, N.P.; Wahl, K.L.; Steenwyk, J.L.; Rokas, A.; Eichman, B.F. Resistance-Guided Mining of Bacterial Genotoxins Defines a Family of DNA Glycosylases. mBio 2022, 13, e0329721. [Google Scholar] [CrossRef]
Figure 1.
Chemical structures of spirocyclopropylcyclohexadienone family compounds.

Figure 2.
(A) DNA modification by YTM, and excision of DNA-drug complex by YtkR2. (B) Hydrolysis of the cyclopropyl moiety in YTM and CC-1065 by YtkR2 and C10R6, respectively.
Figure 2.
(A) DNA modification by YTM, and excision of DNA-drug complex by YtkR2. (B) Hydrolysis of the cyclopropyl moiety in YTM and CC-1065 by YtkR2 and C10R6, respectively.

Figure 3.
Isotopic labelling patterns with serine, methionine, and tyrosine.

Figure 4.
Proposed biosynthetic pathway of CC-1065.

Figure 5.
Proposed enzymatic mechanism of the cyclopropane moiety formation catalyzed by C10P and C10Q.
Figure 5.
Proposed enzymatic mechanism of the cyclopropane moiety formation catalyzed by C10P and C10Q.

Figure 6.
(A) Chemical structures of pluramycins. (B) Alkylation of DNA by hedamycin.

Figure 7.
Proposed biosynthetic pathway of hedamycin. KS, ketosynthase; KR, ketoreductase; ACP, acyl carrier protein; DH, dehydratase; AT, acyl transferase.
Figure 7.
Proposed biosynthetic pathway of hedamycin. KS, ketosynthase; KR, ketoreductase; ACP, acyl carrier protein; DH, dehydratase; AT, acyl transferase.

Figure 8.
(A) Chemical structures of trioxacarcin A, LL-D49194α1 and their derivates. (B) Alkylation of DNA by trioxacarcins.
Figure 8.
(A) Chemical structures of trioxacarcin A, LL-D49194α1 and their derivates. (B) Alkylation of DNA by trioxacarcins.

Figure 9.
Proposed biosynthetic pathway of trioxacarcin A and LL-D49194α1. CLF, chain length factor.
Figure 9.
Proposed biosynthetic pathway of trioxacarcin A and LL-D49194α1. CLF, chain length factor.

Figure 10.
(A) Chemical structures of mitomycins. (B) Proposed mechanism of DNA cross-linking by mitomycin C and FR-900482.
Figure 10.
(A) Chemical structures of mitomycins. (B) Proposed mechanism of DNA cross-linking by mitomycin C and FR-900482.

Figure 11.
Proposed biosynthetic pathway of mitomycins.

Figure 12.
Proposed mechanism of DNA cross-linking by azinomycins.

Figure 13.
Origins of azinomycins revealed by isotopic labelling experiments.

Figure 14.
Proposed biosynthetic pathway of azinomycins. PCP, peptidyl carrier protein; A, adenylation; C, condensation; RE, reduction. Proposed pathway of constructing 3-methoxy-5-methyl-NPA moiety (A), epoxy intermediate (B), azabicyclic fragment (C), and incorporating building blocks (D).
Figure 14.
Proposed biosynthetic pathway of azinomycins. PCP, peptidyl carrier protein; A, adenylation; C, condensation; RE, reduction. Proposed pathway of constructing 3-methoxy-5-methyl-NPA moiety (A), epoxy intermediate (B), azabicyclic fragment (C), and incorporating building blocks (D).

Figure 15.
(A) Origins of pyrrolobenzodiazepines revealed by isotopic labeling experiments. (B) Chemical structure of SJG-136. (C) Proposed mechanism of DNA alkylating by pyrrolobenzodiazapines.
Figure 15.
(A) Origins of pyrrolobenzodiazepines revealed by isotopic labeling experiments. (B) Chemical structure of SJG-136. (C) Proposed mechanism of DNA alkylating by pyrrolobenzodiazapines.

Figure 16.
Proposed biosynthetic pathway of dihydropyrrole moieties (A), anthranilic acid moieties (B), and assembling two building blocks (C).
Figure 16.
Proposed biosynthetic pathway of dihydropyrrole moieties (A), anthranilic acid moieties (B), and assembling two building blocks (C).

Figure 17.
(A) Chemical structures of tetrahydroisoquinoline family. (B) Proposed mechanism of DNA alkylating by saframycin A.
Figure 17.
(A) Chemical structures of tetrahydroisoquinoline family. (B) Proposed mechanism of DNA alkylating by saframycin A.

Figure 18.
Origins of saframycins revealed by isotopic precursors labeling experiments.

Figure 19.
Proposed biosynthetic pathway of quinone moiety (A) and forming the skeleton of saframycins (B).
Figure 19.
Proposed biosynthetic pathway of quinone moiety (A) and forming the skeleton of saframycins (B).

Figure 20.
Post-modification of saframycins.

Figure 21.
Proposed mechanisms of DNA alkylating by leinamycin and leinamycin E1.

Figure 22.
Proposed biosynthetic pathway of leinamycin. CP, carrier protein; CY, condensation/cyclization; MT, transferase; ER, enoyl reductase; TE, thioesterase; DUF, the domain of unknown function; SH, PLP-dependent cysteine lyase domain.
Figure 22.
Proposed biosynthetic pathway of leinamycin. CP, carrier protein; CY, condensation/cyclization; MT, transferase; ER, enoyl reductase; TE, thioesterase; DUF, the domain of unknown function; SH, PLP-dependent cysteine lyase domain.

Figure 23.
(A) Chemical structure of gilvocarcin V, chrysomycin A, and ravidomycin. (B) Mode of action of vinyl-containing gilvocarcin-type natural products.
Figure 23.
(A) Chemical structure of gilvocarcin V, chrysomycin A, and ravidomycin. (B) Mode of action of vinyl-containing gilvocarcin-type natural products.

Figure 24.
Proposed biosynthetic pathway of gilvocarcin V.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Nie, Q.-Y.; Hu, Y.; Hou, X.-F.; Tang, G.-L. Biosynthesis of DNA-Alkylating Antitumor Natural Products. Molecules 2022, 27, 6387. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27196387
AMA Style
Nie Q-Y, Hu Y, Hou X-F, Tang G-L. Biosynthesis of DNA-Alkylating Antitumor Natural Products. Molecules. 2022; 27(19):6387. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27196387
Chicago/Turabian StyleNie, Qiu-Yue, Yu Hu, Xian-Feng Hou, and Gong-Li Tang. 2022. "Biosynthesis of DNA-Alkylating Antitumor Natural Products" Molecules 27, no. 19: 6387. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27196387