
Molecular Engineering of Quinone-Based Nickel Complexes and Polymers for All-Organic Li-Ion Batteries

Abstract
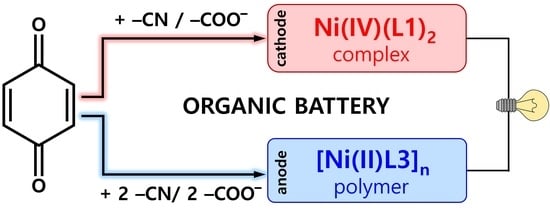
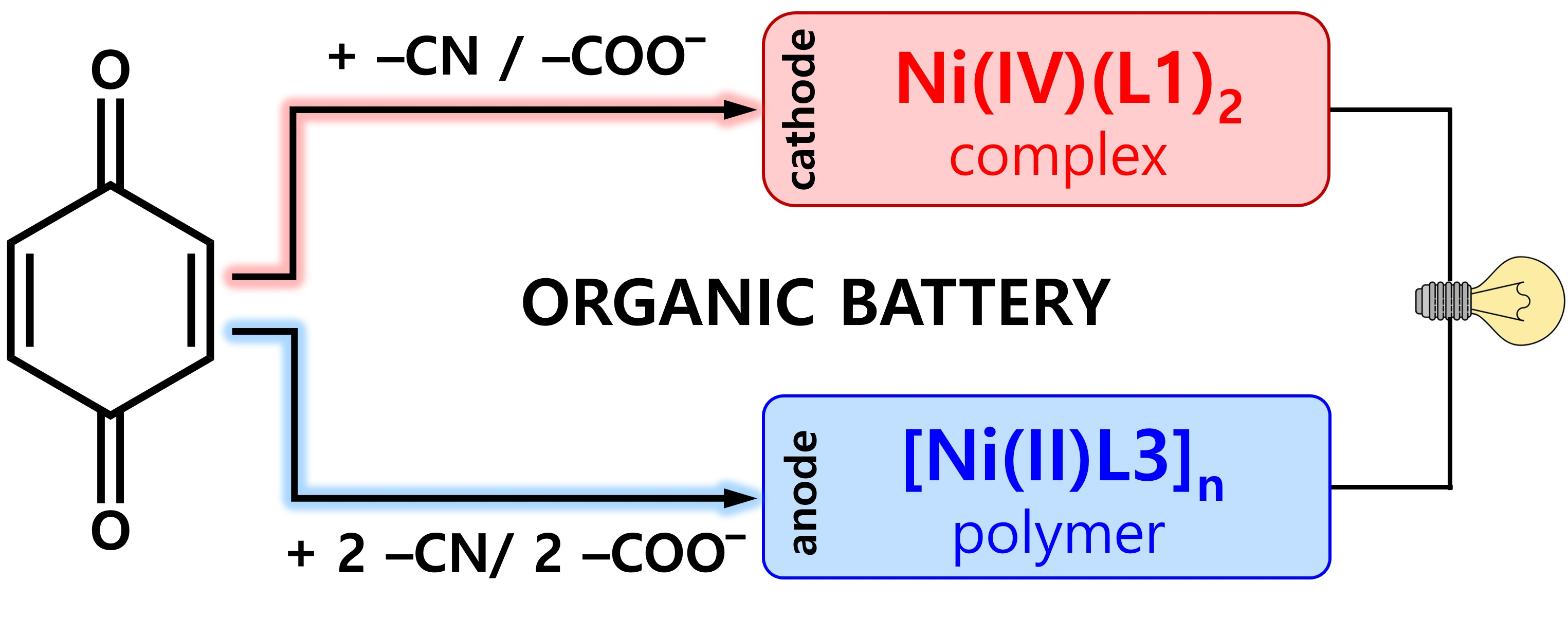
:
1. Introduction
2. Results and Discussion
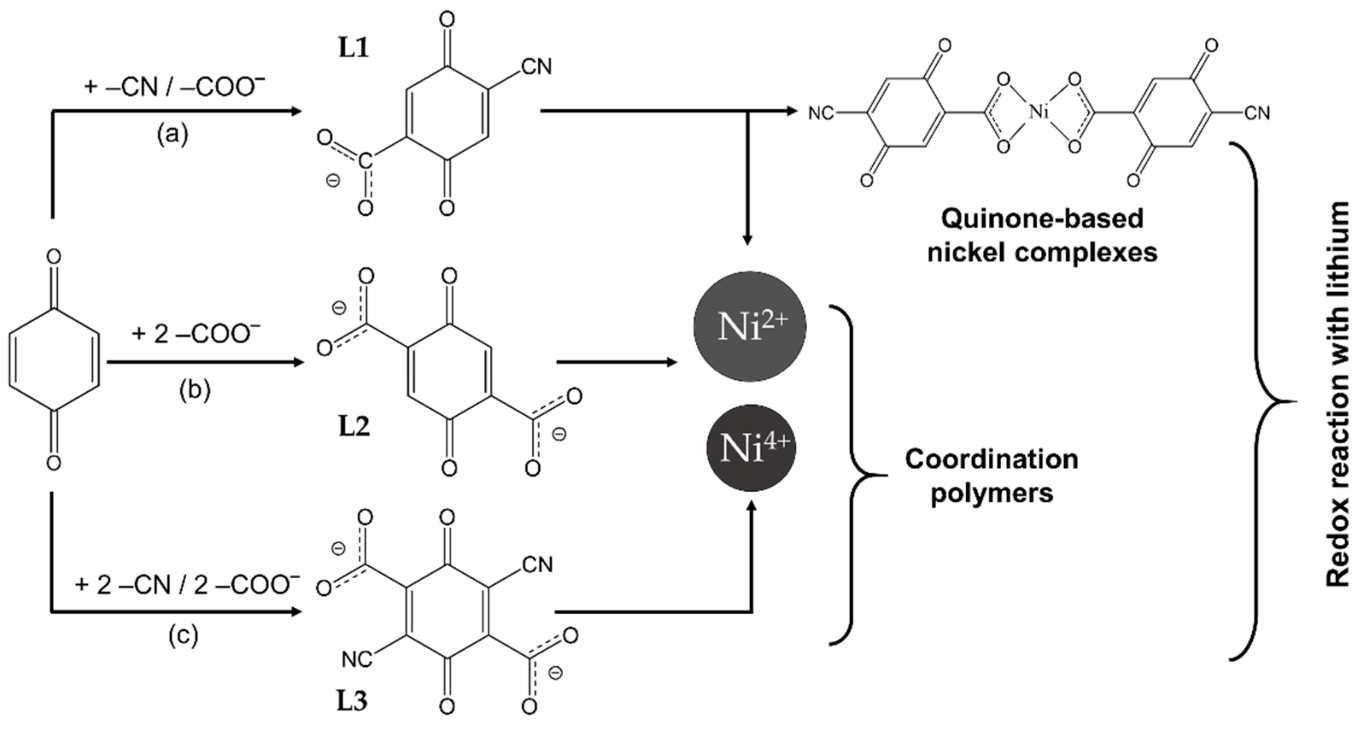
2.1. Transition Metal Complexes
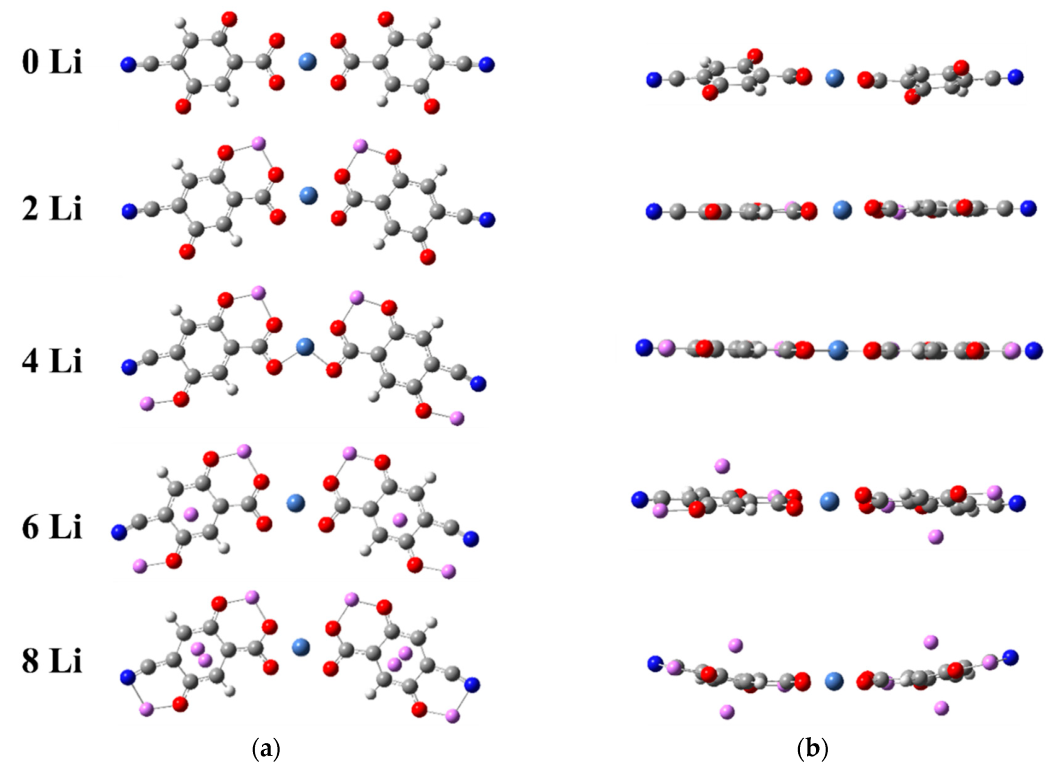
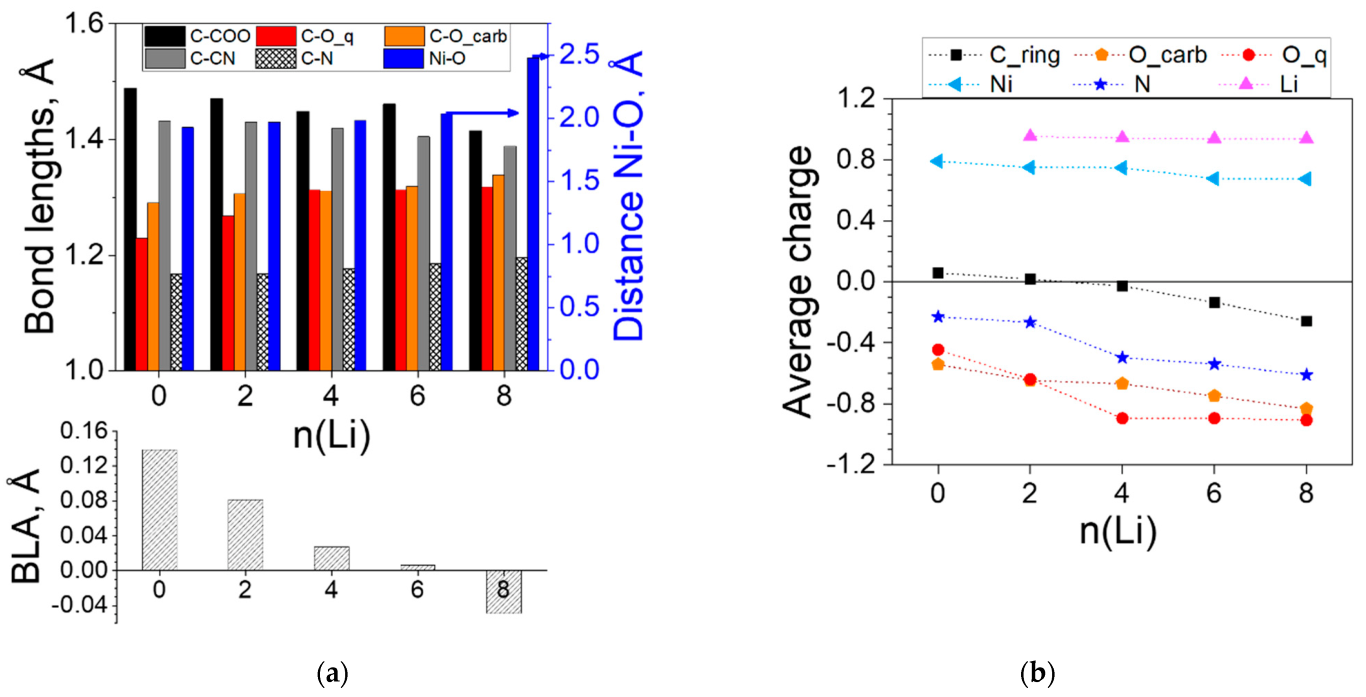
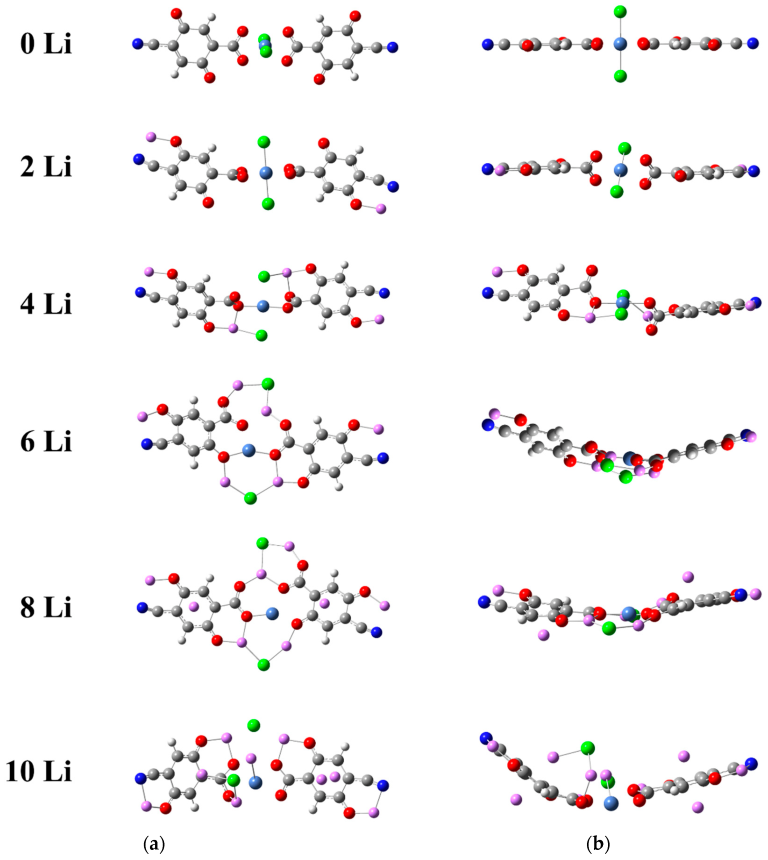
2.1.1. Lithiation of Ni(II)(L1)2
2.1.2. Lithiation of Ni(IV)(L1)2
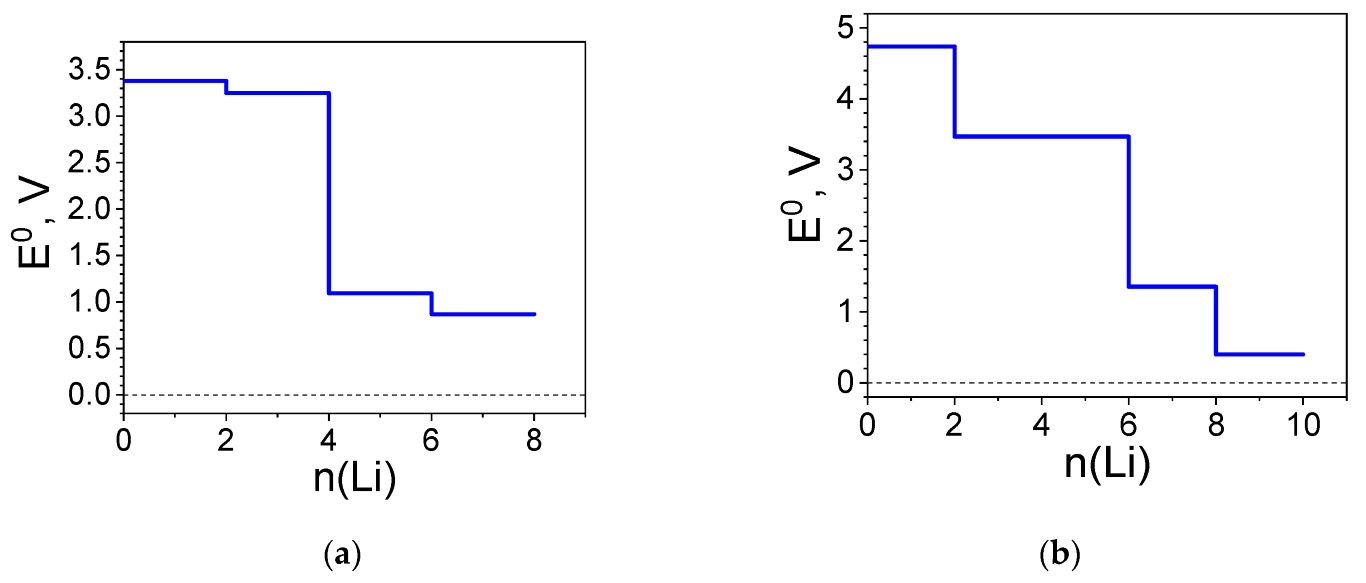
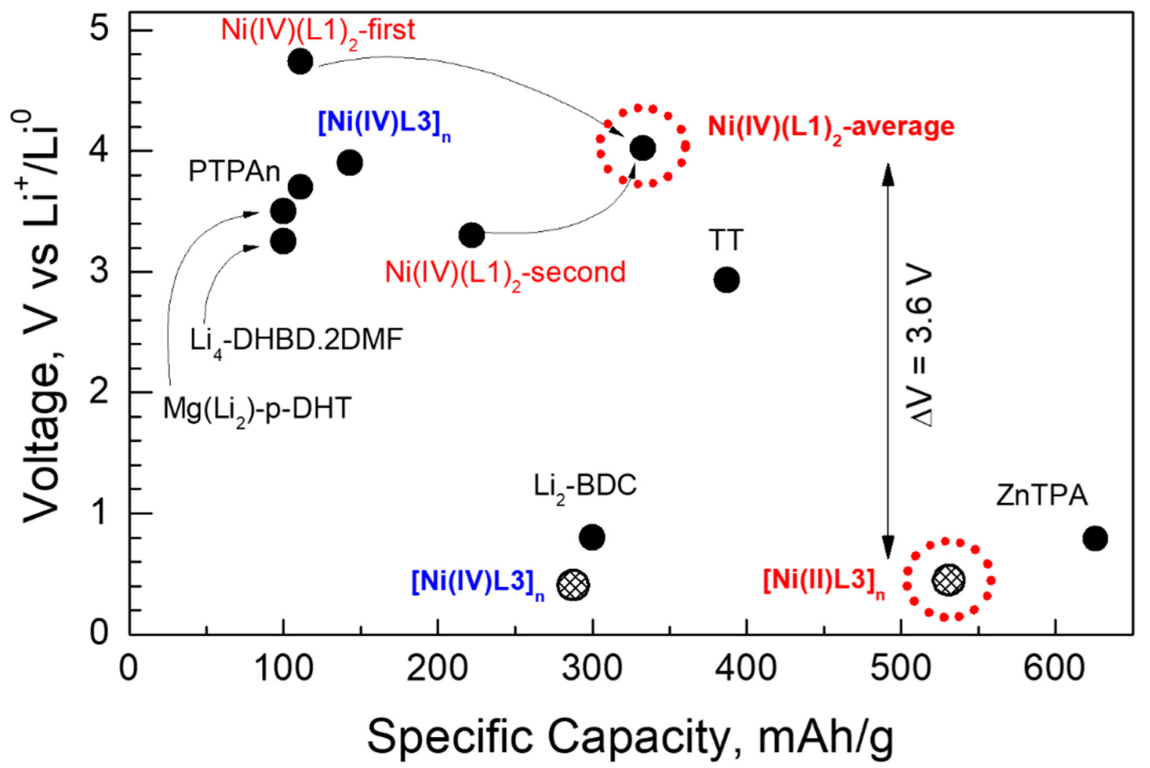
2.1.3. Redox Potentials of the Complexes Versus Li+/Li0
2.2. Coordination Polymers
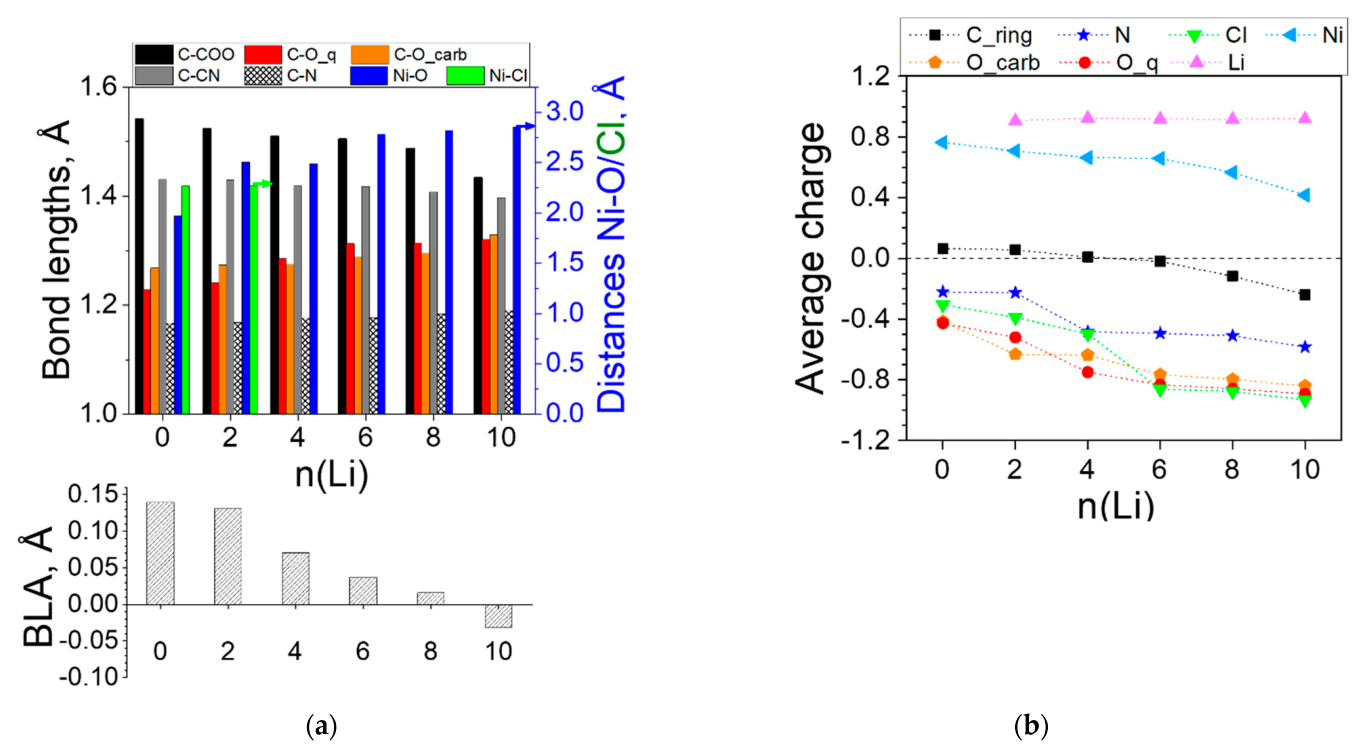
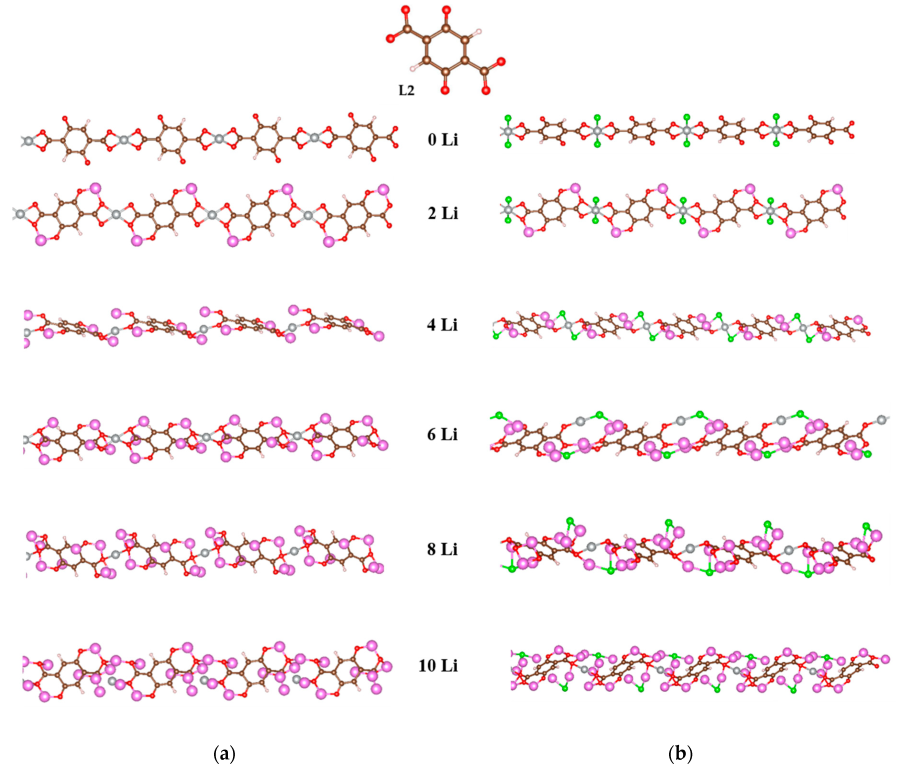
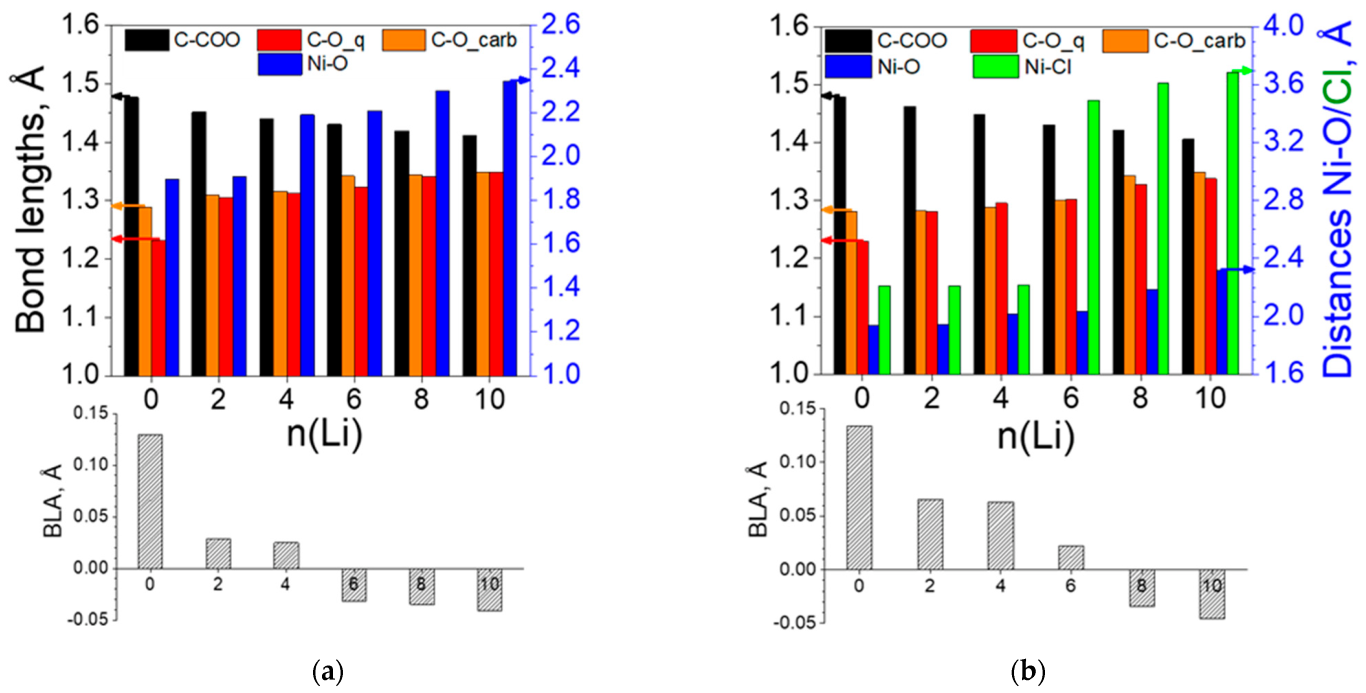
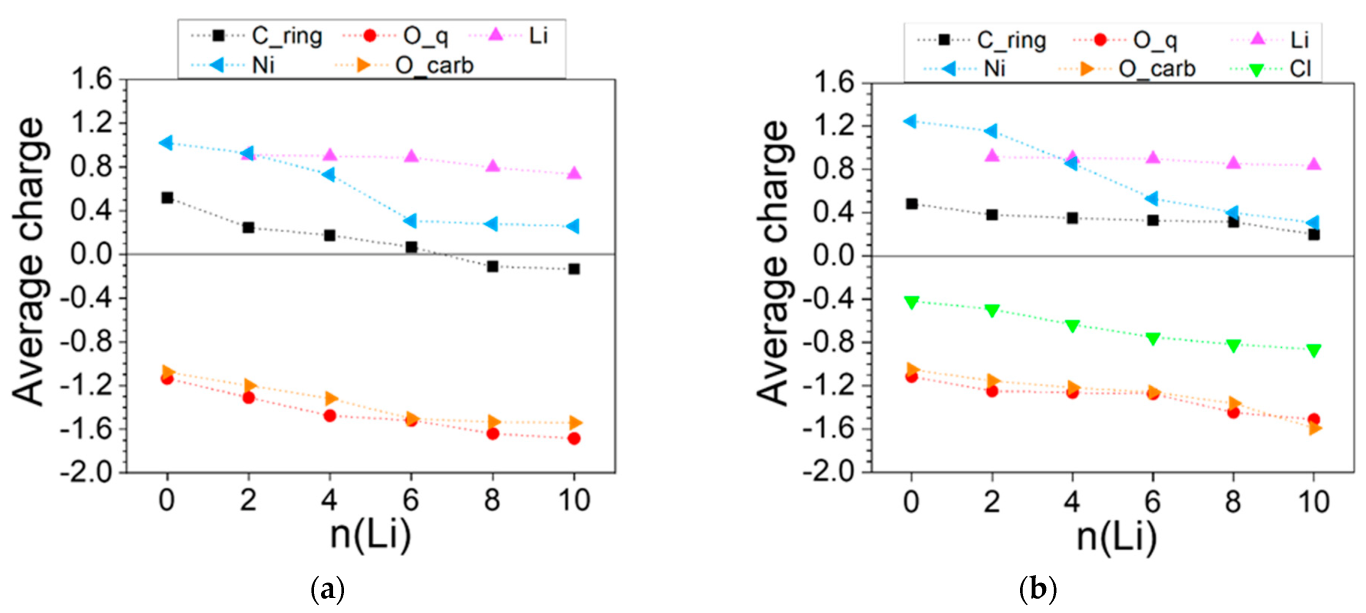
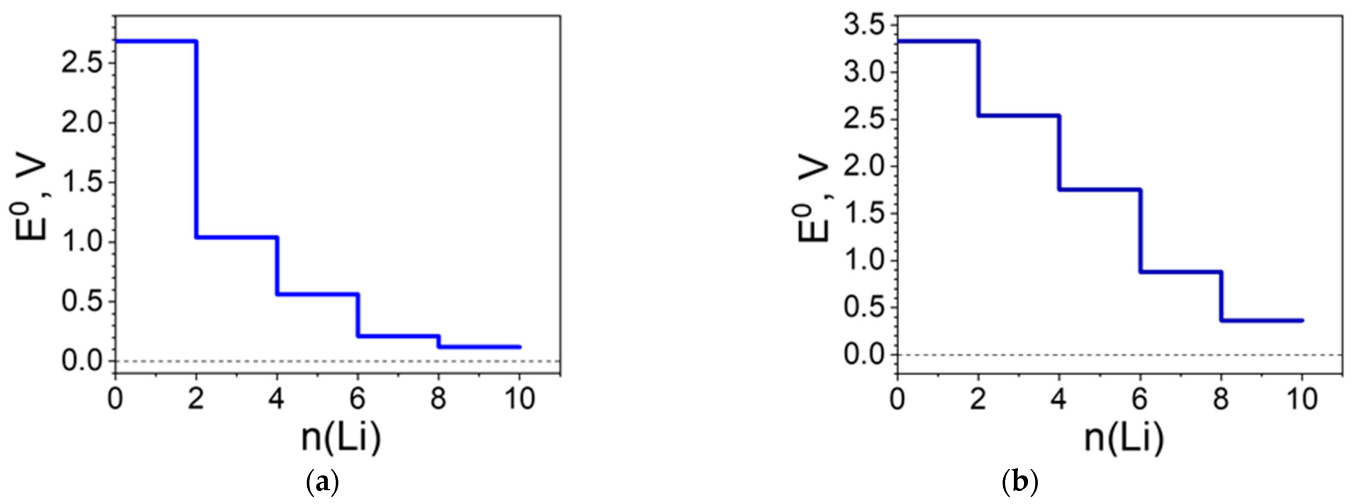
2.2.1. Coordination Polymers of Ni with 2,5-Dicarboxylato-1,4-benzoquinone (L2)
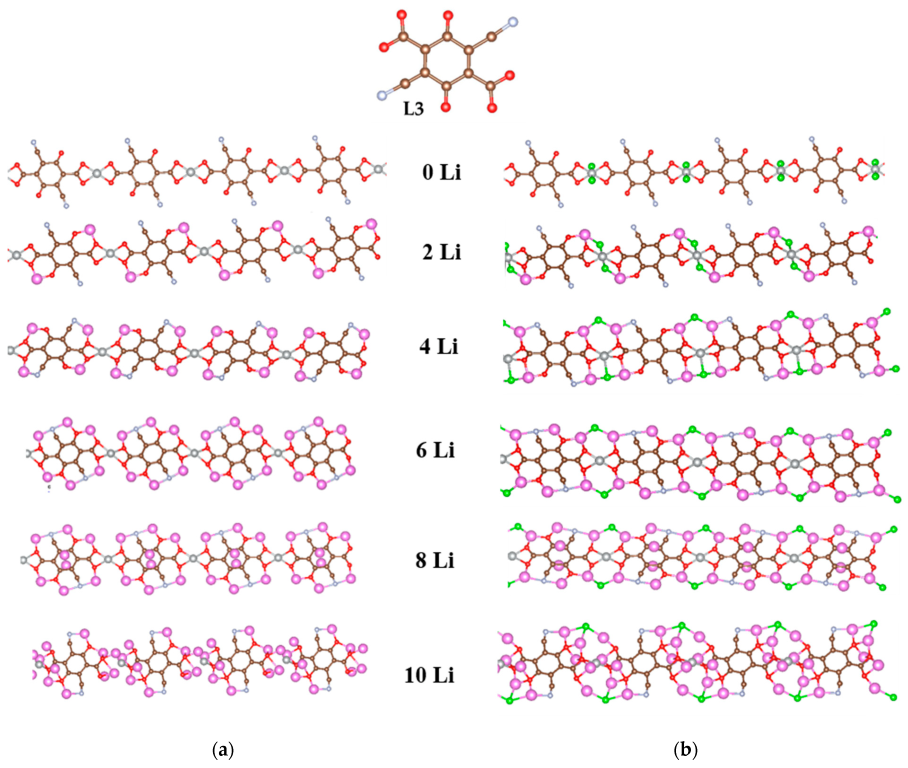
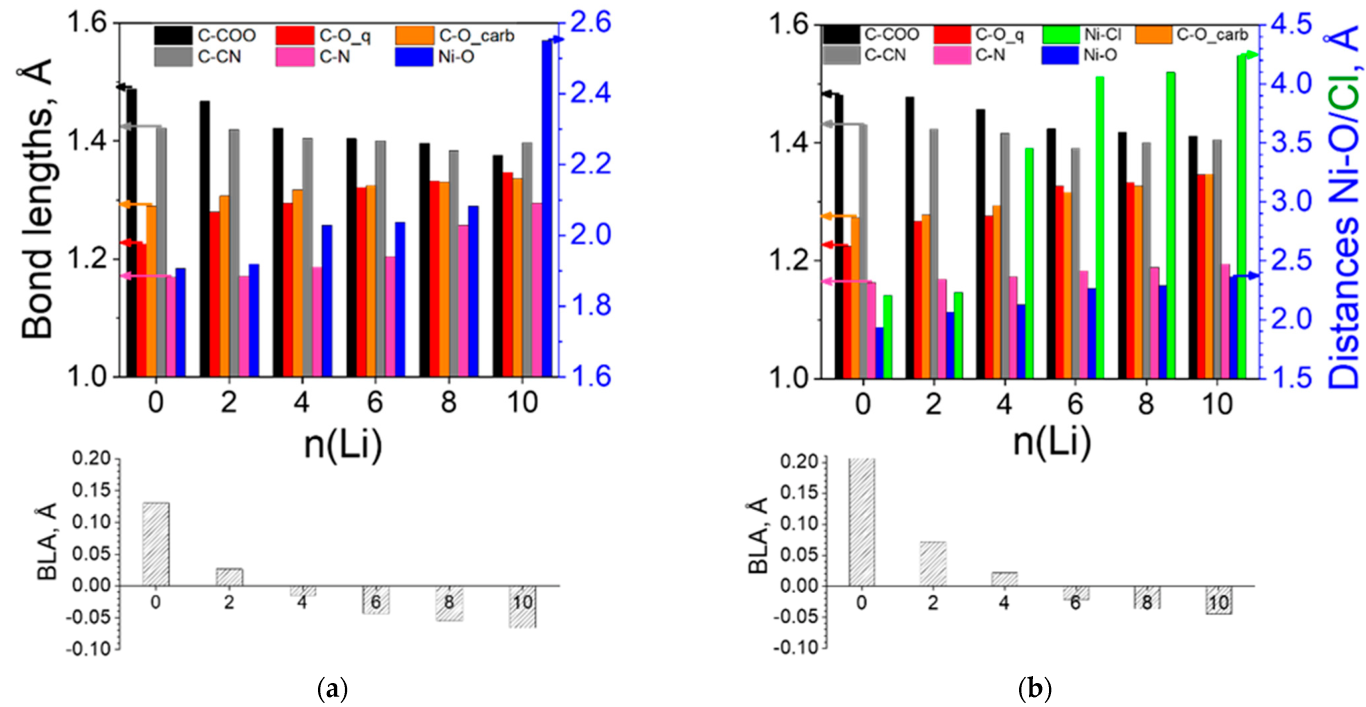
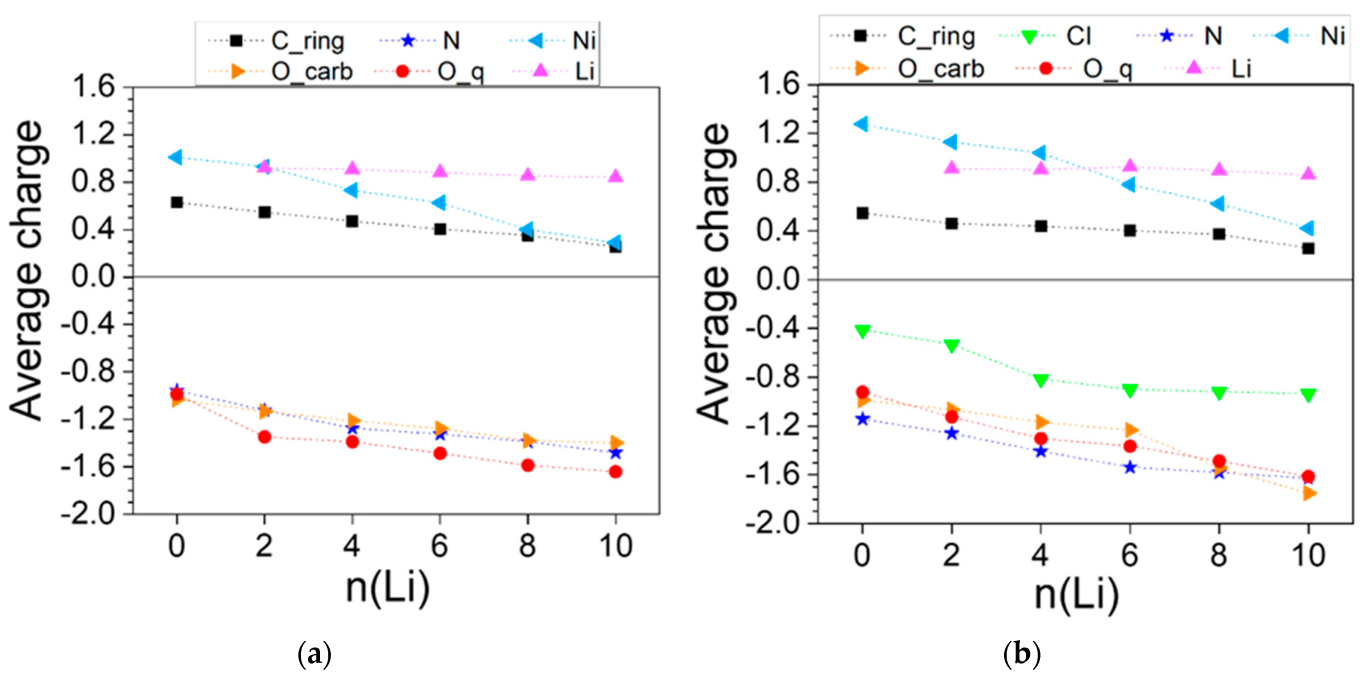
2.2.2. Coordination Polymers of Ni with 2,5-Dicarboxylato-3,6-dicyano-1,4-benzoquinone (L3)
2.3. Construction of an All-Organic Li-Ion Battery
3. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Larcher, D.; Tarascon, J.M. Towards greener and more sustainable batteries for electrical energy storage. Nat. Chem. 2015, 7, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Poizot, P.; Gaubicher, J.; Renault, S.; Dubois, L.; Liang, Y.; Yao, Y. Opportunities and Challenges for Organic Electrodes in Electrochemical Energy Storage. Chem. Rev. 2020, 120, 6490–6557. [Google Scholar] [CrossRef] [PubMed]
- Schon, T.B.; McAllister, B.T.; Li, P.-F.; Seferos, D.S. The rise of organic electrode materials for energy storage. Chem. Soc. Rev. 2016, 45, 6345–6404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Zhang, Q.; Li, L.; Niu, Z.; Chen, J. Design Strategies toward Enhancing the Performance of Organic Electrode Materials in Metal-Ion Batteries. Chemistry 2018, 4, 2786–2813. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Chen, J. Prospects of organic electrode materials for practical lithium batteries. Nat. Rev. Chem. 2020, 4, 127–142. [Google Scholar] [CrossRef]
- Esser, B.; Dolhem, F.; Becuwe, M.; Poizot, P.; Vlad, A.; Brandell, D. A perspective on organic electrode materials and technologies for next generation batteries. J. Power Sources 2021, 482, 228814. [Google Scholar] [CrossRef]
- Whittingham, M.S. Lithium Batteries and Cathode Materials. Chem. Rev. 2004, 104, 4271–4302. [Google Scholar] [CrossRef]
- Li, Q.; Zhou, D.; Zhang, L.; Ning, D.; Chen, Z.; Xu, Z.; Gao, R.; Liu, X.; Xie, D.; Schumacher, G.; et al. Tuning Anionic Redox Activity and Reversibility for a High-Capacity Li-Rich Mn-Based Oxide Cathode via an Integrated Strategy. Adv. Funct. Mater. 2019, 29, 1806706. [Google Scholar] [CrossRef]
- Gottis, S.; Barrès, A.-L.; Dolhem, F.; Poizot, P. Voltage Gain in Lithiated Enolate-Based Organic Cathode Materials by Isomeric Effect. ACS. Appl. Mater. Interfaces 2014, 6, 10870–10876. [Google Scholar] [CrossRef]
- Friebe, C.; Lex-Balducci, A.; Schubert, U.S. Sustainable Energy Storage: Recent Trends and Developments toward Fully Organic Batteries. ChemSusChem 2019, 12, 4093–4115. [Google Scholar] [CrossRef]
- Lakraychi, A.E.; Dolhem, F.; Vlad, A.; Becuwe, M. Organic Negative Electrode Materials for Metal-Ion and Molecular-Ion Batteries: Progress and Challenges from a Molecular Engineering Perspective. Adv. Energy Mater. 2021, 11, 2101562. [Google Scholar] [CrossRef]
- Wang, D.-Y.; Liu, R.; Guo, W.; Li, G.; Fu, Y. Recent advances of organometallic complexes for rechargeable batteries. Coord. Chem. Rev. 2021, 429, 213650. [Google Scholar] [CrossRef]
- Hogue, R.W.; Toghill, K.E. Metal Coordination Complexes in Non-Aqueous Redox Flow Batteries. Curr. Opin. Electrochem. 2019, 18, 37–45. [Google Scholar] [CrossRef]
- Hirao, T. Conjugated systems composed of transition metals and redox-active π-conjugated ligands. Coord. Chem. Rev. 2002, 226, 81–91. [Google Scholar] [CrossRef]
- Kaim, W.; Schwederski, B. Non-innocent ligands in bioinorganic chemistry—An overview. Coord. Chem. Rev. 2010, 254, 1580–1588. [Google Scholar] [CrossRef]
- Broere, D.L.J.; Plessius, R.; der Vlugt, J.I. New avenues for ligand-mediated processes–expanding metal reactivity by the use of redox-active catechol, o-aminophenol and o-phenylenediamine ligands. Chem. Soc. Rev. 2015, 44, 6886–6915. [Google Scholar] [CrossRef]
- Pierpont, C.G.; Buchanan, R.M. Transition metal complexes of o-benzoquinone, o-semiquinone, and catecholate ligands. Coord. Chem. Rev. 1981, 38, 45–87. [Google Scholar] [CrossRef]
- Kaim, W. The Transition Metal Coordination Chemistry of anion radicals. Coord. Chem. Rev. 1987, 76, 187–235. [Google Scholar] [CrossRef]
- Pierpont, C.G. Unique properties of transition metal quinone complexes of the MQ3 series. Coord. Chem. Rev. 2001, 219–221, 415–433. [Google Scholar] [CrossRef]
- Poddel’sky, A.I.; Cherkasov, V.K.; Abakumov, G.A. Transition metal complexes with bulky 4,6-di-tert-butyl-N-aryl(alkyl)-o-iminobenzoquinonato ligands: Structure, EPR and magnetism. Coord. Chem. Rev. 2009, 253, 291–324. [Google Scholar] [CrossRef]
- Kaim, W. Manifestations of Noninnocent Ligand Behavior. Inorg. Chem. 2011, 50, 9752–9765. [Google Scholar] [CrossRef]
- Tezgerevska, T.; Alley, K.G.; Boskovic, C. Valence tautomerism in metal complexes: Stimulated and reversible intramolecular electron transfer between metal centers and organic ligands. Coord. Chem. Rev. 2014, 268, 23–40. [Google Scholar] [CrossRef]
- Kaim, W.; Paretzki, A. Interacting metal and ligand based open shell systems: Challenges for experiment and theory. Coord. Chem. Rev. 2017, 344, 345–354. [Google Scholar] [CrossRef]
- Pashanova, K.I.; Poddel’sky, A.I.; Piskunov, A.V. Complexes of “late” transition metals of the 3d row based on functionalized o-iminobenzoquinone type ligands: Interrelation of molecular and electronic structure, magnetic behaviour. Coord. Chem. Rev. 2022, 459, 214399. [Google Scholar] [CrossRef]
- Liu, Q.; Sleightholme, A.; Shinkle, A.; Li, Y.; Thompson, L. Non-aqueous vanadium acetylacetonate electrolyte for redox flow batteries. Electrochem. Commun. 2009, 11, 2312–2315. [Google Scholar] [CrossRef]
- Liu, Q.; Shinkle, A.; Li, Y.; Monroe, C.; Thompson, L.; Sleightholme, A. Nonaqueous chromium acetylacetonate electrolyte for redox flow batteries. Electrochem. Commun. 2010, 12, 1634–1637. [Google Scholar] [CrossRef]
- Mun, J.; Lee, M.-J.; Park, J.; Oh, D.-J.; Lee, D.-Y.; Doo, S.-G. Non-Aqueous Redox Flow Batteries with Nickel and Iron Tris(2,2′-bipyridine) Complex Electrolyte. Electrochem. Solid-State Lett. 2012, 15, A80–A82. [Google Scholar] [CrossRef]
- Armstrong, C.; Toghill, K. Cobalt(II) complexes with azole-pyridine type ligands for non-aqueous redox-flow batteries: Tunable electrochemistry via structural modification. J. Power Sources 2017, 349, 121–129. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-H.; Kim, K.J.; Park, M.-S.; Lee, N.J.; Hwang, U.; Kim, H.; Kim, Y.-J. Development of metal-based electrodes for non-aqueous redox flow batteries. Electrochem. Commun. 2011, 13, 997–1000. [Google Scholar] [CrossRef]
- Burnea, F.; Shi, H.; Ko, K.; Lee, J. Reduction potential tuning of first row transition metal MIII/MII (M = Cr, Mn, Fe, Co, Ni) hexadentate complexes for viable aqueous redox flow battery catholytes: A DFT study. Electrochim. Acta 2017, 246, 156–164. [Google Scholar] [CrossRef]
- Hwang, S.; Kim, H.; Ryu, J.H.; Oh, S.M. Ni(II)-chelated thio-crown complex as a single redox couple for non-aqueous flow batteries. Electrochem. Commun. 2017, 85, 36–39. [Google Scholar] [CrossRef]
- Alt, H.; Binder, H.; Kőhling, A.; Sandstede, G. Investigation into the use of quinone compounds-for battery cathodes. Electrochim. Acta 1972, 17, 873–887. [Google Scholar] [CrossRef]
- Häupler, B.; Wild, A.; Schubert, U. Carbonyls: Powerful Organic Materials for Secondary Batteries. Adv. Energy Mater. 2015, 5, 1402034. [Google Scholar] [CrossRef]
- Araujo, R.B.; Banerjee, A.; Panigrahi, P.; Yang, L.; Strømme, M.; Sjödin, M.; Araujo, C.M.; Ahuja, R. Designing strategies to tune reduction potential of organic molecules for sustainable high capacity battery application. J. Mater. Chem. A 2017, 5, 4430–4454. [Google Scholar] [CrossRef]
- Lee, S.; Hong, J.; Kang, K. Redox-Active Organic Compounds for Future Sustainable Energy Storage System. Adv. Energy Mater. 2020, 10, 2001445. [Google Scholar] [CrossRef]
- Lin, Z.; Shi, H.; Lin, L.; Yang, X.; Wu, W.; Sun, X. A high capacity small molecule quinone cathode for rechargeable aqueous zinc-organic batteries. Nat. Commun. 2021, 12, 4424. [Google Scholar] [CrossRef]
- Miao, L.; Liu, L.; Shang, Z.; Li, Y.; Lu, Y.; Cheng, F.; Chen, J. The structure–electrochemical property relationship of quinone electrodes for lithium-ion batteries. Phys. Chem. Chem. Phys. 2018, 20, 13478–13484. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zeng, R.; Nan, J.; Shu, D.; Qiu, Y.; Chou, S.-L. Quinone Electrode Materials for Rechargeable Lithium/Sodium Ion Batteries. Adv. Energy Mater. 2017, 20, 1700278. [Google Scholar] [CrossRef]
- Emanuelsson, R.; Sterby, M.; Strømme, M.; Sjödin, M. An All-Organic Proton Battery. J. Am. Chem. Soc. 2017, 139, 4828–4834. [Google Scholar] [CrossRef]
- Oka, K.; Strietzel, C.; Emanuelsson, R.; Nishide, H.; Oyaizu, K.; Strømme, M.; Sjödin, M. Characterization of PE-DOT-Quinone conducting redox polymers in water-in-salt electrolytes for safe and high-energy Li-ion batteries. Electrochem. Commun. 2019, 105, 106489. [Google Scholar] [CrossRef]
- Ding, B.; Solomon, M.; Leong, C.; D’Alessandro, D. Redox-active ligands: Recent advances towards their incorporation into coordination polymers and metal-organic frameworks. Coord. Chem. Rev. 2021, 439, 213891. [Google Scholar] [CrossRef]
- Baumann, A.E.; Burns, D.A.; Liu, B.; Thoi, V.S. Metal-organic framework functionalization and design strategies for advanced electrochemical energy storage devices. Commun. Chem. 2019, 2, 86. [Google Scholar] [CrossRef] [Green Version]
- Rasheev, H.; Seremak, A.; Stoyanova, R.; Tadjer, A. Redox Hyperactive MOF for Li+, Na+ and Mg2+ Storage. Molecules 2022, 27, 586. [Google Scholar] [CrossRef]
- Manecke, G.; Storck, W. Polyvinylanthraquinone redox resins (electron exchange polymers). J. Polym. Sci. Part C Polym. Symp. 1963, 4, 1457–1466. [Google Scholar] [CrossRef]
- Xiang, J.; Chang, C.; Li, M.; Wu, S.; Yuan, L.; Sun, J. A Novel Coordination Polymer as Positive Electrode Material for Lithium Ion Battery. Cryst. Growth Des. 2008, 8, 280–282. [Google Scholar] [CrossRef]
- Chang, C.-H.; Li, A.-C.; Popovs, I.; Kaveevivitchai, W.; Chen, J.-L.; Chou, K.-C.; Kuo, T.-S.; Chen, T.-H. Elucidating Metal and Ligand Redox Activities of Copper-Benzoquinoid Coordination Polymer as Cathode for Lithium-Ion Batteries. J. Mater. Chem. A 2019, 7, 23770–23774. [Google Scholar] [CrossRef]
- Lakraychi, A.E.; Deunf, E.; Fahsi, K.; Jimenez, P.; Bonnet, J.-P.; Djedaini-Pilard, F.; Bécuwe, M.; Poizot, P.; Dolhem, F. An air-stable lithiated cathode material based on a 1,4-benzenedisulfonate backbone for organic Li-ion batteries. J. Mater. Chem. A 2018, 6, 19182–19189. [Google Scholar] [CrossRef]
- Xie, J.; Lu, Y. Towards practical organic batteries. Nat. Mater. 2021, 20, 581–583. [Google Scholar] [CrossRef]
- Kwon, J.E.; Hyun, C.-S.; Ryu, Y.J.; Lee, J.; Min, D.J.; Park, M.J.; An, B.-K.; Park, S.Y. Triptycene-Based Quinone Molecules Showing Multi-Electron Redox Reactions for Large Capacity and High Energy Organic Cathode Materials in Li-Ion Batteries. J. Mater. Chem. A 2018, 6, 3134–3140. [Google Scholar] [CrossRef]
- Cao, K.; Jing, T.; Yang, L.; Jiao, L. Recent progress in conversion reaction metal oxide anodes for Li-ion batteries. Mater. Chem. Front. 2017, 1, 2213–2242. [Google Scholar] [CrossRef]
- Qin, J.; Lan, Q.; Liu, N.; Men, F.; Wang, X.; Song, Z.; Zhan, H. A Metal-Free Battery with Pure Ionic Liquid Electrolyte. iScience 2019, 15, 16–27. [Google Scholar] [CrossRef] [Green Version]
- Jouhara, A.; Dupré, N.; Gaillot, A.-C.; Guyomard, D.; Dolhem, F.; Poizot, P. Raising the redox potential in carboxyphenolate-based positive organic materials via cation substitution. Nat. Commun. 2018, 9, 4401. [Google Scholar] [CrossRef] [Green Version]
- Armand, M.; Grugeon, S.; Vezin, H.; Laruelle, S.; Ribiere, P.; Poizot, P.; Tarascon, J.M. Conjugated dicarboxylate anodes for Li-ion batteries. Nat. Mater. 2009, 8, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Wild, A.; Strumpf, M.; Häupler, B.; Hager, M.; Schubert, U. All-Organic Battery Composed of Thianthrene- and TCAQ-Based Polymers. Adv. Energy Mater. 2016, 7, 1601415. [Google Scholar] [CrossRef]
- Wang, L.; Zou, J.; Chen, S.; Yang, J.; Qing, F.; Gao, P.; Li, J. Zinc Terephthalates ZnC8H4O4 as Anodes for Lithium Ion Batteries. Electrochim. Acta 2017, 235, 304–310. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision, B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian-basis sets for molecular calculations. 1. 2nd row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Raghavachari, K.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. 20. Basis set for correlated wave-functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Wachters, A.J.H. Gaussian basis set for molecular wavefunctions containing third-row atoms. J. Chem. Phys. 1970, 52, 1033. [Google Scholar] [CrossRef]
- Hay, P.J. Gaussian basis sets for molecular calculations–representation of 3D orbitals in transition-metal atoms. J. Chem. Phys. 1977, 66, 4377–4384. [Google Scholar] [CrossRef]
- Weinhold, F.; Foster, J. Natural hybrid orbitals. J. Am. Chem. Soc. 1980, 102, 7211–7218. [Google Scholar] [CrossRef]
- Glendening, E.; Reed, A.; Carpenter, J.; Weinhold, F. NBO; Version 3.1; Gaussian Inc.: Pittsburgh, PA, USA, 2003. [Google Scholar]
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B Condens. Matter Mater. Phys. 1993, 47, 558. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B Condens. Matter Mater. Phys. 1996, 54, 11169. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mat. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865, Erratum in Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef] [Green Version]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Tang, W.; Sanville, E.; Henkelman, G. A Grid-Based Bader Analysis Algorithm Without Lattice Bias. J. Phys. Condens. Matter 2009, 21, 084204. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules. Acc. Chem. Res. 1985, 18, 9–15. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Aydinol, M.K.; Kohan, A.F.; Ceder, G.; Cho, K.; Joannopoulos, J. Ab Initio Study of Lithium Intercalation in Metal Oxides and Metal Dichalcogenides. Phys. Rev. B Condens. Matter Mater. Phys. 1997, 56, 1354–1365. [Google Scholar] [CrossRef] [Green Version]
- Araujo, R.B.; Banerjee, A.; Ahuja, R. Divulging the Hidden Capacity and Sodiation Kinetics of NaxC6Cl4O2: A High Voltage Organic Cathode for Sodium Rechargeable Batteries. J. Phys. Chem. C 2017, 121, 14027–14036. [Google Scholar] [CrossRef]
- Xu, D.; Liang, M.; Qi, S.; Sun, W.; Lv, L.-P.; Du, F.-H.; Wang, B.; Chen, S.; Wang, Y.; Yu, Y. The Progress and Prospect of Tunable Organic Molecules for Organic Lithium-Ion Batteries. ACS Nano 2021, 15, 47–80. [Google Scholar] [CrossRef]
- Lee, J.; Kim, H.; Park, M.J. Long-life, high-rate lithium-organic batteries based on naphthoquinone derivatives. Chem. Mater. 2016, 28, 2408–2416. [Google Scholar] [CrossRef]
- Yao, Z.; Tang, W.; Wang, X.; Wang, C.; Yang, C.; Fan, C. Synthesis of 1,4-benzoquinone dimer as a high-capacity (501 mAhg−1) and high-energy-density (>1000 Whkg−1) organic cathode for organic Li-Ion full batteries. J. Power Sources 2020, 448, 227456. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| n(Li) | 0 | 2 | 4 | 6 | 8 |
|---|---|---|---|---|---|
| multiplicity | S | T | S | S | S |
| l mol, Å | 18.6 | 18.5 | 18.2 | 18.1 | 18.0 |
| n(Li) | 0 | 2 | 4 | 6 | 8 | 10 |
|---|---|---|---|---|---|---|
| multiplicity | S | T | S | S | S | S |
| lmol, Å | 19.0 | 19.0 | 18.8 | 18.0 | 17.3 | 17.0 |
| n(Li) | 0 | 2 | 4 | 6 | 8 | 10 |
|---|---|---|---|---|---|---|
| [Ni(II)L2]n leu, Å | 8.86 | 8.87 | 8.91 | 8.56 | 8.56 | 8.73 |
| [Ni(II)L2]n leu, Å | 8.91 | 8.83 | 8.59 | 8.69 | 8.70 | 8.70 |
| n(Li) | 0 | 2 | 4 | 6 | 8 | 10 |
|---|---|---|---|---|---|---|
| [Ni(II)L3]n leu, Å | 8.98 | 8.97 | 8.93 | 8.95 | 8.87 | 8.24 |
| [Ni(IV)L3]n leu, Å | 8.93 | 8.92 | 8. 96 | 8.94 | 8.95 | 8.97 |
| Coordination Compounds | ΔE0 (Initial), V | nmax | Capacity, mA.h.g−1 | Energy Density, W.h.g−1 |
|---|---|---|---|---|
| Ni(II)(L1)2 | 3.38 | 8 | 522 | 1121 |
| Ni(IV)(L1)2 | 4.74 | 10 | 556 | 1495 |
| [Ni(II)L2]n | 2.68 | 10 | 1 061 | 977 |
| [Ni(IV)L2]n | 3.33 | 10 | 828 | 1467 |
| [Ni(II)L3]n | 3.17 | 10 | 885 | 1073 |
| [Ni(IV)L3]n | 3.95 | 10 | 717 | 1469 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Danchovski, Y.; Rasheev, H.; Stoyanova, R.; Tadjer, A. Molecular Engineering of Quinone-Based Nickel Complexes and Polymers for All-Organic Li-Ion Batteries. Molecules 2022, 27, 6805. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27206805
Danchovski Y, Rasheev H, Stoyanova R, Tadjer A. Molecular Engineering of Quinone-Based Nickel Complexes and Polymers for All-Organic Li-Ion Batteries. Molecules. 2022; 27(20):6805. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27206805
Chicago/Turabian StyleDanchovski, Yanislav, Hristo Rasheev, Radostina Stoyanova, and Alia Tadjer. 2022. "Molecular Engineering of Quinone-Based Nickel Complexes and Polymers for All-Organic Li-Ion Batteries" Molecules 27, no. 20: 6805. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27206805