
Antileishmanial Anthracene Endoperoxides: Efficacy In Vitro, Mechanisms and Structure-Activity Relationships

 , , and
, , and
Abstract
:
1. Introduction
2. Results
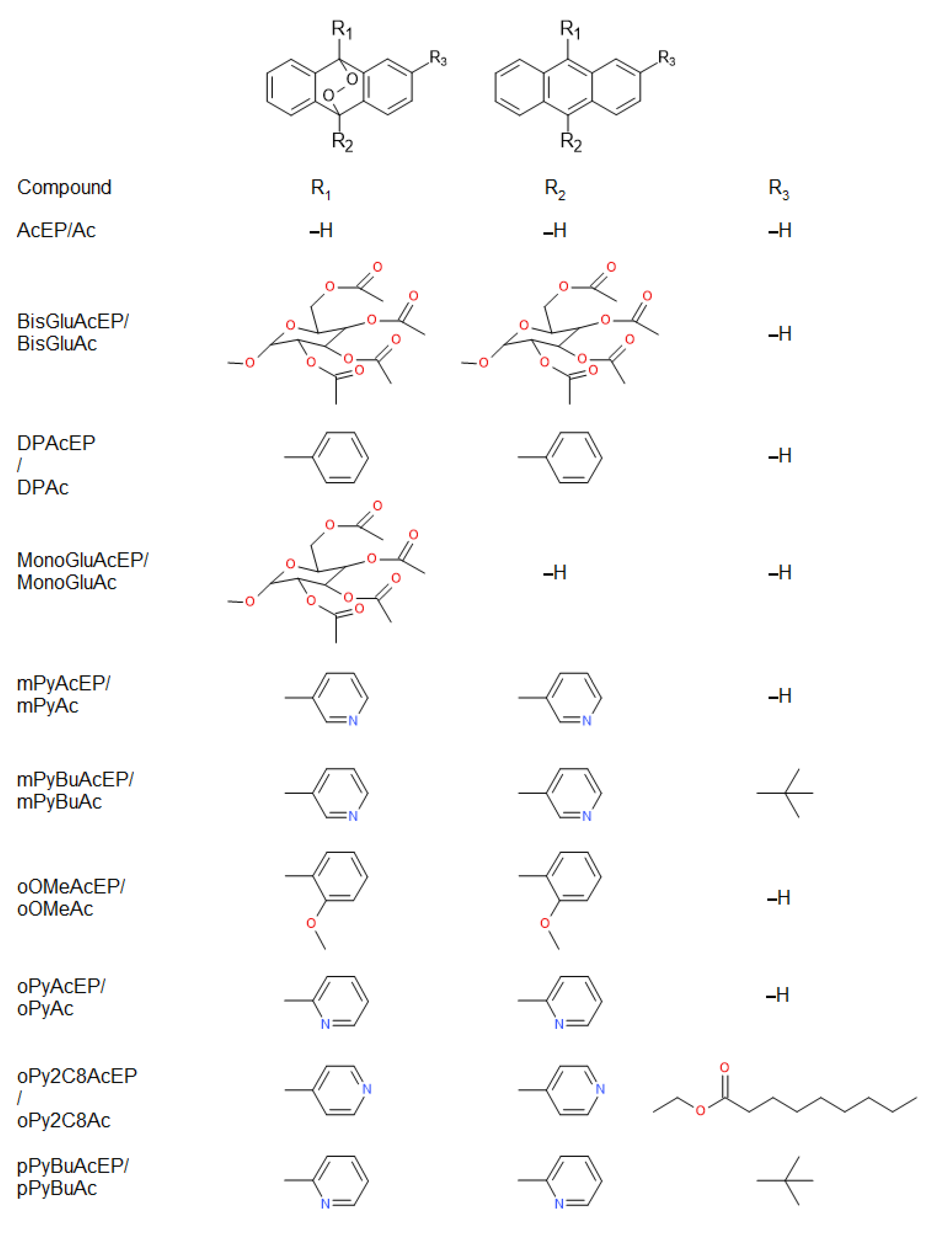
2.1. Compounds and Structural Properties
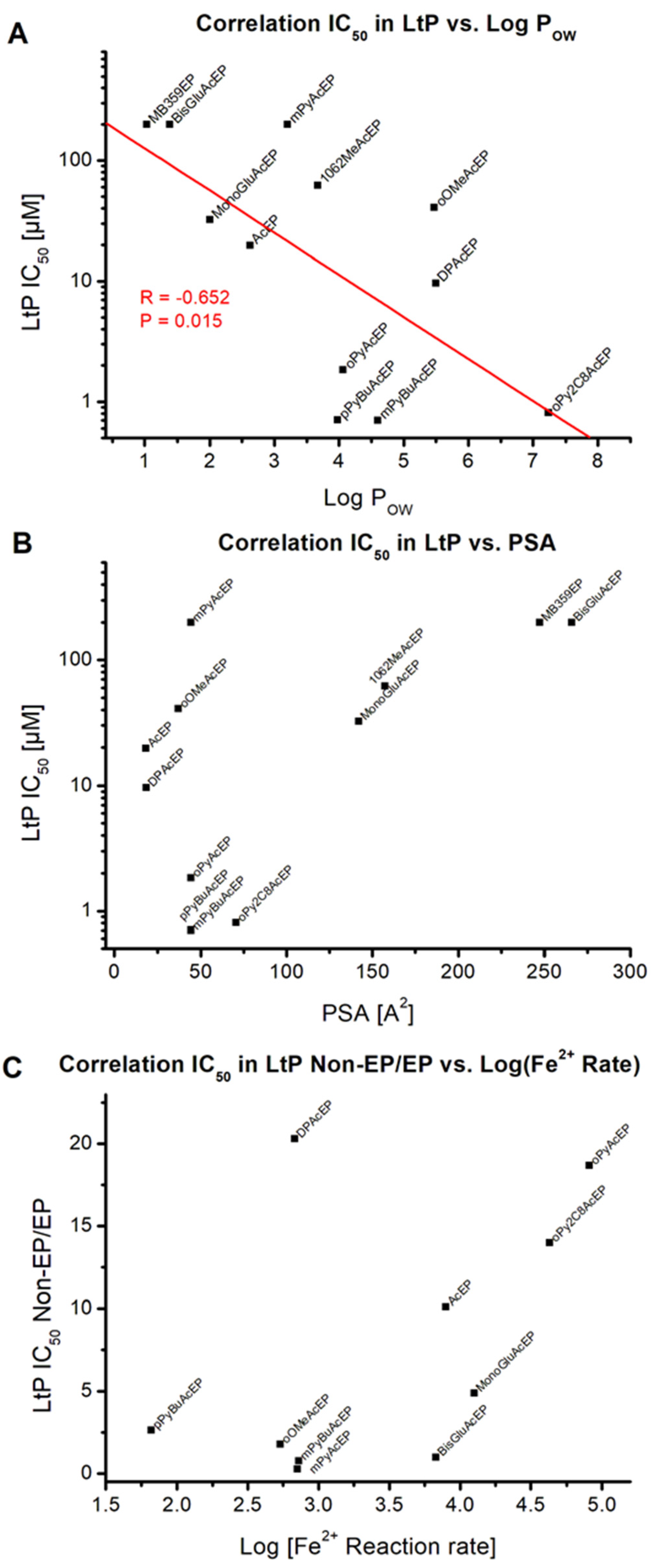
2.2. Influence on LtP and J774 Viability
2.3. Activation of AcEP by Iron in Chemical Systems
2.4. Activation of AcEP by Cycloreversion
2.5. Role of Thiols and Iron in AcEP Actions in LtP
2.6. Influence on Oxygen Consumption and Superoxide Formation
3. Discussion
4. Materials and Methods
4.1. Compounds and Reagents
4.2. Cell Culture
4.2.1. Leishmania Tarentolae Promastigotes (LtP) Cell Culture
4.2.2. J774 Macrophage Cell Culture
4.3. Viability Assays
4.3.1. Viability Assay for LtP
4.3.2. Viability Assay for J774 Macrophages
4.4. Formation of the Xylenol Orange/Fe3+ Complex by EP
4.5. UV Detection of Cycloreversion in AcEP
4.6. Electron Paramagnetic Resonance (EPR) Spectroscopy
4.6.1. Spin Trapping
4.6.2. Detection of Superoxide Radicals by CMH
4.7. O2 consumption of Cells
4.8. Structures
4.9. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Kaye, P.; Scott, P. Leishmaniasis: Complexity at the host-pathogen interface. Nat. Rev. Microbiol. 2011, 9, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Torres-Guerrero, E.; Quintanilla-Cedillo, M.R.; Ruiz-Esmenjaud, J.; Arenas, R. Leishmaniasis: A review. F1000Research 2017, 6, 750. [Google Scholar] [CrossRef]
- Akhoundi, M.; Kuhls, K.; Cannet, A.; Votypka, J.; Marty, P.; Delaunay, P.; Sereno, D. A historical overview of the classification, evolution, and dispersion of Leishmania parasites and sandflies. PLoS Negl. Trop. Dis. 2016, 10, e0004349. [Google Scholar] [CrossRef]
- WHO. Global leishmaniasis update, 2006–2015: A turning point in leishmaniasis surveillance. Wkly. Epid. Rec. 2017, 92, 557–572. [Google Scholar]
- Alvar, J.; Arana, B. Leishmaniasis, impact and therapeutic needs. In Drug Discovery for Leishmaniasis; Rivas, L., Gil, C., Eds.; The Royal Society of Chemistry: London, UK, 2018; pp. 3–23. [Google Scholar]
- WHO. Control of the Leishmaniasis; WHO Technical Report Series 949; WHO: Geneva, Switzerland, 2010; pp. 1–186. [Google Scholar]
- Mann, S.; Frasca, K.; Scherrer, S.; Henao-Martinez, A.F.; Newman, S.; Ramanan, P.; Suarez, J.A. A review of leishmaniasis: Current knowledge and future directions. Curr. Trop. Med. Rep. 2021, 8, 121–132. [Google Scholar] [CrossRef]
- Briones Nieva, C.A.; Cid, A.G.; Romero, A.I.; Garcia-Bustos, M.F.; Villegas, M.; Bermudez, J.M. An appraisal of the scientific current situation and new perspectives in the treatment of cutaneous leishmaniasis. Acta Trop. 2021, 221, 105988. [Google Scholar] [CrossRef]
- Zulfiqar, B.; Shelper, T.B.; Avery, V.M. Leishmaniasis drug discovery: Recent progress and challenges in assay development. Drug Discov. Today 2017, 22, 1516–1531. [Google Scholar] [CrossRef]
- Hendrickx, S.; Van Bockstal, L.; Caljon, G.; Maes, L. In-depth comparison of cell-based methodological approaches to determine drug susceptibility of visceral Leishmania isolates. PLoS Negl. Trop. Dis. 2019, 13, e0007885. [Google Scholar] [CrossRef] [Green Version]
- Dighal, A.; De Sarkar, S.; Gille, L.; Chatterjee, M. Can the iron content of culture media impact on the leishmanicidal effect of artemisinin? Free Radic. Res. 2021, 55, 282–295. [Google Scholar] [CrossRef]
- Dong, Y.; Vennerstrom, J.L. Mechanisms of in situ activation for peroxidic antimalarials. Redox Rep. 2003, 8, 284–288. [Google Scholar] [CrossRef]
- Sen, R.; Chatterjee, M. Plant derived therapeutics for the treatment of Leishmaniasis. Phytomedicine 2011, 18, 1056–1069. [Google Scholar] [CrossRef]
- Machin, L.; Napoles, R.; Gille, L.; Monzote, L. Leishmania amazonensis response to artemisinin and derivatives. Parasitol. Int. 2021, 80, 102218. [Google Scholar] [CrossRef]
- Loo, C.S.; Lam, N.S.; Yu, D.; Su, X.Z.; Lu, F. Artemisinin and its derivatives in treating protozoan infections beyond malaria. Pharmacol. Res. 2017, 117, 192–217. [Google Scholar] [CrossRef] [Green Version]
- Geroldinger, G.; Tonner, M.; Hettegger, H.; Bacher, M.; Monzote, L.; Walter, M.; Staniek, K.; Rosenau, T.; Gille, L. Mechanism of ascaridole activation in Leishmania. Biochem. Pharmacol. 2017, 132, 48–62. [Google Scholar] [CrossRef]
- Geroldinger, G.; Tonner, M.; Quirgst, J.; Walter, M.; De Sarkar, S.; Machin, L.; Monzote, L.; Stolze, K.; Duvigneau, J.C.; Staniek, K.; et al. Activation of artemisinin and heme degradation in Leishmania tarentolae promastigotes: A possible link. Biochem. Pharmacol. 2020, 173, 113737. [Google Scholar] [CrossRef]
- Mukanganyama, S.; Naik, Y.S.; Widersten, M.; Mannervik, B.; Hasler, J.A. Proposed reductive metabolism of artemisinin by glutathione transferases in vitro. Free Radic. Res. 2001, 35, 427–434. [Google Scholar] [CrossRef]
- Creek, D.J.; Ryan, E.; Charman, W.N.; Chiu, F.C.; Prankerd, R.J.; Vennerstrom, J.L.; Charman, S.A. Stability of peroxide antimalarials in the presence of human hemoglobin. Antimicrob. Agents Chemother. 2009, 53, 3496–3500. [Google Scholar] [CrossRef] [Green Version]
- Cortes, S.; Albuquerque, A.; Cabral, L.I.; Lopes, L.; Campino, L.; Cristiano, M.L. In vitro susceptibility of Leishmania infantum to artemisinin derivatives and selected trioxolanes. Antimicrob. Agents Chemother. 2015, 59, 5032–5035. [Google Scholar] [CrossRef] [Green Version]
- Fudickar, W.; Linker, T. Novel anthracene materials for applications in lithography and reversible photoswitching by light and air. Langmuir 2010, 26, 4421–4428. [Google Scholar] [CrossRef]
- Fudickar, W.; Linker, T. Structural motives controlling the binding affinity of 9,10-bis(methylpyridinium)anthracenes towards DNA. Bioorg. Med. Chem. 2020, 28, 115432. [Google Scholar] [CrossRef]
- Geroldinger, G.; Tonner, M.; Fudickar, W.; De Sarkar, S.; Dighal, A.; Monzote, L.; Staniek, K.; Linker, T.; Chatterjee, M.; Gille, L. Activation of anthracene endoperoxides in Leishmania and impairment of mitochondrial functions. Molecules 2018, 23, 1680. [Google Scholar] [CrossRef] [PubMed]
- Caridha, D.; Vesely, B.; van Bocxlaer, K.; Arana, B.; Mowbray, C.E.; Rafati, S.; Uliana, S.; Reguera, R.; Kreishman-Deitrick, M.; Sciotti, R. Route map for the discovery and pre-clinical development of new drugs and treatments for cutaneous leishmaniasis. Int. J. Parasitol. Drugs Drug Resist. 2019, 11, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Taylor, V.M.; Munoz, D.L.; Cedeno, D.L.; Velez, I.D.; Jones, M.A.; Robledo, S.M. Leishmania tarentolae: Utility as an in vitro model for screening of antileishmanial agents. Exp. Parasitol. 2010, 126, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Rudrapal, M.; Chetia, D. Endoperoxide antimalarials: Development, structural diversity and pharmacodynamic aspects with reference to 1,2,4-trioxane-based structural scaffold. Drug Des. Dev. Ther. 2016, 10, 3575–3590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fugi, M.A.; Wittlin, S.; Dong, Y.; Vennerstrom, J.L. Probing the antimalarial mechanism of artemisinin and OZ277 (arterolane) with nonperoxidic isosteres and nitroxyl radicals. Antimicrob. Agents Chemother. 2010, 54, 1042–1046. [Google Scholar] [CrossRef] [Green Version]
- Flannery, A.R.; Renberg, R.L.; Andrews, N.W. Pathways of iron acquisition and utilization in Leishmania. Curr. Opin. Microbiol. 2013, 16, 716–721. [Google Scholar] [CrossRef] [Green Version]
- Miguel, D.C.; Flannery, A.R.; Mittra, B.; Andrews, N.W. Heme uptake mediated by LHR1 is essential for Leishmania amazonensis virulence. Infect. Immun. 2013, 81, 3620–3626. [Google Scholar] [CrossRef] [Green Version]
- Hartwig, C.L.; Rosenthal, A.S.; D’Angelo, J.; Griffin, C.E.; Posner, G.H.; Cooper, R.A. Accumulation of artemisinin trioxane derivatives within neutral lipids of Plasmodium falciparum malaria parasites is endoperoxide-dependent. Biochem. Pharmacol. 2009, 77, 322–336. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, M.; Wittlin, S.; Nehrbass-Stuedli, A.; Dong, Y.; Wang, X.; Hemphill, A.; Matile, H.; Brun, R.; Vennerstrom, J.L. Peroxide bond-dependent antiplasmodial specificity of artemisinin and OZ277 (RBx11160). Antimicrob. Agents Chemother. 2007, 51, 2991–2993. [Google Scholar] [CrossRef] [Green Version]
- Pollack, Y.; Segal, R.; Golenser, J. The effect of ascaridole on the in vitro development of Plasmodium falciparum. Parasitol. Res. 1990, 76, 570–572. [Google Scholar] [CrossRef]
- Benz, S.; Notzli, S.; Siegel, J.S.; Eberli, D.; Jessen, H.J. Controlled oxygen release from pyridone endoperoxides promotes cell survival under anoxic conditions. J. Med. Chem. 2013, 56, 10171–10182. [Google Scholar] [CrossRef] [PubMed]
- Dembitsky, V.M. Bioactive peroxides as potential therapeutic agents. Eur. J. Med. Chem. 2008, 43, 223–251. [Google Scholar] [CrossRef] [PubMed]
- Monzote, L.; Pastor, J.; Scull, R.; Gille, L. Antileishmanial activity of essential oil from Chenopodium ambrosioides and its main components against experimental cutaneous leishmaniasis in BALB/c mice. Phytomedicine 2014, 21, 1048–1052. [Google Scholar] [CrossRef]
- Leliebre-Lara, V.; Monzote, F.L.; Pferschy-Wenzig, E.M.; Kunert, O.; Nogueiras, L.C.; Bauer, R. In vitro antileishmanial activity of sterols from Trametes versicolor (Bres. Rivarden). Molecules 2016, 21, 1045. [Google Scholar] [CrossRef] [Green Version]
- Sen, R.; Bandyopadhyay, S.; Dutta, A.; Mandal, G.; Ganguly, S.; Saha, P.; Chatterjee, M. Artemisinin triggers induction of cell-cycle arrest and apoptosis in Leishmania donovani promastigotes. J. Med. Microbiol. 2007, 56, 1213–1218. [Google Scholar] [CrossRef] [Green Version]
- Bodor, N.; Buchwald, P. Recent advances in the brain targeting of neuropharmaceuticals by chemical delivery systems. Adv. Drug Deliv. Rev. 1999, 36, 229–254. [Google Scholar] [CrossRef]
- Pajouhesh, H.; Lenz, G.R. Medicinal chemical properties of successful central nervous system drugs. NeuroRx 2005, 2, 541–553. [Google Scholar] [CrossRef] [Green Version]
- Aubry, J.M.; Pierlot, C.; Rigaudy, J.; Schmidt, R. Reversible binding of oxygen to aromatic compounds. Acc. Chem. Res. 2003, 36, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Kraeva, N.; Horakova, E.; Kostygov, A.Y.; Koreny, L.; Butenko, A.; Yurchenko, V.; Lukeš, J. Catalase in Leishmaniinae: With me or against me? Infect. Genet. Evol. 2017, 50, 121–127. [Google Scholar] [CrossRef]
- Sarkar, A.; Mandal, G.; Singh, N.; Sundar, S.; Chatterjee, M. Flow cytometric determination of intracellular non-protein thiols in Leishmania promastigotes using 5-chloromethyl fluorescein diacetate. Exp. Parasitol. 2009, 122, 299–305. [Google Scholar] [CrossRef]
- Pal, S.; Dolai, S.; Yadav, R.K.; Adak, S. Ascorbate peroxidase from Leishmania major controls the virulence of infective stage of promastigotes by regulating oxidative stress. PLoS ONE 2010, 5, e11271. [Google Scholar] [CrossRef] [PubMed]
- Monzote, L.; Lackova, A.; Staniek, K.; Steinbauer, S.; Pichler, G.; Jäger, W.; Gille, L. The antileishmanial activity of xanthohumol is mediated by mitochondrial inhibition. Parasitology 2017, 144, 747–759. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Shaha, C. Apoptotic death in Leishmania donovani promastigotes in response to respiratory chain inhibition: Complex II inhibition results in increased pentamidine cytotoxicity. J. Biol. Chem. 2004, 279, 11798–11813. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Jones, D.P. Superoxide in apoptosis. Mitochondrial generation triggered by cytochrome c loss. J. Biol. Chem. 1998, 273, 11401–11404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klaper, M.; Wessig, P.; Linker, T. Base catalysed decomposition of anthracene endoperoxide. Chem. Commun. 2016, 52, 1210–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donkers, R.L.; Workentin, M.S. Elucidation of the electron transfer reduction mechanism of anthracene endoperoxides. J. Am. Chem. Soc. 2004, 126, 1688–1698. [Google Scholar] [CrossRef]
- Fudickar, W.; Linker, T. Synthesis of pyridylanthracenes and their reversible reaction with singlet oxygen to endoperoxides. J. Org. Chem. 2017, 82, 9258–9262. [Google Scholar] [CrossRef]
- Fudickar, W.; Linker, T. Imaging by sensitized oxygenations of photochromic anthracene films: Examination of effects that improve performance and reversibility. Chemistry 2006, 12, 9276–9283. [Google Scholar] [CrossRef]
- Bauch, M.; Fudickar, W.; Linker, T. Stereoselective [4+2] cycloaddition of singlet oxygen to naphthalenes controlled by carbohydrates. Molecules 2021, 26, 804. [Google Scholar] [CrossRef]
- Fudickar, W.; Linker, T. Release of singlet oxygen from aromatic endoperoxides by chemical triggers. Angew. Chem. Int. Ed. Engl. 2018, 57, 12975. [Google Scholar] [CrossRef]
- Fritsche, C.L.E. Untersuchungen zur Optimalen Kultivierung von Leishmania tarentolae. Ph.D Thesis, Martin-Luther-Universität Halle-Wittenberg, Halle, Germany, 2008. [Google Scholar]
- Corral, M.J.; Gonzalez, E.; Cuquerella, M.; Alunda, J.M. Improvement of 96-well microplate assay for estimation of cell growth and inhibition of Leishmania with Alamar Blue. J. Microbiol. Methods 2013, 94, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Kulshrestha, A.; Bhandari, V.; Mukhopadhyay, R.; Ramesh, V.; Sundar, S.; Maes, L.; Dujardin, J.C.; Roy, S.; Salotra, P. Validation of a simple resazurin-based promastigote assay for the routine monitoring of miltefosine susceptibility in clinical isolates of Leishmania donovani. Parasitol. Res. 2013, 112, 825–828. [Google Scholar] [CrossRef] [PubMed]
- Wolff, S.P. Ferrous ion oxidation in presence of ferric ion indicator xylenol orange for measurement of hydroperoxides. Methods Enzymol. 1994, 233, 182–189. [Google Scholar]
- Duling, D.R. Simulation of multiple isotropic spin-trap EPR spectra. J. Magn. Reson. B 1994, 104, 105–110. [Google Scholar] [CrossRef]
- Dikalov, S.I.; Kirilyuk, I.A.; Voinov, M.; Grigor’ev, I.A. EPR detection of cellular and mitochondrial superoxide using cyclic hydroxylamines. Free Radic Res. 2011, 45, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Müllebner, A.; Patel, A.; Stamberg, W.; Staniek, K.; Rosenau, T.; Netscher, T.; Gille, L. Modulation of the mitochondrial cytochrome bc1 complex activity by chromanols and related compounds. Chem. Res. Toxicol. 2010, 23, 193–202. [Google Scholar] [CrossRef]
- Katsuno, K.; Burrows, J.N.; Duncan, K.; van Huijsduijnen, R.H.; Kaneko, T.; Kita, K.; Mowbray, C.E.; Schmatz, D.; Warner, P.; Slingsby, B.T. Hit and lead criteria in drug discovery for infectious diseases of the developing world. Nat. Rev. Drug Discov. 2015, 14, 751–758. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
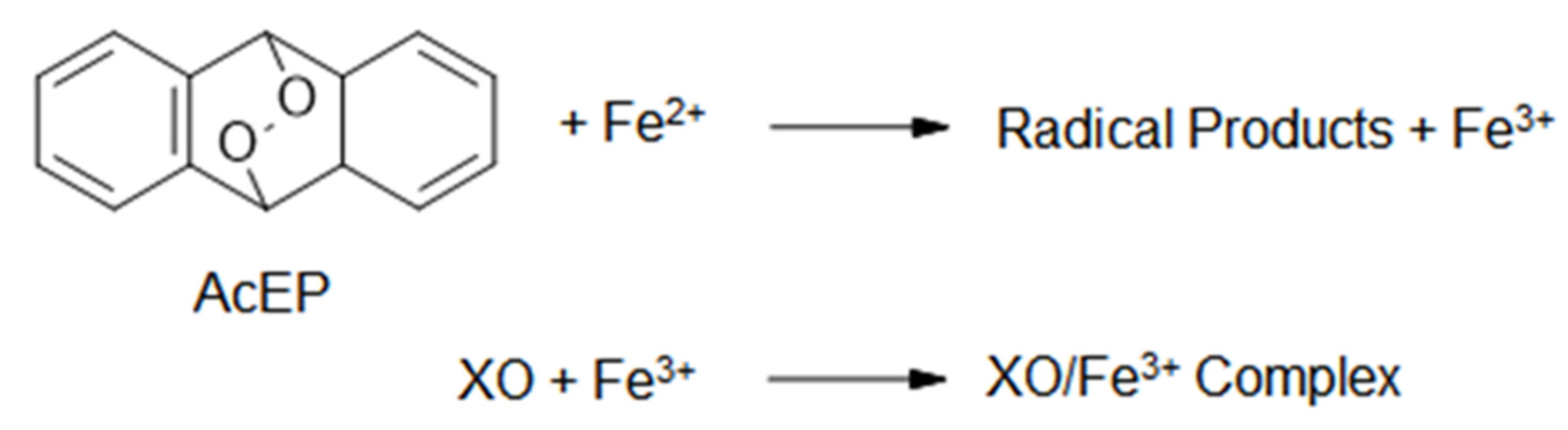{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Sum Formula | MW (g/mol) | LogPOW | HAccept | Stereo Centers | PSA (Å2) |
|---|---|---|---|---|---|---|
| Ac | C14H10 | 178.2 | 3.64 | 0 | 0 | 0 |
| AcEP | C14H10O2 | 210.2 | 2.62 | 2 | 0 | 18.4 |
| 1062MeAcEP | C26H22N2O2 × 2CF3O3S | 692.6 | 3.67 | 8 | 0 | 157.3 |
| BisGluAc | C42H46O20 | 870.8 | 2.33 | 20 | 10 | 247.3 |
| BisGluAcEP | C42H46O22 | 902.8 | 1.38 | 22 | 10 | 265.7 |
| DPAc | C26H18 | 330.4 | 6.68 | 0 | 0 | 0 |
| DPAcEP | C26H18O2 | 362.4 | 5.50 | 2 | 0 | 18.4 |
| MB359EP | C42H52O20 | 876.8 | 1.03 | 20 | 12 | 247.3 |
| MonoGluAc | C28H28O10 | 524.5 | 2.99 | 10 | 5 | 123.6 |
| MonoGluAcEP | C28H28O12 | 556.5 | 2.00 | 12 | 5 | 142.1 |
| mPyAc | C24H16N2 | 332.3 | 4.38 | 2 | 0 | 25.7 |
| mPyAcEP | C24H16N2O2 | 364.3 | 3.20 | 4 | 0 | 44.2 |
| mPyBuAc | C28H24N2 | 388.5 | 5.78 | 2 | 0 | 25.7 |
| mPyBuAcEP | C28H24N2O2 | 420.5 | 4.60 | 4 | 2 | 44.2 |
| oOMeAc | C28H22O2 | 390.4 | 6.65 | 2 | 0 | 18.4 |
| oOMeAcEP | C28H22O4 | 422.4 | 5.47 | 4 | 0 | 36.9 |
| oPyAc | C24H16N2 | 332.3 | 5.23 | 2 | 0 | 25.7 |
| oPyAcEP | C24H16N2O2 | 364.3 | 4.06 | 4 | 0 | 44.2 |
| oPy2C8Ac | C34H34N2O2 | 502.6 | 8.41 | 4 | 0 | 52.0 |
| oPy2C8AcEP | C34H34N2O4 | 534.6 | 7.24 | 6 | 2 | 70.5 |
| pPyBuAc | C28H24N2 | 388.5 | 5.78 | 2 | 0 | 25.7 |
| pPyBuAcEP | C28H24N2O2 | 420.5 | 3.98 | 4 | 4 | 44.2 |
Compound | LtP IC50 (µM) | J774 IC50 (µM) | Selectivity IC50,J774/IC50,LtP |
|---|---|---|---|
| Pen | 0.52 ± 0.16 | 12.27 ± 3.96 | 23.6 |
| Ac | >200 | >200 | n.d. |
| AcEP | 19.7 ± 14.1 | 1.21 ± 0.32 | 0.06 |
| 1062MeAcEP | 62.0 ± 34.2 | >200 | >3.2 |
| BisGluAc | >200 | >200 | n.d. |
| BisGluAcEP | >200 | >200 | n.d. |
| DPAc | 197 ± 92 | 136 ± 35 | 0.7 |
| DPAcEP | 9.67 ± 2.15 | 197 ± 35 | 20 |
| MB359EP | >200 | >200 | n.d. |
| MonoGluAc | 158.3 ± 45.1 | 101.4 ± 7.8 | 0.6 |
| MonoGluAcEP | 32.3 ± 23.4 | 152.6 ± 82.1 | 4.7 |
| mPyAc | 52.5 ± 3.3 | 63.7 ± 7.6 | 1.2 |
| mPyAcEP | >200 | >200 | n.d. |
| mPyBuAc | 0.54 ± 0.09 | 74.2 ± 45.9 | 139 |
| mPyBuAcEP | 0.70 ± 0.31 | 24.6 ± 7.3 | 35 |
| oOMeAc | 73.3 ± 25.2 | >200 | >2.7 |
| oOMeAcEP | 40.7 ± 12.3 | >200 | >4.9 |
| oPyAc | 34.5 ± 12.8 | 70.0 ± 10.5 | 2.0 |
| oPyAcEP | 1.84 ± 0.13 | 2.24 ± 0.08 | 1.2 |
| oPy2C8Ac | 11.3 ± 3.4 | 34.8 ± 10.5 | 3.0 |
| oPy2C8AcEP | 0.81 ± 0.37 | 1.28 ± 0.46 | 1.6 |
| pPyBuAc | 1.86 ± 1.10 | 107 ± 46 | 57 |
| pPyBuAcEP | 0.707 ± 0.662 | 26.1 ± 10.6 | 37 |
Compound | XO/Fe3+ Formation Rate (nmol/min) | Log10 (Formation Rate) | IC50,non-EP/IC50,EP |
|---|---|---|---|
| AcEP | 8110 ± 4197 | 3.90 | >10.1 |
| 1062MeAcEP | 609 ± 27 | 2.78 | - |
| BisGluAcEP | 6800 ± 326 | 3.83 | 1 |
| DPAcEP | 677 ± 86 | 2.83 | 20.3 |
| MB359EP | 18,533 ± 1147 | 4.26 | - |
| MonoGluAcEP | 12,697 ± 1500 | 4.10 | 4.89 |
| mPyAcEP | 708 ± 37 | 2.85 | <0.26 |
| mPyBuAcEP | 731 ± 111 | 2.86 | 0.77 |
| oOMeAcEP | 545 ± 17 | 2.73 | 1.79 |
| oPyAcEP | 81,700 ± 11,921 | 4.91 | 18.7 |
| oPy2C8AcEP | 42,667 ± 6302 | 4.63 | 14.0 |
| pPyBuAcEP | 67 ± 3 | 1.82 | 2.63 |
| Compound | Red. | Most Abundant Species | ||||
|---|---|---|---|---|---|---|
| Int. (%) | aN (G) | aH (G) | aN/aH | Assignment | ||
| MonoGluAcEP | Fe2+ | 75 | 15.8 | 23.9 | 0.66 | DMPO/●C |
| 25 | 15.0 | 14.5 | 1.03 | DMPO/●OH | ||
| oPy2C8AcEP | Fe2+ | 100 | 15.7 | 23.3 | 0.67 | DMPO/●C |
| oPyAcEP | Fe2+ | 100 | 15.7 | 23.3 | 0.67 | DMPO/●C |
| MB359EP | Fe2+ | 100 | 16.1 | 22.8 | 0.70 | DMPO/●C |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Machin, L.; Piontek, M.; Todhe, S.; Staniek, K.; Monzote, L.; Fudickar, W.; Linker, T.; Gille, L. Antileishmanial Anthracene Endoperoxides: Efficacy In Vitro, Mechanisms and Structure-Activity Relationships. Molecules 2022, 27, 6846. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27206846
Machin L, Piontek M, Todhe S, Staniek K, Monzote L, Fudickar W, Linker T, Gille L. Antileishmanial Anthracene Endoperoxides: Efficacy In Vitro, Mechanisms and Structure-Activity Relationships. Molecules. 2022; 27(20):6846. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27206846
Chicago/Turabian StyleMachin, Laura, Martin Piontek, Sara Todhe, Katrin Staniek, Lianet Monzote, Werner Fudickar, Torsten Linker, and Lars Gille. 2022. "Antileishmanial Anthracene Endoperoxides: Efficacy In Vitro, Mechanisms and Structure-Activity Relationships" Molecules 27, no. 20: 6846. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27206846