
Insight on the Interaction between the Camptothecin Derivative and DNA Oligomer Mimicking the Target of Topo I Inhibitors
 , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
2. Results and Discussion
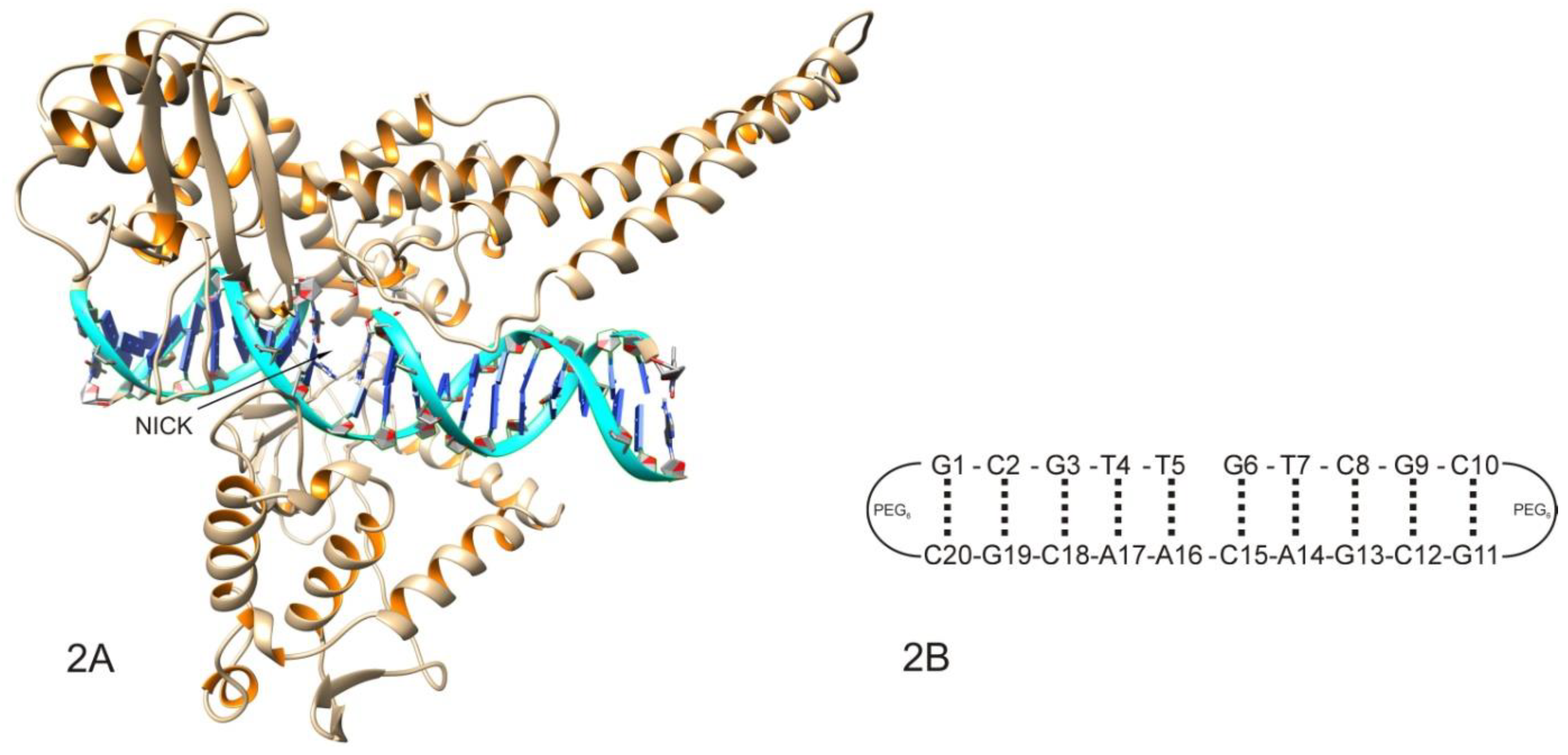
2.1. Molecular Docking Analysis
2.2. MM-PBSA and MM-GBSA Calculations of the Ligand/DNA/Topo I Ternary Complex
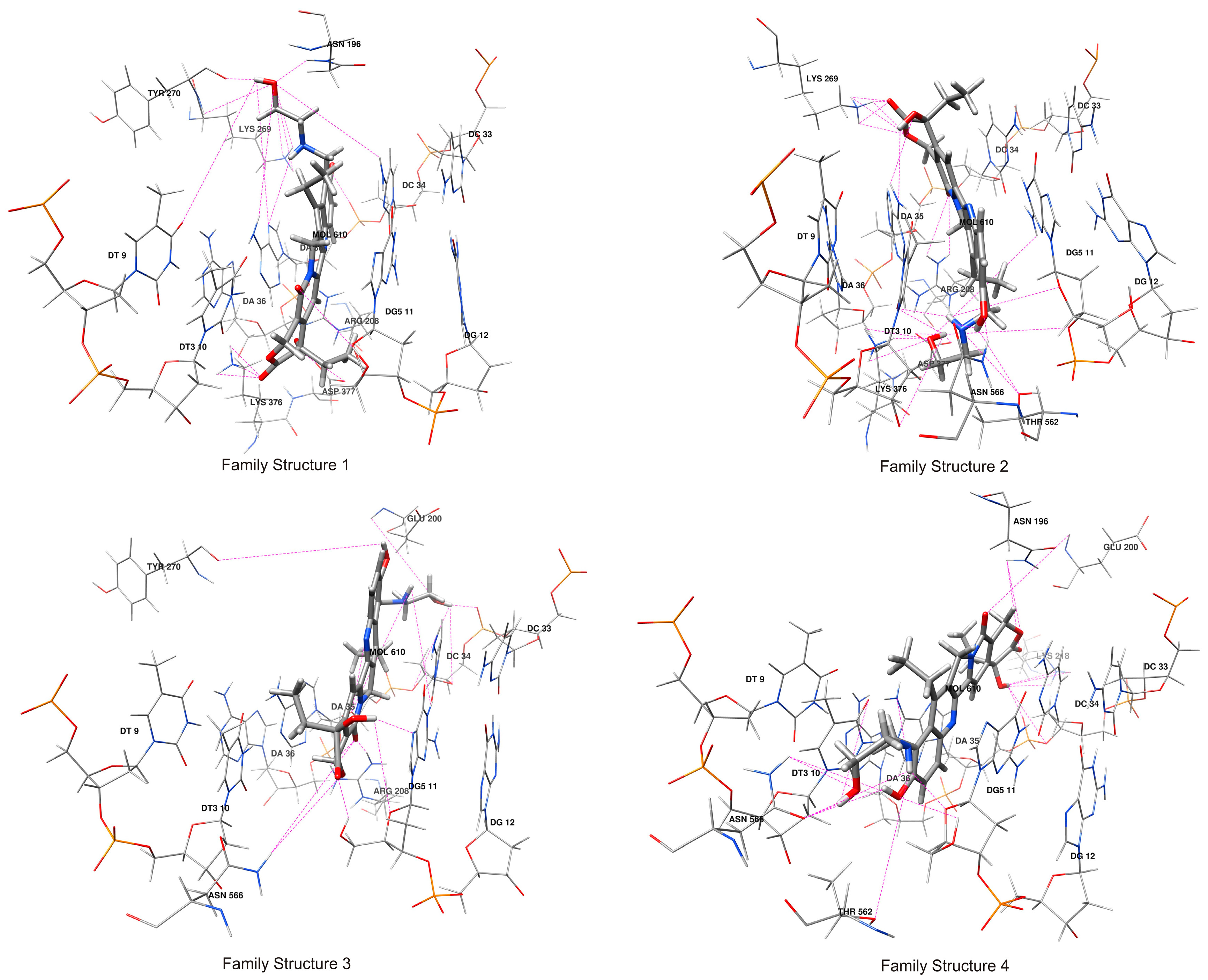
2.3. Hydrogen Bonds Analysis of Compound 1/DNA/Topo I Ternary Complex
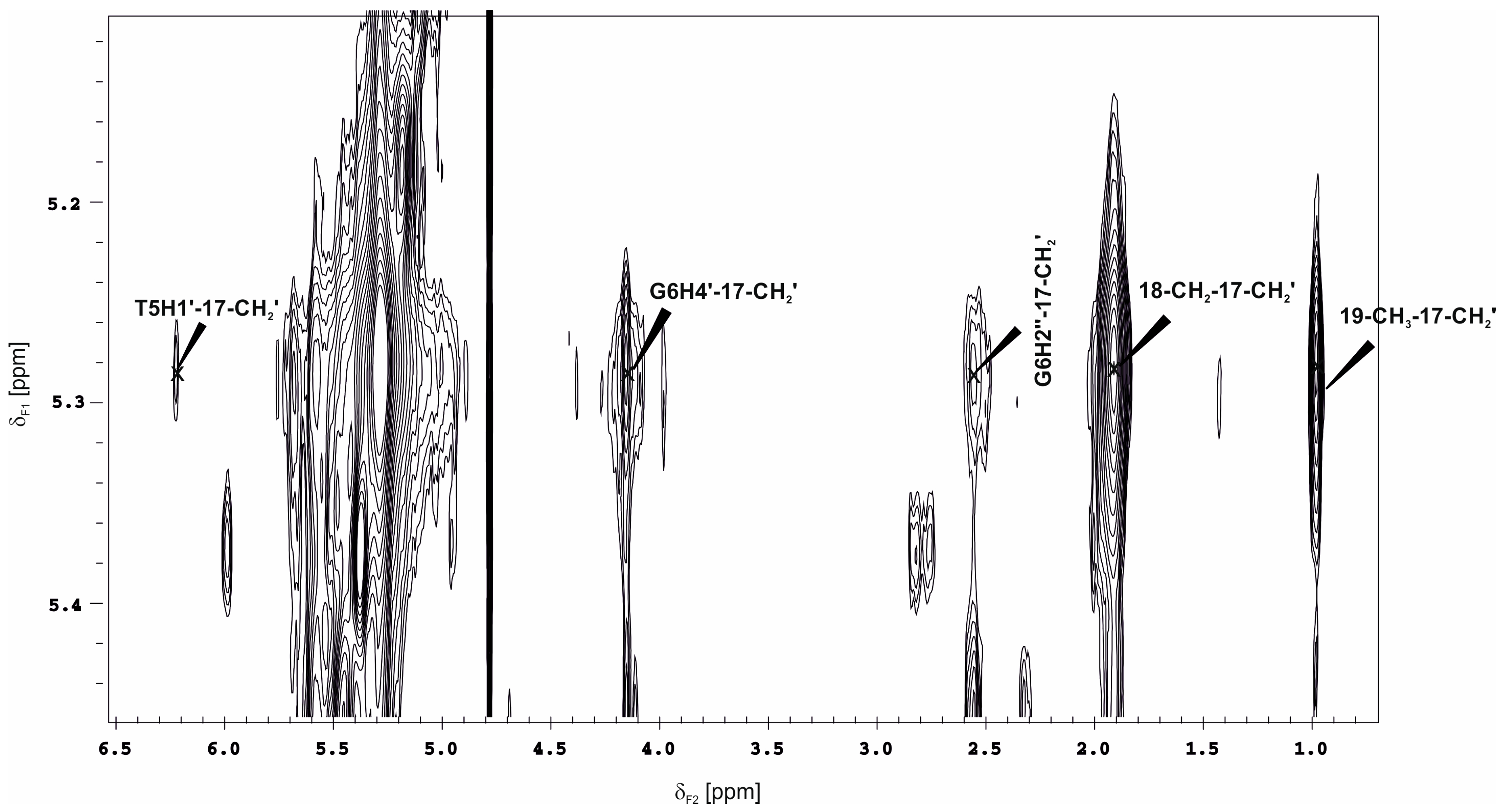
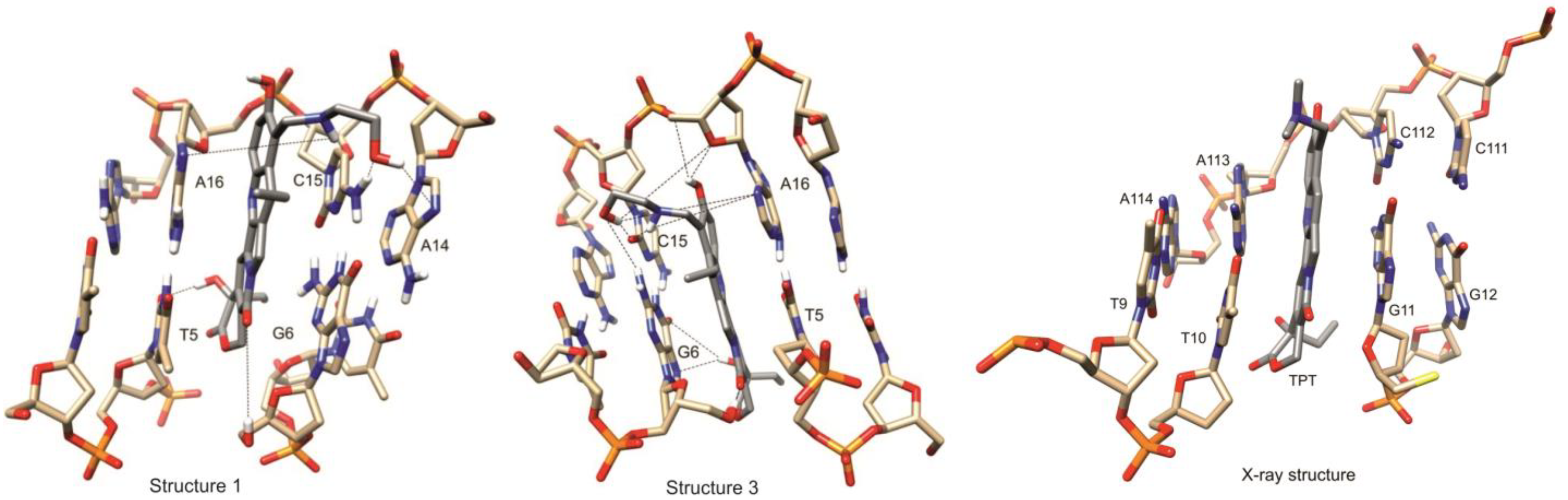
2.4. The NMR Results of TOCSY and NOESY Experiments for the Binary Compound 1/DNA Complex
2.5. MM-PBSA and MM-GBSA Calculations of the Compound 1/DNA Binary Complex
2.6. PM7 Semi-Empirical Calculations of the Truncated Compound 1/DNA Binary Complex
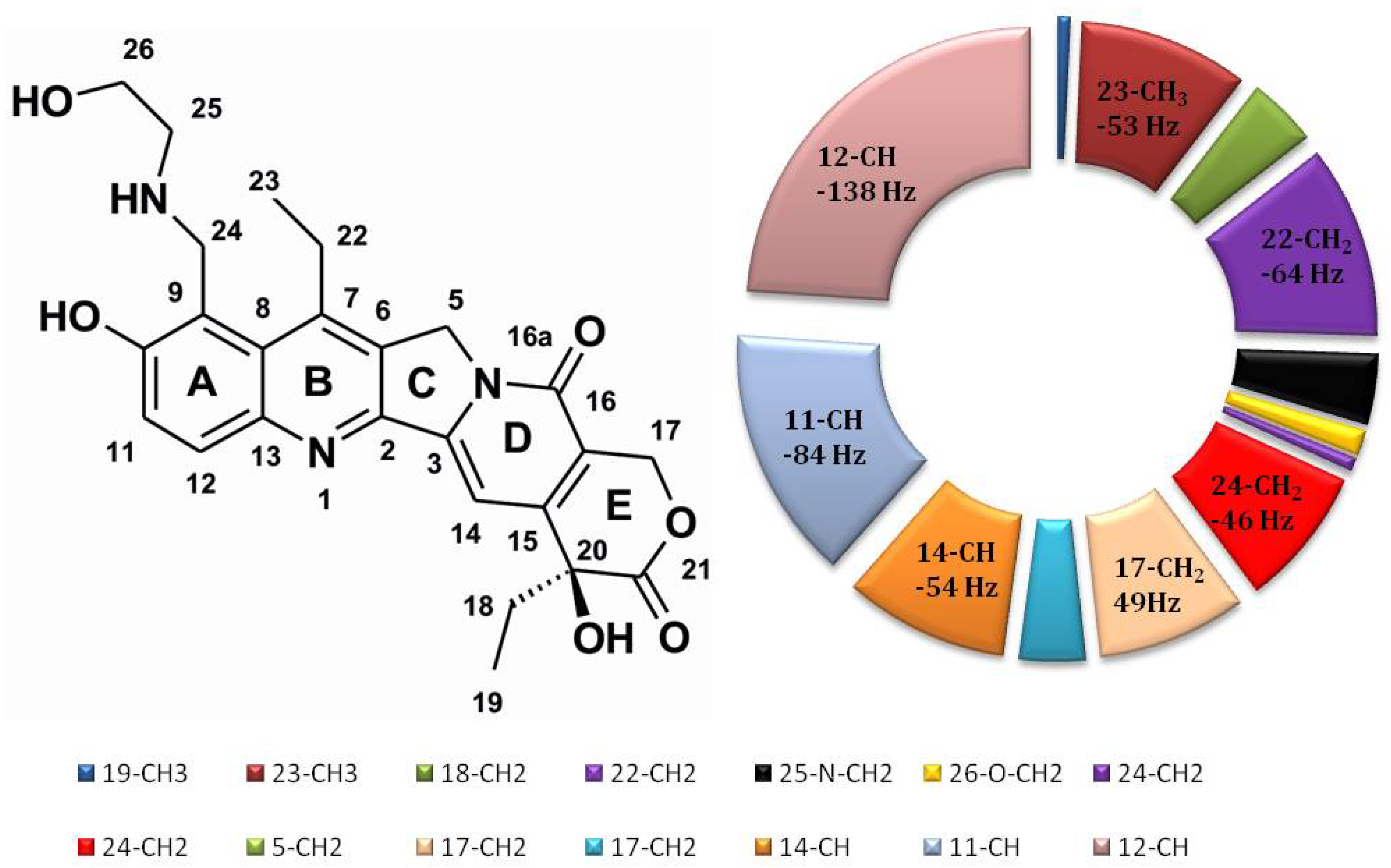
2.7. Comparison of NOE Effects with MD Simulations for the Compound 1/DNA Binary Complex
2.8. DOSY Experiments of Molecular Complexes
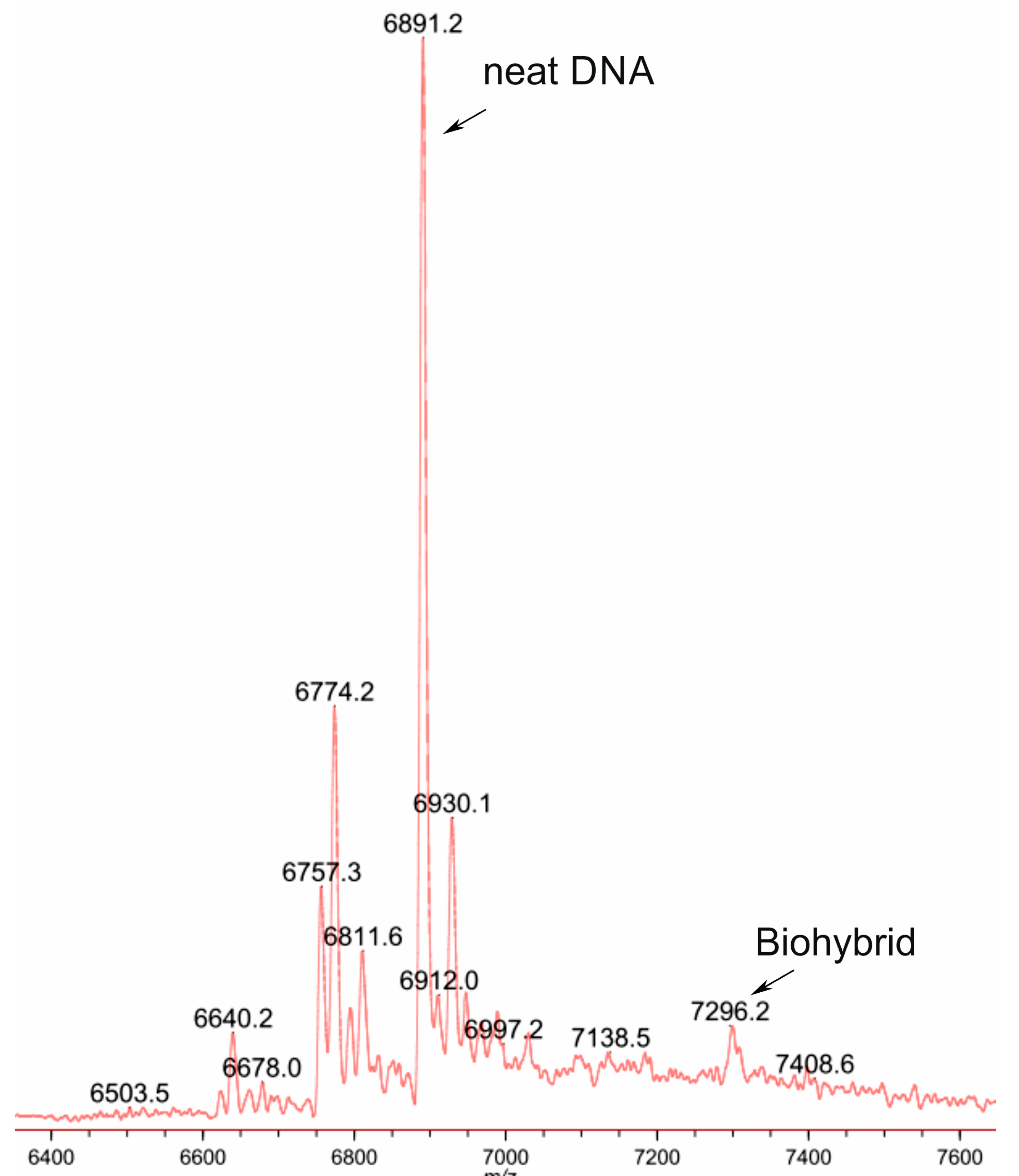
2.9. MALDI Experiment
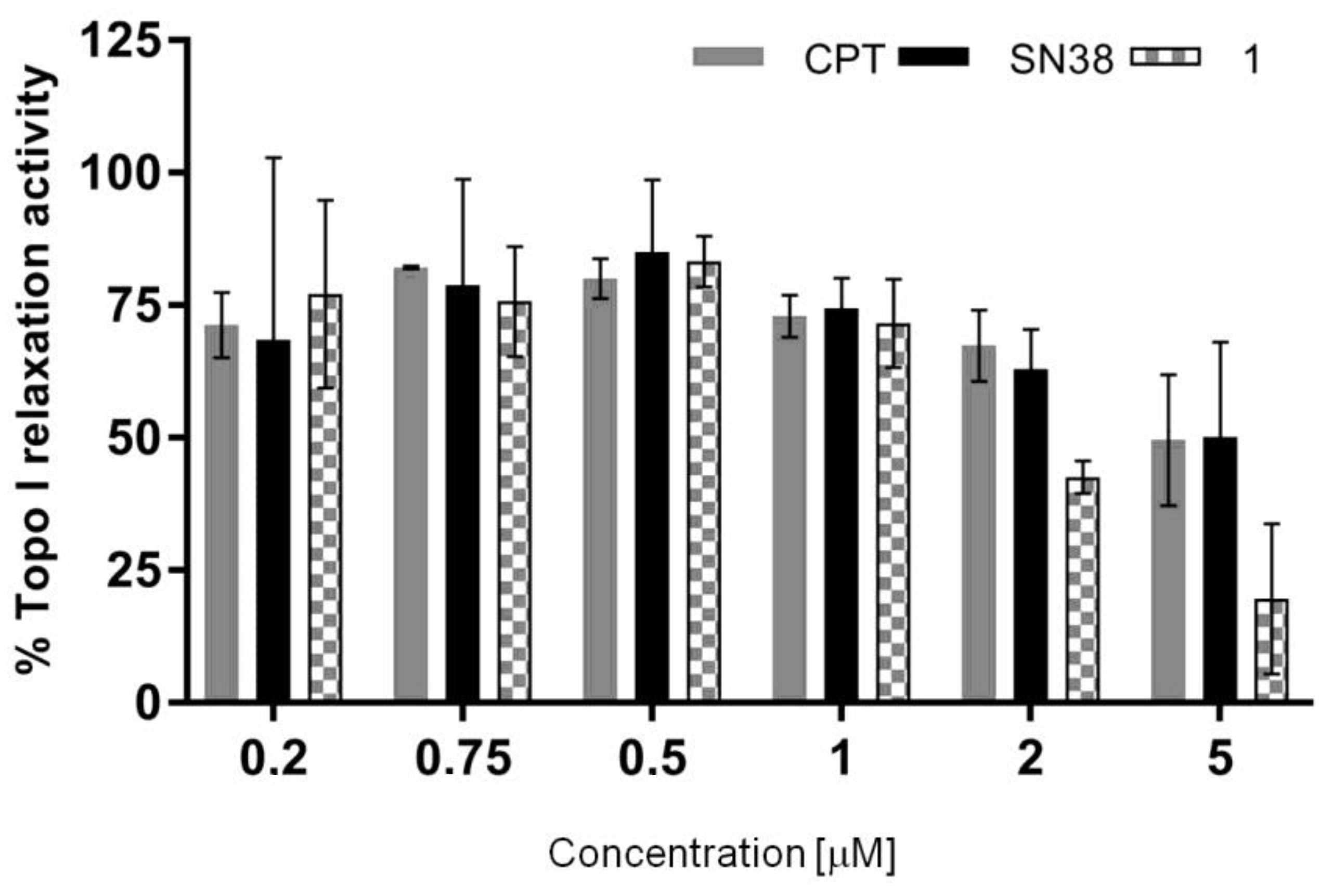
2.10. Topoisomerase Inhibition
3. Materials and Methods
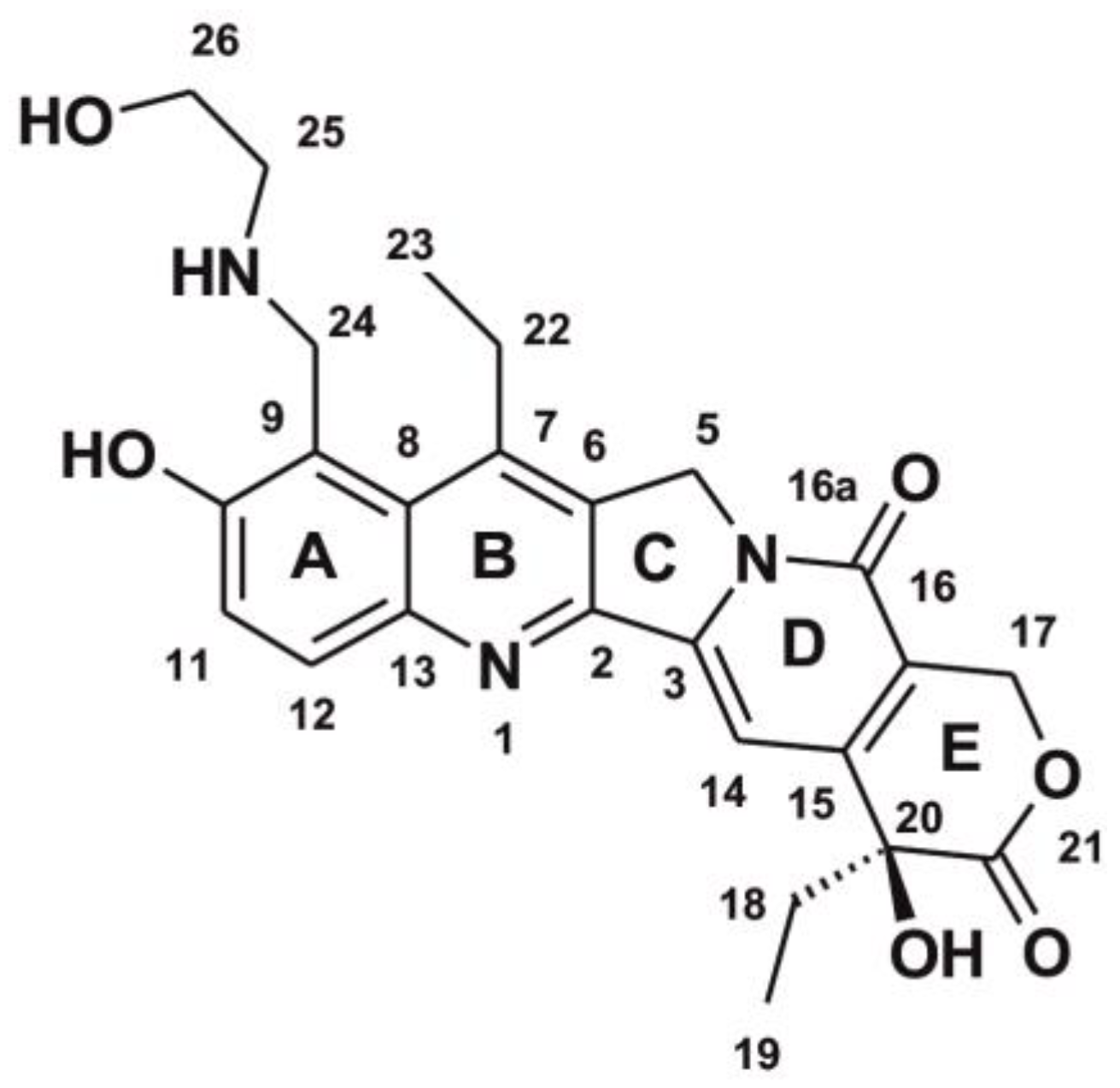
3.1. Chemical Substrates
3.2. Sample Preparation
3.3. NMR Experiments
3.4. MALDI-ToF MS Experiments
3.5. Calculating Binding Constants from the Diffusion Coefficients
3.6. Molecular Dynamics Calculations
3.7. Calculating Binding Free Energies (Enthalpies) Using the MM-PBSA and MM-GBSA Methods [32]
3.8. PM7 Semi-Empirical Calculations
3.9. Cluster Analysis
3.10. Docking Calculations
3.11. The Hydrogen Bonds Analysis
3.12. The Topo I Relaxation Activity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Han, S.; Lim, K.S.; Blackburn, B.J.; Yun, J.; Putnam, C.W.; Bull, D.A.; Won, Y.W. The Potential of Topoisomerase Inhibitor-Based Antibody-Drug Conjugates. Pharmaceutics 2022, 14, 1707. [Google Scholar] [CrossRef]
- Staker, B.L.; Hjerrild, K.; Feese, M.D.; Behnke, C.A.; Burgin, A.B., Jr.; Stewart, L. The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc. Natl. Acad. Sci. USA 2002, 99, 15387–15392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hevenern, K.E.; Verstak, T.A.; Lutat, K.E.; Riggsbee, D.L.; Mooney, J.W. Recent developments in topoisomerase-targeted cancer chemotherapy. Acta Pharm. Sin. B 2018, 8, 844–861. [Google Scholar]
- Stewart, L.; Redinbo, M.R.; Qiu, X.; Hol, W.G.; Champoux, J.J. A model for the mechanism of human topoisomerase I. Science 1998, 279, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
- Dallavalle, S.; Merlini, L. Camptothecin and Analogs: Structure and Synthetic Efforts. In Modern Alkaloids; Fattorusso, E., Taglialatela-Scafati, O., Eds.; John and Wiley and Sons: Hoboken, NJ, USA, 2007; pp. 503–520. [Google Scholar]
- Liu, Y.-Q.; Li, W.-Q.; Morris-Natschke, S.L.; Qian, K.; Yang, L.; Zhu, G.-X.; Wu, X.-B.; Chen, A.-L.; Zhang, S.-Y.; Nan, X.; et al. Perspectives on biologically active camptothecin derivatives. Med. Res. Rev. 2015, 35, 753–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, R.P.; Hansch, C. Camptothecins: A SAR/QSAR study. Chem. Rev. 2009, 109, 213–235. [Google Scholar] [CrossRef]
- Redinbo, M.R.; Stewart, L.; Kuhn, P.; Champoux, J.J.; Hol, W.G. Crystal structures of human topoisomerase I in covalent and noncovalent complexes with DNA. Science 1998, 279, 1504–1513. [Google Scholar] [CrossRef]
- Bocian, W.; Kawęcki, R.; Bednarek, E.; Sitkowski, J.; Williamson, M.P.; Hansen, P.E.; Kozerski, L. Binding of Topotecan to a Nicked DNA Oligomer in Solution. Chemistry 2008, 14, 2788–2794. [Google Scholar] [CrossRef]
- Pommier, Y.; Marchand, C. Interfacial inhibitors: Targeting macromolecular complexes. Nat. Rev. Drug Discov. 2011, 11, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Coletta, A.; Desideri, A. Role of a protein in the DNA sequence specificity of the cleavage site stabilized by the camptothecin topoisomerase IB inhibitor: A metadynamic study. Nucleic Acids Res. 2013, 41, 9986–9997. [Google Scholar] [CrossRef]
- Naumczuk, B.; Hyz, K.; Kawęcki, R.; Bocian, W.; Bednarek, E.; Sitkowski, J.; Wielgus, E.; Kozerski, L. DOSY NMR and MALDI-TOF evidence of covalent binding the DNA duplex by trimethylammonium salts of topotecan upon near UV irradiation. Magn. Reson. Chem. 2015, 53, 565–571. [Google Scholar] [CrossRef]
- Naumczuk, B.; Kawęcki, R.; Sitkowski, J.; Bocian, W.; Bednarek, E.; Kozerski, L. Spontaneous 2′-deoxyguanosine alkylation by a new generation of topoisomerase I inhibitors of the camptothecin family. New J. Chem. 2016, 40, 3010–3013. [Google Scholar] [CrossRef]
- Naumczuk, B.; Kawęcki, R.; Bocian, W.; Bednarek, E.; Sitkowski, J.; Kozerski, L. Regioselective alkylation reaction of the 2′deoxycitidine with 9-aminomethyl derivatives of SN38. J. Mol. Struct. 2019, 1176, 298–302. [Google Scholar] [CrossRef]
- Naumczuk, B.; Bocian, W.; Sitkowski, J.; Kawęcki, R.; Kozerski, L. Solvent-dependent regioselectivity of 2′-deoxyadenosine alkylation by 9-aminomethyl derivatives of SN38. New J. Chem. 2019, 43, 18975–18978. [Google Scholar] [CrossRef]
- Naumczuk, B.; Wiktorska, K.; Lubelska, K.; Kawęcki, R.; Bocian, W.; Bednarek, E.; Sitkowski, J.; Chilmonczyk, Z.; Kozerski, L. New generation of camptothecin derivatives spontaneously alkylating DNA. New J. Chem. 2016, 40, 7978–7985. [Google Scholar] [CrossRef]
- Naumczuk, B.; Górecki, M.; Wiktorska, K.; Urbanowicz, M.; Sitkowski, J.; Lubelska, K.; Milczrek, M.; Bednarek, E.; Bocian, W.; Kozerski, L. New camptothecin derivatives for generalized oncological chemotherapy: Synthesis, stereochemistry and biology. Bioorg. Med. Chem. Lett. 2021, 46, 128146. [Google Scholar] [CrossRef]
- Naumczuk, B.; Sitkowski, J.; Bocian, W.; Stolarska, M.; Bednarek, E.; Kozerski, L. A NMR Study of Binding the Metabolite of SN38 derivatives to a Model Nicked DNA Decamer Mimicking Target of Topo I Inhibitors. Bioorg. Chem. 2021, 107, 104631. [Google Scholar] [CrossRef] [PubMed]
- Bocian, W.; Naumczuk, B.; Urbanowicz, M.; Sitkowski, J.; Bierczyńska-Krzysik, A.; Bednarek, E.; Wiktorska, K.; Milczrek, M.; Kozerski, L. The Mode of SN38 Derivatives Interacting with Nicked DNA Mimics Biological Targeting of Topo I Poisons. Int. J. Mol. Sci. 2021, 22, 7471. [Google Scholar] [CrossRef]
- Santos-Martins, D.; Solis-Vasquez, L.; Tillack, A.F.; Sanner, M.F.; Koch, A.; Forli, S. Accelerating AutoDock4 with GPUs and Gradient-Based Local Search. J. Chem. Theory Comput. 2021, 17, 1060–1073. [Google Scholar] [CrossRef]
- Staker, B.L.; Feese, M.D.; Cushman, M.; Pommier, Y.; Zembower, D.; Stewart, L.; Burgin, A.B. Structures of three classes of anticancer agents bound to the human topoisomerase I-DNA covalent complex. J. Med. Chem. 2005, 48, 2336–2345. [Google Scholar] [CrossRef]
- Ruvinsky, A.M.; Kozintsev, A.V. New and fast statistical-thermodynamic method for computation of protein-ligand binding entropy substantially improves docking accuracy. J. Comput. Chem. 2005, 26, 1089–1095. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Oezguen, N.; Urvil, P.; Ferguson, C.; Dann, S.M.; Savidge, T.C. Regulation of protein-ligand binding affinity by hydrogen bond pairing. Sci. Adv. 2016, 2, e1501240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, M.P. Applications of the NOE in Molecular Biology. In Annual Report NMR Spectroscopy; Elsevier: Amsterdam, The Netherlands, 2009; Volume 65, pp. 77–109. [Google Scholar] [CrossRef]
- Antałek, B. Using Pulsed Gradient Spin Echo NMR for Chemical Mixture Analysis: How to Obtain Optimum Results. Concepts Magn. Reson. 2002, 14, 225–258. [Google Scholar] [CrossRef]
- Antałek, B. Accounting for spin relaxation in quantitative pulse gradient spin echo NMR mixture analysis. J. Am. Chem. Soc. 2006, 128, 8402–8403. [Google Scholar] [CrossRef] [PubMed]
- Pelta, M.D.; Morris, G.A.; Stchedroff, M.J.; Hammond, S.J. A one-shot sequence for high-resolution diffusion-ordered spectroscopy. Magn. Reson. Chem. 2002, 40, S147–S152. [Google Scholar] [CrossRef]
- Kozerski, L.; Mazurek, A.P.; Kawęcki, R.; Bocian, W.; Krajewski, P.; Bednarek, E.; Sitkowski, J.; Williamson, M.P.; Moir, A.J.; Hansen, P.E. A nicked duplex decamer DNA with a PEG(6) tether. Nucleic Acids Res. 2001, 29, 1132–1143. [Google Scholar] [CrossRef] [Green Version]
- Case, D.A.; Berryman, J.T.; Betz, R.M.; Cerutti, D.S.; Cheatham III, T.E.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; et al. Amber 2015; University of California: San Francisco, CA, USA, 2015. [Google Scholar]
- Frisch, J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.01; M. Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Luo, R.; David, L.; Gilson, M.K. Accelerated Poisson-Boltzmann calculations for static and dynamic systems. J. Comput. Chem. 2002, 23, 1244–1253. [Google Scholar] [CrossRef]
- Stewart, J. Mopac2016; Version 17181L; Stewart Computational Chemistry: Colorado Springs, CO, USA, 2016; Available online: http://OpenMOPAC.net (accessed on 12 September 2022).
- Klamt, A.; Schuurmann, G. Cosmo—A New Approach to Dielectric Screening in Solvents with Explicit Expressions for the Screening Energy and Its Gradient. J. Chem. Soc. Perkin Trans. 1993, 2, 799–805. [Google Scholar] [CrossRef]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein-ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef]
- Goodsell, D.S.; Olson, A.J. Automated docking of substrates to proteins by simulated annealing. Proteins 1990, 8, 195–202. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxwell, A.; Burton, N.P.; O’Hagan, N. High-throughput assays for DNA gyrase and other topoisomerases. Nucleic Acids Res. 2006, 34, e104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seol, Y.; Zhang, H.; Agama, K.; Lorence, N.; Pommier, Y.; Neuman, K.C. Single-Molecule Supercoil Relaxation Assay as a Screening Tool to Determine the Mechanism and Efficacy of Human Topoisomerase IB Inhibitors. Mol. Cancer Ther. 2015, 14, 2552–2559. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Calculated Free Energy of Binding FE [kcal/mol] and Inhibition Constant Ki [nM] | |||||||
|---|---|---|---|---|---|---|---|---|
| X-ray Single Structure | 1000 MD-Derived Structures | |||||||
| 1K4T | 1T8I | 1K4T | 1T8I | |||||
| FE | Ki | FE | Ki | FE | Ki | FE | Ki | |
| Compound 1 | −12.15 | 1.24 | −11.59 | 3.22 | −10.98 ± 0.57 | 8.92 | −11.88 ± 0.88 | 1.95 |
| TPT | −11.81 | 2.22 | −11.47 | 3.89 | −10.15 ± 0.49 | 36.25 | −10.96 ± 0.79 | 9.22 |
| CPT | −11.57 | 3.29 | −11.20 | 6.19 | −10.38 ± 0.47 | 24.48 | −10.95 ± 0.77 | 9.38 |
| SN38 | −11.26 | 5.57 | −10.69 | 14.61 | −10.36 ± 0.48 | 25.42 | −11.04 ± 0.76 | 8.08 |
| Ligand | Calculated Free Energy of Binding [kcal/mol], Inhibition Constant Ki [nM], and Cluster Size CS | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Family Structure 1 | Family Structure 2 | Family Structure 3 | Family Structure 4 | |||||||||
| Energy | Ki | CS | Energy | Ki | CS | Energy | Ki | CS | Energy | Ki | CS | |
| Compound 1 | −10.97 ± 0.46 | 9.17 | 1883 | −11.27 ± 0.75 | 5.46 | 2125 | −10.73 ± 0.06 | 13.56 | 82 | −10.65 ± 0.04 | 15.50 | 63 |
| TPT | −10.27 ± 0.40 | 23.60 | 1956 | −10.49 ± 0.47 | 25.36 | 1196 | −10.04 ± 0.01 | 44.12 | 11 | −9.98 ± 0.03 | 48.44 | 93 |
| CPT | −10.33 ± 0.41 | 27.03 | 2844 | −10.06 ± 0.05 | 42.20 | 168 | −9.96 ± 0.01 | 50.49 | 22 | −10.01 ± 0.01 | 46.19 | 19 |
| SN38 | −10.38 ± 0.41 | 24.54 | 2407 | −10.40 ± 0.35 | 27.79 | 1219 | −10.25 ± 0.10 | 30.80 | 203 | −10.04 ± 0.02 | 44.04 | 36 |
| Ligand/Family Structure | Free Energy of Binding [kcal/mol] | |||||
|---|---|---|---|---|---|---|
| MM-PBSA | MM-GBSA | |||||
| Trajectory A | Trajectory B | (A + B)/2 | Trajectory A | Trajectory B | (A + B)/2 | |
| Compound 1/1 | −42.19 ± 2.03 | −38.02 ± 3.46 | −40.10 ± 2.75 | −51.81 ± 1.99 | −51.75 ± 2.01 | −51.78 ± 2.00 |
| Compound 1/2 | −33.37 ± 1.71 | −41.32 ± 4.84 | −37.35 ± 3.28 | −38.80 ± 1.68 | −51.53 ± 5.34 | −45.16 ± 3.51 |
| Compound 1/3 | −32.46 ± 3.02 | −34.34 ± 1.49 | −33.40 ± 2.36 | −39.34 ± 3.93 | −39.60 ± 2.62 | −39.47 ± 3.28 |
| Compound 1/4 | −22.94 ± 2.38 | −30.85 ± 1.50 | −26.89 ± 1.94 | −33.21 ± 1.29 | −38.60 ± 2.48 | −35.91 ± 1.89 |
| TPT/1 | −39.21 ± 1.62 | −48.03 ± 0.58 | ||||
| CPT/1 | −38.69 ± 3.81 | −48.89 ± 3.73 | ||||
| Atom Names | Docking Family Structure 1 | MD Family Structure 1 | |||
|---|---|---|---|---|---|
| Receptor | Compound 1 | HB Length [Å] | Population | HB Length [Å] | Population |
| DA 35 H62 | 9-CH2NHC2H4OH | 1.99 ± 0.18 | 57.39% | 2.69 ± 0.63 | 1.20% |
| DA 35 N7 | 9-CH2NHC2H4OH | 2.06 ± 0.12 | 50.85% | - | - |
| DA 35 N7 | 9-CH2NHC2H4OH | 2.35 ± 0.29 | 48.65% | 2.67 ± 0.88 | 0.50% |
| LYS 376 HZ1, 2, 3 | 21 >C=O | 2.34 ± 0.45 | 48.50% | 2.10 ± 0.43 | 75.55% |
| ARG 208 HH12 | 1-N | 2.47 ± 0.36 | 28.73% | 2.87 ± 0.32 | 39.80% |
| ASP 377 OD1, 2 | 20 -OH | 2.13 ± 0.44 | 27.85% | 2.47 ± 0.79 | 59.05% |
| LYS 269 HZ1, 2, 3 | 9-CH2NHC2H4OH | 2.02 ± 0.37 | 20.73% | 2.24 ± 0.53 | 4.90% |
| DA 35 OP2 | 10 -OH | 2.52 ± 0.35 | 19.65% | - | - |
| DT 9 O4 | 9-CH2NHC2H4OH | 2.10 ± 0.23 | 19.00% | 2.29 ± 0.89 | 0.75% |
| TYR 270 O | 9-CH2NHC2H4OH | 2.07 ± 0.20 | 16.50% | 1.88 ± 0.37 | 23.20% |
| TYR 270 H | 9-CH2NHC2H4OH | 2.00 ± 0.21 | 12.62% | 2.25 ± 0.35 | 11.25% |
| DG5 11 HO5′ | 16 >=O | 2.72 ± 0.58 | 8.50% | 2.67 ± 0.68 | 11.50% |
| DC 34 H42 | 9-CH2NHC2H4OH | 2.05 ± 0.38 | 1.50% | 2.18 ± 0.34 | 34.55% |
| ASN 196 HD22 | 9-CH2NHC2H4OH | 2.09 ± 0.40 | 1.54% | 2.16 ± 0.33 | 13.05% |
| Average H-bond number per residue in family structure 1 | 3.62 | 2.75 | |||
| Docking Family Structure 2 | MD Family Structure 2 | ||||
| ARG 208 HH11, 12 | 16 >C=O | 2.57 ± 0.67 | 59.57% | 2.51 ± 0.71 | 29.45% |
| LYS 376 HZ1, 2, 3 | 9-CH2NHC2H4OH | 2.00 ± 0.24 | 55.30% | 2.17 ± 0.43 | 34.65% |
| ASN 566 OD1 | 10 -OH | 2.02 ± 0.20 | 45.13% | 1.84 ± 0.20 | 61.00% |
| DT3 10 O2 | 9-CH2NHC2H4OH | 1.99 ± 0.13 | 44.70% | 2.28 ± 0.46 | 7.05% |
| LYS 269 HZ1, 2, 3 | 21 >C=O | 2.31 ± 0.52 | 42.35% | 2.58 ± 0.58 | 34.60% |
| ASN 566 HD21 | 10 -OH | 2.17 ± 0.26 | 39.87% | 2.09 ± 0.20 | 35.15% |
| DA 35 H62 | 20 -OH | 2.98 ± 0.29 | 29.48% | 2.99 ± 0.32 | 30.90% |
| THR 562 OG1 | 9-CH2NHC2H4OH | 2.12 ± 0.21 | 29.30% | 2.47 ± 0.35 | 16.75% |
| LYS 269 HZ1, 2, 3 | 21 >O | 2.57 ± 0.58 | 28.39% | 2.44 ± 0.42 | 22.80% |
| DT3 10 O3′ | 9-CH2NHC2H4OH | 2.09 ± 0.29 | 17.70% | 2.45 ± 0.55 | 4.35% |
| DG5 11 O3′ | 9-CH2NHC2H4OH | 2.00 ± 0.23 | 15.17% | 2.26 ± 0.60 | 3.85% |
| DG5 11 H21 | 16 >=O | 3.33 ± 0.35 | 14.04% | 3.01 ± 0.85 | 0.75% |
| DG5 11 O4′ | 9-CH2NHC2H4OH | 2.02 ± 0.21 | 13.74% | 2.56 ± 0.66 | 1.05% |
| DT3 10 O2 | 9-CH2NHC2H4OH | 2.39 ± 0.31 | 11.61% | 2.50 ± 0.28 | 34.30% |
| THR 562 OG1 | 10 -OH | 2.13 ± 0.21 | 11.04% | 2.79 ± 0.40 | 8.75% |
| ASP 377 OD1, 2 | 9-CH2NHC2H4OH | 2.28 ± 0.55 | 10.65% | 2.41 ± 0.29 | 18.65% |
| DA 35 N6 | 20 -OH | - | - | 3.14 +/−0.27 | 23.900 |
| Average H-bond number per residue in family structure 2 | 4.68 | 3.68 | |||
| Docking Family Structure 3 | MD Family Structure 3 | ||||
| DA 35 O4′ | 10 -OH | 2.13 ± 0.34 | 39.54% | 2.39 ± 1.69 | 0.10% |
| ARG 208 HH12, 22 | 16 >C=O | 2.22 ± 0.75 | 27.13% | 2.34 ± 0.58 | 97.80% |
| DC 34 O2 | 9-CH2NHC2H4OH | 2.07 ± 0.38 | 25.58% | - | - |
| DC 34 O3′ | 9-CH2NHC2H4OH | 2.11 ± 0.58 | 16.28% | 2.80 ± 2.01 | 0.10% |
| TYR 270 O | 10 -OH | 2.03 ± 0.50 | 15.50% | 1.82 ± 0.20 | 22.00% |
| DC 34 O4′ | 9-CH2NHC2H4OH | 2.02 ± 0.54 | 13.95% | - | - |
| DC 34 OP2 | 9-CH2NHC2H4OH | 2.16 ± 0.65 | 13.95% | 1.80 ± 0.25 | 18.20% |
| ASN 566 HD21 | 16 >=O | 2.56 ± 0.91 | 12.40% | - | - |
| DA 35 O4′ | 9-CH2NHC2H4OH | 2.48 ± 0.72 | 11.63% | - | - |
| DG5 11 N7 | 20 -OH | 3.05 ± 0.93 | 9.30% | 2.12 ± 0.33 | 43.70% |
| DG5 11 HO5′ | 21 >C=O | 2.24 ± 0.79 | 8.53% | 2.34 ± 0.54 | 19.15% |
| DG5 11 O4′ | 20 -OH | 2.40 ± 1.01 | 6.20% | 2.20 ± 0.48 | 17.30% |
| GLU 200 H | 9-CH2NHC2H4OH | 1.87 ± 0.71 | 5.43% | 2.28 ± 0.29 | 32.00% |
| ASN 566 HD21 | 21 >O | 2.06 ± 0.93 | 3.88% | 2.86 ± 0.44 | 22.05% |
| Average H-bond number per residue in family structure 3 | 2.09 | 2.73 | |||
| Docking Family Structure 4 | MD Family Structure 4 | ||||
| ASN 566 OD1 | 9-CH2NHC2H4OH | 2.05 ± 0.43 | 42.68% | 1.79 ± 0.21 | 14.60% |
| ASN 196 HD22 | 21 >C=O | 2.17 ± 0.50 | 30.49% | 2.28 ± 0.34 | 19.95% |
| ASN 196 HD22 | 21 >O | 2.55 ± 0.69 | 29.27% | 2.41 ± 0.41 | 20.00% |
| DC 34 O3′ | 20 -OH | 2.66 ± 0.77 | 25.61% | 2.63 ± 0.60 | 3.15% |
| DG5 11 O4′ | 9-CH2NHC2H4OH | 2.04 ± 0.55 | 18.29% | 2.36 ± 0.34 | 15.75% |
| ASN 566 HD21 | 9-CH2NHC2H4OH | 2.11 ± 0.63 | 18.29% | 2.50 ± 0.52 | 1.70% |
| ASN 566 OD1 | 9-CH2NHC2H4OH | 2.13 ± 0.60 | 15.85% | - | - |
| ASN 566 HD21 | 10 -OH | 1.98 ± 0.70 | 14.63% | - | - |
| THR 562 OG1 | 9-CH2NHC2H4OH | 2.05 ± 0.67 | 13.42% | 2.86 ± 1.09 | 0.35% |
| DA 35 OP2 | 20 -OH | 2.38 ± 0.78 | 13.42% | 2.80 ± 0.41 | 5.45% |
| ASN 566 OD1 | 10 -OH | 2.07 ± 0.67 | 12.20% | - | - |
| DG5 11 HO5′ | 10 -OH | 2.00 ± 0.75 | 12.20% | 2.16 ± 0.40 | 4.65% |
| LYS 218 HZ1, 2, 3 | 20 -OH | 2.25 ± 1.53 | 10.98% | 1.94 ± 0.29 | 47.30% |
| GLU 200 H | 16 >C=O | 2.01 ± 0.76 | 9.76% | 2.25 ± 0.26 | 49.65% |
| DA 35 N6 | 9-CH2NHC2H4OH | - | - | 2.45 ± 0.31 | 23.60% |
| DT3 10 O4 | 9-CH2NHC2H4OH | - | - | 1.96 ± 0.38 | 18.00% |
| Average H-bond number per residue in family structure 4 | 2.67 | 2.24 | |||
| Base | H1′ | H2′ | H2″ | H3′ | H4′ | H5′, H5″ | H6/8 | H2/5/Me |
|---|---|---|---|---|---|---|---|---|
| T4 | 0.00 | −0.013 | −0.061 | 0.012 | −0.041 | - | 0 | −0.022 |
| T5 | 0.01 | 0.045 | −0.062 | −0.005 | −0.022 | - | 0.069 | −0.020 |
| nick | ----------- | ---------- | ---------- | ----------- | ---------- | ------------------ | ----------- | ----------- |
| G6 | −0.36 | −0.326 | −0.081 | 0.02 | −0.053 | −0.08/−0.26 | 0.009 | - |
| T7 | −0.06 | −0.029 | −0.051 | −0.025 | −0.115 | - | −0.236 | −0.027 |
| A14 | −0.02 | 0.009 | −0.15 | −0.122 | −0.059 | - | −0.094 | −0.06 |
| C15 | −0.09 | 0.238 | 0.142 | - | 0.02 | - | 0.035 | −0.265 |
| A16 | 0.057 | 0.020 | −0.107 | −0.045 | - | - | −0.247 | −0.218 |
| A17 | −0.02 | −0.056 | −0.045 | −0.034 | −0.029 | - | −0.088 | −0.101 |
| Family Structure | Free Energy of Binding [kcal/mol] | |||||
|---|---|---|---|---|---|---|
| MM-PBSA | MM-GBSA | |||||
| Trajectory A | Trajectory B | (A + B)/2 | Trajectory A | Trajectory B | (A + B)/2 | |
| 1 | −32.29 ± 4.35 | −32.23 ± 4.34 | −32.26 ± 4.40 | −33.09 ± 3.17 | −32.71 ± 3.33 | −32.91 ± 3.28 |
| 2 | −32.99 ± 3.41 | −27.10 ± 5.12 | −30.05 ± 4.56 | −30.63 ± 2.86 | −24.76 ± 4.76 | −27.69 ± 3.85 |
| 3 | −37.52 ± 3.47 | −35.74 ± 3.80 | −36.63 ± 3.93 | −32.80 ± 2.99 | −31.46 ± 3.03 | −32.13 ± 3.16 |
| 4 | −34.04 ± 3.05 | −34.20 ± 3.15 | −34.12 ± 3.12 | −33.44 ± 2.37 | −33.45 ± 2.44 | −33.45 ± 2.40 |
| Family Structure | PM7 Energy [kcal/mol] | ||
|---|---|---|---|
| Trajectory A | Trajectory B | (A + B)/2 | |
| 1 | −2378.18 ± 14.84 | −2378.04 ± 16.51 | −2378.11 ± 15.69 |
| 2 | −2379.80 ± 18.12 | −2378.37 ± 23.54 | −2379.09 ± 20.88 |
| 3 | −2385.59 ± 16.02 | −2384.05 ± 21.30 | −2384.82 ± 20.60 |
| 4 | −2376.71 ± 19.07 | −2377.87 ± 15.68 | −2377.29 ± 15.62 |
| Comp. 1 | DNA | NOE a | Family Structure 1 b | Family Structure 2 | Family Structure 3 | Family Structure 4 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <6 Å | <5 Å | <4 Å | <3 Å | <6 Å | <5 Å | <4 Å | <3 Å | <6 Å | <5 Å | <4 Å | <3 Å | <6 Å | <5 Å | <4 Å | <3 Å | |||
| 14 | A16H2 | 68% | ++++ | ++++ | ++++ | +++ | ++ | o | ++++ | ++++ | ++++ | + | ||||||
| 17a | G6H2″ | 90% | ++++ | +++ | + | o | +++ | |||||||||||
| 17b | ++++ | o | o | ++++ | +++ | + | ||||||||||||
| 17a | G6H4′ | 75% | ++++ | ++++ | ++++ | +++ | ++ | + | ||||||||||
| 17b | ++++ | ++++ | ++++ | ++ | +++ | ++ | ++ | o | ||||||||||
| 17a | T5H1′ | 70% | ++++ | ++ | + | o | ||||||||||||
| 17b | ++++ | ++++ | ++ | + | o | |||||||||||||
| 19 | G6H2″ | 14% | ++ | + | o | |||||||||||||
| 19 | G6H4′ | 45% | +++ | ++ | + | |||||||||||||
| 19 | T7H4′ | 43% | ++++ | ++++ | ++++ | + | o | |||||||||||
| 19 | T7H5′ | 60% | ++++ | +++ | +++ | + | o | |||||||||||
| 19 | T7H6 | 18% | ++ | |||||||||||||||
| 23 | C15H6 | 8% | o | |||||||||||||||
| Sample | Concentration (mM) | 1 (19-CH3) Integral | 2B (T7-CH3) Integral | Di of 1 (10−10 m2 s−1) | Di of 2B (10−10 m2 s−1) | Ka (mM−1) | % of Complex | |
|---|---|---|---|---|---|---|---|---|
| 1 | 2B | |||||||
| Complex | 0.33 b | 0.33 b | 1 | 1 | 1.23 | 1.13 | 746.0 | 94% 6% |
| Compound 1 | 0.7 a | - | - | - | 2.75 | - | - | - |
| Decamer 2B | - | 0.96 a | - | - | - | 1.13 | - | - |
| Compound | CPT | SN38 | 1 |
|---|---|---|---|
| IC50 [µM] (mean ± SD) | 11.92 ± 9.77 | 7.37 ± 7.10 | 1.73 ± 0.48 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bocian, W.; Naumczuk, B.; Urbanowicz, M.; Sitkowski, J.; Bednarek, E.; Wiktorska, K.; Pogorzelska, A.; Wielgus, E.; Kozerski, L. Insight on the Interaction between the Camptothecin Derivative and DNA Oligomer Mimicking the Target of Topo I Inhibitors. Molecules 2022, 27, 6946. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27206946
Bocian W, Naumczuk B, Urbanowicz M, Sitkowski J, Bednarek E, Wiktorska K, Pogorzelska A, Wielgus E, Kozerski L. Insight on the Interaction between the Camptothecin Derivative and DNA Oligomer Mimicking the Target of Topo I Inhibitors. Molecules. 2022; 27(20):6946. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27206946
Chicago/Turabian StyleBocian, Wojciech, Beata Naumczuk, Magdalena Urbanowicz, Jerzy Sitkowski, Elżbieta Bednarek, Katarzyna Wiktorska, Anna Pogorzelska, Ewelina Wielgus, and Lech Kozerski. 2022. "Insight on the Interaction between the Camptothecin Derivative and DNA Oligomer Mimicking the Target of Topo I Inhibitors" Molecules 27, no. 20: 6946. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules27206946