Alzheimer’s Disease: An Overview of Major Hypotheses and Therapeutic Options in Nanotechnology

 and
and
Abstract
:1. Introduction
2. Pathophysiology of the Disease
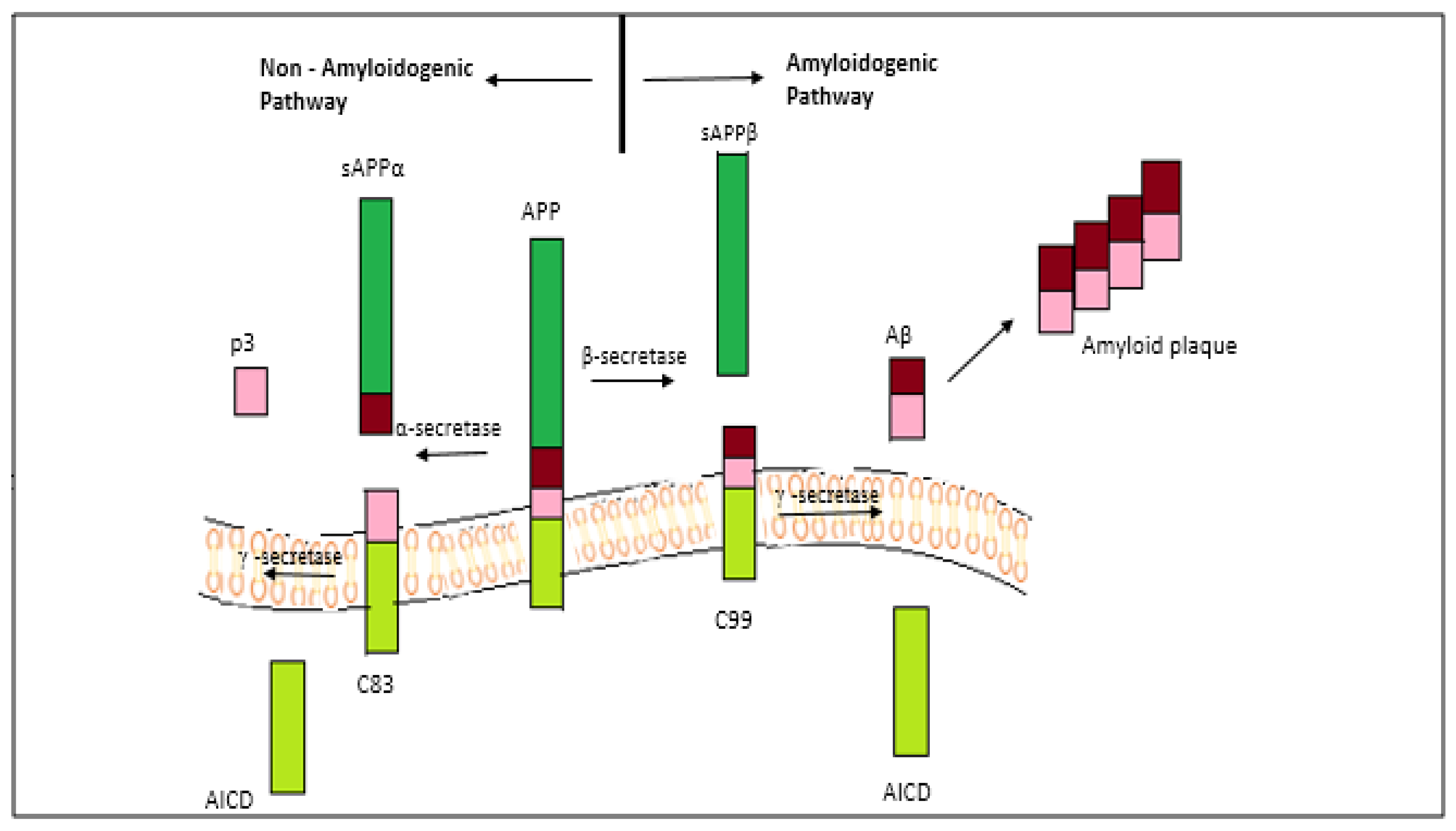
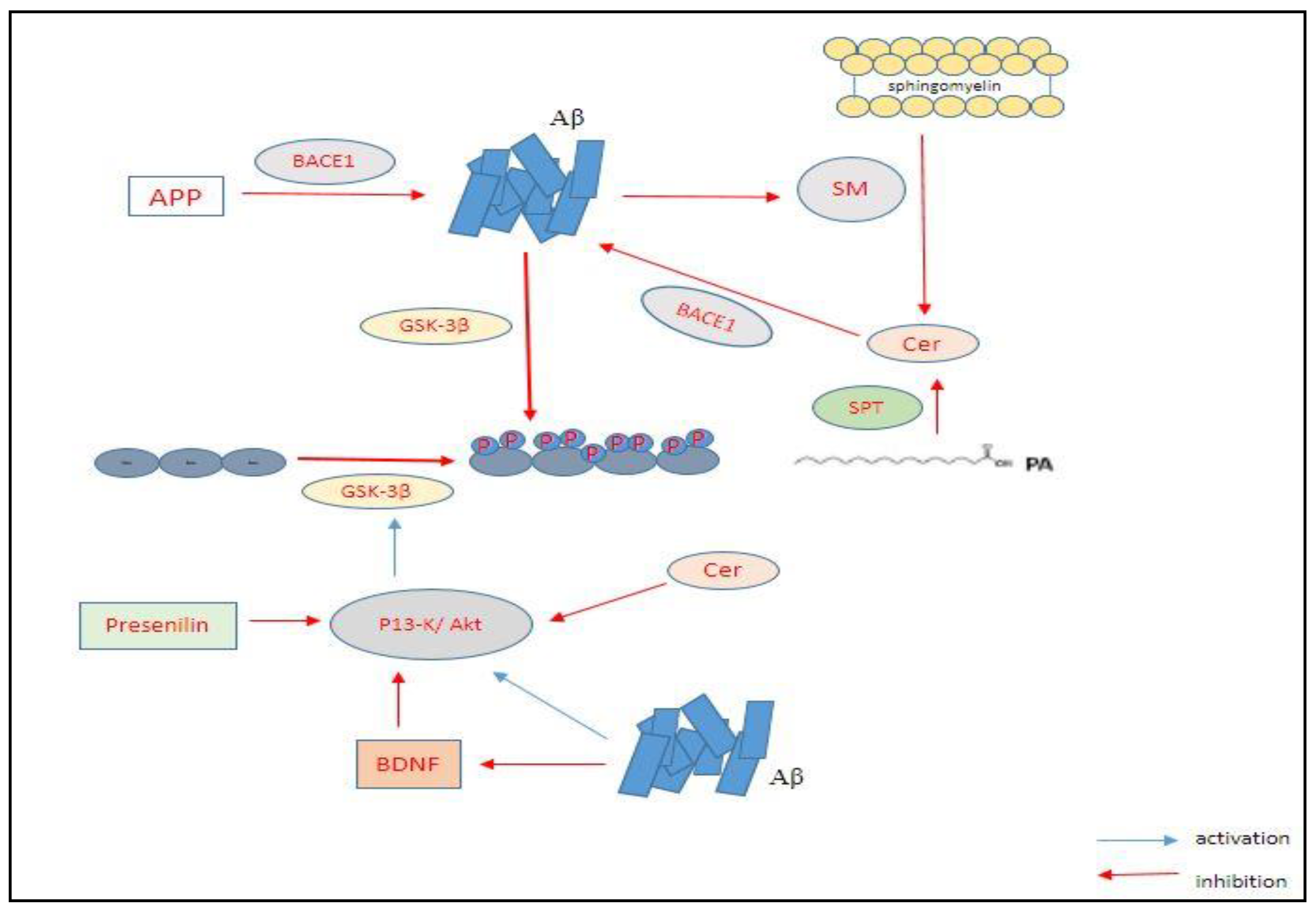
2.1. The Amyloid-Beta Hypothesis
2.2. The Tau Hypothesis
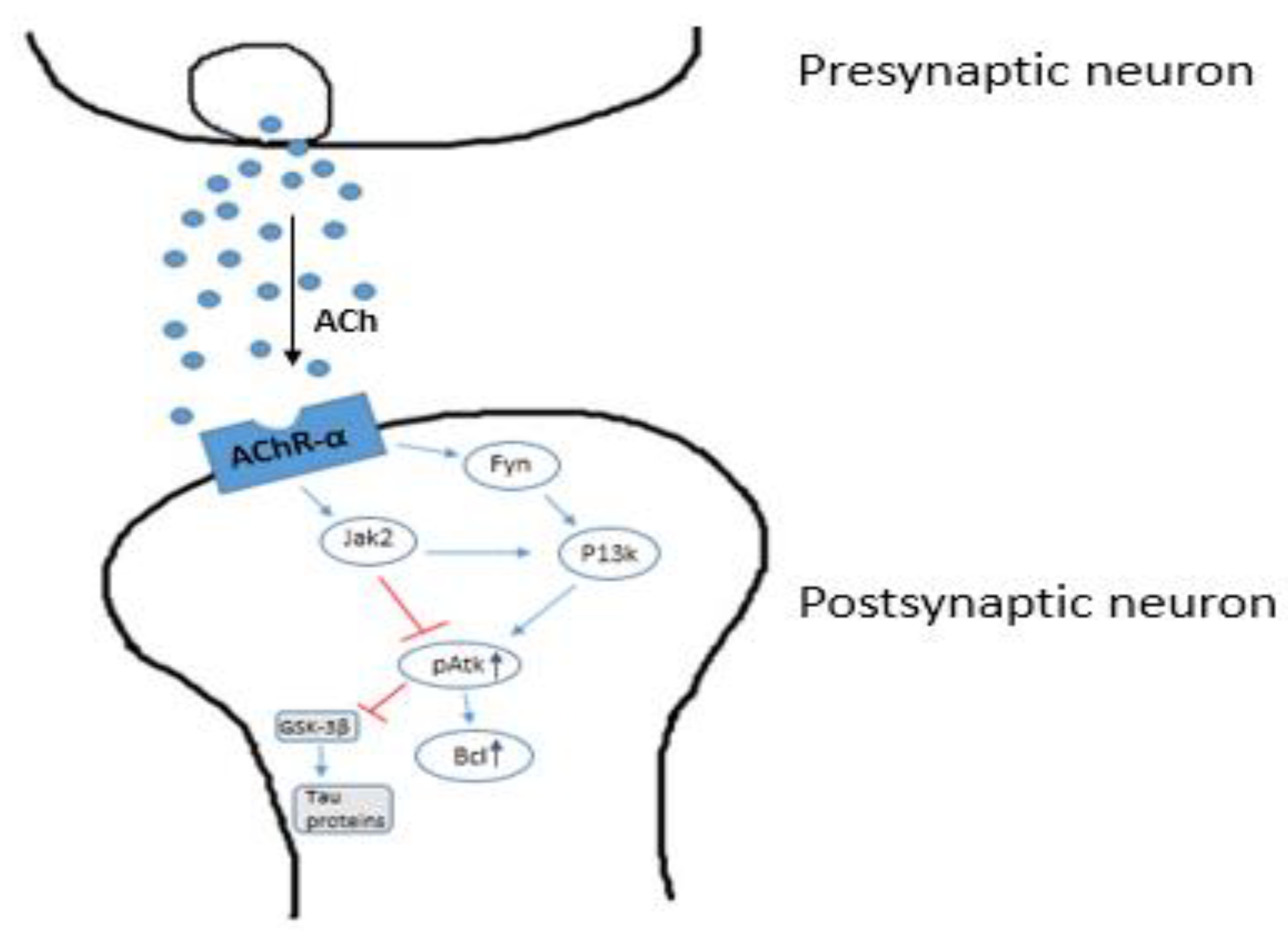
2.3. The Cholinergic Hypothesis
2.4. The Dendritic Hypothesis
2.4.1. Wnt/Beta-Catenin Signaling
2.4.2. GSK3-β Activity
2.5. The 5-HT6 Receptor Hypothesis
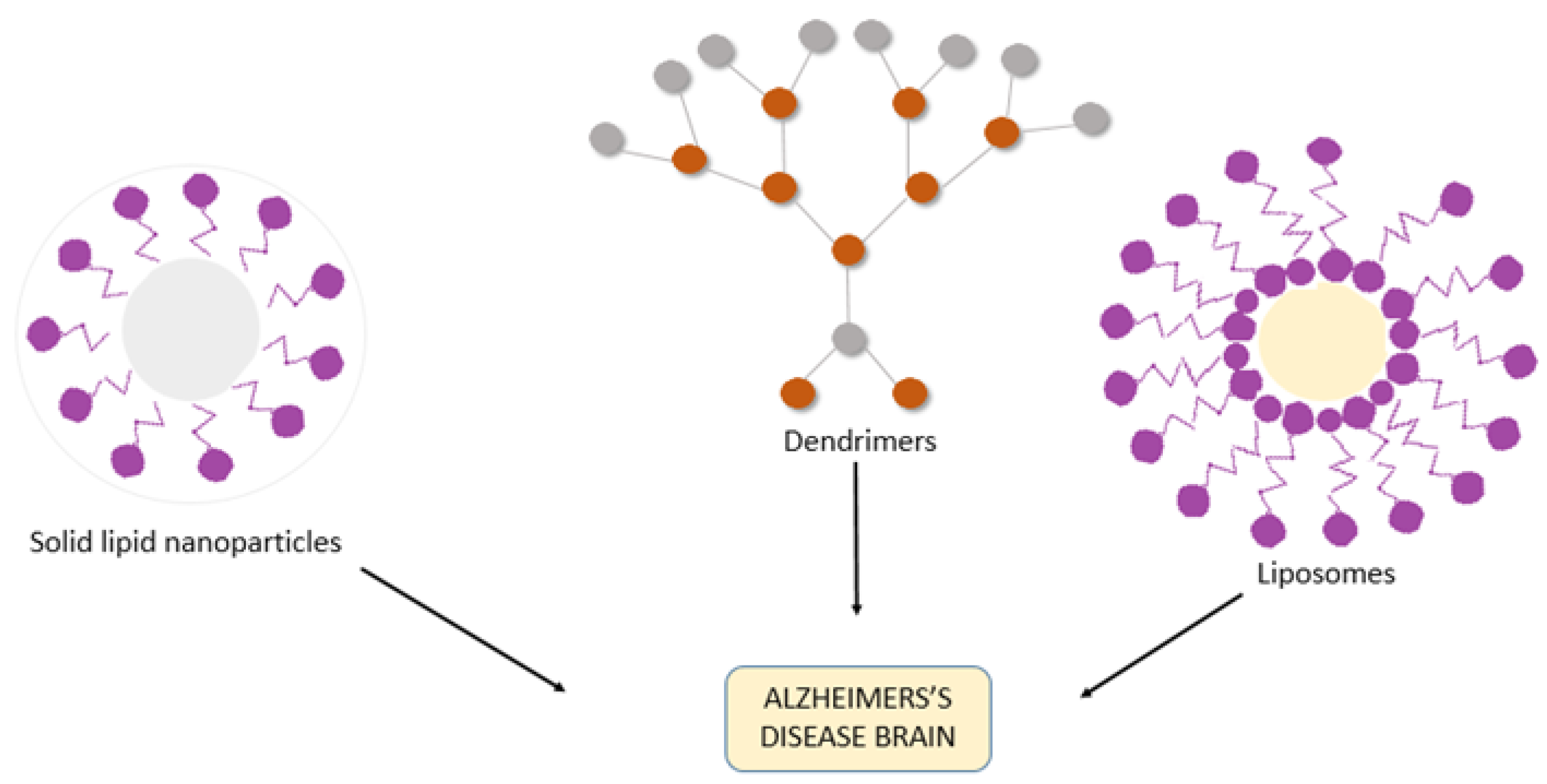
3. Nanotechnology-Assisted Drug Delivery Strategies for AD
3.1. Liposomes
3.2. Polymeric Nanoparticles
3.3. Micro- and Nanoemulsions
3.4. Dendrimers
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prince, M.J. World Alzheimer Report 2015: The Global Impact of Dementia: An Analysis of Prevalence, Incidence, Cost and Trends; Alzheimer’s Disease International: London, UK, 2015. [Google Scholar]
- Inouye, K.; Pedrazzani, E.S.; Pavarini, S.C.I. Alzheimer’s Disease Influence on the Perception of Quality of life from the Elderly People [Influência da Doença de Alzheimer Na Percepção de Qualidade de Vida do Idoso]. Rev. Esc. Enferm. 2010, 44, 1093–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciobica, A.; Popescu, R.; Haulica, I.; Bild, W. Aspects regarding the neurobiology of psycho-affective functions. J. Med. Biochem. 2012, 31, 83–87. [Google Scholar] [CrossRef] [Green Version]
- Selkoe, D.J. Clearing the brain’s amyloid cobwebs. Neuron 2001, 32, 177–180. [Google Scholar] [CrossRef] [Green Version]
- Zlokovic, B.V.; Yamada, S.; Holtzman, D.; Ghiso, J.; Frangione, B. Clearance of amyloid β-peptide from brain: Transport or metabolism? Nat. Med. 2000, 6, 718. [Google Scholar] [CrossRef]
- Tanzi, R.; Moir, R.; Wagner, S. Clearance of Alzheimer’s Aβ peptide: The many roads to perdition. Neuron 2004, 43, 605–608. [Google Scholar] [PubMed] [Green Version]
- Zlokovic, B.V. New therapeutic targets in the neurovascular pathway in Alzheimer’s disease. Neurotherapeutics 2008, 5, 409–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karran, E.; Mercken, M.; De Strooper, B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov. 2011, 10, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Lilly, E. Eli Lilly and Company Announces Top-Line Results on Solanezumab Phase 3 Clinical Trials in Patients with Alzheimer’s Disease. Acquir. Media 2012, 24. Available online: http://newsroom.lilly.com/releasedetail.cfm?releaseid=702211 (accessed on 28 December 2020).
- Johnson, J. Johnson & Johnson Announces Discontinuation Of Phase 3 Development of Bapineuzumab Intravenous (IV). Mild-To-Moderate Alzheimer’s Disease 2012. Available online: http://www.jnj.com/connect/news/all/johnson-and-johnson-announcesdiscontinuation-of-phase-3-development-of-bapineuzumab-intravenous-iv-inmild-to-moderate-alzheimers-disease (accessed on 28 December 2020).
- Chen, X.; Pan, W. The treatment strategies for neurodegenerative diseases by integrative medicine. Integr. Med. Int. 2014, 1, 223–225. [Google Scholar] [CrossRef]
- Aisen, P.S.; Cummings, J.; Jack, C.R.; Morris, J.C.; Sperling, R.; Frölich, L.; Jones, R.W.; Dowsett, S.A.; Matthews, B.R.; Raskin, J. On the path to 2025: Understanding the Alzheimer’s disease continuum. Alzheimer’s Res. Ther. 2017, 9, 60. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.; Lee, G.; Ritter, A.; Sabbagh, M.; Zhong, K. Alzheimer’s disease drug development pipeline: 2019. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2019, 5, 272–293. [Google Scholar] [CrossRef]
- Hung, S.-Y.; Fu, W.-M. Drug candidates in clinical trials for Alzheimer’s disease. J. Biomed. Sci. 2017, 24, 47. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2019 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Bondi, M.W.; Edmonds, E.C.; Salmon, D.P. Alzheimer’s disease: Past, present, and future. J. Int. Neuropsychol. Soc. 2017, 23, 818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alzheimer, A. Allgemeine Zeitschrift für Psychiatrie und Psychisch-Gerichtliche Medizin. E. Schultze Und O. Snell. Vol. 1907, 64, 146–148. [Google Scholar]
- Sanabria-Castro, A.; Alvarado-Echeverría, I.; Monge-Bonilla, C. Molecular pathogenesis of Alzheimer’s disease: An update. Ann. Neurosci. 2017, 24, 46–54. [Google Scholar] [CrossRef]
- Slot, R.E.; Sikkes, S.A.; Berkhof, J.; Brodaty, H.; Buckley, R.; Cavedo, E.; Dardiotis, E.; Guillo-Benarous, F.; Hampel, H.; Kochan, N.A. Subjective cognitive decline and rates of incident Alzheimer’s disease and non-Alzheimer’s disease dementia. Alzheimer’s Dement. 2019, 15, 465–476. [Google Scholar] [CrossRef]
- Cacace, R.; Sleegers, K.; Van Broeckhoven, C. Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimer’s Dement. 2016, 12, 733–748. [Google Scholar] [CrossRef] [Green Version]
- Grothe, M.J.; Sepulcre, J.; Gonzalez-Escamilla, G.; Jelistratova, I.; Schöll, M.; Hansson, O.; Teipel, S.J.; Initiative, A.S.D.N. Molecular properties underlying regional vulnerability to Alzheimer’s disease pathology. Brain 2018, 141, 2755–2771. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–186. [Google Scholar] [CrossRef]
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Karlawish, J.; Jack, C.R., Jr.; Rocca, W.A.; Snyder, H.M.; Carrillo, M.C. Alzheimer’s disease: The next frontier—Special Report 2017. Alzheimer’s Dement. 2017, 13, 374–380. [Google Scholar] [CrossRef]
- Qiang, W.; Yau, W.-M.; Lu, J.-X.; Collinge, J.; Tycko, R. Structural variation in amyloid-β fibrils from Alzheimer’s disease clinical subtypes. Nature 2017, 541, 217–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, T.; Iwata, N.; Tsubuki, S.; Takaki, Y.; Takano, J.; Huang, S.-M.; Suemoto, T.; Higuchi, M.; Saido, T.C. Somatostatin regulates brain amyloid β peptide Aβ 42 through modulation of proteolytic degradation. Nat. Med. 2005, 11, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Arya, M.; Kumar, M.K.M.; Sabitha, M.; Menon, K.N.; Nair, S.C. Nanotechnology approaches for enhanced CNS delivery in treating Alzheimer’s disease. J. Drug Deliv. Sci. Technol. 2019, 51, 297–309. [Google Scholar] [CrossRef]
- Willem, M.; Tahirovic, S.; Busche, M.A.; Ovsepian, S.V.; Chafai, M.; Kootar, S.; Hornburg, D.; Evans, L.D.; Moore, S.; Daria, A. η-Secretase processing of APP inhibits neuronal activity in the hippocampus. Nature 2015, 526, 443–447. [Google Scholar] [CrossRef]
- Puzzo, D.; Arancio, O. Amyloid-β peptide: Dr. Jekyll or Mr. Hyde? J. Alzheimer’s Dis. 2013, 33, S111–S120. [Google Scholar] [CrossRef] [Green Version]
- Barber, R.C. The genetics of Alzheimer’s disease. Scientifica 2012, 23, 213–227. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; North, B.J.; Zhang, T.; Dai, X.; Tao, K.; Guo, J.; Wei, W. The emerging roles of protein homeostasis-governing pathways in Alzheimer’s disease. Aging Cell 2018, 17, e12801. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, A.; Kaneko, N.; Villemagne, V.L.; Kato, T.; Doecke, J.; Doré, V.; Fowler, C.; Li, Q.-X.; Martins, R.; Rowe, C. High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature 2018, 554, 249–254. [Google Scholar] [CrossRef]
- Eckman, C.B.; Mehta, N.D.; Crook, R.; Perez-tur, J.; Prihar, G.; Pfeiffer, E.; Graff-Radford, N.; Hinder, P.; Yager, D.; Zenk, B. A new pathogenic mutation in the APP gene (I716V) increases the relative proportion of Aβ42 (43). Hum. Mol. Genet. 1997, 6, 2087–2089. [Google Scholar] [CrossRef] [PubMed]
- Ancolio, K.; Dumanchin, C.; Barelli, H.; Warter, J.; Brice, A.; Campion, D.; Frebourg, T.; Checler, F. Unusual phenotypic alteration of β amyloid precursor protein (βAPP) maturation by a new Val-715→ Met βAPP-770 mutation responsible for probable early-onset Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 1999, 96, 4119–4124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar-Singh, S.; De Jonghe, C.; Cruts, M.; Kleinert, R.; Wang, R.; Mercken, M.; De Strooper, B.; Vanderstichele, H.; Löfgren, A.; Vanderhoeven, I. Nonfibrillar diffuse amyloid deposition due to a γ 42-secretase site mutation points to an essential role for N-truncated A β 42 in Alzheimer’s disease. Hum. Mol. Genet. 2000, 9, 2589–2598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murrell, J.R.; Hake, A.M.; Quaid, K.A.; Farlow, M.R.; Ghetti, B. Early-onset Alzheimer disease caused by a new mutation (V717L) in the amyloid precursor protein gene. Arch. Neurol. 2000, 57, 885–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwok, J.B.; Li, Q.X.; Hallupp, M.; Whyte, S.; Ames, D.; Beyreuther, K.; Masters, C.L.; Schofield, P.R. Novel Leu723Pro amyloid precursor protein mutation increases amyloid β42 (43) peptide levels and induces apoptosis. Ann. Neurol. 2000, 47, 249–253. [Google Scholar] [CrossRef]
- Steeland, S.; Vandenbroucke, R.E. Choroid plexus tumor necrosis factor receptor 1: A new neuroinflammatory piece of the complex Alzheimer’s disease puzzle. Neural Regen. Res. 2019, 14, 1144. [Google Scholar]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Sawda, C.; Moussa, C.; Turner, R.S. Resveratrol for Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2017, 1403, 142. [Google Scholar] [CrossRef]
- Tobinick, E. Tumour necrosis factor modulation for treatment of Alzheimer’s disease. CNS Drugs 2009, 23, 713–725. [Google Scholar] [CrossRef]
- Bachurin, S.O.; Bovina, E.V.; Ustyugov, A.A. Drugs in clinical trials for Alzheimer’s disease: The major trends. Med. Res. Rev. 2017, 37, 1186–1225. [Google Scholar] [CrossRef]
- Zhao, Q.-F.; Tan, L.; Wang, H.-F.; Jiang, T.; Tan, M.-S.; Tan, L.; Xu, W.; Li, J.-Q.; Wang, J.; Lai, T.-J. The prevalence of neuropsychiatric symptoms in Alzheimer’s disease: Systematic review and meta-analysis. J. Affect. Disord. 2016, 190, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Egan, M.F.; Kost, J.; Voss, T.; Mukai, Y.; Aisen, P.S.; Cummings, J.L.; Tariot, P.N.; Vellas, B.; van Dyck, C.H.; Boada, M. Randomized trial of verubecestat for prodromal Alzheimer’s disease. N. Engl. J. Med. 2019, 380, 1408–1420. [Google Scholar] [CrossRef] [PubMed]
- Kadavath, H.; Hofele, R.V.; Biernat, J.; Kumar, S.; Tepper, K.; Urlaub, H.; Mandelkow, E.; Zweckstetter, M. Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc. Natl. Acad. Sci. USA 2015, 112, 7501–7506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlyle, B.C.; Nairn, A.C.; Wang, M.; Yang, Y.; Jin, L.E.; Simen, A.A.; Ramos, B.P.; Bordner, K.A.; Craft, G.E.; Davies, P. cAMP-PKA phosphorylation of tau confers risk for degeneration in aging association cortex. Proc. Natl. Acad. Sci. USA 2014, 111, 5036–5041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, D.; Gutmann, J.M.; Götz, J. Mobility and subcellular localization of endogenous, gene-edited Tau differs from that of over-expressed human wild-type and P301L mutant Tau. Sci. Rep. 2016, 6, 29074. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K. Spreading of tau pathology in sporadic Alzheimer’s disease along cortico-cortical top-down connections. Cereb. Cortex 2018, 28, 3372–3384. [Google Scholar] [CrossRef] [Green Version]
- Ksiezak-Reding, H.; Liu, W.-K.; Yen, S.-H. Phosphate analysis and dephosphorylation of modified tau associated with paired helical filaments. Brain Res. 1992, 597, 209–219. [Google Scholar] [CrossRef]
- Köpke, E.; Tung, Y.-C.; Shaikh, S.; Alonso, A.d.C.; Iqbal, K.; Grundke-Iqbal, I. Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J. Biol. Chem. 1993, 268, 24374–24384. [Google Scholar]
- Kametani, F.; Hasegawa, M. Reconsideration of amyloid hypothesis and tau hypothesis in Alzheimer’s disease. Front. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef] [Green Version]
- Bejanin, A.; Schonhaut, D.R.; La Joie, R.; Kramer, J.H.; Baker, S.L.; Sosa, N.; Ayakta, N.; Cantwell, A.; Janabi, M.; Lauriola, M. Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer’s disease. Brain 2017, 140, 3286–3300. [Google Scholar] [CrossRef] [Green Version]
- Mohandas, E.; Rajmohan, V.; Raghunath, B. Neurobiology of Alzheimer’s disease. Indian J. Psychiatry 2009, 51, 55. [Google Scholar] [CrossRef] [PubMed]
- Medina, M.; Castro, A. Glycogen synthase kinase-3 (GSK-3) inhibitors reach the clinic. Curr. Opin. Drug Discov. Dev. 2008, 11, 533–543. [Google Scholar]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 hypothesis of Alzheimer’s disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzpatrick, A.W.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakota, L.; Brandt, R. Tau biology and tau-directed therapies for Alzheimer’s disease. Drugs 2016, 76, 301–313. [Google Scholar] [CrossRef] [Green Version]
- Jack Jr, C.R.; Wiste, H.J.; Schwarz, C.G.; Lowe, V.J.; Senjem, M.L.; Vemuri, P.; Weigand, S.D.; Therneau, T.M.; Knopman, D.S.; Gunter, J.L. Longitudinal tau PET in ageing and Alzheimer’s disease. Brain 2018, 141, 1517–1528. [Google Scholar] [CrossRef] [Green Version]
- Buée, L.; Bussière, T.; Buée-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef]
- Himmler, A.; Drechsel, D.; Kirschner, M.W.; Martin, D.W. Tau consists of a set of proteins with repeated C-terminal microtubule-binding domains and variable N-terminal domains. Mol. Cell. Biol. 1989, 9, 1381–1388. [Google Scholar] [CrossRef] [Green Version]
- Hanger, D.P.; Byers, H.L.; Wray, S.; Leung, K.-Y.; Saxton, M.J.; Seereeram, A.; Reynolds, C.H.; Ward, M.A.; Anderton, B.H. Novel phosphorylation sites in tau from Alzheimer brain support a role for casein kinase 1 in disease pathogenesis. J. Biol. Chem. 2007, 282, 23645–23654. [Google Scholar] [CrossRef] [Green Version]
- Collins, M.O.; Yu, L.; Coba, M.P.; Husi, H.; Campuzano, I.; Blackstock, W.P.; Choudhary, J.S.; Grant, S.G. Proteomic analysis of in vivo phosphorylated synaptic proteins. J. Biol. Chem. 2005, 280, 5972–5982. [Google Scholar] [CrossRef] [Green Version]
- Hanger, D.P.; Betts, J.C.; Loviny, T.L.; Blackstock, W.P.; Anderton, B.H. New phosphorylation sites identified in hyperphosphorylated tau (paired helical filament-tau) from Alzheimer’s disease brain using nanoelectrospray mass spectrometry. J. Neurochem. 1998, 71, 2465–2476. [Google Scholar] [CrossRef] [PubMed]
- Morishima-Kawashima, M.; Hasegawa, M.; Takio, K.; Suzuki, M.; Yoshida, H.; Watanabe, A.; Titani, K.; Ihara, Y. Hyperphosphorylation of tau in PHF. Neurobiol. Aging 1995, 16, 365–371. [Google Scholar] [CrossRef]
- Guo, C.; Jeong, H.-H.; Hsieh, Y.-C.; Klein, H.-U.; Bennett, D.A.; De Jager, P.L.; Liu, Z.; Shulman, J.M. Tau activates transposable elements in Alzheimer’s disease. Cell Rep. 2018, 23, 2874–2880. [Google Scholar] [CrossRef] [PubMed]
- Francis, P.T.; Palmer, A.M.; Snape, M.; Wilcock, G.K. The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J. Neurol. Neurosurg. Psychiatry 1999, 66, 137–147. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.-M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef]
- Fine, A.; Hoyle, C.; Maclean, C.; Levatte, T.; Baker, H.; Ridley, R. Learning impairments following injection of a selective cholinergic immunotoxin, ME20. 4 IgG-saporin, into the basal nucleus of Meynert in monkeys. Neuroscience 1997, 81, 331–343. [Google Scholar] [CrossRef]
- Miranda, M.I.; Bermúdez-Rattoni, F. Reversible inactivation of the nucleus basalis magnocellularis induces disruption of cortical acetylcholine release and acquisition, but not retrieval, of aversive memories. Proc. Natl. Acad. Sci. USA 1999, 96, 6478–6482. [Google Scholar] [CrossRef] [Green Version]
- Dunnett, S.B.; Everitt, B.J.; Robbins, T.W. The basal forebrain-cortical cholinergic system: Interpreting the functional consequences of excitotoxic lesions. Trends Neurosci. 1991, 14, 494–501. [Google Scholar] [CrossRef]
- Hasselmo, M.E.; Anderson, B.P.; Bower, J.M. Cholinergic modulation of cortical associative memory function. J. Neurophysiol. 1992, 67, 1230–1246. [Google Scholar] [CrossRef]
- Sarter, M.; Bruno, J.P. Cognitive functions of cortical acetylcholine: Toward a unifying hypothesis. Brain Res. Rev. 1997, 23, 28–46. [Google Scholar] [CrossRef]
- Whitehouse, P.J.; Price, D.L.; Clark, A.W.; Coyle, J.T.; DeLong, M.R. Alzheimer disease: Evidence for selective loss of cholinergic neurons in the nucleus basalis. Ann. Neurol. 1981, 10, 122–126. [Google Scholar] [CrossRef]
- Whitehouse, P.J.; Price, D.L.; Struble, R.G.; Clark, A.W.; Coyle, J.T.; Delon, M.R. Alzheimer’s disease and senile dementia: Loss of neurons in the basal forebrain. Science 1982, 215, 1237–1239. [Google Scholar] [CrossRef]
- Bell, K.F.; Ducatenzeiler, A.; Ribeiro-da-Silva, A.; Duff, K.; Bennett, D.A.; Cuello, A.C. The amyloid pathology progresses in a neurotransmitter-specific manner. Neurobiol. Aging 2006, 27, 1644–1657. [Google Scholar] [CrossRef] [PubMed]
- Wilcock, G.; Esiri, M.; Bowen, D.; Smith, C. Alzheimer’s disease: Correlation of cortical choline acetyltransferase activity with the severity of dementia and histological abnormalities. J. Neurol. Sci. 1982, 57, 407–417. [Google Scholar] [CrossRef]
- Oda, Y. Choline acetyltransferase: The structure, distribution and pathologic changes in the central nervous system. Pathol. Int. 1999, 49, 921–937. [Google Scholar] [CrossRef] [PubMed]
- Schliebs, R.; Arendt, T. The significance of the cholinergic system in the brain during aging and in Alzheimer’s disease. J. Neural Transm. 2006, 113, 1625–1644. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the cholinergic system. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Blennow, K.; Hampel, H.; Weiner, M.; Zetterberg, H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat. Rev. Neurol. 2010, 6, 131–144. [Google Scholar] [CrossRef]
- Drummond, E.; Wisniewski, T. Alzheimer’s disease: Experimental models and reality. Acta Neuropathol. 2017, 133, 155–175. [Google Scholar] [CrossRef]
- Birks, J.S.; Harvey, R.J. Donepezil for dementia due to Alzheimer’s disease. Cochrane Database Syst. Rev. 2018, 6, CD001190. [Google Scholar] [CrossRef]
- Martorana, A.; Esposito, Z.; Koch, G. Beyond the cholinergic hypothesis: Do current drugs work in Alzheimer’s disease? CNS Neurosci. Ther. 2010, 16, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Asztély, F.; Gustafsson, B. Ionotropic glutamate receptors. Mol. Neurobiol. 1996, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Dingledine, R.; Borges, K.; Bowie, D.; Traynelis, S.F. The glutamate receptor ion channels. Pharmacol. Rev. 1999, 51, 7–62. [Google Scholar] [PubMed]
- Lau, C.G.; Zukin, R.S. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat. Rev. Neurosci. 2007, 8, 413–426. [Google Scholar] [CrossRef]
- Paoletti, P.; Bellone, C.; Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 2013, 14, 383–400. [Google Scholar] [CrossRef]
- Twomey, E.C.; Sobolevsky, A.I. Structural mechanisms of gating in ionotropic glutamate receptors. Biochemistry 2018, 57, 267–276. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Reddy, P.H. Role of glutamate and NMDA receptors in Alzheimer’s disease. J. Alzheimer’s Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [Green Version]
- Lüscher, C.; Malenka, R.C. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb. Perspect. Biol. 2012, 4, a005710. [Google Scholar] [CrossRef] [Green Version]
- Ivanova, O.Y.; Dobryakova, Y.V.; Salozhin, S.V.; Aniol, V.A.; Onufriev, M.V.; Gulyaeva, N.V.; Markevich, V.A. Lentiviral modulation of Wnt/β-catenin signaling affects in vivo LTP. Cell. Mol. Neurobiol. 2017, 37, 1227–1241. [Google Scholar] [CrossRef]
- Abramov, E.; Dolev, I.; Fogel, H.; Ciccotosto, G.D.; Ruff, E.; Slutsky, I. Amyloid-β as a positive endogenous regulator of release probability at hippocampal synapses. Nat. Neurosci. 2009, 12, 1567. [Google Scholar] [CrossRef] [PubMed]
- Mota, S.I.; Ferreira, I.L.; Rego, A.C. Dysfunctional synapse in Alzheimer’s disease–A focus on NMDA receptors. Neuropharmacology 2014, 76, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.-J.; Han, H.-S.; Kim, M.-J.; Koo, S.-H. CREB and FoxO1: Two transcription factors for the regulation of hepatic gluconeogenesis. BMB Rep. 2013, 46, 567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanke, M.L.; VanDongen, A.M. Activation Mechanisms of the NMDA Receptor. In Biology of the NMDA Receptor; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2008; Chapter 13. [Google Scholar]
- Schmidt, A.P.; Tort, A.B.; Silveira, P.P.; Böhmer, A.E.; Hansel, G.; Knorr, L.; Schallenberger, C.; Dalmaz, C.; Elisabetsky, E.; Crestana, R.H. The NMDA antagonist MK-801 induces hyperalgesia and increases CSF excitatory amino acids in rats: Reversal by guanosine. Pharmacol. Biochem. Behav. 2009, 91, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Wegner, C.; Esiri, M.M.; Chance, S.; Palace, J.; Matthews, P. Neocortical neuronal, synaptic, and glial loss in multiple sclerosis. Neurology 2006, 67, 960–967. [Google Scholar] [CrossRef]
- Nakanishi, N.; Ryan, S.D.; Zhang, X.; Khan, A.; Holland, T.; Cho, E.-G.; Huang, X.; Liao, F.-F.; Xu, H.; Lipton, S.A. Synaptic protein α1-takusan mitigates amyloid-β-induced synaptic loss via interaction with tau and postsynaptic density-95 at postsynaptic sites. J. Neurosci. 2013, 33, 14170–14183. [Google Scholar] [CrossRef]
- Wang, H.; Ren, C.H.; Gunawardana, C.G.; Schmitt-Ulms, G. Overcoming barriers and thresholds–signaling of oligomeric Aβ through the prion protein to Fyn. Mol. Neurodegener. 2013, 8, 24. [Google Scholar] [CrossRef] [Green Version]
- Joiner, D.M.; Ke, J.; Zhong, Z.; Xu, H.E.; Williams, B.O. LRP5 and LRP6 in development and disease. Trends Endocrinol. Metab. 2013, 24, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Z.; Biechele, T.; Wei, Z.; Morrone, S.; Moon, R.T.; Wang, L.; Xu, W. Crystal structures of the extracellular domain of LRP6 and its complex with DKK1. Nat. Struct. Mol. Biol. 2011, 18, 1204. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/β-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [Green Version]
- Kuwabara, T.; Hsieh, J.; Muotri, A.; Yeo, G.; Warashina, M.; Lie, D.C.; Moore, L.; Nakashima, K.; Asashima, M.; Gage, F.H. Wnt-mediated activation of NeuroD1 and retro-elements during adult neurogenesis. Nat. Neurosci. 2009, 12, 1097–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, A.R.; Godoy, J.A.; Mullendorff, K.; Olivares, G.H.; Bronfman, M.; Inestrosa, N.C. Wnt-3a overcomes β-amyloid toxicity in rat hippocampal neurons. Exp. Cell Res. 2004, 297, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Piña-Crespo, J.; Li, Y. Restoring Wnt/β-catenin signaling is a promising therapeutic strategy for Alzheimer’s disease. Mol. Brain 2019, 12, 104. [Google Scholar] [CrossRef] [PubMed]
- Haughey, N.J.; Nath, A.; Chan, S.L.; Borchard, A.; Rao, M.S.; Mattson, M.P. Disruption of neurogenesis by amyloid β-peptide, and perturbed neural progenitor cell homeostasis, in models of Alzheimer’s disease. J. Neurochem. 2002, 83, 1509–1524. [Google Scholar] [CrossRef] [PubMed]
- Mardones, M.D.; Andaur, G.A.; Varas-Godoy, M.; Henriquez, J.F.; Salech, F.; Behrens, M.I.; Couve, A.; Inestrosa, N.C.; Varela-Nallar, L. Frizzled-1 receptor regulates adult hippocampal neurogenesis. Mol. Brain 2016, 9, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Luo, J.; Xu, L.; Zhou, H.; Zhang, W. Harnessing low-density lipoprotein receptor protein 6 (LRP6) genetic variation and Wnt signaling for innovative diagnostics in complex diseases. Pharm. J. 2018, 18, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Buechler, J.; Salinas, P.C. Deficient Wnt signaling and synaptic vulnerability in Alzheimer’s disease: Emerging roles for the LRP6 receptor. Front. Synaptic Neurosci. 2018, 10, 38. [Google Scholar] [CrossRef]
- Ly, P.T.; Wu, Y.; Zou, H.; Wang, R.; Zhou, W.; Kinoshita, A.; Zhang, M.; Yang, Y.; Cai, F.; Woodgett, J. Inhibition of GSK3β-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J. Clin. Investig. 2012, 123, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ghosal, K.; Vogt, D.L.; Liang, M.; Shen, Y.; Lamb, B.T.; Pimplikar, S.W. Alzheimer’s disease-like pathological features in transgenic mice expressing the APP intracellular domain. Proc. Natl. Acad. Sci. USA 2009, 106, 18367–18372. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.; Wang, Y.; McCarthy, D.; Wen, H.; Borchelt, D.R.; Price, D.L.; Wong, P.C. BACE1 is the major β-secretase for generation of Aβ peptides by neurons. Nat. Neurosci. 2001, 4, 233–234. [Google Scholar] [CrossRef]
- Chow, V.W.; Mattson, M.P.; Wong, P.C.; Gleichmann, M. An overview of APP processing enzymes and products. Neuromol. Med. 2010, 12, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Strooper, B.; Iwatsubo, T.; Wolfe, M.S. Presenilins and γ-secretase: Structure, function, and role in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006304. [Google Scholar] [CrossRef] [PubMed]
- Hernández, F.; de Barreda, E.G.; Fuster-Matanzo, A.; Lucas, J.J.; Avila, J. GSK3: A possible link between beta amyloid peptide and tau protein. Exp. Neurol. 2010, 223, 322–325. [Google Scholar] [CrossRef] [PubMed]
- Illenberger, S.; Zheng-Fischhofer, Q.; Preuss, U.; Stamer, K.; Baumann, K.; Trinczek, B.; Biernat, J.; Godemann, R.; Mandelkow, E.-M.; Mandelkow, E. The endogenous and cell cycle-dependent phosphorylation of tau protein in living cells: Implications for Alzheimer’s disease. Mol. Biol. Cell 1998, 9, 1495–1512. [Google Scholar] [CrossRef] [Green Version]
- Godemann, R.; Biernat, J.; Mandelkow, E.; Mandelkow, E.-M. Phosphorylation of tau protein by recombinant GSK-3β: Pronounced phosphorylation at select Ser/Thr-Pro motifs but no phosphorylation at Ser262 in the repeat domain. FEBS Lett. 1999, 454, 157–164. [Google Scholar] [CrossRef] [Green Version]
- Dewachter, I.; Ris, L.; Jaworski, T.; Seymour, C.; Kremer, A.; Borghgraef, P.; De Vijver, H.; Godaux, E.; Van Leuven, F. GSK3ß, a centre-staged kinase in neuropsychiatric disorders, modulates long term memory by inhibitory phosphorylation at Serine-9. Neurobiol. Dis. 2009, 35, 193–200. [Google Scholar] [CrossRef]
- Barbier, P.; Zejneli, O.; Martinho, M.; Lasorsa, A.; Belle, V.; Smet-Nocca, C.; Tsvetkov, P.O.; Devred, F.; Landrieu, I. Role of Tau as a microtubule associated protein: Structural and functional aspects. Front. Aging Neurosci. 2019, 11, 204. [Google Scholar] [CrossRef] [Green Version]
- Llorens-Marítin, M.; Jurado, J.; Hernández, F.; Ávila, J. GSK-3β, a pivotal kinase in Alzheimer disease. Front. Mol. Neurosci. 2014, 7, 46. [Google Scholar]
- Sengupta, A.; Novak, M.; Grundke-Iqbal, I.; Iqbal, K. Regulation of phosphorylation of tau by cyclin-dependent kinase 5 and glycogen synthase kinase-3 at substrate level. Febs Lett. 2006, 580, 5925–5933. [Google Scholar] [CrossRef] [Green Version]
- Engel, T.; Hernández, F.; Avila, J.; Lucas, J.J. Full reversal of Alzheimer’s disease-like phenotype in a mouse model with conditional overexpression of glycogen synthase kinase-3. J. Neurosci. 2006, 26, 5083–5090. [Google Scholar] [CrossRef] [Green Version]
- Avila, J.; León-Espinosa, G.; García, E.; García-Escudero, V.; Hernández, F.; DeFelipe, J. Tau phosphorylation by GSK3 in different conditions. Int. J. Alzheimer’s Dis. 2012, 2012, 578373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Hersh, L.B. Choline acetyltransferase: Celebrating its fiftieth year. J. Neurochem. 1994, 62, 1653–1663. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Vieyra, R.; Bravo-Patiño, A.; Valdez-Alarcón, J.J.; Juárez, M.C.; Finlay, B.B.; Baizabal-Aguirre, V.M. Role of glycogen synthase kinase-3 beta in the inflammatory response caused by bacterial pathogens. J. Inflamm. 2012, 9, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, W.-Y.; Wang, X.; Wu, Y.; Doble, B.W.; Patel, S.; Woodgett, J.R.; Snider, W.D. GSK-3 is a master regulator of neural progenitor homeostasis. Nat. Neurosci. 2009, 12, 1390–1397. [Google Scholar] [CrossRef] [Green Version]
- Benhamú, B.; Martín-Fontecha, M.; Vázquez-Villa, H.; Pardo, L.; López-Rodríguez, M.L. Serotonin 5-HT6 receptor antagonists for the treatment of cognitive deficiency in Alzheimer’s disease. J. Med. Chem. 2014, 57, 7160–7181. [Google Scholar] [CrossRef]
- Mitchell, E.S.; Neumaier, J.F. 5-HT6 receptors: A novel target for cognitive enhancement. Pharmacol. Ther. 2005, 108, 320–333. [Google Scholar] [CrossRef]
- Ramírez, M.J. 5-HT 6 receptors and Alzheimer’s disease. Alzheimer’s Res. Ther. 2013, 5, 15. [Google Scholar]
- Upton, N.; Chuang, T.T.; Hunter, A.J.; Virley, D.J. 5-HT6 receptor antagonists as novel cognitive enhancing agents for Alzheimer’s disease. Neurotherapeutics 2008, 5, 458–469. [Google Scholar] [CrossRef]
- Lieben, C.K.; Blokland, A.; Şık, A.; Sung, E.; van Nieuwenhuizen, P.; Schreiber, R. The selective 5-HT 6 receptor antagonist Ro4368554 restores memory performance in cholinergic and serotonergic models of memory deficiency in the rat. Neuropsychopharmacology 2005, 30, 2169–2179. [Google Scholar] [CrossRef] [Green Version]
- Gyertyán, I.; Kassai, F.; Kozma, K.; Kitka, T.; Ernyey, A.J. Procognitive profiling of a serotonin 5-HT6 receptor antagonist in a complex model system in rats: A novel translational approach for clinical prediction. Brain Res. Bull. 2020, 165, 238–245. [Google Scholar] [CrossRef]
- Yun, H.-M.; Kim, S.; Kim, H.-J.; Kostenis, E.; Kim, J.I.; Seong, J.Y.; Baik, J.-H.; Rhim, H. The novel cellular mechanism of human 5-HT6 receptor through an interaction with Fyn. J. Biol. Chem. 2007, 282, 5496–5505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folch, J.; Petrov, D.; Ettcheto, M.; Abad, S.; Sánchez-López, E.; García, M.L.; Olloquequi, J.; Beas-Zarate, C.; Auladell, C.; Camins, A. Current research therapeutic strategies for Alzheimer’s disease treatment. Neural Plast. 2016, 2016, 8501693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doody, R.S.; Gavrilova, S.I.; Sano, M.; Thomas, R.G.; Aisen, P.S.; Bachurin, S.O.; Seely, L.; Hung, D. Effect of dimebon on cognition, activities of daily living, behaviour, and global function in patients with mild-to-moderate Alzheimer’s disease: A randomised, double-blind, placebo-controlled study. Lancet 2008, 372, 207–215. [Google Scholar] [CrossRef]
- Jones, R.W. Dimebon disappointment. Alzheimer’s Res. Ther. 2010, 2, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, M.M.; El-Salamouni, N.S.; El-Refaie, W.M.; Hazzah, H.A.; Ali, M.M.; Tosi, G.; Farid, R.M.; Blanco-Prieto, M.J.; Billa, N.; Hanafy, A.S. Nanotechnology-based drug delivery systems for Alzheimer’s disease management: Technical, industrial, and clinical challenges. J. Control. Release 2017, 245, 95–107. [Google Scholar] [CrossRef]
- Zhao, Z.; Nelson, A.R.; Betsholtz, C.; Zlokovic, B.V. Establishment and dysfunction of the blood-brain barrier. Cell 2015, 163, 1064–1078. [Google Scholar] [CrossRef] [Green Version]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef]
- Sood, S.; Jain, K.; Gowthamarajan, K. Intranasal therapeutic strategies for management of Alzheimer’s disease. J. Drug Target. 2014, 22, 279–294. [Google Scholar] [CrossRef]
- Ahmad, M.Z.; Ahmad, J.; Amin, S.; Rahman, M.; Anwar, M.; Mallick, N.; Ahmad, F.J.; Rahman, Z.; Kamal, M.A.; Akhter, S. Role of nanomedicines in delivery of anti-acetylcholinesterase compounds to the brain in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2014, 13, 1315–1324. [Google Scholar] [CrossRef]
- Kumar, N.; Kumar, R. Nanotechnology and Nanomaterials in the Treatment of Life-Threatening Diseases; William Andrew: Norwich, NY, USA, 2013. [Google Scholar]
- Miyake, M.M.; Bleier, B.S. The blood-brain barrier and nasal drug delivery to the central nervous system. Am. J. Rhinol. Allergy 2015, 29, 124–127. [Google Scholar] [CrossRef]
- Wong, Y.C.; Zuo, Z. Intranasal delivery—modification of drug metabolism and brain disposition. Pharm. Res. 2010, 27, 1208–1223. [Google Scholar] [CrossRef] [PubMed]
- Ligade, P.C.; Jadhav, K.R.; Kadam, V.J. Brain drug delivery system: An overview. Curr. Drug Ther. 2010, 5, 105–110. [Google Scholar] [CrossRef]
- Mittal, D.; Ali, A.; Md, S.; Baboota, S.; Sahni, J.K.; Ali, J. Insights into direct nose to brain delivery: Current status and future perspective. Drug Deliv. 2014, 21, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Kozlovskaya, L.; Abou-Kaoud, M.; Stepensky, D. Quantitative analysis of drug delivery to the brain via nasal route. J. Control. Release 2014, 189, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.I.; Beg, S.; Samad, A.; Baboota, S.; Kohli, K.; Ali, J.; Ahuja, A.; Akbar, M. Strategy for effective brain drug delivery. Eur. J. Pharm. Sci. 2010, 40, 385–403. [Google Scholar] [CrossRef] [PubMed]
- Lajoie, J.M.; Shusta, E.V. Targeting receptor-mediated transport for delivery of biologics across the blood-brain barrier. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 613–631. [Google Scholar] [CrossRef] [Green Version]
- Gaillard, P.J.; Visser, C.C.; Appeldoorn, C.C.M.; Rip, J. Targeted blood-to-brain drug delivery–10 key development criteria. Curr. Pharm. Biotechnol. 2012, 13, 2328–2339. [Google Scholar] [CrossRef]
- Spuch, C.; Navarro, C. The therapeutic potential of microencapsulate implants: Patents and clinical trials. Recent Pat. Endocr. Metab. Immune Drug Discov. 2010, 4, 59–68. [Google Scholar] [CrossRef]
- Szoka, F., Jr.; Papahadjopoulos, D. Comparative properties and methods of preparation of lipid vesicles (liposomes). Annu. Rev. Biophys. Bioeng. 1980, 9, 467–508. [Google Scholar] [CrossRef]
- Sigurdsson, E.M. Tau Immunotherapy. In Immunotherapy and Biomarkers in Neurodegenerative Disorders; Springer: Berlin/Heidelberg, Germany, 2016; pp. 109–120. [Google Scholar]
- Sigurdsson, E.M. Tau immunotherapies for Alzheimer’s disease and related tauopathies: Progress and potential pitfalls. J. Alzheimer’s Dis. 2018, 64, S555–S565. [Google Scholar] [CrossRef]
- Lim, S.B.; Banerjee, A.; Önyüksel, H. Improvement of drug safety by the use of lipid-based nanocarriers. J. Control. Release 2012, 163, 34–45. [Google Scholar] [CrossRef]
- Zielińska, A.; Carreiró, F.; Oliveira, A.M.; Neves, A.; Pires, B.; Venkatesh, D.N.; Durazzo, A.; Lucarini, M.; Eder, P.; Silva, A.M. Polymeric nanoparticles: Production, characterization, toxicology and ecotoxicology. Molecules 2020, 25, 3731. [Google Scholar] [CrossRef] [PubMed]
- Carradori, D.; Balducci, C.; Re, F.; Brambilla, D.; Le Droumaguet, B.; Flores, O.; Gaudin, A.; Mura, S.; Forloni, G.; Ordoñez-Gutierrez, L. Antibody-functionalized polymer nanoparticle leading to memory recovery in Alzheimer’s disease-like transgenic mouse model. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Lane, L.A.; Qian, X.; Nie, S. SERS nanoparticles in medicine: From label-free detection to spectroscopic tagging. Chem. Rev. 2015, 115, 10489–10529. [Google Scholar] [CrossRef] [PubMed]
- Stuart, M.A.C.; Huck, W.T.; Genzer, J.; Müller, M.; Ober, C.; Stamm, M.; Sukhorukov, G.B.; Szleifer, I.; Tsukruk, V.V.; Urban, M. Emerging applications of stimuli-responsive polymer materials. Nat. Mater. 2010, 9, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, S.; Parayath, N.N.; Taurin, S.; Greish, K. Polymeric nano-micelles: Versatile platform for targeted delivery in cancer. Ther. Deliv. 2014, 5, 1101–1121. [Google Scholar] [CrossRef] [PubMed]
- McClements, D.J. Nanoemulsions versus microemulsions: Terminology, differences, and similarities. Soft Matter 2012, 8, 1719–1729. [Google Scholar] [CrossRef]
- Kaur, A.; Nigam, K.; Srivastava, S.; Tyagi, A.; Dang, S. Memantine nanoemulsion: A new approach to treat Alzheimer’s disease. J. Microencapsul. 2020, 37, 355–365. [Google Scholar] [CrossRef]
- Chanphai, P.; Bekale, L.; Sanyakamdhorn, S.; Agudelo, D.; Bérubé, G.; Thomas, T.; Tajmir-Riahi, H. PAMAM dendrimers in drug delivery: Loading efficacy and polymer morphology. Can. J. Chem. 2017, 95, 891–896. [Google Scholar] [CrossRef]
- Kojima, C.; Turkbey, B.; Ogawa, M.; Bernardo, M.; Regino, C.A.; Bryant Jr, L.H.; Choyke, P.L.; Kono, K.; Kobayashi, H. Dendrimer-based MRI contrast agents: The effects of PEGylation on relaxivity and pharmacokinetics. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 1001–1008. [Google Scholar] [CrossRef] [Green Version]
- Shcharbin, D.; Klajnert, B.; Bryszewska, M. Dendrimers in gene transfection. Biochemistry 2009, 74, 1070–1079. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MOA | AChE Inhibitors | NMDA-Receptor Antagonist | ||
|---|---|---|---|---|
| Drug | Donepezil | Galantamine | Rivastigmine | Memantine |
| Indication | Moderate and severe AD | Mild and moderate AD | Mild and moderate AD | Moderate and severe AD |
| Initial dose | Tablet: 5 mg qd | Tablet/Oral soln.: 4 mg bid ER capsule: 8 mg qd | Capsule/Oral soln.: 1.5 mg bid Patch: 4.6 mg qd | Tablet/Oral soln.: 5 mg qd |
| Maximal dose | Tablet: 10 mg qd | Tablet/Oral soln.: 12 mg bid ER capsule: 24 mg qd | Capsule/Oral soln.: 6 mg bid Patch: 9.5 mg qd | Tablet/Oral soln.: 10 mg bid |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agarwal, M.; Alam, M.R.; Haider, M.K.; Malik, M.Z.; Kim, D.-K. Alzheimer’s Disease: An Overview of Major Hypotheses and Therapeutic Options in Nanotechnology. Nanomaterials 2021, 11, 59. https://0-doi-org.brum.beds.ac.uk/10.3390/nano11010059
Agarwal M, Alam MR, Haider MK, Malik MZ, Kim D-K. Alzheimer’s Disease: An Overview of Major Hypotheses and Therapeutic Options in Nanotechnology. Nanomaterials. 2021; 11(1):59. https://0-doi-org.brum.beds.ac.uk/10.3390/nano11010059
Chicago/Turabian StyleAgarwal, Mugdha, Mohammad Rizwan Alam, Mohd Kabir Haider, Md. Zubbair Malik, and Dae-Kwang Kim. 2021. "Alzheimer’s Disease: An Overview of Major Hypotheses and Therapeutic Options in Nanotechnology" Nanomaterials 11, no. 1: 59. https://0-doi-org.brum.beds.ac.uk/10.3390/nano11010059