
Engineering Modular Half-Antibody Conjugated Nanoparticles for Targeting CD44v6-Expressing Cancer Cells

 , and
, and
Abstract
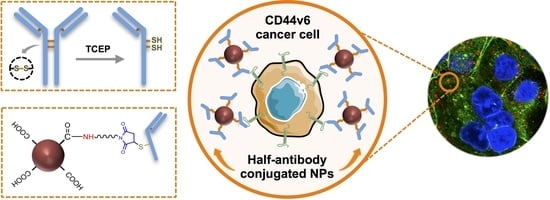
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Preparation of Tumor-Conditioned Media
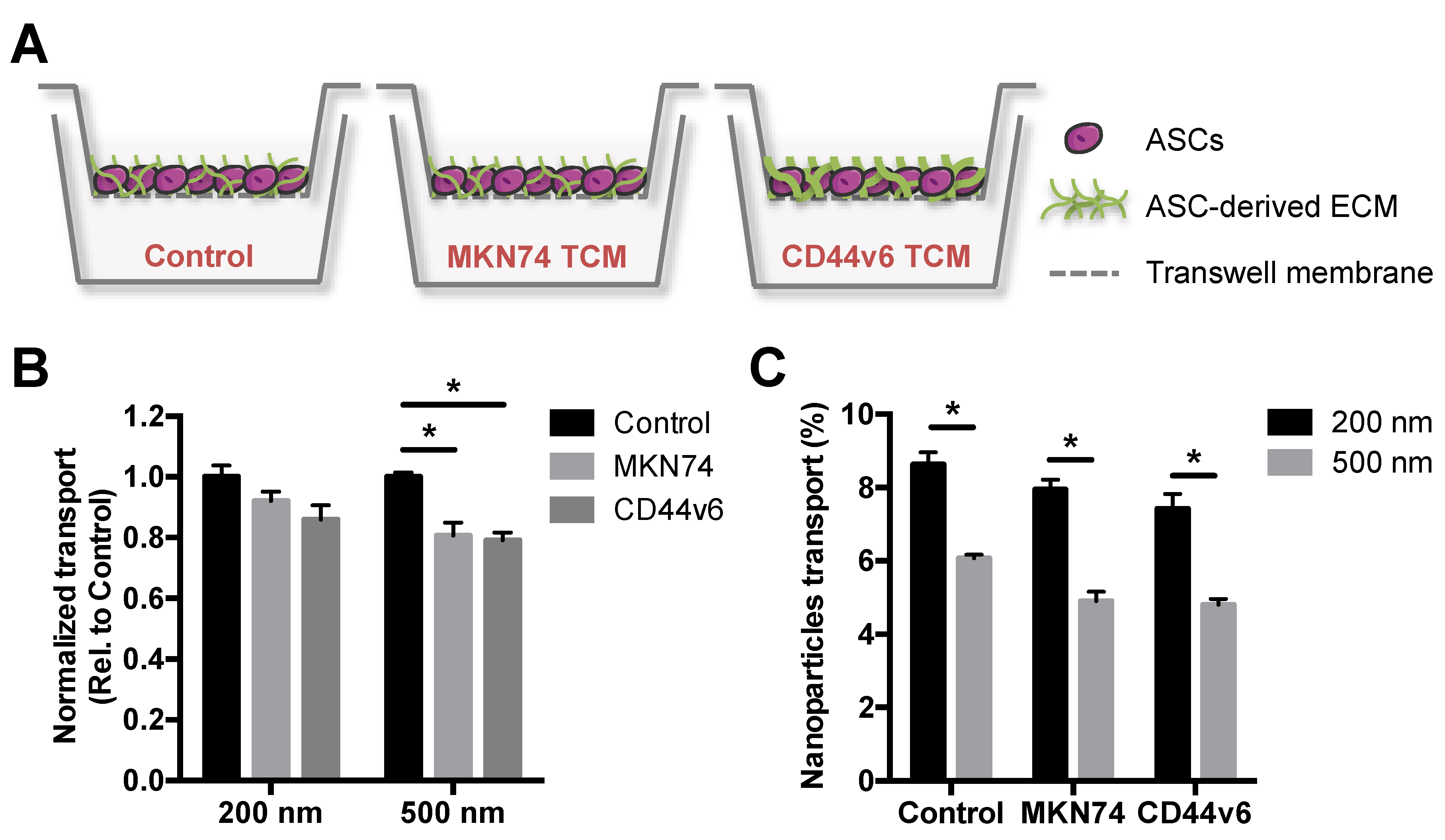
2.3. Establishment of In Vitro GC Stroma Model for Transport Studies
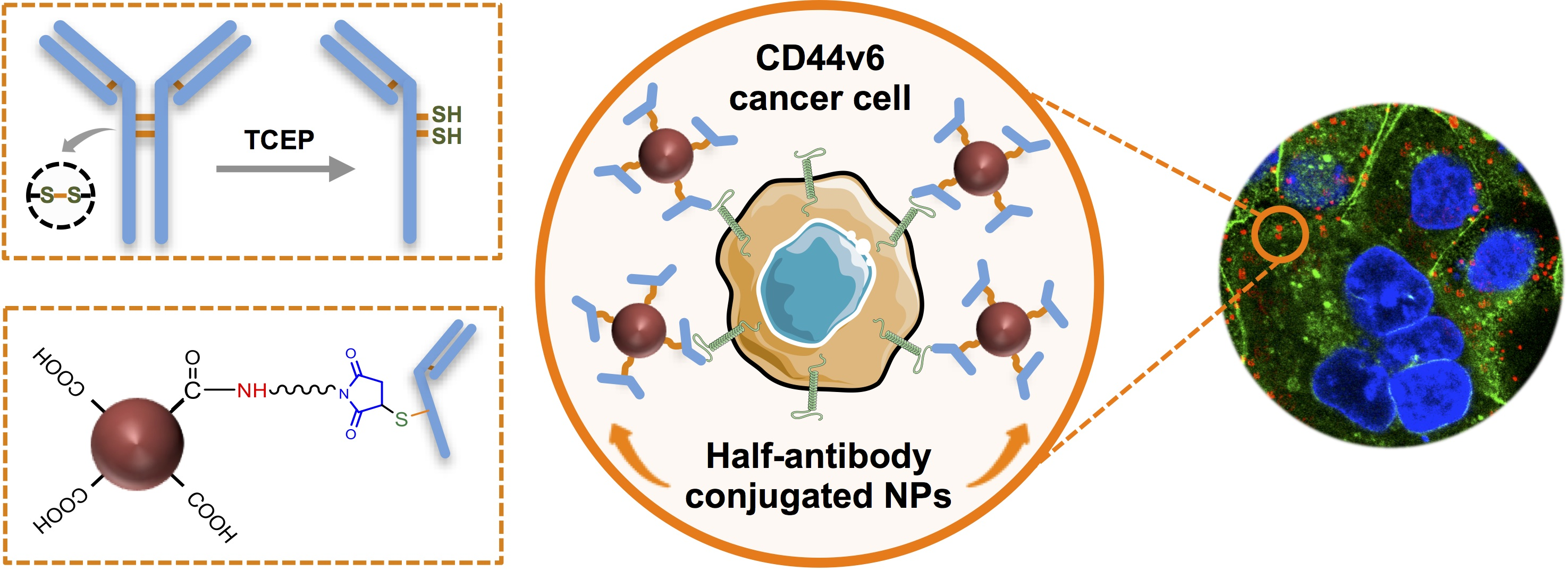
2.4. Half-Antibody Synthesis and Characterization
2.5. Analysis of CD44v6 Half-Antibody Binding Affinity to CD44v6-Expressing GC Cells
2.6. Synthesis of CD44v6 Half-Antibody-Conjugated Fluorescent Nanoparticles
2.7. Nanoparticle Characterization
2.8. In Vitro Binding Studies
2.9. Statistical Analysis
3. Results
3.1. Design of a Modular CD44v6 Half-Antibody Conjugated Nanoscale System Using a Bioorthogonal Strategy
3.2. GC-Mimetic Stroma Remodeling Hinders the Transport of PNPs in a Size-Dependent Manner
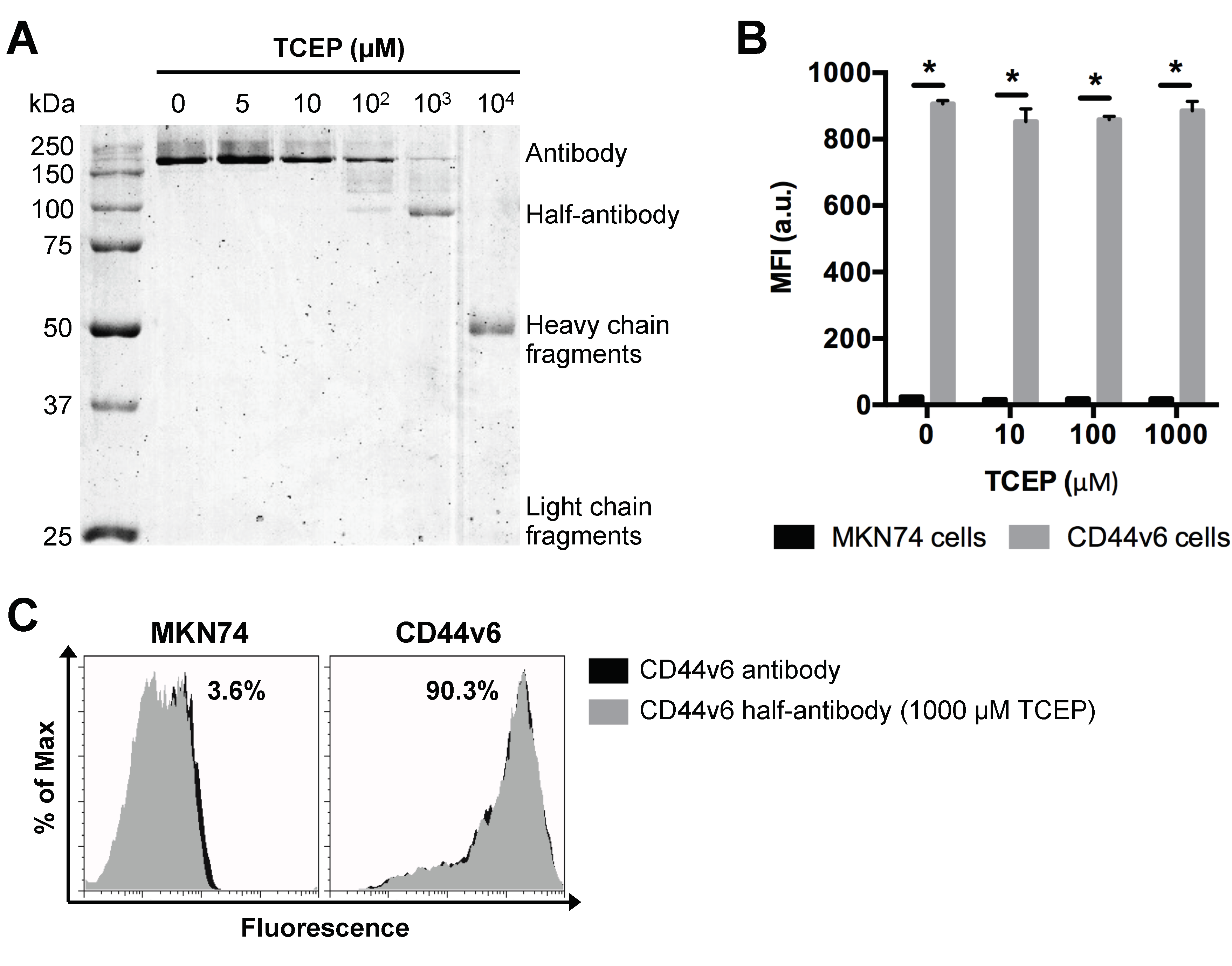
3.3. Selective TCEP-Reduction of CD44v6 Antibody Generates Functional Half-Antibody Fragments
3.4. TCEP-Reduced Half-Antibody Fragments Retain Binding Affinity to CD44v6-Expressing GC Cells without Loss of Selectivity
3.5. Clickable Surface-Engineering Strategy Allows Efficient Ligand Bioconjugation
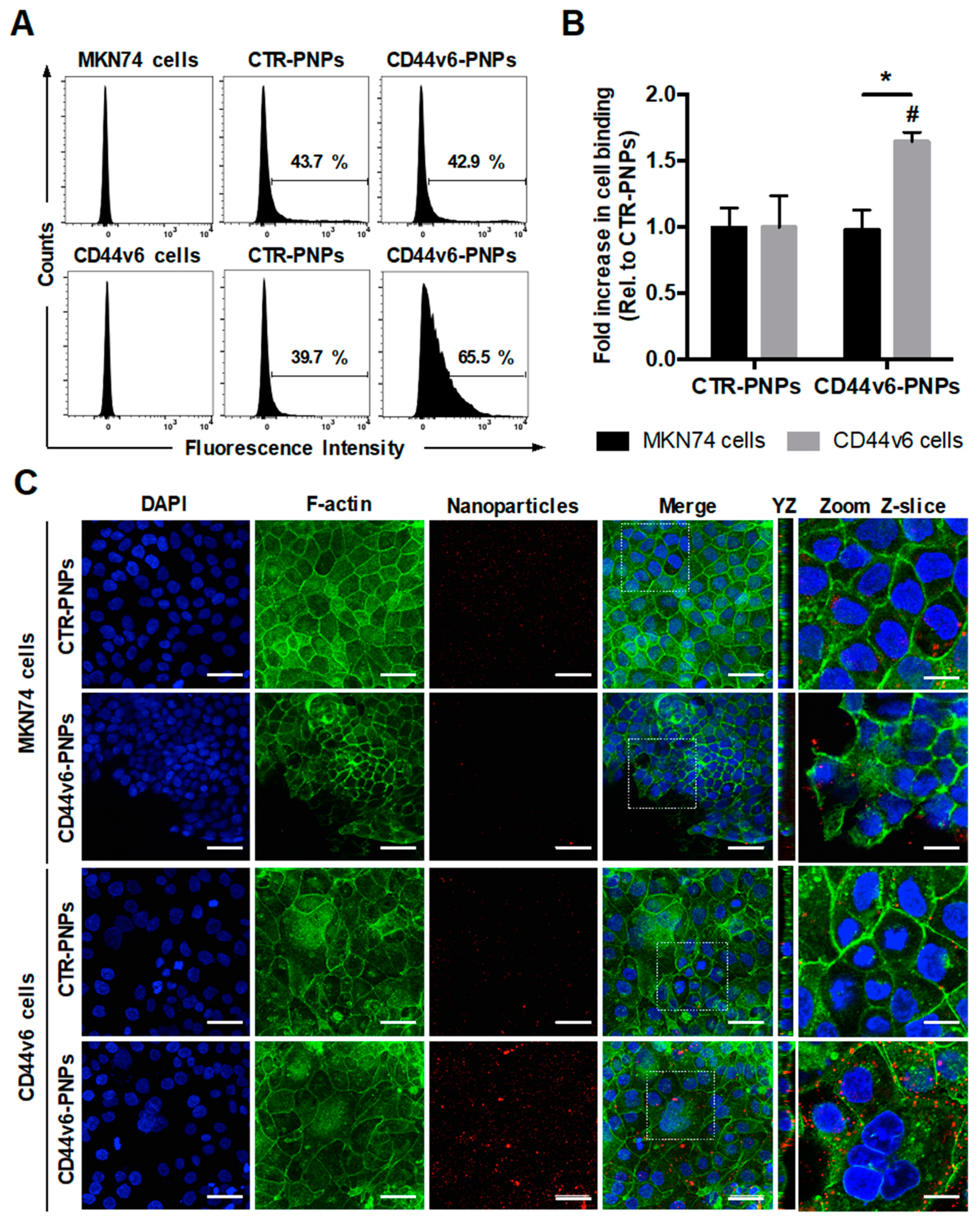
3.6. CD44v6 Half-Antibody Conjugated PNPs Selectively Bind to CD44v6-Expressing GC Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Cutsem, E.; Sagaert, X.; Topal, B.; Haustermans, K.; Prenen, H. Gastric cancer. Lancet 2016, 388, 2654–2664. [Google Scholar] [CrossRef]
- Brigger, I.; Dubernet, C.; Couvreur, P. Nanoparticles in cancer therapy and diagnosis. Adv. Drug Deliv. Rev. 2012, 64, 24–36. [Google Scholar] [CrossRef]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016, 1, 16014. [Google Scholar] [CrossRef]
- Wolfram, J.; Ferrari, M. Clinical cancer nanomedicine. Nano Today 2019, 25, 85–98. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Liu, B.; Gao, J. The application of nanoparticles in diagnosis and theranostics of gastric cancer. Cancer Lett. 2017, 386, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Durães, C.; Almeida, G.M.; Seruca, R.; Oliveira, C.; Carneiro, F. Biomarkers for gastric cancer: Prognostic, predictive or targets of therapy? Virchows Arch. 2014, 464, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Apicella, M.; Corso, S.; Giordano, S. Targeted therapies for gastric cancer: Failures and hopes from clinical trials. Oncotarget 2017, 8, 57654–57669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaufmann, M.; Heider, K.H.; Sinn, H.P.; Von Minckwitz, G.; Ponta, H.; Herrlich, P. CD44 variant exon epitopes in primary breast cancer and length of survival. Lancet 1995, 345, 615–619. [Google Scholar] [CrossRef]
- Miyoshi, T.; Kondo, K.; Hino, N.; Uyama, T.; Monden, Y. The expression of the CD44 variant exon 6 is associated with lymph node metastasis in non-small cell lung cancer. Clin. Cancer Res. 1997, 3, 1289–1297. [Google Scholar] [CrossRef]
- Zeimet, A.; Widschwendter, M.; Uhl-Steidl, M.; Müller-Holzner, E.; Daxenbichler, G.; Marth, C.; Dapunt, O. High serum levels of soluble CD44 variant isoform v5 are associated with favourable clinical outcome in ovarian cancer. Br. J. Cancer 1997, 76, 1646–1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.-J.; Yan, P.-S.; Li, J.; Jia, J.-F. Expression and significance of CD44s, CD44v6, and nm23 mRNA in human cancer. World J. Gastroenterol. 2005, 11, 6601–6606. [Google Scholar] [CrossRef] [PubMed]
- Wielenga, V.J.; Heider, K.H.; Offerhaus, G.J.; Adolf, G.R.; Berg, F.M.V.D.; Ponta, H.; Herrlich, P.; Pals, S.T. Expression of CD44 variant proteins in human colorectal cancer is related to tumor progression. Cancer Res. 1993, 53, 4754–4756. [Google Scholar] [PubMed]
- Cunha, C.B.; Oliveira, C.; Wen, X.; Gomes, B.; Sousa, S.; Suriano, G.; Grellier, M.; Huntsman, D.G.; Carneiro, F.; Granja, P.L.; et al. De novo expression of CD44 variants in sporadic and hereditary gastric cancer. Lab. Investig. 2010, 90, 1604–1614. [Google Scholar] [CrossRef] [Green Version]
- Pereira, C.; Ferreira, D.; Mendes, N.; Granja, P.L.; Almeida, G.M.; Oliveira, C. Expression of CD44v6-Containing Isoforms Influences Cisplatin Response in Gastric Cancer Cells. Cancers 2020, 12, 858. [Google Scholar] [CrossRef] [Green Version]
- Lourenco, B.N.; Springer, N.L.; Ferreira, D.; Oliveira, C.; Granja, P.L.; Fischbach, C. CD44v6 increases gastric cancer malignant phenotype by modulating adipose stromal cell-mediated ECM remodeling. Integr. Biol. 2018, 10, 145–158. [Google Scholar] [CrossRef]
- Bertrand, N.; Wu, J.; Xu, X.; Kamaly, N.; Farokhzad, O.C. Cancer nanotechnology: The impact of passive and active targeting in the era of modern cancer biology. Adv. Drug Deliv. Rev. 2014, 66, 2–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahbazi, M.-A.; Shrestha, N.; Mäkilä, E.; Araújo, F.; Correia, A.; Ramos, T.; Sarmento, B.; Salonen, J.; Hirvonen, J.; Santos, H.A. A prospective cancer chemo-immunotherapy approach mediated by synergistic CD326 targeted porous silicon nanovectors. Nano Res. 2015, 8, 1505–1521. [Google Scholar] [CrossRef]
- Liang, S.; Li, C.; Zhang, C.; Chen, Y.; Xu, L.; Bao, C.; Wang, X.; Liu, G.; Zhang, F.; Cui, D. CD44v6 Monoclonal Antibody-Conjugated Gold Nanostars for Targeted Photoacoustic Imaging and Plasmonic Photothermal Therapy of Gastric Cancer Stem-like Cells. Theranostics 2015, 5, 970–984. [Google Scholar] [CrossRef]
- Shargh, V.H.; Hondermarck, H.; Liang, M. Antibody-targeted biodegradable nanoparticles for cancer therapy. Nanomedicine 2016, 11, 63–79. [Google Scholar] [CrossRef]
- Marques, A.C.; Costa, P.; Velho, S.; Amaral, M. Functionalizing nanoparticles with cancer-targeting antibodies: A comparison of strategies. J. Control. Release 2020, 320, 180–200. [Google Scholar] [PubMed]
- Montenegro, J.M.; Grazu, V.; Sukhanova, A.; Agarwal, S.; Jesus, M.; Nabiev, I.; Greiner, A.; Parak, W.J. Controlled antibody/(bio-) conjugation of inorganic nanoparticles for targeted delivery. Adv. Drug Deliv. Rev. 2013, 65, 677–688. [Google Scholar] [PubMed]
- Kennedy, P.J.; Sousa, F.; Ferreira, D.; Pereira, C.; Nestor, M.; Oliveira, C.; Granja, P.L.; Sarmento, B. Fab-conjugated PLGA nanoparticles effectively target cancer cells expressing human CD44v6. Acta Biomater. 2018, 81, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Greene, M.K.; Nogueira, J.C.F.; Tracey, S.R.; Richards, D.A.; McDaid, W.J.; Burrows, J.F.; Campbell, K.; Longley, D.B.; Chudasama, V.; Scott, C.J. Refined construction of antibody-targeted nanoparticles leads to superior antigen binding and enhanced delivery of an entrapped payload to pancreatic cancer cells. Nanoscale 2020, 12, 11647–11658. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.M.; Kaushal, S.; Tran Cao, H.S.; Aryal, S.; Sartor, M.; Esener, S.; Bouvet, M.; Zhang, L. Half-antibody functionalized lipid-polymer hybrid nanoparticles for targeted drug delivery to carcinoembryonic antigen presenting pancreatic cancer cells. Mol. Pharm. 2010, 7, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.; Wang, Y.; Chen, Y.; Zeng, L.; Zhang, Q.; Shuai, X.; Huang, K. Suppression of pancreatic tumor growth by targeted arsenic delivery with anti-CD44v6 single chain antibody conjugated nanoparticles. Biomaterials 2013, 34, 6175–6184. [Google Scholar] [CrossRef] [PubMed]
- Richards, D.A.; Maruani, A.; Chudasama, V. Antibody fragments as nanoparticle targeting ligands: A step in the right direction. Chem. Sci. 2017, 8, 63–77. [Google Scholar]
- Kennedy, P.J.; Oliveira, C.; Granja, P.L.; Sarmento, B. Antibodies and associates: Partners in targeted drug delivery. Pharmacol. Ther. 2017, 177, 129–145. [Google Scholar]
- Yoon, T.-J.; Yu, K.N.; Kim, E.; Kim, J.S.; Kim, B.G.; Yun, S.-H.; Sohn, B.-H.; Cho, M.-H.; Lee, J.-K.; Park, S.B. Specific Targeting, Cell Sorting, and Bioimaging with Smart Magnetic Silica Core-Shell Nanomaterials. Small 2006, 2, 209–215. [Google Scholar] [CrossRef]
- Xing, Y.; Chaudry, Q.; Shen, C.; Kong, K.Y.; Zhau, H.E.; Chung, L.W.; Petros, J.A.; O’Regan, R.M.; Yezhelyev, M.V.; Simons, J.W.; et al. Bioconjugated quantum dots for multiplexed and quantitative immunohistochemistry. Nat. Protoc. 2007, 2, 1152–1165. [Google Scholar] [CrossRef]
- Cho, Y.-S.; Yoon, T.-J.; Jang, E.-S.; Hong, K.S.; Lee, S.Y.; Kim, O.R.; Park, C.; Kim, Y.-J.; Yi, G.-C.; Chang, K. Cetuximab-conjugated magneto-fluorescent silica nanoparticles for in vivo colon cancer targeting and imaging. Cancer Lett. 2010, 299, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Fiandra, L.; Mazzucchelli, S.; De Palma, C.; Colombo, M.; Allevi, R.; Sommaruga, S.; Clementi, E.; Bellini, M.; Prosperi, D.; Corsi, F. Assessing the In Vivo Targeting Efficiency of Multifunctional Nanoconstructs Bearing Antibody-Derived Ligands. ACS Nano 2013, 7, 6092–6102. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, V.P.; Stylianopoulos, T.; Boucher, Y.; Jain, R.K. Delivery of Molecular and Nanoscale Medicine to Tumors: Transport Barriers and Strategies. Annu. Rev. Chem. Biomol. Eng. 2011, 2, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.R.A.; Weaver, A.M.; Cummings, P.T.; Quaranta, V. Tumor Morphology and Phenotypic Evolution Driven by Selective Pressure from the Microenvironment. Cell 2006, 127, 905–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pluen, A.; Boucher, Y.; Ramanujan, S.; McKee, T.D.; Gohongi, T.; Di Tomaso, E.; Brown, E.B.; Izumi, Y.; Campbell, R.B.; Berk, D.A.; et al. Role of tumor-host interactions in interstitial diffusion of macromolecules: Cranial vs. subcutaneous tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 4628–4633. [Google Scholar] [CrossRef] [Green Version]
- Jain, R.K.; Stylianopoulos, T. Delivering nanomedicine to solid tumors. Nat. Rev. Clin. Oncol. 2010, 7, 653–664. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Yokozaki, H.; Yasui, W.; Nikai, H.; Tahara, E. Silencing of the CD44 gene by CpG methylation in a human gastric carcinoma cell line. Jpn. J. Cancer Res. 1999, 90, 485–489. [Google Scholar] [CrossRef]
- Dos Santos, T.; Varela, J.; Lynch, I.; Salvati, A.; Dawson, K.A. Quantitative assessment of the comparative nanoparticle-uptake efficiency of a range of cell lines. Small 2011, 7, 3341–3349. [Google Scholar] [CrossRef] [Green Version]
- Lourenço, B.N.; Dos Santos, T.; Oliveira, C.; Barrias, C.C.; Granja, P.L. Bioengineering a novel 3D in vitro model of gastric mucosa for stomach permeability studies. Acta Biomater. 2018, 82, 68–78. [Google Scholar] [CrossRef]
- Lunov, O.; Syrovets, T.; Loos, C.; Beil, J.; Delacher, M.; Tron, K.; Nienhaus, G.U.; Musyanovych, A.; Mailänder, V.; Landfester, K.; et al. Differential Uptake of Functionalized Polystyrene Nanoparticles by Human Macrophages and a Monocytic Cell Line. ACS Nano 2011, 5, 1657–1669. [Google Scholar] [CrossRef]
- Zimmermann, J.L.; Nicolaus, T.; Neuert, G.; Blank, K. Thiol-based, site-specific and covalent immobilization of biomolecules for single-molecule experiments. Nat. Protoc. 2010, 5, 975–985. [Google Scholar] [CrossRef] [PubMed]
- Harlow, E.; Lane, D. A Laboratory Manual; Cold Spring Harbor Laboratory: New York, NY, USA, 1988; p. 579. [Google Scholar]
- Nair, D.P.; Podgórski, M.; Chatani, S.; Gong, T.; Xi, W.; Fenoli, C.R.; Bowman, C.N. The Thiol-Michael Addition Click Reaction: A Powerful and Widely Used Tool in Materials Chemistry. Chem. Mater. 2014, 26, 724–744. [Google Scholar] [CrossRef]
- Li, L.; Schmitt, M.; Matzke-Ogi, A.; Wadhwani, P.; Orian-Rousseau, V.; Levkin, P.A. CD44v6-Peptide Functionalized Nanoparticles Selectively Bind to Metastatic Cancer Cells. Adv. Sci. 2016, 4, 1600202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wörn, A.; Plückthun, A. Stability engineering of antibody single-chain Fv fragments. J. Mol. Biol. 2001, 305, 989–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malpiedi, L.P.; Díaz, C.A.; Nerli, B.B.; Pessoa, A., Jr. Single-chain antibody fragments: Purification methodologies. Process. Biochem. 2013, 48, 1242–1251. [Google Scholar] [CrossRef]
- Makaraviciute, A.; Jackson, C.D.; Millner, P.A.; Ramanaviciene, A. Considerations in producing preferentially reduced half-antibody fragments. J. Immunol. Methods 2016, 429, 50–56. [Google Scholar] [CrossRef] [Green Version]
- Hermanson, G.T. Antibody Modification and Conjugation. In Bioconjugate Techniques, 3rd ed.; Academic Press: Boston, MA, USA, 2013; Chapter 20; pp. 867–920. [Google Scholar]
- Lilie, H. Folding of the Fab fragment within the intact antibody. FEBS Lett. 1997, 417, 239–242. [Google Scholar] [CrossRef] [Green Version]
- Pereira, R.F.; Sousa, A.; Barrias, C.C.; Bártolo, P.; Granja, P.L. A single-component hydrogel bioink for bioprinting of bioengineered 3D constructs for dermal tissue engineering. Mater. Horiz. 2018, 5, 1100–1111. [Google Scholar] [CrossRef]
- Kolate, A.; Baradia, D.; Patil, S.; Vhora, I.; Kore, G.; Misra, A. PEG—A versatile conjugating ligand for drugs and drug delivery systems. J. Control. Release 2014, 192, 67–81. [Google Scholar] [CrossRef]
- Begg, G.; Speicher, D.W. Mass spectrometry detection and reduction of disulfide adducts between reducing agents and recombinant proteins with highly reactive cysteines. J. Biomol. Tech. 1999, 10, 17–20. [Google Scholar]
- Getz, E.B.; Xiao, M.; Chakrabarty, T.; Cooke, R.; Selvin, P.R. A comparison between the sulfhydryl reductants tris (2-carboxyethyl) phosphine and dithiothreitol for use in protein biochemistry. Anal. Biochem. 1999, 273, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.; Lin, C.M.; Huang, L. Stromal barriers and strategies for the delivery of nanomedicine to desmoplastic tumors. J. Control. Release 2015, 219, 192–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schädlich, A.; Caysa, H.; Mueller, T.; Tenambergen, F.; Rose, C.; Göpferich, A.; Kuntsche, J.; Mäder, K. Tumor Accumulation of NIR Fluorescent PEG–PLA Nanoparticles: Impact of Particle Size and Human Xenograft Tumor Model. ACS Nano 2011, 5, 8710–8720. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, D.; Joshi, N.; Tao, W.; Karp, J.M.; Peer, D. Progress and challenges towards targeted delivery of cancer therapeutics. Nat. Commun. 2018, 9, 1–12. [Google Scholar]
- Lee, D.-E.; Koo, H.; Sun, I.-C.; Ryu, J.H.; Kim, K.; Kwon, I.C. Multifunctional nanoparticles for multimodal imaging and theragnosis. Chem. Soc. Rev. 2012, 41, 2656–2672. [Google Scholar] [CrossRef]
- Kuhn, S.J.; Finch, S.K.; Hallahan, D.E.; Giorgio, T.D. Proteolytic surface functionalization enhances in vitro magnetic nanoparticle mobility through extracellular matrix. Nano Lett. 2006, 6, 306–312. [Google Scholar] [CrossRef]
- Goodman, T.T.; Olive, P.L.; Pun, S.H. Increased nanoparticle penetration in collagenase-treated multicellular spheroids. Int. J. Nanomed. 2007, 2, 265–274. [Google Scholar]
- Parodi, A.; Haddix, S.G.; Taghipour, N.; Scaria, S.; Taraballi, F.; Cevenini, A.; Yazdi, I.K.; Corbo, C.; Palomba, R.; Khaled, S.Z.; et al. Bromelain Surface Modification Increases the Diffusion of Silica Nanoparticles in the Tumor Extracellular Matrix. ACS Nano 2014, 8, 9874–9883. [Google Scholar] [CrossRef]
- Seo, B.R.; Delnero, P.; Fischbach, C. In vitro models of tumor vessels and matrix: Engineering approaches to investigate transport limitations and drug delivery in cancer. Adv. Drug Deliv. Rev. 2014, 70, 205–216. [Google Scholar]
- Santos, T.D.; Lourenço, B.N.; Coentro, J.; Granja, P.L. Cell-based in vitro models for gastric permeability studies. In Concepts and Models for Drug Permeability Studies; Sarmento, B., Ed.; Woodhead Publishing: Sawston, UK, 2016; pp. 41–56. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nanoparticle | Composition | Mean Size (nm) | PDI | Zeta Potencial (mV) |
|---|---|---|---|---|
| Bare-PNPs | PNP | 237 ± 2 | 0.05 ± 0.01 | −45.3 ± 1.1 |
| CTR-PNPs | PNP-PEG-MAL | 309 ± 50 * | 0.19 ± 0.09 * | −31.4 ± 3.5 * |
| CD44v6-PNPs | PNP-PEG-MAL-CD44v6 | 352 ± 72 * | 0.25 ± 0.10 * | −25.1 ± 7.2 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lourenço, B.N.; Pereira, R.F.; Barrias, C.C.; Fischbach, C.; Oliveira, C.; Granja, P.L. Engineering Modular Half-Antibody Conjugated Nanoparticles for Targeting CD44v6-Expressing Cancer Cells. Nanomaterials 2021, 11, 295. https://0-doi-org.brum.beds.ac.uk/10.3390/nano11020295
Lourenço BN, Pereira RF, Barrias CC, Fischbach C, Oliveira C, Granja PL. Engineering Modular Half-Antibody Conjugated Nanoparticles for Targeting CD44v6-Expressing Cancer Cells. Nanomaterials. 2021; 11(2):295. https://0-doi-org.brum.beds.ac.uk/10.3390/nano11020295
Chicago/Turabian StyleLourenço, Bianca N., Rúben F. Pereira, Cristina C. Barrias, Claudia Fischbach, Carla Oliveira, and Pedro L. Granja. 2021. "Engineering Modular Half-Antibody Conjugated Nanoparticles for Targeting CD44v6-Expressing Cancer Cells" Nanomaterials 11, no. 2: 295. https://0-doi-org.brum.beds.ac.uk/10.3390/nano11020295