
Targeted Immuno-Antiretroviral to Promote Dual Protection against HIV: A Proof-of-Concept Study

 ,
,
Abstract
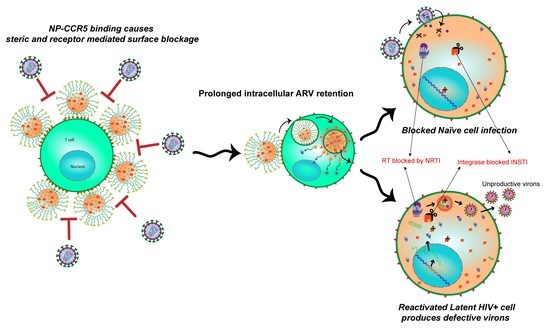
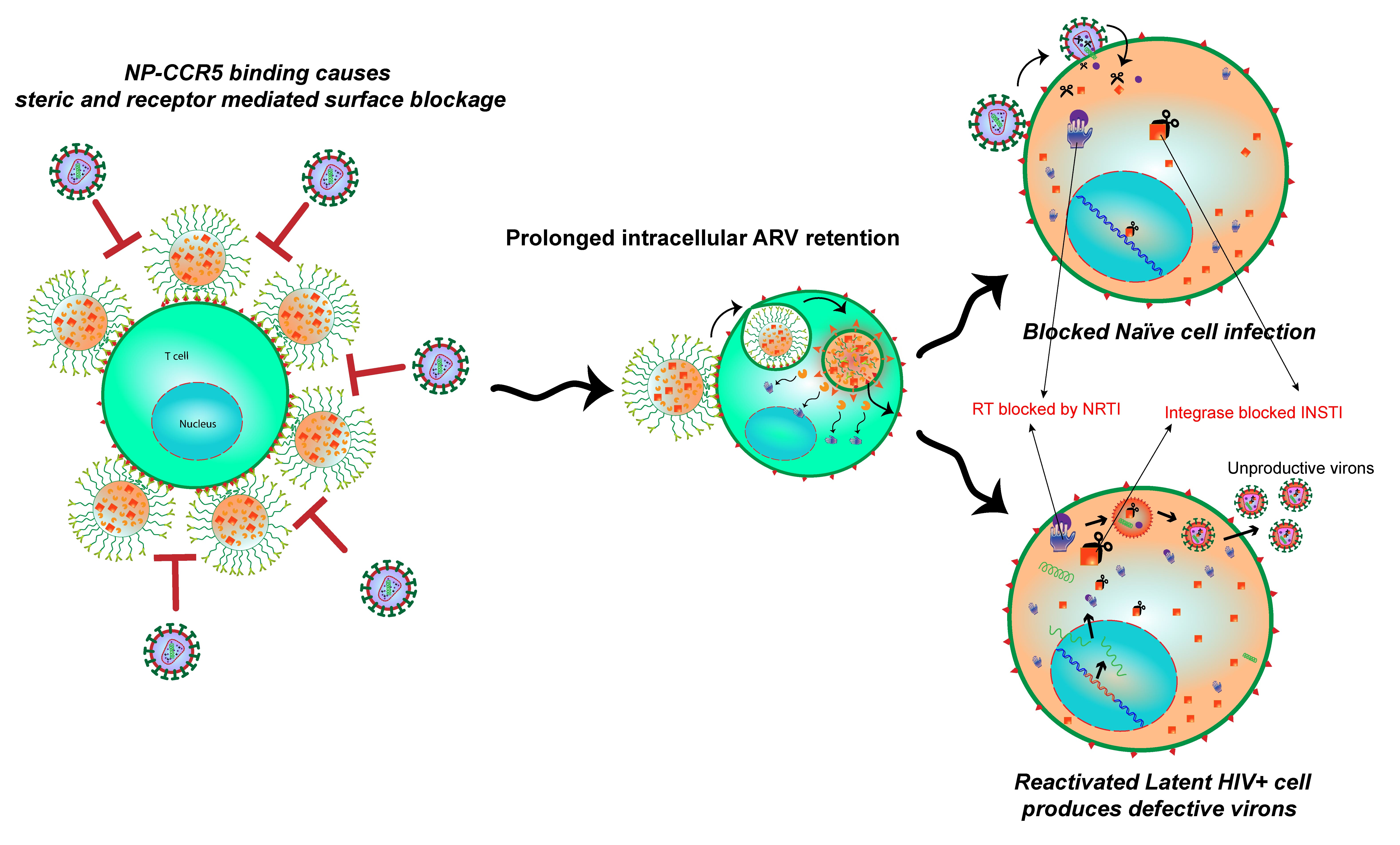
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Primary Cells and Cell Lines
2.3. HIV Strain
2.4. Production, Purification, and Characterization of xfR5 mAb
2.5. ARV-Loaded NPs Formulation
2.6. ARV NP Functionalization
2.7. CCR5 Targeted ARV NP Characterization
2.8. Antibody Binding and Binding Affinity Evaluation
2.9. Immunophenotype Study
2.10. Intracellular Kinetics Experiments
2.11. In Vitro Cytotoxicity Experiments
2.12. In Vitro Protection Experiments
2.13. Statistical Analysis
3. Results
3.1. CCR5 Targeted cARV-Loaded NPs Characterization
3.2. Binding Affinity
3.3. Intracellular Drug Kinetics
3.4. Cytotoxicity and HIV Protection Study
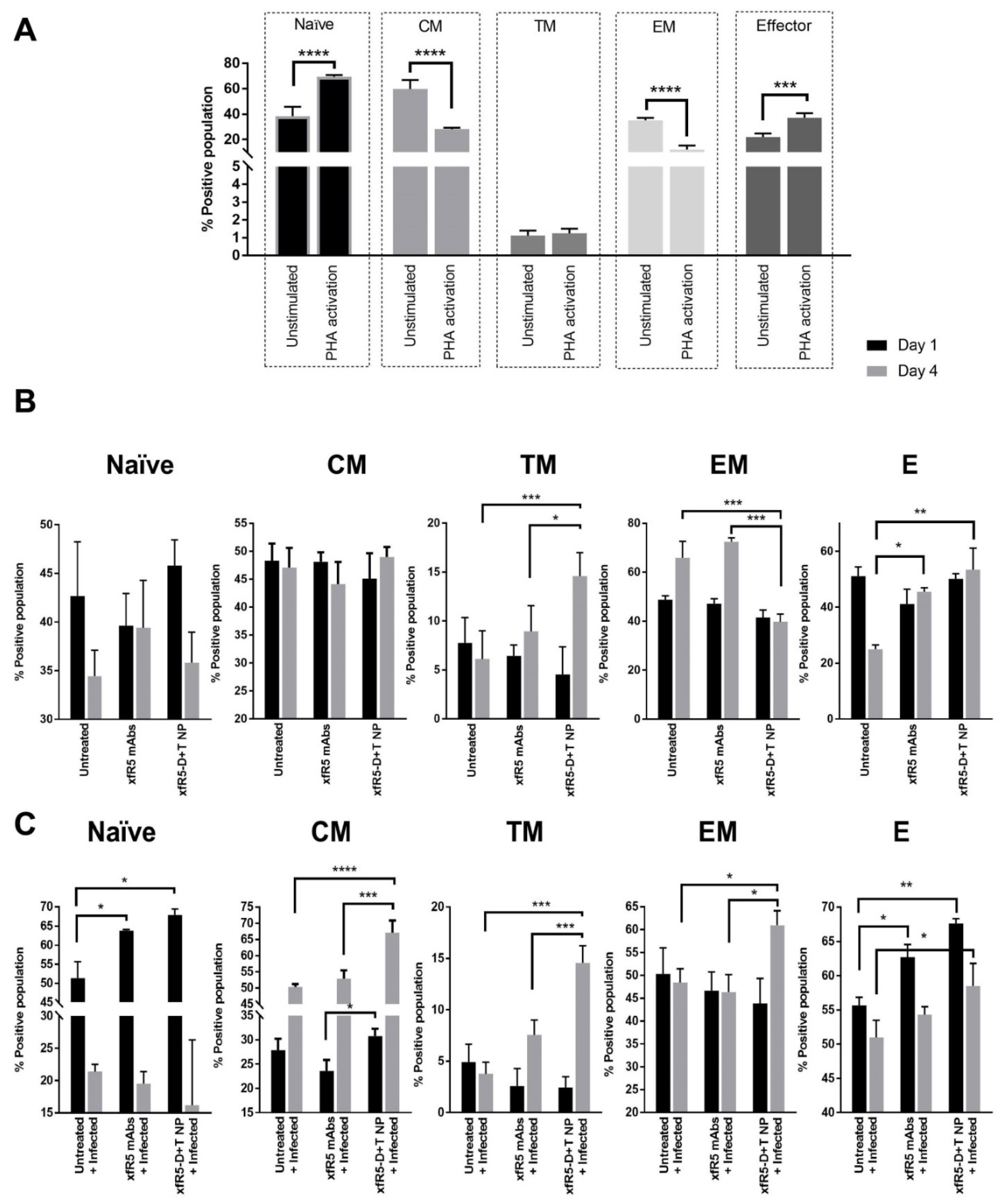
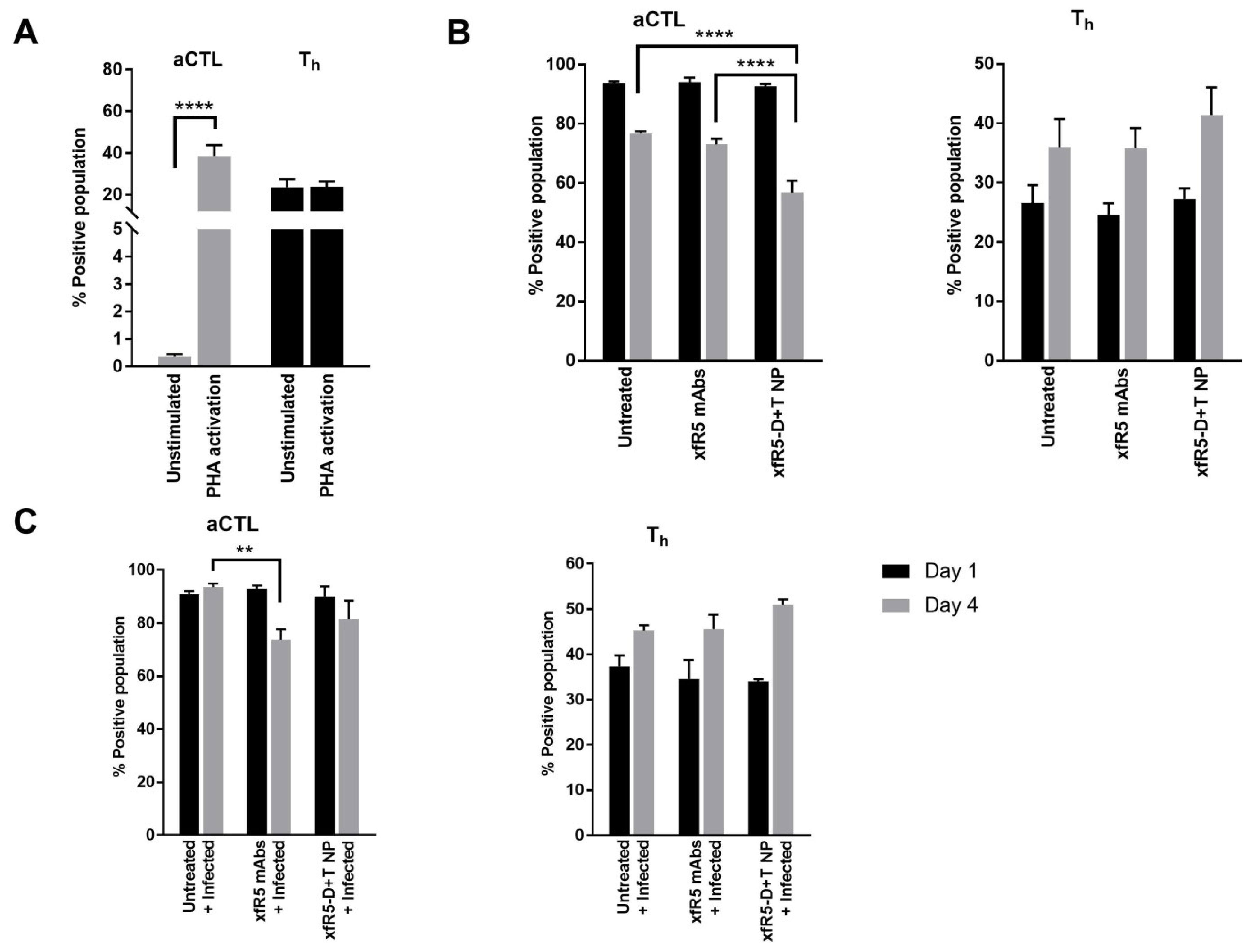
3.5. Immunophenotype during In Vitro Short-Term Prophylaxis and HIV Treatment Study
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pitman, M.C.; Lau, J.S.Y.; McMahon, J.H.; Lewin, S.R. Barriers and strategies to achieve a cure for HIV. Lancet HIV 2018, 5, e317–e328. [Google Scholar] [CrossRef]
- Berger, E.A.; Murphy, P.M.; Farber, J.M. Chemokine receptors as HIV-1 coreceptors: Roles in viral entry, tropism, and disease. Annu. Rev. Immunol. 1999, 17, 657–700. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Zhu, Y.; Li, J.; Chen, Z.; Han, G.W.; Kufareva, I.; Li, T.; Ma, L.; Fenalti, G.; Li, J.; et al. Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science 2013, 341, 1387–1390. [Google Scholar] [CrossRef] [Green Version]
- Ji, H.; Lu, P.; Liu, B.; Qu, X.; Wang, Y.; Jiang, Z.; Yang, X.; Zhong, Y.; Yang, H.; Pan, H.; et al. Zinc-Finger Nucleases Induced by HIV-1 Tat Excise HIV-1 from the Host Genome in Infected and Latently Infected Cells. Mol. Ther. Nucleic Acids 2018, 12, 67–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarborough, R.J.; Gatignol, A. HIV and Ribozymes. In Gene Therapy for HIV and Chronic Infections; Berkhout, B., Ertl, H.C.J., Weinberg, M.S., Eds.; Springer: New York, NY, USA, 2015; pp. 97–116. [Google Scholar]
- Cornu, T.I.; Mussolino, C.; Cathomen, T. Refining strategies to translate genome editing to the clinic. Nat. Med. 2017, 23, 415–423. [Google Scholar] [CrossRef]
- Swamy, M.N.; Wu, H.; Shankar, P. Recent advances in RNAi-based strategies for therapy and prevention of HIV-1/AIDS. Adv. Drug Deliv. Rev. 2016, 103, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Q.; Guo, D.; Chen, S. Application of CRISPR/Cas9-Based Gene Editing in HIV-1/AIDS Therapy. Front. Cell. Infect. Microbiol. 2019, 9, 69. [Google Scholar] [CrossRef]
- Mehlotra, R.K. CCR5 Promoter Polymorphism −2459G > A: Forgotten or Ignored? Cells 2019, 8, 651. [Google Scholar] [CrossRef] [Green Version]
- Steenhuysen, J. First Woman Reported Cured of HIV after Stem Cell Transplant. Available online: https://www.reuters.com/business/healthcare-pharmaceuticals/first-woman-reported-cured-hiv-after-bone-marrow-transplant-2022-02-15/ (accessed on 29 May 2022).
- Gao, Y.; Kraft, J.C.; Yu, D.; Ho, R.J.Y. Recent developments of nanotherapeutics for targeted and long-acting, combination HIV chemotherapy. Eur. J. Pharm. Biopharm. 2019, 138, 75–91. [Google Scholar] [CrossRef]
- Hobson, J.J.; Al-Khouja, A.; Curley, P.; Meyers, D.; Flexner, C.; Siccardi, M.; Owen, A.; Meyers, C.F.; Rannard, S.P. Semi-solid prodrug nanoparticles for long-acting delivery of water-soluble antiretroviral drugs within combination HIV therapies. Nat. Commun. 2019, 10, 1413. [Google Scholar] [CrossRef]
- Holec, A.D.; Mandal, S.; Prathipati, P.K.; Destache, C.J. Nucleotide Reverse Transcriptase Inhibitors: A Thorough Review, Present Status and Future Perspective as HIV Therapeutics. Curr. HIV Res. 2017, 15, 411–421. [Google Scholar] [CrossRef]
- Mandal, S.; Prathipati, P.K.; Kang, G.; Zhou, Y.; Yuan, Z.; Fan, W.; Li, Q.; Destache, C.J. Tenofovir alafenamide and elvitegravir loaded nanoparticles for long-acting prevention of HIV-1 vaginal transmission. AIDS 2017, 31, 469–476. [Google Scholar] [CrossRef] [Green Version]
- Mandal, S.; Kang, G.; Prathipati, P.K.; Zhou, Y.; Fan, W.; Li, Q.; Destache, C.J. Nanoencapsulation introduces long-acting phenomenon to tenofovir alafenamide and emtricitabine drug combination: A comparative pre-exposure prophylaxis efficacy study against HIV-1 vaginal transmission. J. Control. Release 2019, 294, 216–225. [Google Scholar] [CrossRef]
- Shao, J.; Kraft, J.C.; Li, B.; Yu, J.; Freeling, J.; Koehn, J.; Ho, R.J. Nanodrug formulations to enhance HIV drug exposure in lymphoid tissues and cells: Clinical significance and potential impact on treatment and eradication of HIV/AIDS. Nanomedicine 2016, 11, 545–564. [Google Scholar] [CrossRef] [Green Version]
- United States DHHS. Limitations to Treatment Safety and Efficacy: Adverse Effects of Antiretroviral Agents. Available online: https://aidsinfo.nih.gov/guidelines/html/1/adult-and-adolescent-arv/31/adverse-effects-of-arv (accessed on 29 May 2022).
- Margolis, D.A.; Gonzalez-Garcia, J.; Stellbrink, H.J.; Eron, J.J.; Yazdanpanah, Y.; Podzamczer, D.; Lutz, T.; Angel, J.B.; Richmond, G.J.; Clotet, B.; et al. Long-acting intramuscular cabotegravir and rilpivirine in adults with HIV-1 infection (LATTE-2): 96-week results of a randomised, open-label, phase 2b, non-inferiority trial. Lancet 2017, 390, 1499–1510. [Google Scholar] [CrossRef]
- Vasan, S.; Wansom, T.; Schuetz, A.; Krebs, S.; Thomas, R.; Kijak, G.; Polyak, C. Highlights from the HIV Research for Prevention Conference (R4P),: 17–21 October 2016, Chicago, IL, USA. J. Virus Erad. 2017, 3, 92–96. [Google Scholar] [CrossRef]
- Ford, S.L.; Stancil, B.S.; Markowitz, M.; Frank, I.; Grant, R.M.; Mayer, K.H.; Elion, R.; Goldstein, D.; Fisher, C.; Sobieszczyk, M.E.; et al. ECLAIR Study of Cabotegravir (CAB) LA Injections: Characterization of Safety and PK during the ‘PK Tail’ Phase. In Proceedings of the HIV Research for Prevention (HIVR4P) Conference, Chicago, IL, USA, 17–21 October 2016. [Google Scholar]
- Spreen, W.R.; Margolis, D.A.; Pottage, J.C., Jr. Long-acting injectable antiretrovirals for HIV treatment and prevention. Curr. Opin. HIV AIDS 2013, 8, 565–571. [Google Scholar] [CrossRef]
- Passmore, J.-A.S.; Jaspan, H.B.; Masson, L. Genital inflammation, immune activation and risk of sexual HIV acquisition. Curr. Opin. HIV AIDS 2016, 11, 156–162. [Google Scholar] [CrossRef]
- Boettler, T.; Cunha-Neto, E.; Kalil, J.; von Herrath, M. Can an immune-regulatory vaccine prevent HIV infection? Expert Rev. Anti-Infect. Ther. 2012, 10, 299–305. [Google Scholar] [CrossRef] [Green Version]
- Tomaras, G.D.; Plotkin, S.A. Complex immune correlates of protection in HIV-1 vaccine efficacy trials. Immunol. Rev. 2017, 275, 245–261. [Google Scholar] [CrossRef] [Green Version]
- Lewis George, K.; DeVico Anthony, L.; Gallo Robert, C. Antibody persistence and T-cell balance: Two key factors confronting HIV vaccine development. Proc. Natl. Acad. Sci. USA 2014, 111, 15614–15621. [Google Scholar] [CrossRef] [Green Version]
- Promadej-Lanier, N.; Srinivasan, P.; Curtis, K.; Adams, D.R.; Kim, C.; Luo, W.; Jia, H.; Subbarao, S.; Otten, R.A.; Butera, S. Systemic and mucosal immunological responses during repeated mucosal SHIV162P3 challenges prior to and following infection in pigtailed macaques. Virology 2008, 375, 492–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecuroux, C.; Girault, I.; Urrutia, A.; Doisne, J.M.; Deveau, C.; Goujard, C.; Meyer, L.; Sinet, M.; Venet, A. Identification of a particular HIV-specific CD8+ T-cell subset with a CD27+ CD45RO-/RA+ phenotype and memory characteristics after initiation of HAART during acute primary HIV infection. Blood 2009, 113, 3209–3217. [Google Scholar] [CrossRef] [Green Version]
- McMichael, A.J.; Koff, W.C. Vaccines that stimulate T cell immunity to HIV-1: The next step. Nat. Immunol. 2014, 15, 319–322. [Google Scholar] [CrossRef] [Green Version]
- Perdomo-Celis, F.; Taborda, N.A.; Rugeles, M.T. CD8+ T-Cell Response to HIV Infection in the Era of Antiretroviral Therapy. Front. Immunol. 2019, 10, 1896. [Google Scholar] [CrossRef] [PubMed]
- Capjak, I.; Goreta, S.S.; Jurasin, D.D.; Vrcek, I.V. How protein coronas determine the fate of engineered nanoparticles in biological environment. Arch. Ind. Hyg. Toxicol. 2017, 68, 245–253. [Google Scholar] [CrossRef] [Green Version]
- Venuti, A.; Pastori, C.; Pennisi, R.; Riva, A.; Sciortino, M.T.; Lopalco, L. Class B beta-arrestin2-dependent CCR5 signalosome retention with natural antibodies to CCR5. Sci. Rep. 2016, 6, 39382. [Google Scholar] [CrossRef] [Green Version]
- Roschke, V.; Rosen, C.A.; Ruben, S.M. Human G-Protein Chemokine Receptor (CCR5) Hdgnr10. U.S. Patent US20060111559A2, 25 May 2006. [Google Scholar]
- Ablashi, D.V.; Handy, M.; Bernbaum, J.; Chatlynne, L.G.; Lapps, W.; Kramarsky, B.; Berneman, Z.N.; Komaroff, A.L.; Whitman, J.E. Propagation and characterization of human herpesvirus-7 (HHV-7) isolates in a continuous T-lymphoblastoid cell line (SupT1). J. Virol. Methods 1998, 73, 123–140. [Google Scholar] [CrossRef]
- Meijerink, H.; Indrati, A.R.; van Crevel, R.; Joosten, I.; Koenen, H.; van der Ven, A.J.A.M. The number of CCR5 expressing CD4+ T lymphocytes is lower in HIV-infected long-term non-progressors with viral control compared to normal progressors: A cross-sectional study. BMC Infect. Dis. 2014, 14, 683. [Google Scholar] [CrossRef] [Green Version]
- OARAC. Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in HIV-1-Infected Adults and Adolescents; Department of Health and Human Services: Bethesda, MD, USA, 2018.
- Wassner, C.; Bradley, N.; Lee, Y. A Review and Clinical Understanding of Tenofovir: Tenofovir Disoproxil Fumarate versus Tenofovir Alafenamide. J. Int. Assoc. Provid. AIDS Care 2020, 19, 2325958220919231. [Google Scholar] [CrossRef] [Green Version]
- Katlama, C.; Murphy, R. Dolutegravir for the treatment of HIV. Expert Opin. Investig. Drugs 2012, 21, 523–530. [Google Scholar] [CrossRef]
- Nanda, J.S.; Lorsch, J.R. Chapter Eight—Labeling a Protein with Fluorophores Using NHS Ester Derivitization. In Methods in Enzymology; Lorsch, J., Ed.; Academic Press: Cambridge, MA, USA, 2014; Volume 536, pp. 87–94. [Google Scholar]
- Yang, J.-Y.; Bae, J.; Jung, A.; Park, S.; Chung, S.; Seok, J.; Roh, H.; Han, Y.; Oh, J.-M.; Sohn, S.; et al. Surface functionalization-specific binding of coagulation factors by zinc oxide nanoparticles delays coagulation time and reduces thrombin generation potential in vitro. PLoS ONE 2017, 12, e0181634. [Google Scholar] [CrossRef]
- Mandal, S.; Zhou, Y.; Shibata, A.; Destache, C.J. Confocal fluorescence microscopy: An ultra-sensitive tool used to evaluate intracellular antiretroviral nano-drug delivery in HeLa cells. AIP Adv. 2015, 5, 084803. [Google Scholar] [CrossRef] [Green Version]
- Mandal, S.; Kang, G.; Prathipati, P.K.; Fan, W.; Li, Q.; Destache, C.J. Long-acting parenteral combination antiretroviral loaded nano-drug delivery system to treat chronic HIV-1 infection: A humanized mouse model study. Antivir. Res. 2018, 156, 85–91. [Google Scholar] [CrossRef]
- Mandal, S.; Khandalavala, K.; Pham, R.; Bruck, P.; Varghese, M.; Kochvar, A.; Monaco, A.; Prathipati, K.P.; Destache, C.; Shibata, A. Cellulose Acetate Phthalate and Antiretroviral Nanoparticle Fabrications for HIV Pre-Exposure Prophylaxis. Polymers 2017, 9, 423. [Google Scholar] [CrossRef] [Green Version]
- Prathipati, P.K.; Mandal, S.; Destache, C.J. Simultaneous quantification of tenofovir, emtricitabine, rilpivirine, elvitegravir and dolutegravir in mouse biological matrices by LC-MS/MS and its application to a pharmacokinetic study. J. Pharm. Biomed. Anal. 2016, 129, 473–481. [Google Scholar] [CrossRef] [Green Version]
- FDA. Bioanalytical Method Validation Guidance for Industry; FDA: Bethesda, MD, USA, 2018.
- Mandal, S.; Belshan, M.; Holec, A.; Zhou, Y.; Destache, C.J. An Enhanced Emtricitabine-Loaded Long-Acting Nanoformulation for Prevention or Treatment of HIV Infection. Antimicrob. Agents Chemother. 2017, 61, e01475-16. [Google Scholar] [CrossRef] [Green Version]
- Wiercigroch, E.; Szafraniec, E.; Czamara, K.; Pacia, M.Z.; Majzner, K.; Kochan, K.; Kaczor, A.; Baranska, M.; Malek, K. Raman and infrared spectroscopy of carbohydrates: A review. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2017, 185, 317–335. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Gothwal, A.; Sharma, A.K.; Qayum, A.; Singh, S.K.; Gupta, U. Biodegradable nano-architectural PEGylated approach for the improved stability and anticancer efficacy of bendamustine. Int. J. Biol. Macromol. 2016, 92, 1242–1251. [Google Scholar] [CrossRef]
- Okwundu, C.I.; Uthman, O.A.; Okoromah, C.A. Antiretroviral pre-exposure prophylaxis (PrEP) for preventing HIV in high-risk individuals. Cochrane Database Syst. Rev. 2012, 7, Cd007189. [Google Scholar] [CrossRef]
- Boncompain, G.; Herit, F.; Tessier, S.; Lescure, A.; Del Nery, E.; Gestraud, P.; Staropoli, I.; Fukata, Y.; Fukata, M.; Brelot, A.; et al. Targeting CCR5 trafficking to inhibit HIV-1 infection. Sci. Adv. 2019, 5, eaax0821. [Google Scholar] [CrossRef] [Green Version]
- Platt, E.J.; Wehrly, K.; Kuhmann, S.E.; Chesebro, B.; Kabat, D. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J. Virol. 1998, 72, 2855–2864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desmetz, C.; Lin, Y.L.; Mettling, C.; Portalès, P.; Rabesandratana, H.; Clot, J.; Corbeau, P. The strength of the chemotactic response to a CCR5 binding chemokine is determined by the level of cell surface CCR5 density. Immunology 2006, 119, 551–561. [Google Scholar] [CrossRef]
- Lin, Y.L.; Mettling, C.; Portalès, P.; Rouzier, R.; Clot, J.; Reynes, J.; Corbeau, P. The chemokine CCL5 regulates the in vivo cell surface expression of its receptor, CCR5. AIDS 2008, 22, 430–432. [Google Scholar] [CrossRef]
- Lee, M.; Kim, H.; Kim, E.; Yi, S.Y.; Hwang, S.G.; Yang, S.; Lim, E.-K.; Kim, B.; Jung, J.; Kang, T. Multivalent Antibody–Nanoparticle Conjugates To Enhance the Sensitivity of Surface-Enhanced Raman Scattering-Based Immunoassays. ACS Appl. Mater. Interfaces 2018, 10, 37829–37834. [Google Scholar] [CrossRef]
- Mandal, S.; Eksteen-Akeroyd, Z.H.; Jacobs, M.J.; Hammink, R.; Koepf, M.; Lambeck, A.J.A.; van Hest, J.C.M.; Wilson, C.J.; Blank, K.; Figdor, C.G.; et al. Therapeutic nanoworms: Towards novel synthetic dendritic cells for immunotherapy. Chem. Sci. 2013, 4, 4168–4174. [Google Scholar] [CrossRef] [Green Version]
- Sallusto, F.; Lanzavecchia, A.; Mackay, C.R. Chemokines and chemokine receptors in T-cell priming and Th1/Th2-mediated responses. Immunol. Today 1998, 19, 568–574. [Google Scholar] [CrossRef]
- Fukada, K.; Sobao, Y.; Tomiyama, H.; Oka, S.; Takiguchi, M. Functional expression of the chemokine receptor CCR5 on virus epitope-specific memory and effector CD8+ T cells. J. Immunol. 2002, 168, 2225–2232. [Google Scholar] [CrossRef] [Green Version]
- Crawford, K.; Gabuzda, D.; Pantazopoulos, V.; Xu, J.; Clement, C.; Reinherz, E.; Alper, C.A. Circulating CD2+ monocytes are dendritic cells. J. Immunol. 1999, 163, 5920–5928. [Google Scholar] [PubMed]
- Appay, V.; Rowland-Jones, S.L. RANTES: A versatile and controversial chemokine. Trends Immunol. 2001, 22, 83–87. [Google Scholar] [CrossRef]
- Kettenmann, H.; Burton, G.; Moenning, U. Neuroinflammation—From Bench to Bedside; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Briggs, J.A.G.; Wilk, T.; Welker, R.; Kräusslich, H.-G.; Fuller, S.D. Structural organization of authentic, mature HIV-1 virions and cores. EMBO J. 2003, 22, 1707–1715. [Google Scholar] [CrossRef] [Green Version]
- Barmania, F.; Pepper, M.S. C–C chemokine receptor type five (CCR5): An emerging target for the control of HIV infection. Appl. Transl. Genom. 2013, 2, 3–16. [Google Scholar] [CrossRef] [Green Version]
- Signoret, N.; Pelchen-Matthews, A.; Mack, M.; Proudfoot, A.E.; Marsh, M. Endocytosis and recycling of the HIV coreceptor CCR5. J. Cell Biol. 2000, 151, 1281–1294. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P. Long term non-progressor (LTNP) HIV infection. Indian J. Med. Res. 2013, 138, 291–293. [Google Scholar]
- Zhang, S.; Gao, H.; Bao, G. Physical Principles of Nanoparticle Cellular Endocytosis. ACS Nano 2015, 9, 8655–8671. [Google Scholar] [CrossRef] [Green Version]
- Di Mascio, M.; Sereti, I.; Matthews, L.T.; Natarajan, V.; Adelsberger, J.; Lempicki, R.; Yoder, C.; Jones, E.; Chow, C.; Metcalf, J.A.; et al. Naïve T-cell dynamics in human immunodeficiency virus type 1 infection: Effects of highly active antiretroviral therapy provide insights into the mechanisms of naive T-cell depletion. J. Virol. 2006, 80, 2665–2674. [Google Scholar] [CrossRef] [Green Version]
- Kohlmeier, J.E.; Miller, S.C.; Smith, J.; Lu, B.; Gerard, C.; Cookenham, T.; Roberts, A.D.; Woodland, D.L. The chemokine receptor CCR5 plays a key role in the early memory CD8+ T cell response to respiratory virus infections. Immunity 2008, 29, 101–113. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.L.; Crumpacker, C. Eradication of HIV and Cure of AIDS, Now and How? Front. Immunol. 2013, 4, 337. [Google Scholar] [CrossRef] [Green Version]
- Pennock, N.D.; White, J.T.; Cross, E.W.; Cheney, E.E.; Tamburini, B.A.; Kedl, R.M. T cell responses: Naïve to memory and everything in between. Adv. Physiol. Educ. 2013, 37, 273–283. [Google Scholar] [CrossRef] [Green Version]
- Chahroudi, A.; Silvestri, G.; Lichterfeld, M. T memory stem cells and HIV: A long-term relationship. Curr. HIV/AIDS Rep. 2015, 12, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Potter, S.J.; Lacabaratz, C.; Lambotte, O.; Perez-Patrigeon, S.; Vingert, B.; Sinet, M.; Colle, J.-H.; Urrutia, A.; Scott-Algara, D.; Boufassa, F.; et al. Preserved Central Memory and Activated Effector Memory CD4+ T-Cell Subsets in Human Immunodeficiency Virus Controllers: An ANRS EP36 Study. J. Virol. 2007, 81, 13904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidya Vijayan, K.K.; Karthigeyan, K.P.; Tripathi, S.P.; Hanna, L.E. Pathophysiology of CD4+ T-Cell Depletion in HIV-1 and HIV-2 Infections. Front. Immunol. 2017, 8, 580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vingert, B.; Benati, D.; Lambotte, O.; de Truchis, P.; Slama, L.; Jeannin, P.; Galperin, M.; Perez-Patrigeon, S.; Boufassa, F.; Kwok, W.W.; et al. HIV controllers maintain a population of highly efficient Th1 effector cells in contrast to patients treated in the long term. J. Virol. 2012, 86, 10661–10674. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
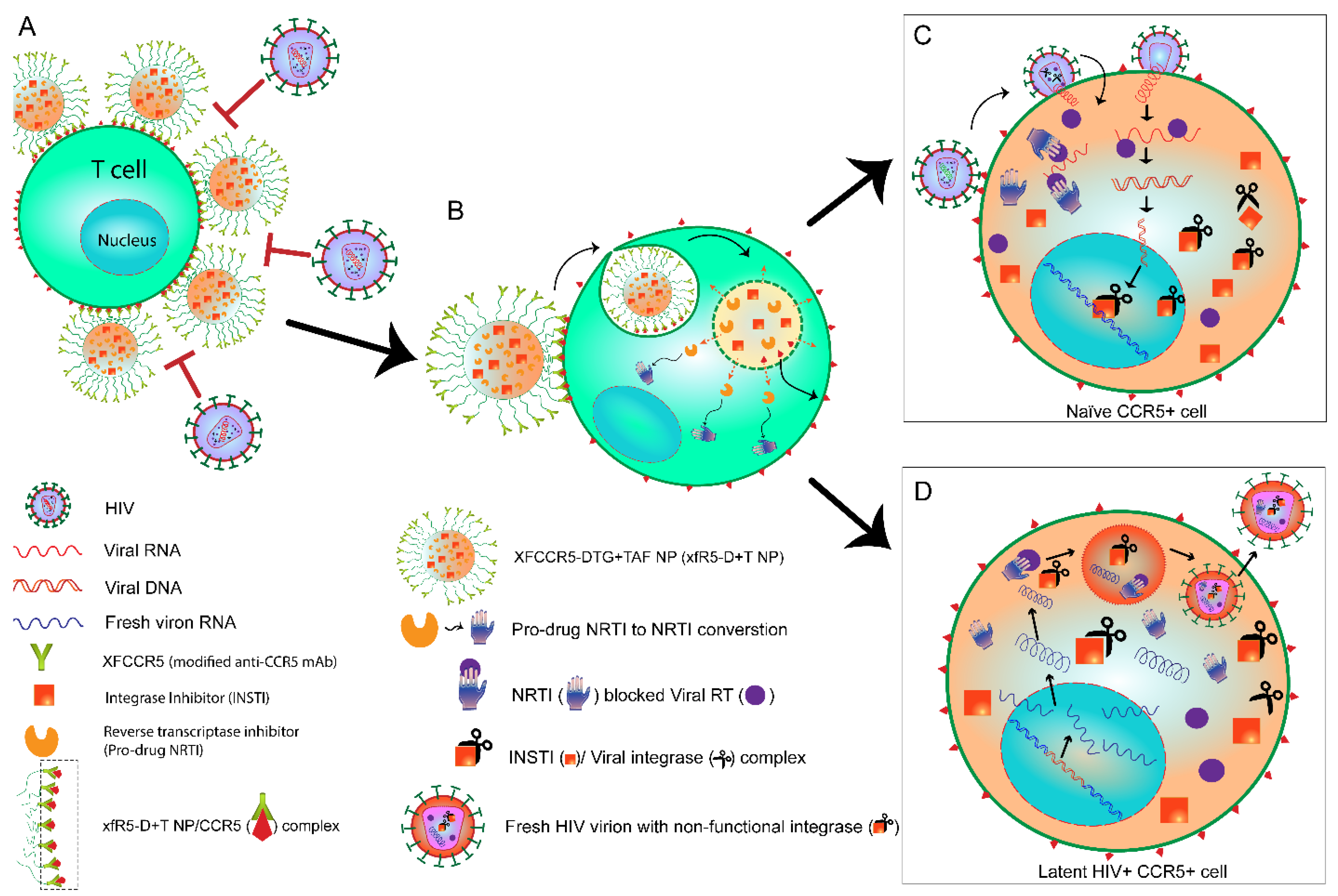{kind=link}
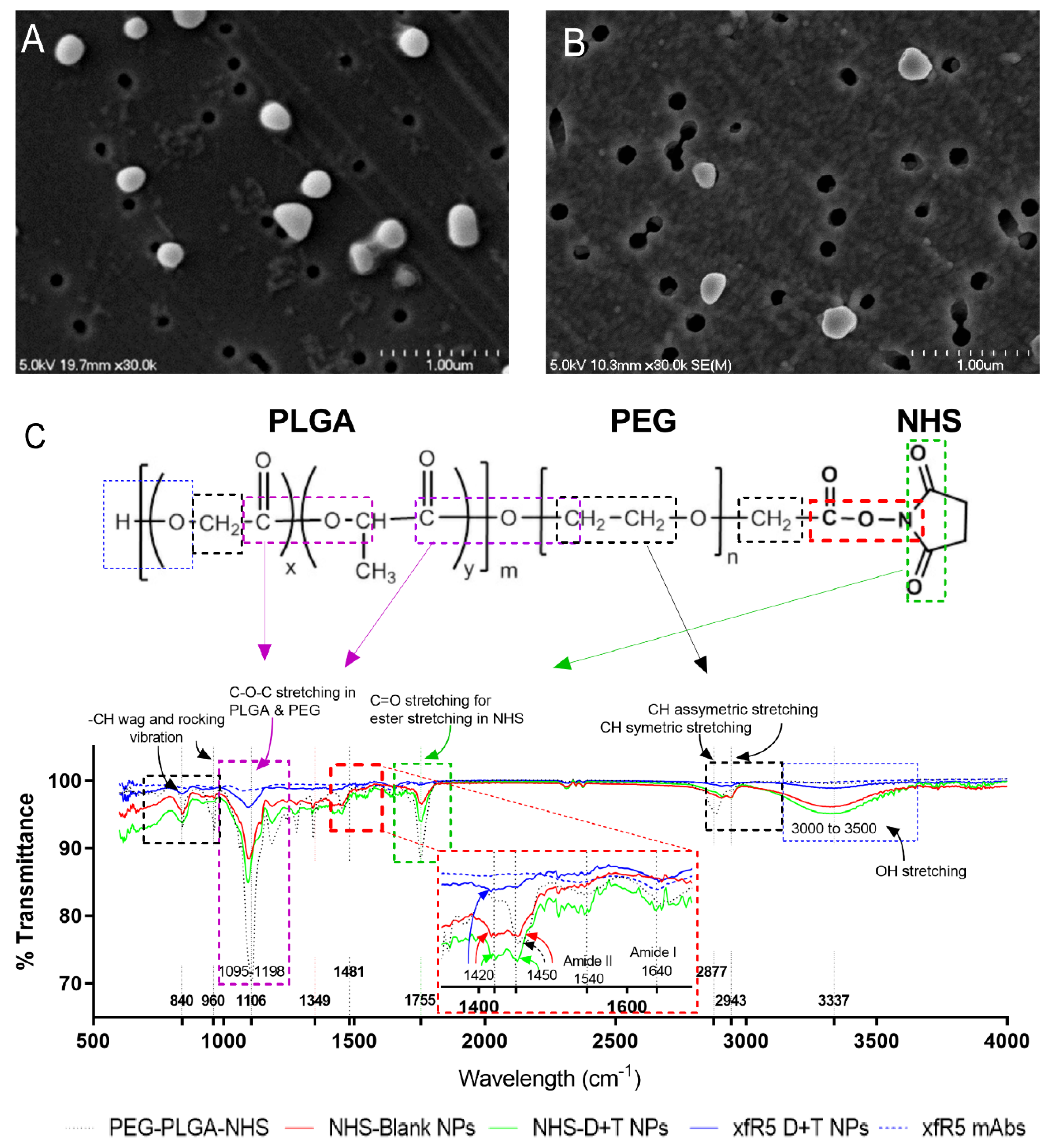{kind=link}
{kind=link}
{kind=link}
| T-Lymphocyte Target | Marker | Fluorescence Dye Conjugated | Monoclonal Antibody Type | Company |
|---|---|---|---|---|
| T lymphocytes | CD3 | Alexa Flour 594 | Mouse anti-human | BioLegend |
| Helper T-cells | CD4 | PerCP-Cy5.5 | Mouse anti-human | TONBO biosciences |
| Alexa Flour 700 | Mouse anti-human | BioLegend | ||
| Cytotoxic T-cells | CD8 | PE/Cy7 | Mouse anti-human | BioLegend |
| Monocytes | CD68 | APC | Mouse anti-human | Invitrogen |
| HIV target on T-cell | xfR5 | Cy3 | Modified anti-human | ATCC |
| Transition T-cells | CCR7 | CCR7-APC eFlour 780 | Mouse anti-human | Invitrogen |
| Memory T-cells | CD45RO | Pacific Blue | Mouse anti-human | BioLegend |
| Activated T-cells | CD69 | Alexa Flour 488 | Mouse anti-human | BioLegend |
| HIV maker on T-cells | CCR5 | APC | Mouse anti-human | BioLegend |
| Intermediate memory T-cells | CD27 | Alexa Flour 700 | Mouse anti-human | BioLegend |
| DC or latently infected T-cells | CD2 | Pacific Blue | Mouse anti-human | BioLegend |
| Type | Size (nm) | Surface Charge (mV) | Polydispersity Index (PDI) | %Drug Entrapment Efficiency (%EE) | xfR5 mAb Bound per mg D+T NP |
|---|---|---|---|---|---|
| NHS-D+T NP | 198.7 ± 10 | −28.15 ± 1.9 | 0.15 ± 0.01 | DTG: 58.8 ± 10 TAF: 60.5 ± 10.3 | N/A |
| xfR5-D+T NP | 212.6 ± 20.7 | −17.77 ± 1.9 | 0.20 ± 0.018 | DTG: 58.5 ± 9 TAF: 60.4 ± 10.6 | 3.7 ± 0.52 µg |
| Type | Binding Affinity (Kd, nM) | ||||
|---|---|---|---|---|---|
| TZM-bl Cells | CD4+ T-Cells | CD8+ T-Cells | CD68+ T-Cells | CD2+ T-Cells | |
| xfR5-D+T NPs | 0.038 ± 0.020 | 4.31 ± 1.47 | 0.99 ± 0.38 | 0.11 ± 0.066 | 1.5 ± 1.1 |
| xfR5 mAbs | 0.25 ± 0.15 | 20.87 ± 10.65 | 25.02 ± 14.3 | 0.21 ± 0.10 | 2.6 ± 2.5 |
| Wild-type anti-CCR5 mAb | 2.02 ± 0.66 | ND | ND | ND | ND |
| Parameter | Units | NP | Solution | ||||||
|---|---|---|---|---|---|---|---|---|---|
| TAF | TFV | DTG | TFV-dp | TAF | TFV | DTG | TFV-dp | ||
| Cmax | (pmole/106 cells) | 18.1 ± 3.3 | 1250.2 ± 269.1 | 37,027.8 ± 5401.0 | 29.7 ±13.1 | 1.4 ± 0.7 | 1428.4 ± 580.4 | 334.0 ± 197.4 | 12.9 ± 4.1 |
| AUCall | h*(pmole/106 cells) | 825.4 ± 119.5 | 52,384.8 ± 4613.2 | 2,240,490.8 ± 240,106.3 | 827.5 ± 125.5 | 11.4 ± 4.4 | 47,465.0 ± 9641.7 | 8805.0 ± 1934.1 | 585.9 ± 94.6 |
| t1/2 | h | 75.5 | 21.6 | 79.2 | 32.3 | 6.5 | 22.3 | 18.0 | 25.4 |
| Cell Type | Treatment Type | CC50 (nM) | IC50 (nM) | SI |
|---|---|---|---|---|
| TZM-bl | xfR5-D+T NPs | 2910 ± 134.9 | 0.035 ± 0.01 | 82,670 |
| xfR5 NPs | 998.7 ± 122.2 | 0.055 ± 0.01 | 18,254 | |
| D+T NPs | 3095 ± 102.3 | 0.2 ± 0.16 | 15,872 | |
| xfR5 mAb | 1464 ± 35.5 | 18.53 ± 2.85 | 79 | |
| PBMCs | xfR5 D+T NPs | 690.6 ± 114.7 | 9.77 ± 2.03 | 71 |
| xfR5 NPs | 1428 ± 83.5 | 53.34 ± 2.34 | 26 | |
| D+T NPs | 1009 ± 88.7 | 33.95 ± 1.91 | 30 | |
| xfR5 mAb | 816.7 ± 93 | 115.9 ± 1.61 | 7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mandal, S.; Sunagawa, S.W.; Prathipati, P.K.; Belshan, M.; Shibata, A.; Destache, C.J. Targeted Immuno-Antiretroviral to Promote Dual Protection against HIV: A Proof-of-Concept Study. Nanomaterials 2022, 12, 1942. https://0-doi-org.brum.beds.ac.uk/10.3390/nano12111942
Mandal S, Sunagawa SW, Prathipati PK, Belshan M, Shibata A, Destache CJ. Targeted Immuno-Antiretroviral to Promote Dual Protection against HIV: A Proof-of-Concept Study. Nanomaterials. 2022; 12(11):1942. https://0-doi-org.brum.beds.ac.uk/10.3390/nano12111942
Chicago/Turabian StyleMandal, Subhra, Shawnalyn W. Sunagawa, Pavan Kumar Prathipati, Michael Belshan, Annemarie Shibata, and Christopher J. Destache. 2022. "Targeted Immuno-Antiretroviral to Promote Dual Protection against HIV: A Proof-of-Concept Study" Nanomaterials 12, no. 11: 1942. https://0-doi-org.brum.beds.ac.uk/10.3390/nano12111942