

Anti-EGFR Targeted Multifunctional I-131 Radio-Nanotherapeutic for Treating Osteosarcoma: In Vitro 3D Tumor Spheroid Model

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Cell Culture
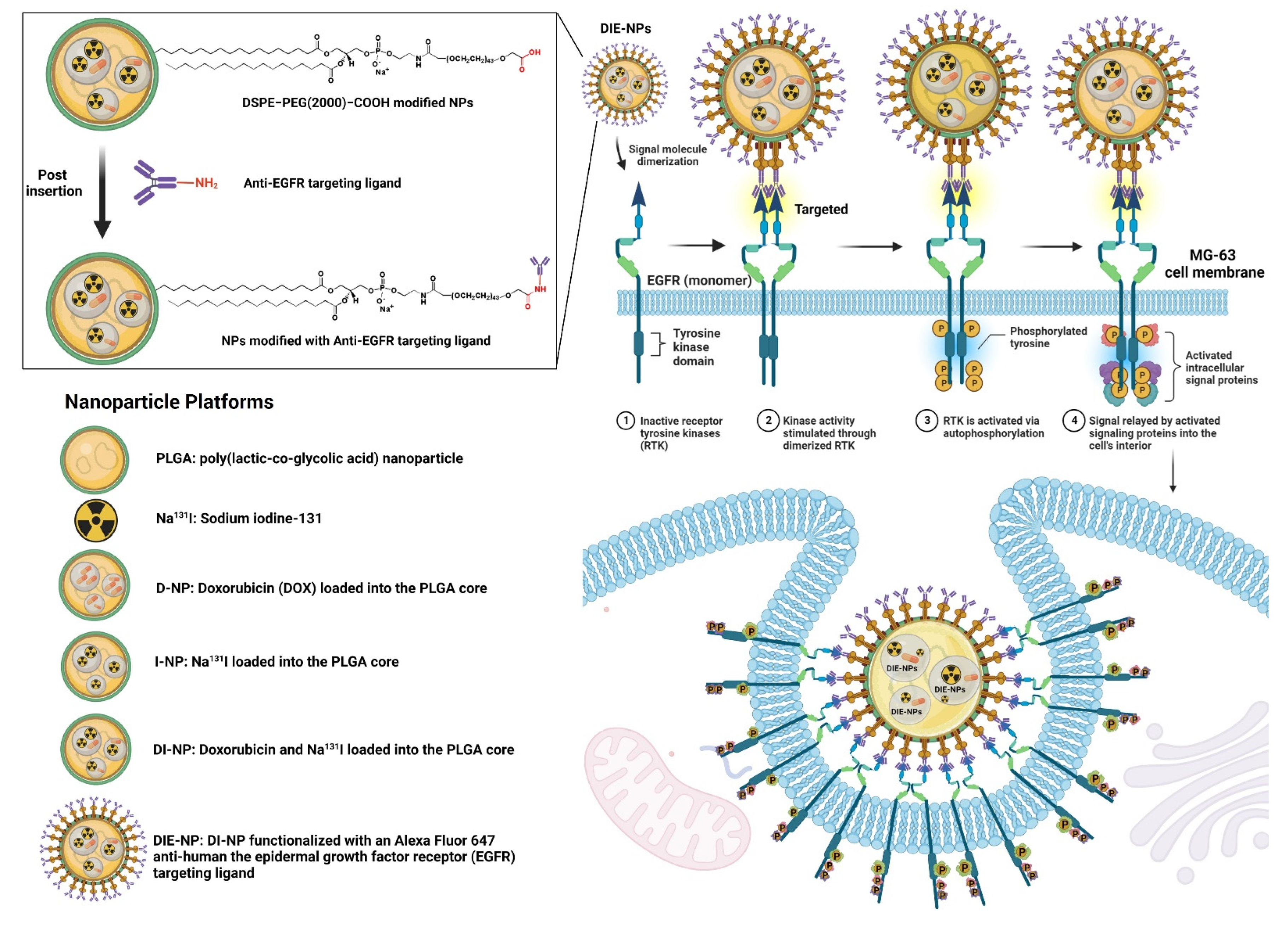
2.2. Preparation and Characterization of Nanoparticles
- Control: Dulbecco’s Modified Eagle Medium (DMEM) cell culture media.
- PLGA: PLGA nanoparticles were synthesized by dissolving the pellets in dichloromethane organic solvent (DCM), and adding FITC (fluorescence isothiocyanate) dye. This was added dropwise to an aqueous solution of 1 PBS, and agitated gently for 4 h to aid evaporation in a fume hood.
- Na131I: sodium iodide (Na131I) with an activity of 3.70 MBq (100 µCi), dissolved in 1 PBS.
- D-NPs: doxorubicin (DOX) loaded into the PLGA cores, prepared by the double emulsion process.
- I-NPs: Na131I (activity of 3.70 MBq) loaded into the PLGA cores, prepared by the double emulsion process. Briefly, for the inner phase Na131I was diffused with 25 μL of 500 mM 1 PBS at pH 8, and sonicated for 2 min at 70% pulsed power (2 s on/1 s off) with 500 μL of PLGA in 10 mg/mL dichloromethane (DCM). The solution was then added to 5 mL of 10 mM 1 PBS at pH 8 and sonicated for 2 min. Then 10 mL of 10 mM 1 PBS at pH 8 was added to assist evaporation and stirred for 4 h in a fume hood.
- DI-NPs: DOX and Na131I (activity of 3.70 MBq) were loaded into the PLGA cores prepared by double emulsion as described in D-NPs and I-NPs (above).
- DIE-NPs: To fabricate the targeted DIE-NPs, the DI-NPs were functionalized with an Alexa Fluor 647 anti-human EGFR antibody targeting ligand. 500 µL of DI-NPs (10 mg/mL) were suspended in 4 mL 1 PBS, incubated in 0.5 mg DSPE−PEG(2000) carboxylic acid and mixed at 400 g for 60 min. They were then rinsed twice with 1 PBS and mixed for 5 min at 1000 g, and suspended in 1 PBS. Afterward, 1 µg of Alexa Fluor 647 anti-human EGFR antibody targeting ligand was added and stirred gently for 120 min at 4 °C. Lastly, the DIE-NPs were washed with 1 PBS twice and stored at 4 °C.
2.3. Structural Characterization and Stability of Nanoparticles
2.4. Quantification of Ligand Surface Coverage
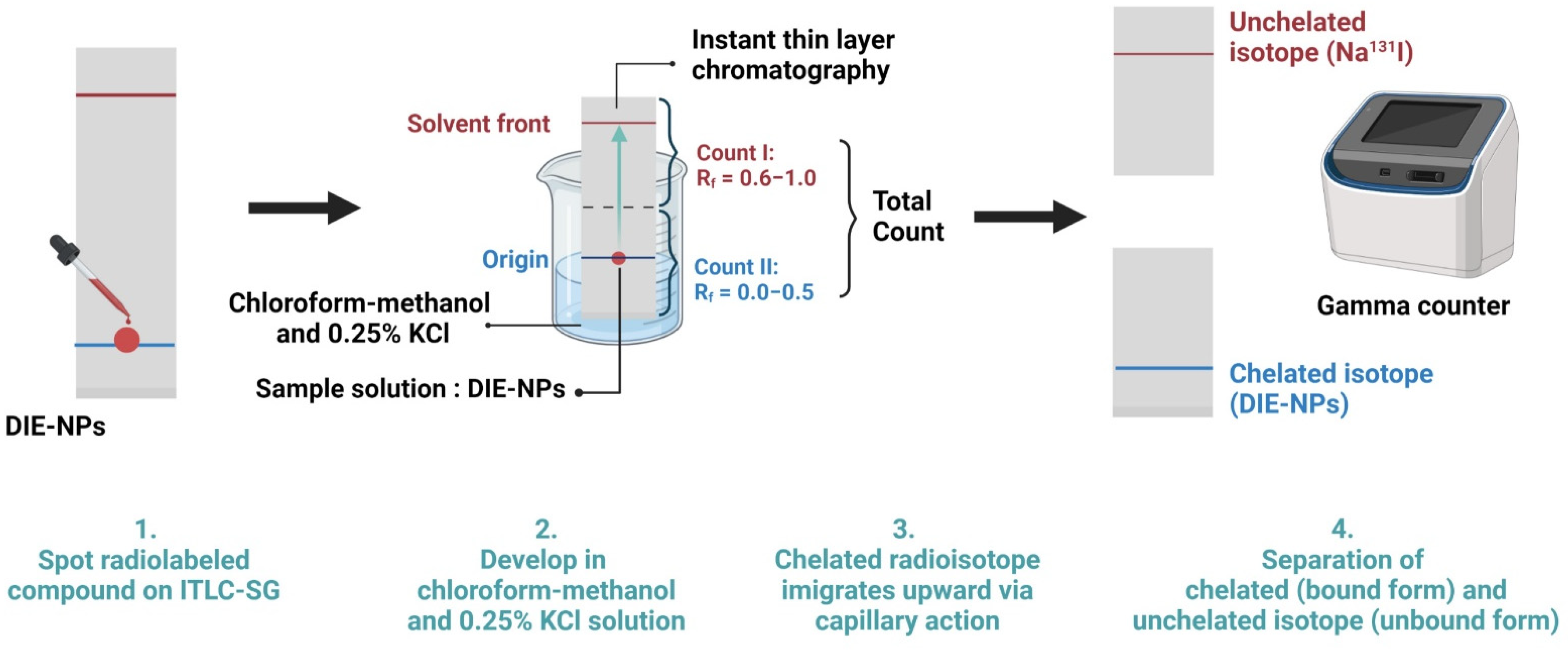
2.5. Sodium Iodine-131 Radiochemical Purity and Radioactive Stability of DIE-NPs
2.6. Radioactive Labeling Yield
2.7. Calculation of Na131I Radiation Dose Delivered to Each Well
2.8. The Two-Dimensional (2D) In Vitro Therapeutic I-131 Dose Optimization
2.9. Radioactive Encapsulation Efficiency
2.10. Two-Dimensional (2D) In Vitro Therapeutic DOX Optimization
2.11. Drug Loading and Encapsulation Efficiency
2.12. Cumulative Amount of Drug Release
2.13. Three-Dimensional (3D) Human MG-63 Tumor Spheroid In Vitro Cytotoxicity
2.14. In Vitro Surface Immunofluorescence Cellular Binding/Uptake Targeting Efficiency
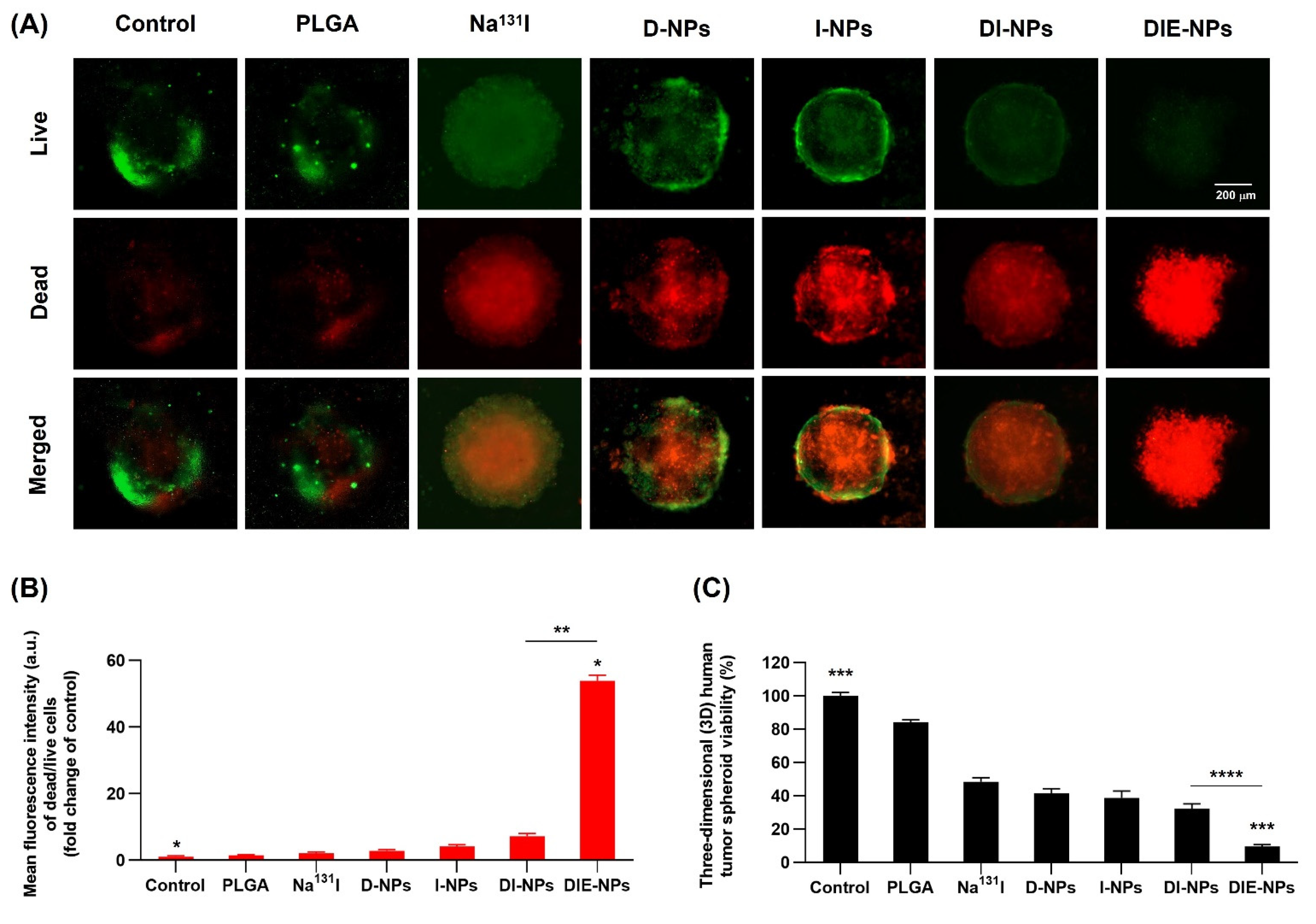
2.15. Human MG-63 Tumor Spheroid Three-Dimensional (3D) In Vitro Live/Dead Cell Imaging
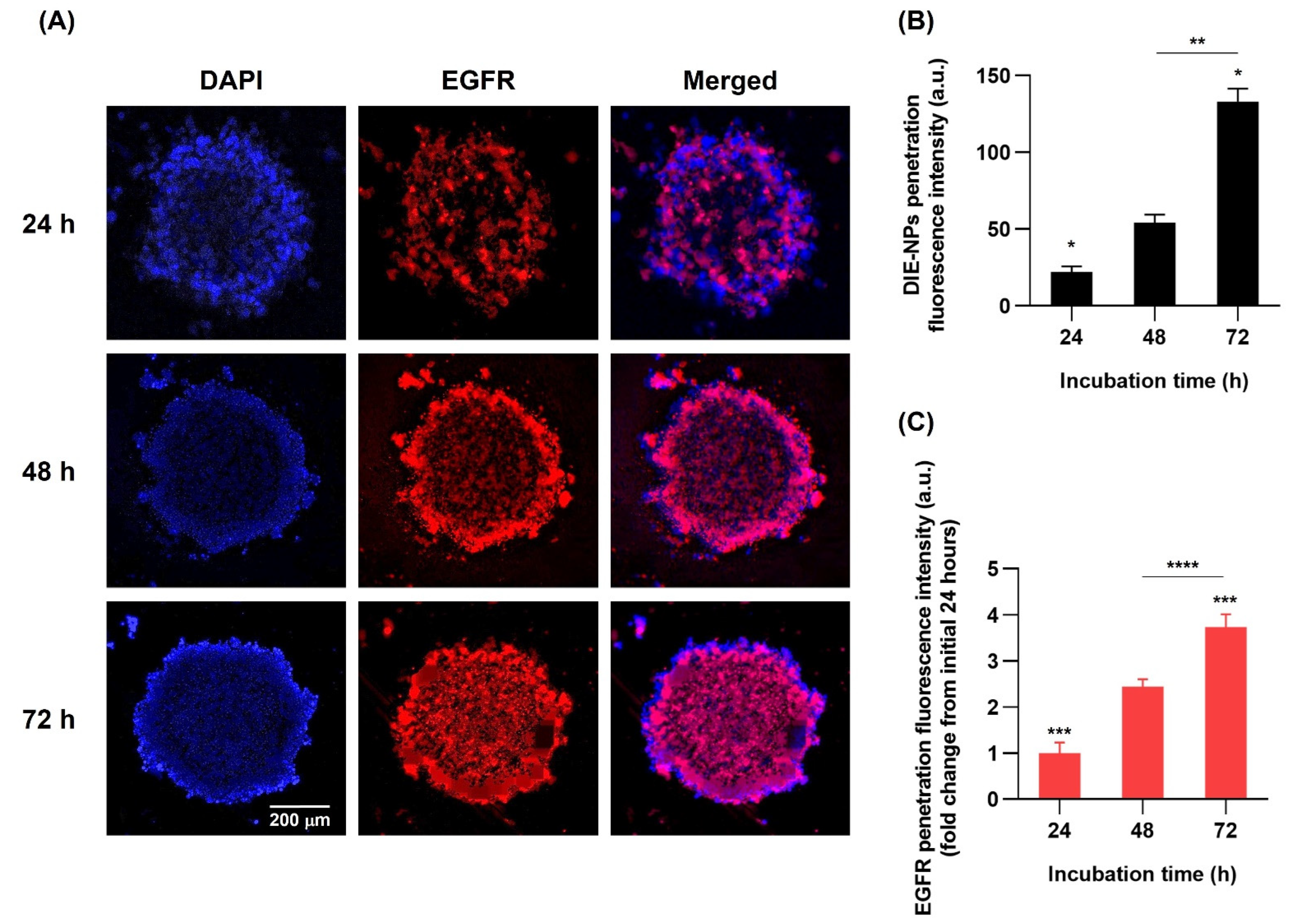
2.16. Three-Dimensional (3D) Human MG-63 Tumor Spheroid DIE-NPs Penetration
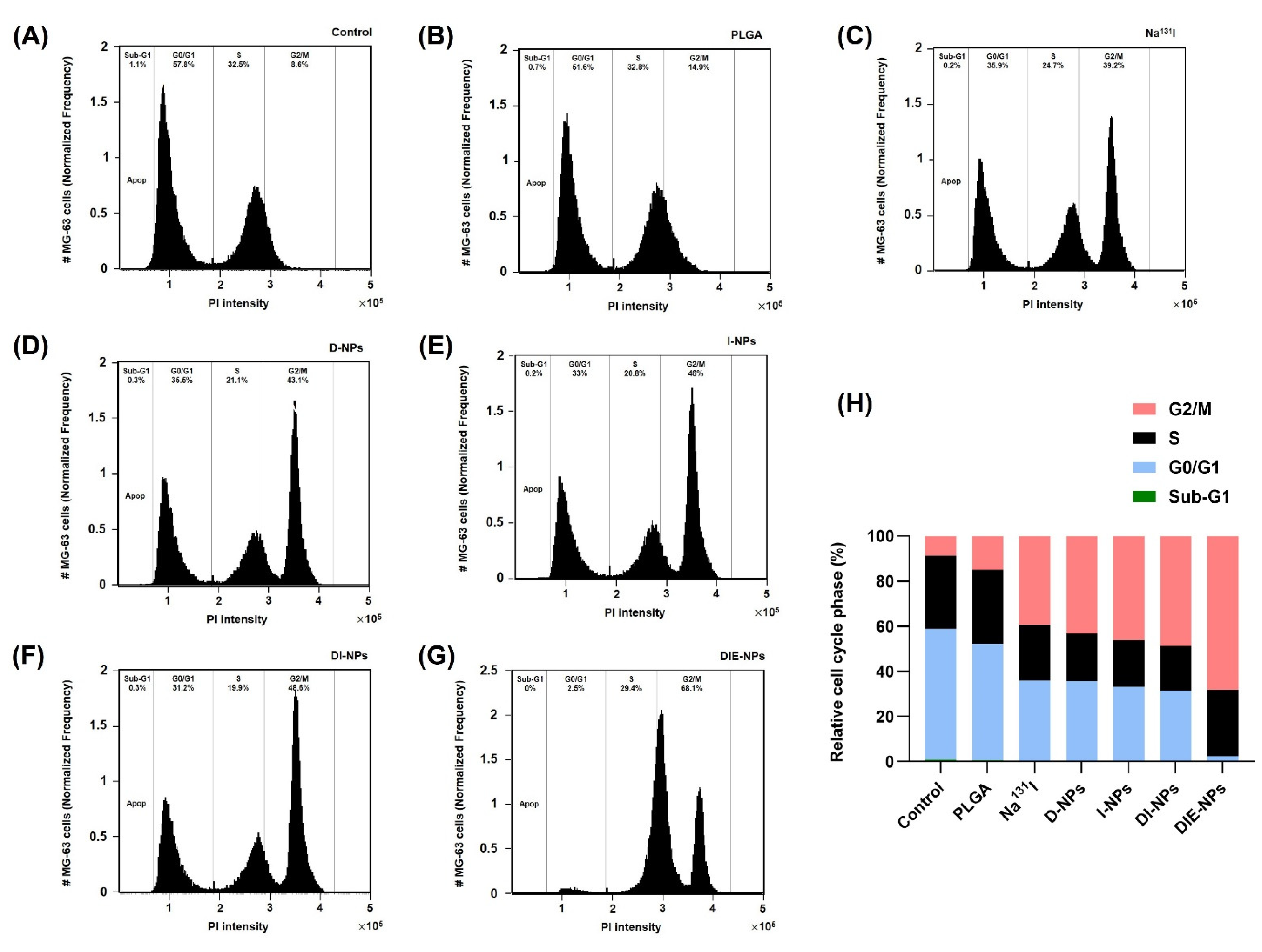
2.17. Cell Cycle
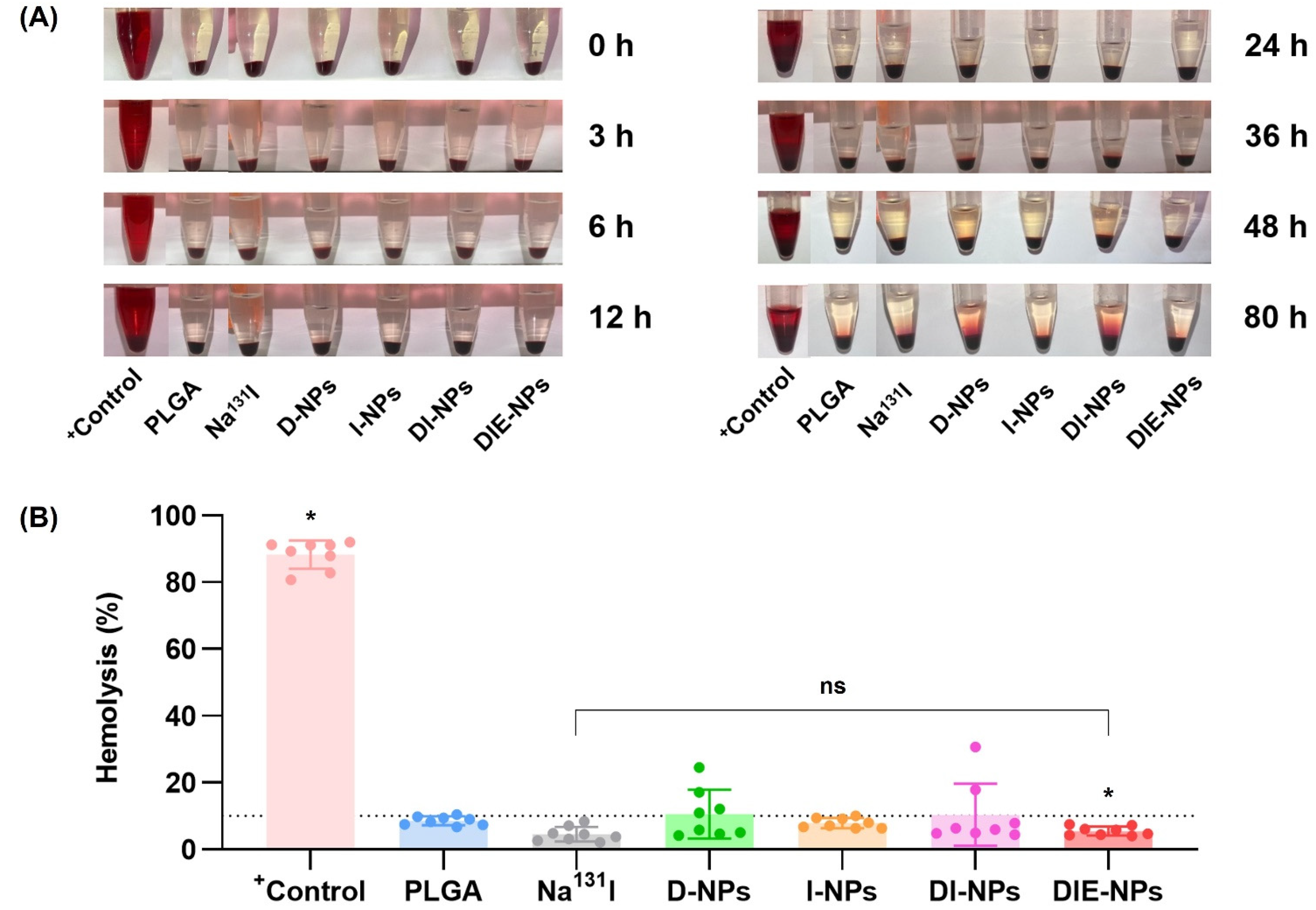
2.18. Blood Compatibility of Samples: Hemolysis Assay
2.19. Cellular Uptake by Macrophage Cells
2.20. Statistical Analysis
3. Results
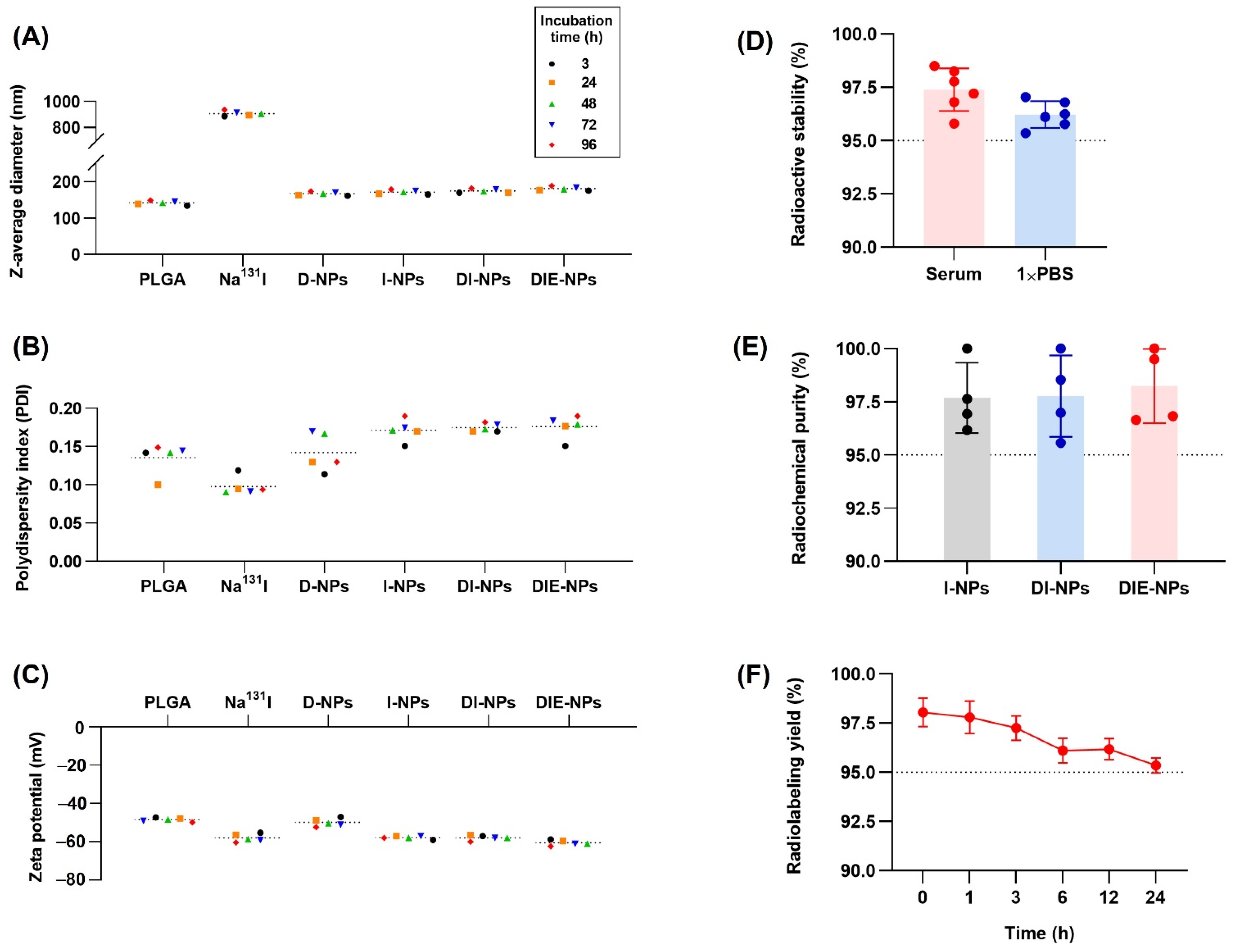
3.1. Nanoparticle Physicochemical Properties and Characterization
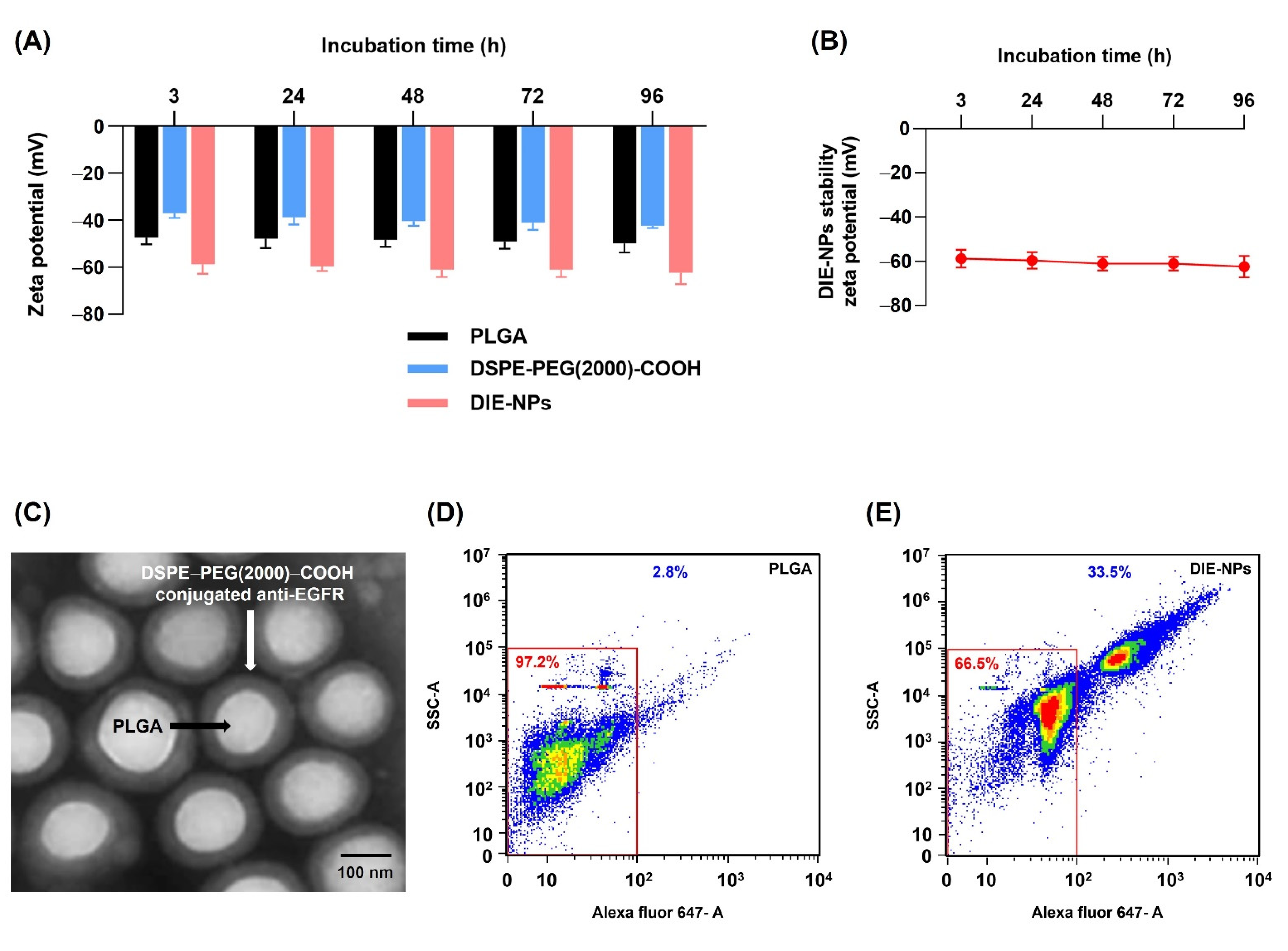
3.2. Structural Characterization and Stability of Nanoparticles
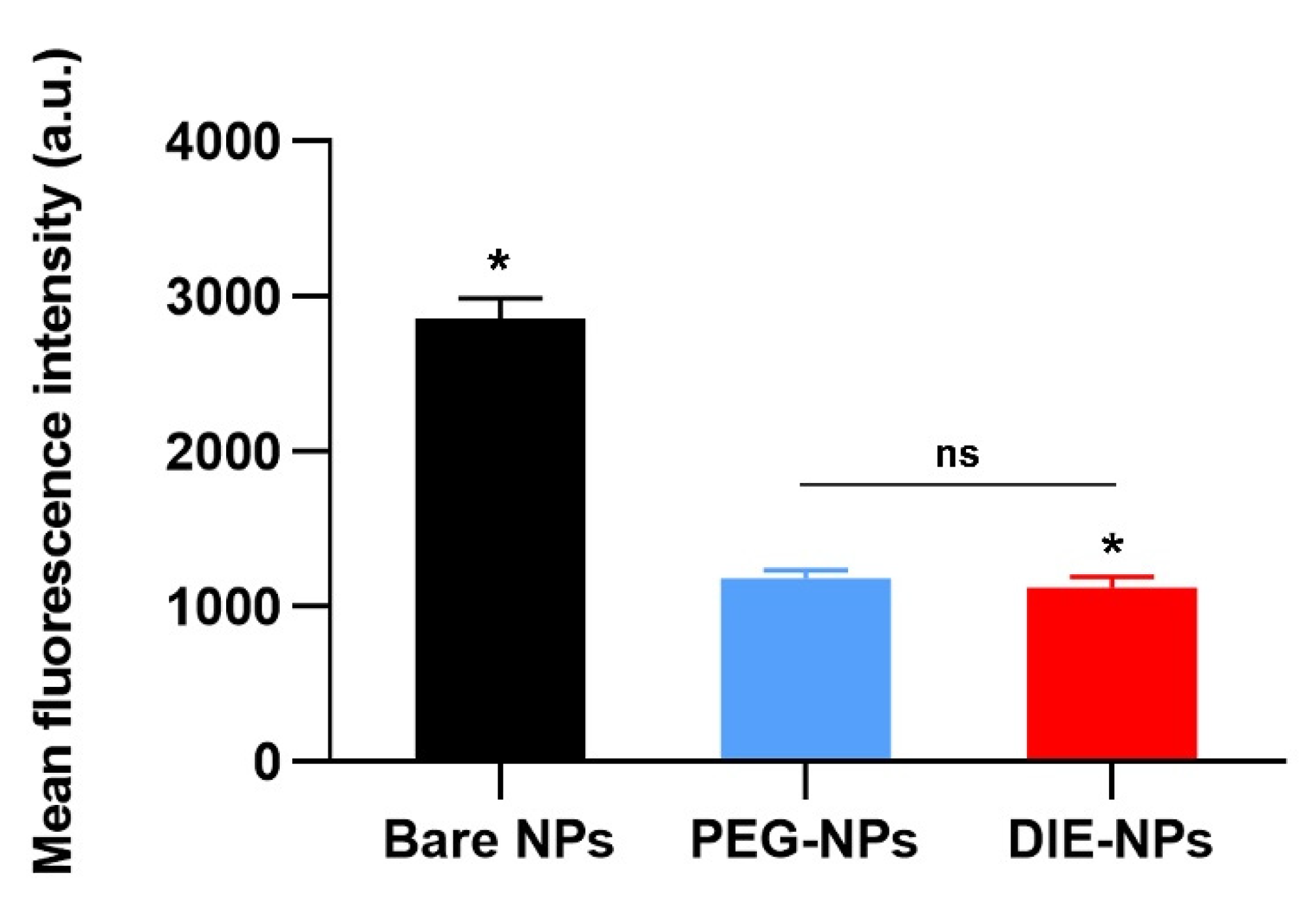
3.3. Quantification of Ligand Surface Coverage
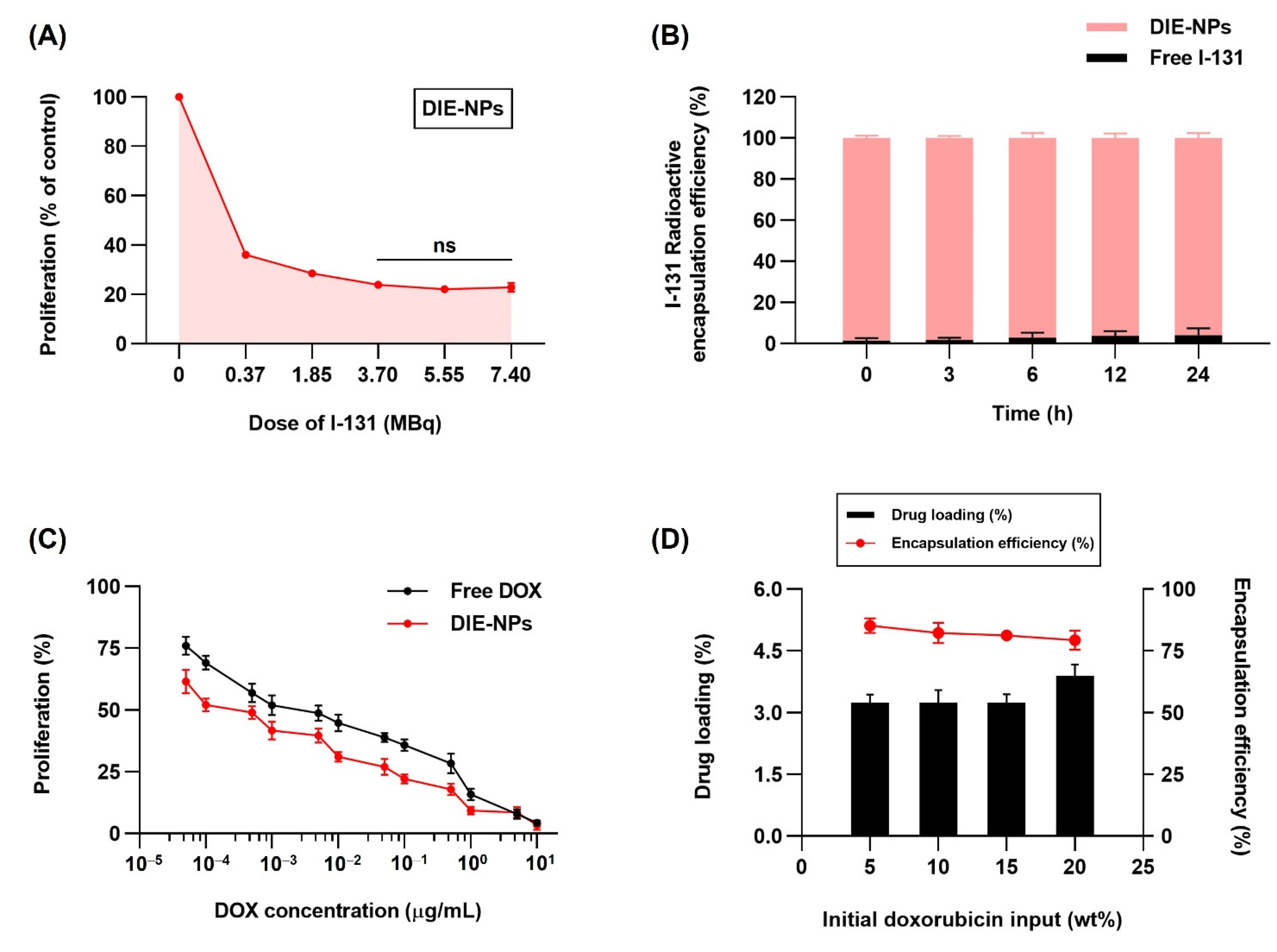
3.4. In Vitro Two-Dimensional (2D) Therapeutic I-131 Dose Optimization
3.5. Radioactive I-131 Encapsulation Efficiency
3.6. In Vitro Therapeutic DOX Optimization, Drug Loading and Encapsulation Efficiency of the DIE-NPs
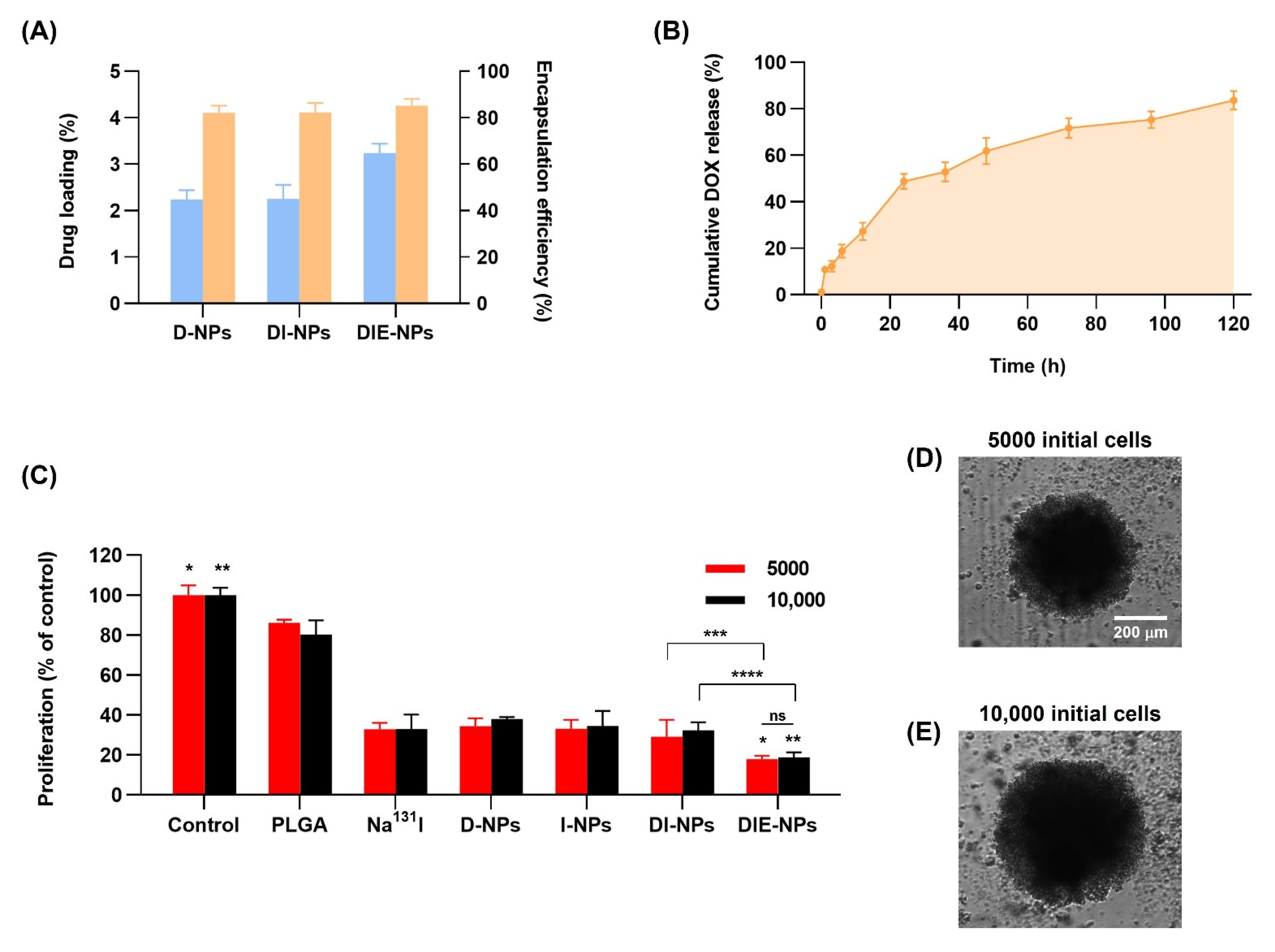
3.7. Drug Loading and Encapsulation Efficiency of Nanoparticles
3.8. Cumulative Amount of DOX Release
3.9. Three-Dimensional (3D) Human MG-63 Tumor Spheroid In Vitro Cytotoxicity
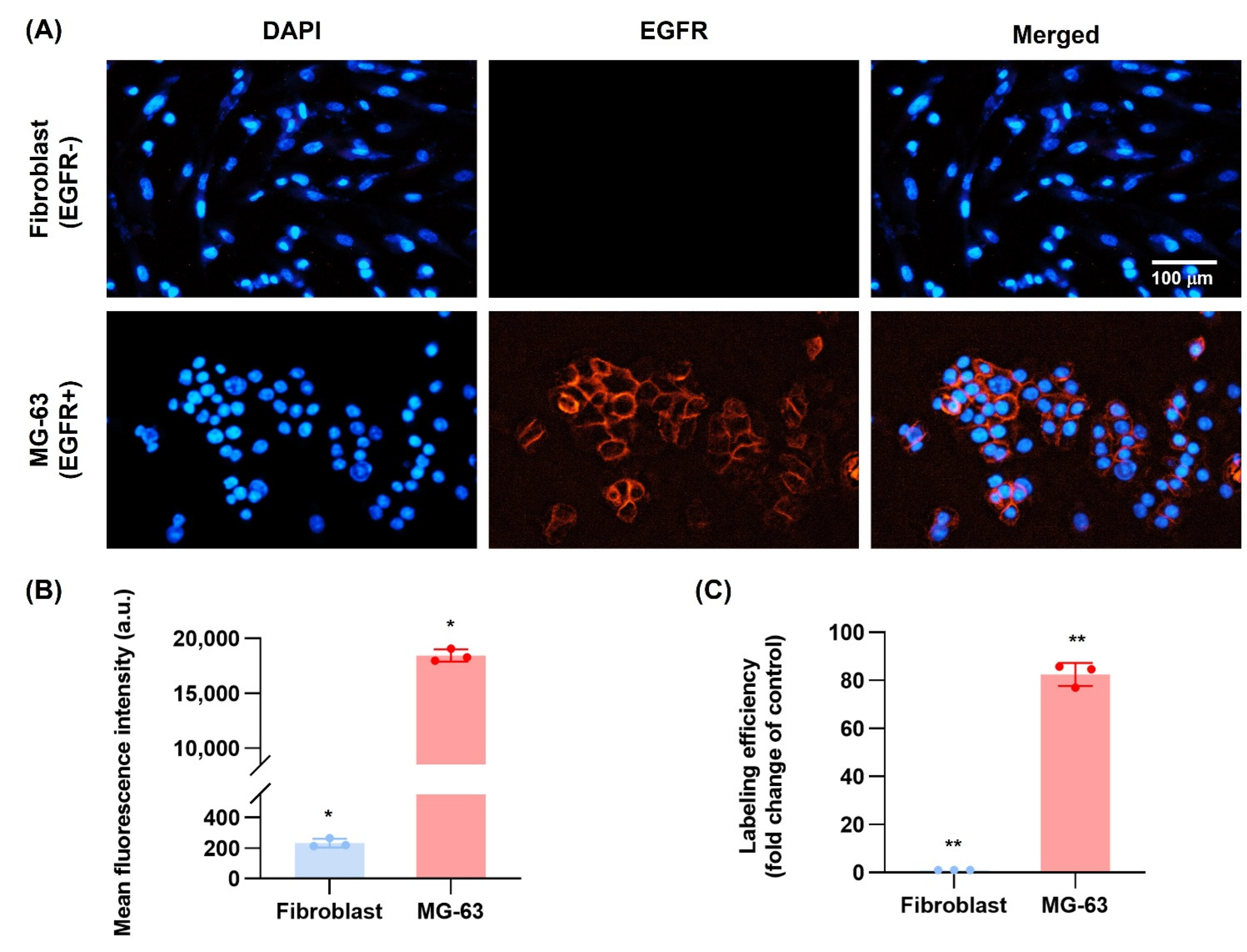
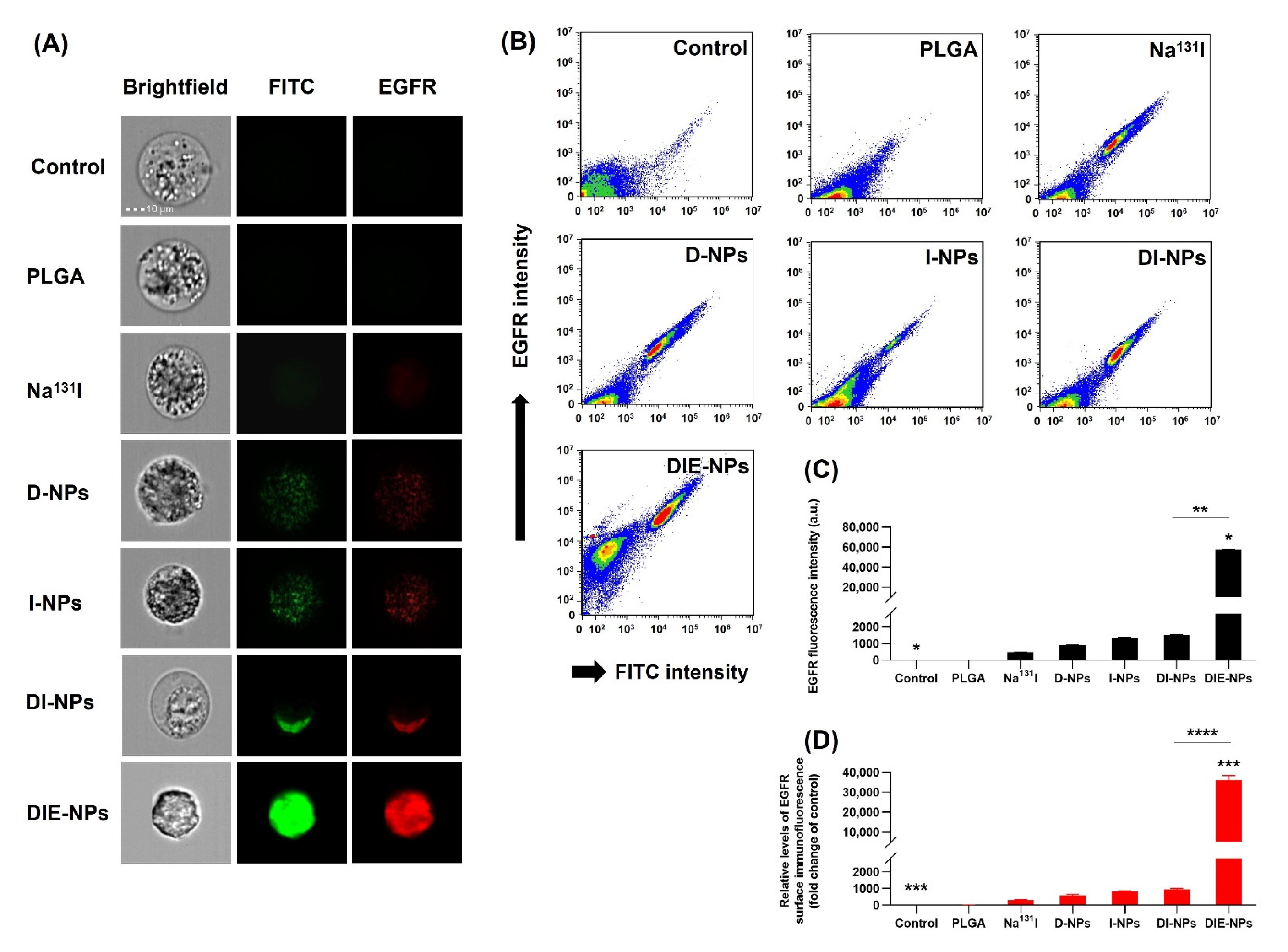
3.10. EGFR Antibody Cellular Binding/Uptake In Vitro Targeting Efficiency
3.11. Cytotoxicity Assay of 3D Spheroid MG-63 Osteosarcoma Cells, Live/Dead Cell Assay Method Using LionHeart FX Automated Live Cell Imager
3.12. Three-Dimensional (3D) Human MG-63 Tumor Spheroid DIE-NPs Penetration
3.13. Cell Cycle
3.14. Blood Compatibility of Samples: Hemolysis Assay
3.15. Cellular Uptake by Macrophage Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vasquez, L.; Tarrillo, F.; Oscanoa, M.; Maza, I.; Geronimo, J.; Paredes, G.; Silva, J.M.; Sialer, L. Analysis of Prognostic Factors in High-Grade Osteosarcoma of the Extremities in Children: A 15-Year Single-Institution Experience. Front. Oncol. 2016, 6, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ries, L.A.; Harkins, D.; Krapcho, M.; Mariotto, A.; Miller, B.A.; Feuer, E.J.; Clegg, L.X.; Eisner, M.P.; Horner, M.J.; Howlader, N.; et al. SEER Cancer Statistics Review 1975–2017; National Cancer Institute: Bethesda, MD, USA, 2020. Available online: https://seer.cancer.gov/csr/1975_2017/ (accessed on 24 June 2022).
- Schwartz, C.L.; Gorlick, R.; Teot, L.; Krailo, M.; Chen, Z.; Goorin, A.; Grier, H.E.; Bernstein, M.L.; Meyers, P. Multiple Drug Resistance in Osteogenic Sarcoma: INT0133 From the Children’s Oncology Group. J. Clin. Oncol. 2007, 25, 2057–2062. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Zhao, J.; Bai, J.; Shen, H.; Zhang, B.; Deng, L.; Sun, C.; Liu, Y.; Zhang, J.; Zheng, J. Risk and clinicopathological features of osteosarcoma metastasis to the lung: A population-based study. J. Bone Oncol. 2019, 16, 100230. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, W. New molecular insights into osteosarcoma targeted therapy. Curr. Opin. Oncol. 2013, 25, 398–406. [Google Scholar] [CrossRef]
- Hattinger, C.M.; Fanelli, M.; Tavanti, E.; Vella, S.; Riganti, C.; Picci, P.; Serra, M. Doxorubicin-Resistant Osteosarcoma: Novel Therapeutic Approaches in Sight? Future Oncol. 2017, 13, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 2013, 65, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.A.; Hawkins, C.J. Recent and Ongoing Research into Metastatic Osteosarcoma Treatments. Int. J. Mol. Sci. 2022, 23, 3817. [Google Scholar] [CrossRef]
- Luetke, A.; Meyers, P.A.; Lewis, I.; Juergens, H. Osteosarcoma treatment—Where do we stand? A state of the art review. Cancer Treat. Rev. 2014, 40, 523–532. [Google Scholar] [CrossRef]
- Heldin, C.-H.; Rubin, K.; Pietras, K.; Östman, A. High interstitial fluid pressure—An obstacle in cancer therapy. Nat. Cancer 2004, 4, 806–813. [Google Scholar] [CrossRef]
- Trédan, O.; Galmarini, C.M.; Patel, K.; Tannock, I.F. Drug Resistance and the Solid Tumor Microenvironment. J. Natl. Cancer Inst. 2007, 99, 1441–1454. [Google Scholar] [CrossRef]
- Kadkhoda, J.; Akrami-Hasan-Kohal, M.; Tohidkia, M.R.; Khaledi, S.; Davaran, S.; Aghanejad, A. Advances in antibody nanoconjugates for diagnosis and therapy: A review of recent studies and trends. Int. J. Biol. Macromol. 2021, 185, 664–678. [Google Scholar] [CrossRef] [PubMed]
- Senapati, S.; Mahanta, A.K.; Kumar, S.; Maiti, P. Controlled Drug Delivery Vehicles for Cancer Treatment and Their Performance. Signal Transduct. Target. Ther. 2018, 3, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.-Y.; Hu, H.-Z.; Qing, X.-C.; Zhang, Z.-C.; Shao, Z.-W. Recent Advances of Drug Delivery Nanocarriers in Osteosarcoma Treatment. J. Cancer 2020, 11, 69. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Zeng, Y.; Qi, X.; Chen, Y.; Ge, Z.; Jiang, Z.; Zhang, X.; Dong, Y.; Chen, H.; Yu, Z. Targeted Salinomycin Delivery with EGFR and CD133 Aptamers Based Dual-Ligand Lipid-Polymer Nanoparticles to Both Osteosarcoma Cells and Cancer Stem Cells. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 2115–2127. [Google Scholar] [CrossRef]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Y.; Wang, Y.; Liu, Z.; Sun, Y.; Yao, Y.; Yu, W.; Shen, Z. MAT2B Promotes Proliferation and Inhibits Apoptosis in Osteosarcoma by Targeting Epidermal Growth Factor Receptor and Proliferating Cell Nuclear Antigen. Int. J. Oncol. 2019, 54, 2019–2029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sgouros, G.; Bodei, L.; McDevitt, M.R.; Nedrow, J.R. Radiopharmaceutical Therapy in Cancer: Clinical Advances and Challenges. Nat. Rev. Drug Discov. 2020, 19, 589–608. [Google Scholar] [CrossRef]
- Sou, K.; Goins, B.; Leland, M.M.; Tsuchida, E.; Phillips, W.T. Bone Marrow-Targeted Liposomal Carriers: A Feasibility Study in Nonhuman Primates. Nanomedicine 2010, 5, 41–49. [Google Scholar] [CrossRef]
- Te Beek, E.T.; Burggraaf, J.; Teunissen, J.J.; Vriens, D. Clinical Pharmacology of Radiotheranostics in Oncology. Clin. Pharmacol. Ther. 2022. [Google Scholar] [CrossRef]
- Nag, O.K.; Delehanty, J.B. Active Cellular and Subcellular Targeting of Nanoparticles for Drug Delivery. Pharmaceutics 2019, 11, 543. [Google Scholar] [CrossRef]
- Vauthier, C.; Schmidt, C.; Couvreur, P. Measurement of the Density of Polymeric Nanoparticulate Drug Carriers by Isopycnic Centrifugation. J. Nanopart. Res. 1999, 1, 411–418. [Google Scholar] [CrossRef]
- Marshall, S.K.; Panrak, Y.; Makchuchit, N.; Jaroenpakdee, P.; Saelim, B.; Taweesap, M.; Pachana, V. Anti-EpCAM Functionalized I-131 Radiolabeled Biomimetic Nanocarrier Sodium/Iodide-Symporter-Mediated Breast-Cancer Treatment. Bioengineering 2022, 9, 294. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Li, C.; Baguley, B.C.; Zhou, F.; Zhou, W.; Shaw, J.P.; Wang, Z.; Wu, Z.; Liu, J. Optimization of the Formation of Embedded Multicellular Spheroids of MCF-7 Cells: How to Reliably Produce a Biomimetic 3D Model. Anal. Biochem. 2016, 515, 47–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. The International Pharmacopoeia. WHO Drug Inf. 2013, 27, 35–40. [Google Scholar]
- Hosseinimehr, S.J.; Shafaghati, N.; Hedayati, M. Genotoxicity Induced by Iodine-131 in Human Cultured Lymphocytes. Interdiscip. Toxicol. 2013, 6, 74. [Google Scholar] [CrossRef] [PubMed]
- Aryal, S.; Hu, C.-M.J.; Fang, R.H.; Dehaini, D.; Carpenter, C.; Zhang, D.-E.; Zhang, L. Erythrocyte Membrane-Cloaked Polymeric Nanoparticles for Controlled Drug Loading and Release. Nanomedicine 2013, 8, 1271–1280. [Google Scholar] [CrossRef] [Green Version]
- Hughes, D.P.; Thomas, D.G.; Giordano, T.J.; Baker, L.H.; McDonagh, K.T. Cell Surface Expression of Epidermal Growth Factor Receptor and Her-2 with Nuclear Expression of Her-4 in Primary Osteosarcoma. Cancer Res. 2004, 64, 2047–2053. [Google Scholar] [CrossRef] [Green Version]
- Chow, T.; Wutami, I.; Lucarelli, E.; Choong, P.F.; Duchi, S.; Di Bella, C. Creating in Vitro Three-Dimensional Tumor Models: A Guide for the Biofabrication of a Primary Osteosarcoma Model. Tissue Eng. Part B Rev. 2021, 27, 514–529. [Google Scholar] [CrossRef]
- Dobrovolskaia, M.A.; Clogston, J.D.; Neun, B.W.; Hall, J.B.; Patri, A.K.; McNeil, S.E. Method for Analysis of Nanoparticle Hemolytic Properties in Vitro. Nano Lett. 2008, 8, 2180–2187. [Google Scholar] [CrossRef]
- Amin, K.; Dannenfelser, R. In Vitro Hemolysis: Guidance for the Pharmaceutical Scientist. J. Pharm. Sci. 2006, 95, 1173–1176. [Google Scholar] [CrossRef]
- Krzyzaniak, J.F.; Raymond, D.M.; Yalkowsky, S.H. Lysis of Human Red Blood Cells 1: Effect of Contact Time on Water Induced Hemolysis. PDA J. Pharm. Sci. Technol. 1996, 50, 223–226. [Google Scholar] [PubMed]
- Hu, C.-M.J.; Fang, R.H.; Luk, B.T.; Chen, K.N.; Carpenter, C.; Gao, W.; Zhang, K.; Zhang, L. ‘Marker-of-Self’Functionalization of Nanoscale Particles through a Top-down Cellular Membrane Coating Approach. Nanoscale 2013, 5, 2664–2668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Jin, K.; Luo, M.; Wang, X.; Zhu, X.; Liu, X.; Jiang, T.; Zhang, Q.; Wang, S.; Pang, Z. Size Dependency of Circulation and Biodistribution of Biomimetic Nanoparticles: Red Blood Cell Membrane-Coated Nanoparticles. Cells 2019, 8, 881. [Google Scholar] [CrossRef] [Green Version]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-Co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef]
- Li, W.; Sun, D.; Li, N.; Shen, Y.; Hu, Y.; Tan, J. Therapy of Cervical Cancer Using 131I-Labeled Nanoparticles. J. Int. Med. Res. 2018, 46, 2359–2370. [Google Scholar] [CrossRef] [Green Version]
- Pellico, J.; Gawne, P.J.; de Rosales, R.T. Radiolabelling of Nanomaterials for Medical Imaging and Therapy. Chem. Soc. Rev. 2021, 50, 3355–3423. [Google Scholar] [CrossRef] [PubMed]
- Thorne, B.A.; Plowman, G.D. The Heparin-Binding Domain of Amphiregulin Necessitates the Precursor pro-Region for Growth Factor Secretion. Mol. Cell. Biol. 1994, 14, 1635–1646. [Google Scholar]
- Khalili, L.; Dehghan, G.; Sheibani, N.; Khataee, A. Smart Active-Targeting of Lipid-Polymer Hybrid Nanoparticles for Therapeutic Applications: Recent Advances and Challenges. Int. J. Biol. Macromol. 2022, 213, 166–194. [Google Scholar] [CrossRef]
- Wang, R.; Xiao, R.; Zeng, Z.; Xu, L.; Wang, J. Application of Poly (Ethylene Glycol)–Distearoylphosphatidylethanolamine (PEG-DSPE) Block Copolymers and Their Derivatives as Nanomaterials in Drug Delivery. Int. J. Nanomed. 2012, 7, 4185. [Google Scholar]
- Yu, M.K.; Park, J.; Jon, S. Targeting Strategies for Multifunctional Nanoparticles in Cancer Imaging and Therapy. Theranostics 2012, 2, 3. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.-T.; Lee, J.-Y.; Kim, D.-D.; Yoon, I.-S.; Cho, H.-J. Recent Progress in the Development of Poly (Lactic-Co-Glycolic Acid)-Based Nanostructures for Cancer Imaging and Therapy. Pharmaceutics 2019, 11, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Li, Y.; Tan, X.; Rao, R.; Ren, Y.; Liu, L.; Yang, X.; Liu, W. Multifunctional Hybrid Micelles with Tunable Active Targeting and Acid/Phosphatase-Stimulated Drug Release for Enhanced Tumor Suppression. Biomaterials 2018, 157, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a Strategy for Improving Nanoparticle-Based Drug and Gene Delivery. Adv. Drug Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef]
- Chen, S.; Yang, K.; Tuguntaev, R.G.; Mozhi, A.; Zhang, J.; Wang, P.C.; Liang, X.-J. Targeting Tumor Microenvironment with PEG-Based Amphiphilic Nanoparticles to Overcome Chemoresistance. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 269–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Li, J.; Lykotrafitis, G.; Bao, G.; Suresh, S. Size-dependent Endocytosis of Nanoparticles. Adv. Mater. 2009, 21, 419–424. [Google Scholar] [CrossRef] [Green Version]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Hasanzadeh Davarani, F.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M. Impact of Particle Size and Polydispersity Index on the Clinical Applications of Lipidic Nanocarrier Systems. Pharmaceutics 2018, 10, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tantra, R.; Schulze, P.; Quincey, P. Effect of Nanoparticle Concentration on Zeta-Potential Measurement Results and Reproducibility. Particuology 2010, 8, 279–285. [Google Scholar] [CrossRef]
- Shao, X.; Wei, X.; Song, X.; Hao, L.; Cai, X.; Zhang, Z.; Peng, Q.; Lin, Y. Independent Effect of Polymeric Nanoparticle Zeta Potential/Surface Charge, on Their Cytotoxicity and Affinity to Cells. Cell Prolif. 2015, 48, 465–474. [Google Scholar] [CrossRef]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of Nanoparticle Design for Overcoming Biological Barriers to Drug Delivery. Nat. Biotechnol. 2015, 33, 941. [Google Scholar] [CrossRef]
- He, M.; Jiang, L.; Ren, Z.; Wang, G.; Wang, J. Noscapine Targets EGFRp-Tyr1068 to Suppress the Proliferation and Invasion of MG63 Cells. Sci. Rep. 2016, 6, 37062. [Google Scholar] [CrossRef] [Green Version]
- Gomez, G.G.; Wykosky, J.; Zanca, C.; Furnari, F.B.; Cavenee, W.K. Therapeutic Resistance in Cancer: MicroRNA Regulation of EGFR Signaling Networks. Cancer Biol. Med. 2013, 10, 192. [Google Scholar] [PubMed]
- Jones, S.; King, P.J.; Antonescu, C.N.; Sugiyama, M.G.; Bhamra, A.; Surinova, S.; Angelopoulos, N.; Kragh, M.; Pedersen, M.W.; Hartley, J.A. Targeting of EGFR by a Combination of Antibodies Mediates Unconventional EGFR Trafficking and Degradation. Sci. Rep. 2020, 10, 663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Tian, X.; Zhang, W.; Zhang, Z.; Lazcano, R.; Hingorani, P.; Roth, M.E.; Gill, J.D.; Harrison, D.J.; Xu, Z. Comprehensive Surfaceome Profiling to Identify and Validate Novel Cell-Surface Targets in Osteosarcoma. Mol. Cancer Ther. 2022, 21, 903–913. [Google Scholar] [CrossRef]
- Wang, S.; Wei, H.; Huang, Z.; Wang, X.; Shen, R.; Wu, Z.; Lin, J. Epidermal Growth Factor Receptor Promotes Tumor Progression and Contributes to Gemcitabine Resistance in Osteosarcoma. Acta Biochim. Biophys. Sin. 2021, 53, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Messerschmitt, P.J.; Rettew, A.N.; Brookover, R.E.; Garcia, R.M.; Getty, P.J.; Greenfield, E.M. Specific Tyrosine Kinase Inhibitors Regulate Human Osteosarcoma Cells in Vitro. Clin. Orthop. Relat. Res. 2008, 466, 2168–2175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M. Caspase-Dependent Immunogenicity of Doxorubicin-Induced Tumor Cell Death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Bailly, C.; Thuru, X.; Quesnel, B. Combined Cytotoxic Chemotherapy and Immunotherapy of Cancer: Modern Times. NAR Cancer 2020, 2, zcaa002. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wei, X.; Zhang, C.; Zhang, F.; Liang, W. Nanoparticle Delivery Strategies to Target Doxorubicin to Tumor Cells and Reduce Side Effects. Ther. Deliv. 2010, 1, 273–287. [Google Scholar] [CrossRef]
- Kim, J.; Choi, Y.; Yang, S.; Lee, J.; Choi, J.; Moon, Y.; Kim, J.; Shim, N.; Cho, H.; Shim, M.K. Sustained and Long-Term Release of Doxorubicin from PLGA Nanoparticles for Eliciting Anti-Tumor Immune Responses. Pharmaceutics 2022, 14, 474. [Google Scholar] [CrossRef]
- Rybinski, B.; Yun, K. Addressing Intra-Tumoral Heterogeneity and Therapy Resistance. Oncotarget 2016, 7, 72322. [Google Scholar] [CrossRef] [Green Version]
- Kumskova, N.; Ermolenko, Y.; Osipova, N.; Semyonkin, A.; Kildeeva, N.; Gorshkova, M.; Kovalskii, A.; Kovshova, T.; Tarasov, V.; Kreuter, J. How Subtle Differences in Polymer Molecular Weight Affect Doxorubicin-Loaded PLGA Nanoparticles Degradation and Drug Release. J. Microencapsul. 2020, 37, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Engineer, C.; Parikh, J.; Raval, A. Review on Hydrolytic Degradation Behavior of Biodegradable Polymers from Controlled Drug Delivery System. Trends Biomater. Artif. Organs 2011, 25, 79–85. [Google Scholar]
- Park, T.G. Degradation of Poly (Lactic-Co-Glycolic Acid) Microspheres: Effect of Copolymer Composition. Biomaterials 1995, 16, 1123–1130. [Google Scholar] [CrossRef]
- Wang, J.; Chen, J.-S.; Zong, J.-Y.; Zhao, D.; Li, F.; Zhuo, R.-X.; Cheng, S.-X. Calcium Carbonate/Carboxymethyl Chitosan Hybrid Microspheres and Nanospheres for Drug Delivery. J. Phys. Chem. C 2010, 114, 18940–18945. [Google Scholar] [CrossRef]
- Kemp, J.A.; Shim, M.S.; Heo, C.Y.; Kwon, Y.J. “Combo” Nanomedicine: Co-Delivery of Multi-Modal Therapeutics for Efficient, Targeted, and Safe Cancer Therapy. Adv. Drug Deliv. Rev. 2016, 98, 3–18. [Google Scholar] [CrossRef] [Green Version]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.-L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N. Calreticulin Exposure Dictates the Immunogenicity of Cancer Cell Death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Ho, W.; Zhang, X.; Bertrand, N.; Farokhzad, O. Cancer Nanomedicine: From Targeted Delivery to Combination Therapy. Trends Mol. Med. 2015, 21, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Silberstein, E.B. Radioiodine: The Classic Theranostic Agent; Elsevier: Amsterdam, The Netherlands, 2012; Volume 42, pp. 164–170. [Google Scholar]
- Gao, J.; Fang, L.; Sun, D.; Shen, Y.; Hu, Y.; Li, N.; Chang, J.; Li, W.; Tan, J. 131I-Labeled and DOX-Loaded Multifunctional Nanoliposomes for Radiotherapy and Chemotherapy in Brain Gliomas. Brain Res. 2020, 1739, 145218. [Google Scholar] [CrossRef]
- Li, W.; Liu, Z.; Li, C.; Li, N.; Fang, L.; Chang, J.; Tan, J. Radionuclide Therapy Using 131I-Labeled Anti-Epidermal Growth Factor Receptor-Targeted Nanoparticles Suppresses Cancer Cell Growth Caused by EGFR Overexpression. J. Cancer Res. Clin. Oncol. 2016, 142, 619–632. [Google Scholar] [CrossRef]
- Pozzi, S.; Scomparin, A.; Dangoor, S.I.; Ajamil, D.R.; Ofek, P.; Neufeld, L.; Krivitsky, A.; Vaskovich-Koubi, D.; Kleiner, R.; Dey, P. Meet Me Halfway: Are in Vitro 3D Cancer Models on the Way to Replace in Vivo Models for Nanomedicine Development? Adv. Drug Deliv. Rev. 2021, 175, 113760. [Google Scholar] [CrossRef]
- Duval, K.; Grover, H.; Han, L.-H.; Mou, Y.; Pegoraro, A.F.; Fredberg, J.; Chen, Z. Modeling Physiological Events in 2D vs. 3D Cell Culture. Physiology 2017, 32, 266–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, J.; Sarmento, B.; Pereira, C.L. Osteosarcoma Tumor Microenvironment: The Key for the Successful Development of Biologically Relevant 3D in Vitro Models. Vitr. Models 2022, 1, 5–27. [Google Scholar] [CrossRef]
- Nunes, A.S.; Barros, A.S.; Costa, E.C.; Moreira, A.F.; Correia, I.J. 3D Tumor Spheroids as in Vitro Models to Mimic in Vivo Human Solid Tumors Resistance to Therapeutic Drugs. Biotechnol. Bioeng. 2019, 116, 206–226. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Yang, Y.; Yuan, K.; Yang, S.; Zhang, S.; Li, H.; Tang, T. Multi-Omics Analysis Based on 3D-Bioprinted Models Innovates Therapeutic Target Discovery of Osteosarcoma. Bioact. Mater. 2022, 18, 459–470. [Google Scholar] [CrossRef]
- Nath, S.; Devi, G.R. Three-Dimensional Culture Systems in Cancer Research: Focus on Tumor Spheroid Model. Pharmacol. Ther. 2016, 163, 94–108. [Google Scholar] [CrossRef] [Green Version]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin Pathways: Pharmacodynamics and Adverse Effects. Pharm. Genom. 2011, 21, 440. [Google Scholar] [CrossRef]
- Kim, H.-S.; Lee, Y.-S.; Kim, D.-K. Doxorubicin Exerts Cytotoxic Effects through Cell Cycle Arrest and Fas-Mediated Cell Death. Pharmacology 2009, 84, 300–309. [Google Scholar] [CrossRef]
- Zhao, L.; Pang, A. Iodine-131 Treatment of Thyroid Cancer Cells Leads to Suppression of Cell Proliferation Followed by Induction of Cell Apoptosis and Cell Cycle Arrest by Regulation of B-Cell Translocation Gene 2-Mediated JNK/NF-ΚB Pathways. Braz. J. Med. Biol. Res. 2017, 50, e5933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krzyzaniak, J.F.; Yalkowsky, S.H. Lysis of Human Red Blood Cells 3: Effect of Contact Time on Surfactant-Induced Hemolysis. PDA J. Pharm. Sci. Technol. 1998, 52, 66–69. [Google Scholar]
- Kim, D.; El-Shall, H.; Dennis, D.; Morey, T. Interaction of PLGA Nanoparticles with Human Blood Constituents. Colloids Surf. B Biointerfaces 2005, 40, 83–91. [Google Scholar] [CrossRef]
- Partikel, K.; Korte, R.; Stein, N.C.; Mulac, D.; Herrmann, F.C.; Humpf, H.-U.; Langer, K. Effect of Nanoparticle Size and PEGylation on the Protein Corona of PLGA Nanoparticles. Eur. J. Pharm. Biopharm. 2019, 141, 70–80. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Ligand (µg) | Surface Density (nmol/mg NP) | Surface Coverage (nmol/cm2 NP) | Molecules/mg NP | Molecules/NP |
|---|---|---|---|---|---|
| DSPE−PEG(2000) −COOH | 25 | 9 ± 2 | 7 ± 3 | 1015 | 36 ± 5 |
| 50 | 19 ± 5 | 15 ± 4 | 1016 | 75 ± 8 | |
| 100 | 34 ± 6 | 33 ± 7 | 1016 | 148 ± 18 | |
| 250 | 92 ± 11 | 77 ± 8 | 1016 | 363 ± 17 | |
| 500 | 180 ± 23 | 150 ± 17 | 1017 | 719 ± 11 | |
| 1000 | 360 ± 38 | 300 ± 29 | 1017 | 1439 ± 59 | |
| anti-human EGFR antibody | 1 | 10−3 | 10−3 | 1012 | 1125 ± 23 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marshall, S.K.; Saelim, B.; Taweesap, M.; Pachana, V.; Panrak, Y.; Makchuchit, N.; Jaroenpakdee, P. Anti-EGFR Targeted Multifunctional I-131 Radio-Nanotherapeutic for Treating Osteosarcoma: In Vitro 3D Tumor Spheroid Model. Nanomaterials 2022, 12, 3517. https://0-doi-org.brum.beds.ac.uk/10.3390/nano12193517
Marshall SK, Saelim B, Taweesap M, Pachana V, Panrak Y, Makchuchit N, Jaroenpakdee P. Anti-EGFR Targeted Multifunctional I-131 Radio-Nanotherapeutic for Treating Osteosarcoma: In Vitro 3D Tumor Spheroid Model. Nanomaterials. 2022; 12(19):3517. https://0-doi-org.brum.beds.ac.uk/10.3390/nano12193517
Chicago/Turabian StyleMarshall, Suphalak Khamruang, Boonyisa Saelim, Maneerat Taweesap, Verachai Pachana, Yada Panrak, Naritsara Makchuchit, and Passara Jaroenpakdee. 2022. "Anti-EGFR Targeted Multifunctional I-131 Radio-Nanotherapeutic for Treating Osteosarcoma: In Vitro 3D Tumor Spheroid Model" Nanomaterials 12, no. 19: 3517. https://0-doi-org.brum.beds.ac.uk/10.3390/nano12193517