Genome-Wide Technologies to Study RNA–Chromatin Interactions


Laboratory for Transcriptome Technology, RIKEN Center for Integrative Medical Sciences, Yokohama, Kanagawa 230-0045, Japan
*
Authors to whom correspondence should be addressed.
Non-Coding RNA 2020, 6(2), 20; https://0-doi-org.brum.beds.ac.uk/10.3390/ncrna6020020
Submission received: 3 May 2020
/
Revised: 23 May 2020
/
Accepted: 25 May 2020
/
Published: 27 May 2020
(This article belongs to the Special Issue Non-Coding RNA and Intracellular Structures)
Abstract
:An increasing number of studies have revealed that long non-coding RNAs (lncRNAs) play important roles in gene regulation and nuclear organization. Although the mechanisms are still largely unknown, many lncRNAs have been shown to interact with chromatin. Thus, one approach to understanding the function of these lncRNAs is to identify their sites of genomic interaction. Hybridization capture methods using oligonucleotide probes have been used for years to study chromatin-associated RNA. Recently, several groups have developed novel methods based on proximity ligation to investigate RNA–chromatin interactions at a genome-wide scale. This review discusses these technologies and highlights their advantages and disadvantages for the consideration of potential users.
1. Introduction
Chromatin-associated RNAs (chrRNAs) have important roles in gene regulation, genomic organization, and chromatin structure maintenance. Although the majority of chrRNAs are nascent transcripts attached to chromatin by RNA polymerase II (Pol II) [1], chrRNAs also include many long non-coding RNAs (lncRNAs) that function in specific regions of the genome. The most famous and well-studied example is XIST lncRNA, responsible for X chromosome inactivation in mammals [2,3]. XIST lncRNA is expressed from only one of the two X chromosomes. It coats the X chromosome and recruits multiple complexes including epigenetic modifiers that establish gene silencing and formation of facultative heterochromatin.
ChrRNAs (or lncRNAs) are generally categorized into two groups based on their mode of action. LncRNAs that regulate the expression of nearby genes are defined as cis-acting. However, it is often hard to distinguish the mature RNA molecules that regulate gene expression from transcribed lncRNAs involved in gene regulation independently of their RNA sequence. For example, the Airn lncRNA gene, which overlaps with Igf2r in antisense orientation in the mammalian genome, is required for silencing the paternal genes in the Igf2r cluster. It has been reported that transcription through the Igf2r promoter, rather than the sequence of the mature Airn RNA, leads to Igf2r silencing [4]. Subsequent study has shown that the Airn RNA molecules recruit repressive chromatin modifiers to distant, non-overlapped genes like Slc22a2, Slc22a3, and Pde10a to cause silencing [5]. Thus, the Airn lncRNA gene has two ways to repress gene expression in the Igf2r cluster. Although XIST is considered to be a cis-acting lncRNA, it can act over long genomic distances through travel of XIST RNA to distal regions of the X chromosome. Thus, the mode of action of cis-acting lncRNAs can be further subclassified.
The other group of chrRNAs, which regulate gene expression on a genome-wide scale, consists of trans-acting lncRNAs. HOTAIR is one of the trans-acting lncRNAs reported. HOTAIR, is expressed from the HOXC locus and has been reported to silence HOXD genes by recruiting the repressive chromatin modifiers PRC2, LSD1, and CoREST/REST [6,7,8]. However, later studies raised doubts regarding this initial model. It has been reported that overexpression of HOTAIR in the MDA-MD-231 breast cancer cell line represses several genes independently of PRC2 [9]. In addition, it is still controversial whether HOTAIR trans-regulates HOXD cluster genes owing to inconsistencies in the results of studies using Hotair mutant mice [10,11,12,13].
The functions of chrRNAs (or lncRNAs) have been reviewed comprehensively elsewhere [14,15]. This review focuses on recent technologies to capture RNA–chromatin interactions. Early hybridization capture methods using oligonucleotide probes (so-called “one-to-all” methods because they involve a single target RNA), such as Chromatin Isolation by RNA Purification (ChIRP), Capture Hybridization Analysis of RNA Targets (CHART), and RAP-DNA (RNA antisense purification), have been used extensively for a long time. Using these methods, XIST RNA was found to spread on the X chromosome at an initial stage of X-chromosome inactivation [16,17]. Many sites of lncRNA interaction, including MALAT1 and NEAT1, were also identified by using one-to-all methods [18]. However, these methods might be suitable only for lncRNAs for which the mechanism is known, but for which it has not been possible to identify novel regulators. The FANTOM5 consortium showed that approximately 19,000 human lncRNAs likely to be functional [19] and that ~35% of those found in human dermal fibroblasts (HDFs) are nuclear enriched [20]. In addition, in some cases these one-to-all methods suffer from high background signals [21], preventing researchers from clearly interpreting their results and producing unambiguous results. For these reasons, non-biased, high-throughput methods to non-selectively capture all nuclear RNAs are required. Recently, four groups developed related “all-to-all” methods based on the use of proximity ligation to capture RNA–chromatin interactions on a genome-wide scale. We compare these new technologies to each other and to the one-to-all methods.
2. Technologies to Capture RNA–Chromatin Interactions
2.1. One-to-All Methods
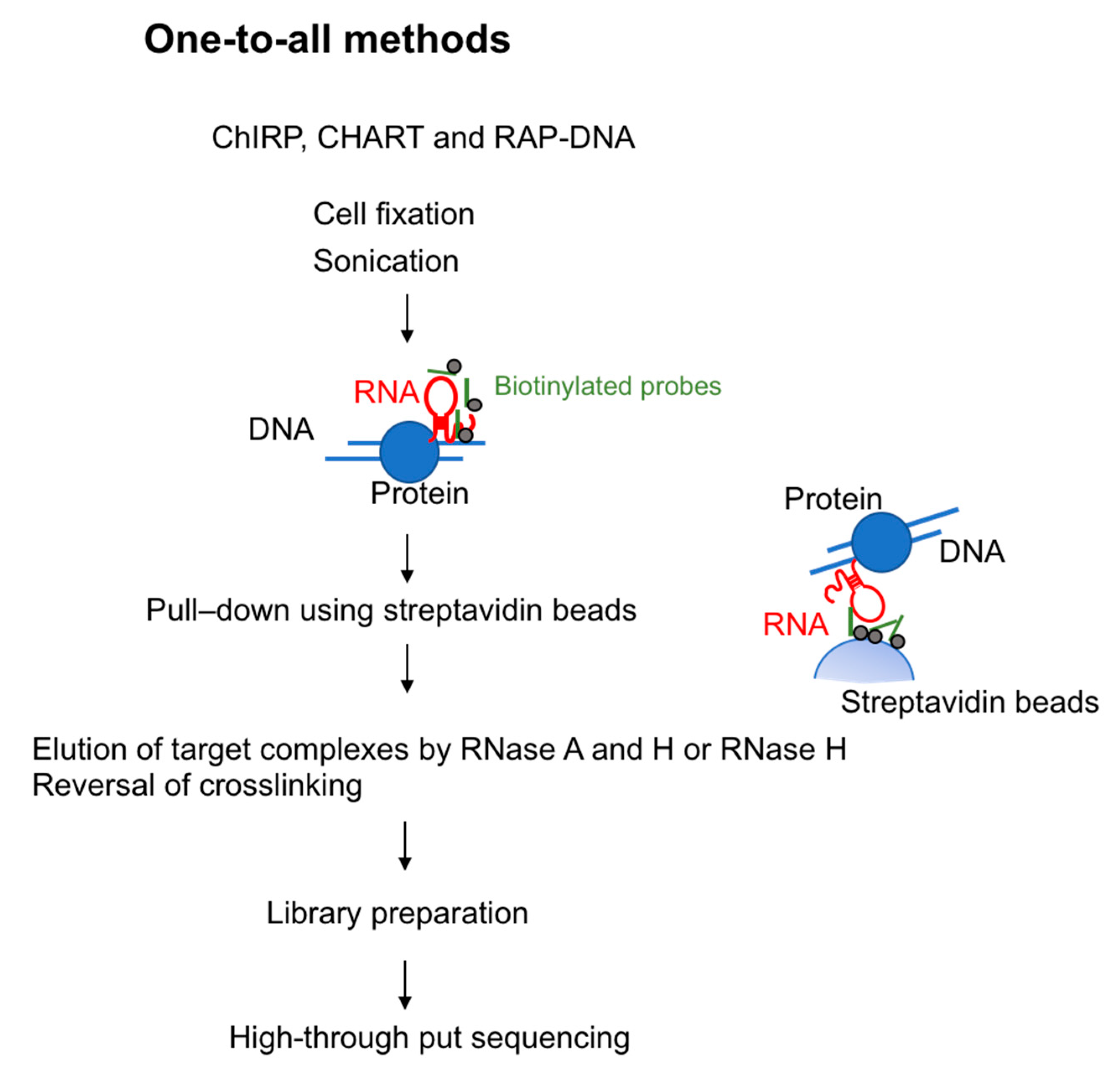
To investigate lncRNA binding sites on chromatin, in 2011 two groups developed methods using biotinylated DNA probes. These methods are termed ChIRP-seq (chromatin isolation by RNA purification) [22] and CHART-seq (capture hybridization analysis of RNA targets) [23]. In 2013, RAP-DNA (RNA antisense purification) was developed: This method uses longer antisense RNA probes to increase specificity [16]. These three methods are similar in that all use hybridization-based strategies. Specifically, they use biotin-conjugated probes that hybridize with target RNA and pull down the chromatin fraction associated with that RNA species (Figure 1).
2.1.1. Chromatin Isolation by RNA Purification (ChIRP-seq)
ChIRP-seq uses 20-mer biotin-conjugated DNA oligonucleotide probes to recognize the target lncRNA. Cells are crosslinked by glutaraldehyde or formaldehyde to stabilize RNA-chromatin interaction, sonicated for DNA fragmentation and then hybridized with the probes. To ensure specificity, multiple probes are designed and split into two separate pools (referred to as the “odd” and “even” pools) that identify common targets. After the reversal of crosslinking, genomic DNA is analyzed by high-throughput sequencing to identify genomic regions associated with the lncRNAs. To test the reliability of ChIRP-seq, the authors performed ChIRP-seq targeting roX2, a gene involved in dosage compensation in Drosophila and associated with many regions on the X chromosome in Drosophila cells [22]. They also isolated HOTAIR-associated DNA, demonstrating that HOTAIR lncRNA preferentially associated with regions containing a GA-rich DNA motif [22]. ChIRP-seq is the most commonly used one-to-all method because the DNA probes can be designed without prior knowledge of the target RNA structure or functional domain.
2.1.2. Capture Hybridization Analysis of RNA Targets (CHART-seq)
CHART-seq, developed by Simon et al., is conceptually similar to ChIRP-seq [23]. This protocol uses 25-mer desthiobiotin-conjugated DNA oligonucleotide probes. An RNase H sensitivity assay is used to identify regions in the target lncRNA that are accessible to the probes. These genomic regions are enriched followed by high-throughput DNA sequencing to identify the loci bound by the targeted lncRNAs. To minimize background, RNase H is used to elute RNA–chromatin complexes bound to the oligonucleotide probes. Any binding of genomic DNA or proteins to the probes and streptavidin magnetic beads is non-specific; thus, in theory only RNAs bound specifically to the probes would be eluted. CHART-seq was first used to investigate the function of roX2 in Drosophila [23], and was later used to study the Xist genome-wide binding regions [17].
2.1.3. RNA Antisense Purification (RAP-DNA)
RAP-DNA is another method that can be used to identify chromatin regions that interact with RNAs [16]. RAP-DNA uses a set of 120-nt antisense RNA probes that have high binding affinity to the target lncRNA (for example, pool of 120-nt probes tiled every 15 nucleotides across the entire Xist transcript (~17 kb)). The genomic regions are isolated followed by high-throughput DNA sequencing. Using RAP-DNA, the authors showed that Xist RNA is initially transferred to chromosome regions of the X chromosome that have high contact frequencies with the Xist transcription site as determined by genome-wide Chromatin Conformation Capture (Hi-C), and then gradually spreads to other accessible locations [16].
2.2. All-to-All Methods
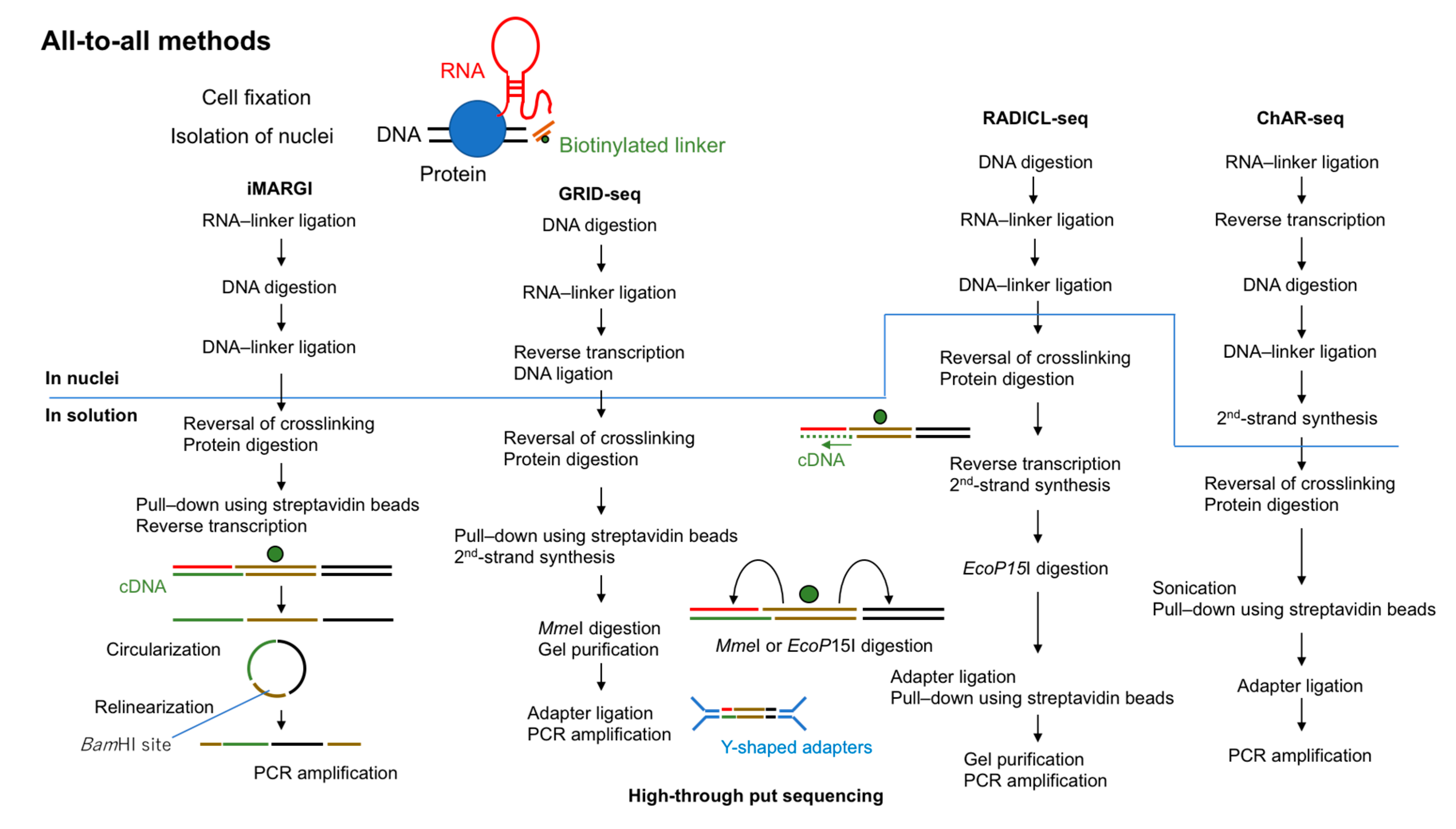
In 2017, two groups reported the first “all-to-all” methods to detect RNA–chromatin interactions genome-wide. These methods were termed MARGI (mapping RNA–genome interactions) and GRID-seq (global RNA interaction with DNA sequencing) [24,25]. In 2018, ChAR-seq (chromatin-associated RNA sequencing) was reported [26]. Very recently (in 2020), RADICL-seq (RNA and DNA interacting complexes ligated and sequenced) was developed [27]. All of these methods are based on proximity ligations using a bivalent linker that can ligate to RNA at one end and to digested DNA at the other end (Figure 2).
2.2.1. MARGI and iMARGI
MARGI was invented by Sridhar et al. [24] in 2017. A derived protocol reported in 2019, iMARGI, reduced the required number of input cells [28]. The original MARGI protocol uses approximately 4 × 108 cells fixed in 1% formaldehyde. After nuclear isolation, all proteins are biotinylated using iodoacetyl PEG2 biotin (IPB) in preparation for subsequent purification using streptavidin beads. This step can ease the numerous buffer changes required in MARGI. In iMARGI, the ligation steps are performed in situ instead of on streptavidin beads. This modification may contribute to the lower number of input cells required for iMARGI (~5 × 106) owing to less diffusion of RNA molecules or to less non-specific ligation in nuclei. Both MARGI and iMARGI use a biotinylated bivalent DNA linker to ligate RNA and DNA in formaldehyde-fixed nuclei. In MARGI, streptavidin beads require to be blocked with D-biotin before RNA ligation. Because both proteins and the linker are biotinylated this is done to prevent free linkers from binding to streptavidin beads. The 5′ end of this linker is pre-adenylated with Mth RNA ligase. T4 RNA ligase 2, truncated KQ is used for RNA ligation, which ligates the pre-adenylated 5′ end of the linker to the 3′ OH end of the RNA. The double-stranded end of the linker ligates with T4 DNA ligase to the digested and end prepared genomic DNA (digested by HaeIII for MARGI and by AluI for iMARGI). It is critical to keep track of the directionality of the linker to identify the RNA or DNA end for subsequent data analysis. As discussed later (Section 2.2.2 and Section 3.1.4), in GRID-seq and RADICL-seq the entire linker must be sequenced to identify the source of each end of the ligation product. To overcome this issue, in MARGI the ligation product is circularized after reverse transcription and then relinearized by cutting at the BamHI site in the middle of linker sequence. The halves of the digested linker are designed to be parts of the two PCR primers which are usable for the Illumina sequencing platform. The relinearized library can be directly amplified by PCR without further linker ligation.
2.2.2. GRID-seq
GRID-seq uses a biotinylated bivalent linker to ligate RNA and DNA in situ in nuclei double-fixed in formaldehyde and disuccinimidyl glutarate. Approximately 2 × 106 mammalian cells or 1 × 107 Drosophila cells are required. RNA ligation is performed, and then the ligated RNA is converted to cDNA by using one of the DNA strands in the linker as a primer. The other end of the linker is ligated to AluI-digested genomic DNA. Although GRID-seq uses a bivalent linker structurally similar to that used for MARGI, the linker for GRID-seq consists of single-stranded RNA (rather than DNA) that is pre-adenylated at the 5′ end. T4 RNA ligase 2, truncated KQ is used for RNA ligation, which can ligate pre-adenylated 5′end of DNA or RNA to the 3′ OH end of the RNA. However, the authors have not reported which linker with pre-adenylated DNA or RNA could be ligated to RNA more efficiently and why they used the linker with pre-adenylated RNA [25,29]. After reversal of crosslinking and protein digestion, linker-ligated products are selectively purified by biotin. MmeI is then used to cut 18–20 bases away from its recognition sites in the linker to generate paired-end tags (cDNA (18–20 bp)—linker-DNA—genomic DNA (18–20 bp)). After separation by PAGE, digested products of the correct size are purified from the gel and then ligated to the Y-shaped sequencing adapters, which preserve the directionality of the library for PCR amplification.
2.2.3. ChAR-seq
ChAR-seq is another method to capture RNA–chromatin interactions based on proximity ligation. First, RNA is ligated to a biotinylated bivalent linker. After second-strand synthesis using one of the DNA strands in the linker as a primer, genomic DNA is digested by DpnII and DNA ligation reactions are performed. Unlike in GRID-seq, the ligated molecules in ChAR-seq are sonicated to generate smaller fragments for library construction. ChAR-seq has been tested initially with Drosophila cells. Later the authors reported the use of human embryonic stem cells (hESCs), human retinal pigmented epithelial cells (hTERT RPE-1) and nuclei isolated from Xenopus laevis embryos in their protocol paper although they did not provide any data [30]. Random ligations may be more common in mammalian cells than in Drosophila cells because the genome size of mammals is much larger than that of Drosophila. If so, mammalian cells will require more high-throughput sequencing reads to generate usable sequence reads.
2.2.4. RADICL-seq
RADICL-seq is the latest all-to-all sequencing method and was published in 2020 [27]. Despite its conceptual similarity to other all-to-all methods, the authors pointed out several key differences that improve its performance relative to other methods for studying RNA–chromatin interactions. The authors used ~2 × 106 mammalian cells (mouse ES cells (mESCs) and mouse oligodendrocyte progenitor cells (mOPCs)) fixed in formaldehyde (1% or 2%). One end of the linker is single-stranded DNA that is pre-adenylated at the 5′ end for RNA ligation and the other end is double-stranded DNA that is ligated to genomic DNA ends digested by DNase I. Similar to the GRID-seq linker, the RADICL-seq linker contains two restriction enzyme recognition sites (in this case, for EcoP15I) that direct cutting within the RNA and DNA ends of the ligation product to generate tags of 25–27 bp each. After separation by PAGE, digested products of the correct size are purified from the gel. The Y-shaped sequencing adapters are then ligated to the biotin-purified fragments for library construction. One unique feature of RADICL-seq is the use of RNase H treatment to reduce abundant nascent transcripts. As an additional control, actinomycin D, which inhibits Pol II, is used before cell fixation to stop Pol II transcription and thus eliminate nascent transcripts.
3. Comparison of Four All-to-All Methods
All four all-to-all methods described above are conceptually similar because they are based on proximity ligations; however, they have some differences in experimental conditions and data processing. In brief, they differ in four main technical aspects: (1) number of cells required, (2) crosslinking conditions, (3) method used to digest genomic DNA, and (4) use of long reads versus reads trimmed by restriction enzyme digestion (Table 1). We discuss each of these in the following sections.
3.1. Experimental Conditions for All-to-All Methods
3.1.1. Cell Systems
MARGI and RADICL-seq have been tested only with mammalian cells [24,27,28], whereas ChAR-seq was initially tested only with Drosophila cells. GRID-seq has been tested with both Drosophila and mammalian cells. Because mESCs were analyzed using both GRID-seq and RADICL-seq, these methods can be compared directly. The original MARGI protocol uses ~4 × 108 cells (human embryonic kidney (HEK) and H9 (human ESC)), which is a fairly large number of cells compared with other methods. Development of iMARGI reduced the required cell quantity to a more treatable number, i.e., ~5 × 106 human foreskin fibroblasts (HFFs). GRID-seq used ~2 × 106 mammalian cells (human MDA-MB-231 (breast cancer cells), MM.1S (multiple myeloma cells) and mouse ESC) or 1 × 107 Drosophila cells (S2). ChAR-seq was successfully used with 1-1.5 × 108 Drosophila cells (CME-W1-cl8+ and Kc167). RADICL-seq was successfully performed with 2 × 106 mammalian cells (mouse ESCs and OPCs). Thus, GRID-seq and RADICL-seq could be achieved with only 2 × 106 cells, whereas MARGI and ChAR-seq require higher numbers of cells (Table 1). It will be possible to achieve with less number of cells after refining of their protocol. However, although sequencing libraries can be made from the cell number indicated or less for each method, the result is more directed by the endogenous expression level of the RNAs. Thus, the amount of initial material required will depend on the expression level of the target RNA to be investigated. In particular, lncRNAs tend to have lower expression than coding RNAs [31]. More over RNA extraction issue has to be dealt with even though the expression level of the RNA is high. For example, it has been reported that extensive needle sharing or heating of cell lysate during RNA extraction could improve the extraction of NEAT1 lncRNA, which forms a paraspeckle nuclear bodies [32]. When choosing the number of cells, one should take the expression level or the solubility of the RNA of interest into account to obtain enough tags for the RNA–chromatin interactions.
3.1.2. Crosslinking Conditions
Among the four methods, all except GRID-seq use formaldehyde to crosslink cells. Testing of RADICL-seq with 1% and 2% formaldehyde fixation gave highly comparable results, so 1% is now commonly used. The GRID-seq method uses a higher concentration of formaldehyde (3%) in the fixation step and additional disuccinimidyl glutarate (DSG), a strong protein–protein crosslinker. Although the authors did not show the data, they mentioned that they found more small RNAs when using 3% formaldehyde + DSG than with weaker crosslinking conditions (i.e., 1% formaldehyde) [29]. The RADICL-seq procedure also includes a condition called “non-protein mediated” in which proteinase K digestion and reversal of crosslinking are done before the RNA ligation reaction. Under this condition, RNA–DNA interactions would be captured only if the binding is direct and not mediated by proteins. Overall, the fixation step is critical for all protocols. Mechanisms of RNA localization are varied (e.g., direct interaction to DNA by R-loops and triple helixes or indirect interaction via proteins or RNA); thus, the choice of fixation method might depend on how the RNAs interact with chromatin and should be further explored during future technology development.
3.1.3. Genomic DNA Digestion
All four protocols begin with the nucleus isolation step by using a lysis buffer containing non-ionic detergent (0.2% NP-40), after which the nuclei are permeabilized by SDS. Except for RADICL-seq, which uses DNase I to digest the genomic DNA, the protocols use restriction enzymes (MARGI, HaeIII (iMARGI: AluI); GRID-seq, AluI; ChAR-seq, DpnII).
Bonetti et al. [27] found that the genomic coverage of DNA regions identified by RADICL-seq was higher than that by GRID-seq data. This could have resulted from the stronger crosslinking condition in GRID-seq and the use of AluI for DNA digestion, which introduces biases in the genome in particular for Alu elements (the enzyme is named AluI because it cleaves Alu repeats very frequently and Alu elements are located in gene-rich regions) [33]. In contrast, it has been shown that DNase I causes intrinsic DNA-cutting biases towards DNase-seq [34]. Thus, digestion conditions for each cell type should be tested before library construction. It is important to keep in mind that every digestion method has its own bias.
3.1.4. Long Reads vs. Reads Trimmed by Restriction Enzyme Digestion
After RNA and DNA ligation to the bivalent linker, the methods differ in how the ligation products are treated. In MARGI, the ligated molecules are circularized for library construction. GRID-seq and RADICL-seq use a linker that contains two recognition sites for type II and III restriction enzymes, respectively (MmeI and EcoP15I, respectively) to digest the ligated products into fixed-size fragments. In ChAR-seq, the ligation products are sonicated to obtain smaller fragments for library construction. One of advantages of iMARGI is that longer sequencing reads can be obtained. On the other hand, in GRID-seq and RADICL-seq the entire linker must be sequenced to identify which end of the sequence corresponds to RNA and which corresponds to DNA. To obtain fixed-size fragments can be sequenced, GRID-seq uses MmeI, which cuts both the RNA and DNA ends to generate tags of 18–20 bp each, and only these 18–20 bp will be used for mapping. RADICL-seq instead uses EcoP15I, which digests 25–27 bp away from its recognition sites. RADICL-seq therefore results in longer reads for mapping and about three times as many uniquely mapped reads than GRID-seq (45% vs. 14% of mappable reads to the mouse genome). The restriction-enzyme-based methods such as GRID-seq and RADICL-seq ensure that all reads contain both RNA and DNA sequences; in contrast, MARGI and ChAR-seq do not produce fixed-size fragments and require more sequencing reads to obtain comparable numbers of RNA and DNA interactions. Future technology development should consider methods to achieve even longer reads to further complete the RNA/DNA interactome with more broadly repeated sequences of the genome, including retrotransposons.
3.2. Data Processing
3.2.1. Sequencing
In the original MARGI report, the library is sequenced with 100 cycles of paired-end sequencing on an Illumina platform. The authors suggest obtaining 300 million or more read pairs [35]. For GRID-seq, a single-end 100-cycle kit was used for sequencing on an Illumina HiSeq 2500 sequencer. The authors sequenced at an average depth of 160 million reads per replicate for mouse ESC libraries [29]. For ChAR-seq, the libraries are typically sequenced with 152 single-read cycles or paired-end 150 reads. The authors sequenced more than 400 million reads for Drosophila cell libraries [26,30].
For RADICL-seq, ~120 million reads were sequenced using the 150-cycle Illumina kit [27]. Usable reads are very limited for any of these protocols after mapping. For example, in the case of ChAR-seq the authors have multiple steps to extract usable reads: (1) Removal of PCR duplicates, (2) extraction of reads containing linker adapters, (3) splitting of RNA and DNA reads, (4) alignment, and (5) removal of rRNA. At the end, only 10% of the total reads are useful for downstream analysis [26]. GRID-seq of mammalian cells returned 26–39% unique mapped read pairs, and RADICL-seq (using 1% FA) returned 13–19%, although the investigators might have used different alignment tools for each other. As a result, the required sequencing depths depend on the question researchers want to address. With MARGI, the interaction between XIST and the X chromosome and the interaction between MALAT1 and the NEAT1 gene locus could be identified with ~30 million total read pairs [29], although XIST and NEAT1 are known to be highly expressed lncRNAs.
3.2.2. Dealing with Background
One of the important challenges during data analysis is to differentiate specific from non-specific RNA–chromatin interactions. In principle, proximity-based methods can capture significant amounts of nascent transcript because transcribing or newly transcribed RNAs are very close to their transcription sites. In addition, transcribed RNAs might interact with all accessible chromatin regions in a non-specific manner; for instance, RNA molecules diffusing in the nucleus may be captured during library construction. In a test of GRID-seq, mixed Drosophila and mammalian cells were used as the input to estimate false positives; the results showed that 6.8% of the RNAs linked to Drosophila DNAs were of mammalian origin [25]. The specificity of ChAR-seq was tested using spike-in RNAs, and the results showed less than 0.5% non-specific ligation [26]. Both GRID-seq and RADICL-seq include data normalization methods for statistical analysis to correct for background. The correction method used in GRID-seq was inspired by the strategy developed for processing Hi-C data [36]. Hi-C is a high-throughput technology to measure the probability of contact between different chromosome loci; however, Hi-C data have many biases due to the ligation efficiencies of different fragment sizes, frequencies of restriction enzyme recognition sites in the genome and GC content. Thus, several normalization methods have been developed for the Hi-C method. In brief, GRID-seq uses trans-acting reads of mRNAs to estimate background and normalize the data [25]. The authors provided the pipeline for data analysis (GridTools) [29]. RADICL-seq uses the one-sided cumulative binomial test to detect significant RNA-chromatin interactions and the Benjamini–Hochberg multiple-testing correction to correct for the false discovery rate. After the test, “significant interactions” were extracted to use their downstream analysis [27]. After background correction, only 1.5% of trans-interaction reads remained in the mESC data of GRID-seq. Similarly, in RADICL-seq only 0.8% of the total significant dataset was attributed to trans interaction. Thus, for GRID-seq and RADICL-seq, the majority of trans interactions in the raw data are considered background. The MARGI and ChAR-seq methods do not include any statistical methods to deal with the background for all RNA–chromatin interactions at once. Instead, statistical tests are performed according to the biological question being addressed [24,26,28,37]. In summary, it is very important to define what to consider as background. Even when RNAs are localized to their usual site of interaction, what is their biological meaning, especially in the case of nascent RNAs? Although many interactions may happen, it is hard to distinguish which ones are meaningful in terms of gene regulation.
4. Comparison of All-to-All Methods with One-to-All Methods
The major advantage of using all-to-all methods is that we can identify de novo RNA-chromatin interactions. For example, a GRID-seq analysis provided information about the strength of enhancers, showing that RNAs interact more frequently at a super-enhancer locus than at a typical enhancer locus [25], and a RADICL-seq study demonstrated that repeat elements were differentially engaged in specific chromatin interactions in different cell types [27].
The major limitation of all-to-all methods is the requirement for a large number of sequencing reads to detect RNA species with low expression and to generate interpretable interaction data. To validate the specificity of the detected biological interactions, each all-to-all method should be compared with one or more one-to-all methods using several RNAs as positive controls. roX2 is a well-known lncRNA involved in dosage compensation in Drosophila. Interestingly, in a comparison of the raw data from ChIRP-seq, CHART-seq and GRID-seq, the percentage of total raw roX2 DNA reads interacting with the X chromosome was the highest with GRID-seq (~70%), moderate with ChIRP-seq (25–30%) and lowest with CHART-seq (10%) [25]. These data suggest that specificity of one-to-all methods is lower than that of all-to-all methods. We need to reevaluate the lncRNA-interacting sites detected by one-to-all methods by comparing the results with those from all-to-all methods (Table 2).
5. Conclusions
Here, we have reviewed several one-to-all and all-to-all technologies used to study RNA–chromatin interactions. In particular, we have argued that the proximity-based all-to-all methods are more powerful for understanding the role of chromatin-associated RNAs and genome organization. Zhou et al. also pointed out that Hi-C was a main technology to study genome organization before developing of the proximity-based all-to-all methods to capture RNA–chromatin interactions. The all-to-all methods have more advantages to study cell-type-specific genomic interactions because RNAs play roles for cell-type-specific gene regulation even though DNA-DNA contacts detected by Hi-C are not varied [29]. As discussed above, the first major limitation of all-to-all methods is the need to distinguish true interactions from background introduced by nascent transcripts. To overcome this problem, we need to develop further strategies. For example, the developers of RADICL-seq introduced RNase H treatment into their protocol to reduce nascent transcripts or actinomycin D treatment before cell fixation to stop Pol II transcription. The second limitation is the sensitivity to low-abundance RNAs. One strategy that should be considered is combining all-to-all methods with a capture strategy for specific RNAs or antibody pull-down to enrich RNAs associated with molecules of interest. For example, using an anti-Pol II antibody may enrich promoter-enhancer RNA (eRNA) interactions, and antibodies against CCCTC-binding factor or some subunits of cohesin may help reveal enrichment of transcription-associated RNAs within topologically associating domain boundaries.
In conclusion, these technologies to study RNA–chromatin interactions have great potential to enable exploration of chrRNAs and identify their functions in gene regulation and genome organization. By building on these current protocols, researchers may develop more advanced protocols, perhaps even capable of analyzing single cells. Recently RIC-seq (RNA in situ conformation sequencing), which is also a technology based on proximity ligation was reported [40]. This technology can generate three-dimensional interaction maps of RNA to study regulatory roles of RNA. More novel lncRNAs will be discovered and their biological functions will be investigated. Additional methods of capturing RNA–chromatin interactions, including one-to-all methods, will always be welcome to help us identify the function of lncRNAs.
Author Contributions
M.K. wrote the original article and designed the figures, P.C. wrote, revised and approved the final version of this review. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by a Research Grant from MEXT to the RIKEN Center for Life Science Technologies and a Research Grant from MEXT to the RIKEN IMS. This work is also supported by JSPS KAKENHI Grant Number 19K06623 (M.K.) We thank Win Hin Yip for comments on the manuscript. We also thank members of the PC lab.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Werner, M.S.; Ruthenburg, A.J. Nuclear Fractionation Reveals Thousands of Chromatin-Tethered Noncoding RNAs Adjacent to Active Genes. Cell Rep. 2015, 12, 1089–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerase, A.; Pintacuda, G.; Tattermusch, A.; Avner, P. Xist localization and function: New insights from multiple levels. Genome Biol. 2015, 16, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Rocha, S.T.; Heard, E. Novel players in X inactivation: Insights into Xist-mediated gene silencing and chromosome conformation. Nat. Struct. Mol. Biol. 2017, 24, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Latos, P.A.; Pauler, F.M.; Koerner, M.V.; Şenergin, H.B.; Hudson, Q.J.; Stocsits, R.R.; Allhoff, W.; Stricker, S.H.; Klement, R.M.; Warczok, K.E.; et al. Airn transcriptional overlap, but not its lncRNA products, induces imprinted Igf2r silencing. Science 2012, 338, 1469–1472. [Google Scholar] [CrossRef] [PubMed]
- Andergassen, D.; Muckenhuber, M.; Bammer, P.C.; Kulinski, T.M.; Theussl, H.C.; Shimizu, T.; Penninger, J.M.; Pauler, F.M.; Hudson, Q.J. The Airn lncRNA does not require any DNA elements within its locus to silence distant imprinted genes. PLoS Genet. 2019, 15, e1008268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef]
- Portoso, M.; Ragazzini, R.; Brenčič, Ž.; Moiani, A.; Michaud, A.; Vassilev, I.; Wassef, M.; Servant, N.; Sargueil, B.; Margueron, R. PRC2 is dispensable for. EMBO J. 2017, 36, 981–994. [Google Scholar] [CrossRef]
- Amândio, A.R.; Necsulea, A.; Joye, E.; Mascrez, B.; Duboule, D. Hotair Is Dispensible for Mouse Development. PLoS Genet. 2016, 12, e1006232. [Google Scholar] [CrossRef]
- Li, L.; Liu, B.; Wapinski, O.L.; Tsai, M.C.; Qu, K.; Zhang, J.; Carlson, J.C.; Lin, M.; Fang, F.; Gupta, R.A.; et al. Targeted disruption of Hotair leads to homeotic transformation and gene derepression. Cell Rep. 2013, 5, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Helms, J.A.; Chang, H.Y. Comment on “Hotair Is Dispensable for Mouse Development”. PLoS Genet. 2016, 12, e1006406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selleri, L.; Bartolomei, M.S.; Bickmore, W.A.; He, L.; Stubbs, L.; Reik, W.; Barsh, G.S. A Hox-Embedded Long Noncoding RNA: Is It All Hot Air? PLoS Genet. 2016, 12, e1006485. [Google Scholar] [CrossRef]
- Li, X.; Fu, X.D. Chromatin-associated RNAs as facilitators of functional genomic interactions. Nat. Rev. Genet. 2019, 20, 503–519. [Google Scholar] [CrossRef]
- Mishra, K.; Kanduri, C. Understanding Long Noncoding RNA and Chromatin Interactions: What We Know So Far. Noncoding RNA 2019, 5, 54. [Google Scholar] [CrossRef] [Green Version]
- Engreitz, J.M.; Pandya-Jones, A.; McDonel, P.; Shishkin, A.; Sirokman, K.; Surka, C.; Kadri, S.; Xing, J.; Goren, A.; Lander, E.S.; et al. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science 2013, 341, 1237973. [Google Scholar] [CrossRef] [Green Version]
- Simon, M.D.; Pinter, S.F.; Fang, R.; Sarma, K.; Rutenberg-Schoenberg, M.; Bowman, S.K.; Kesner, B.A.; Maier, V.K.; Kingston, R.E.; Lee, J.T. High-resolution Xist binding maps reveal two-step spreading during X-chromosome inactivation. Nature 2013, 504, 465–469. [Google Scholar] [CrossRef] [Green Version]
- West, J.A.; Davis, C.P.; Sunwoo, H.; Simon, M.D.; Sadreyev, R.I.; Wang, P.I.; Tolstorukov, M.Y.; Kingston, R.E. The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites. Mol. Cell 2014, 55, 791–802. [Google Scholar] [CrossRef] [Green Version]
- Hon, C.C.; Ramilowski, J.A.; Harshbarger, J.; Bertin, N.; Rackham, O.J.; Gough, J.; Denisenko, E.; Schmeier, S.; Poulsen, T.M.; Severin, J.; et al. An atlas of human long non-coding RNAs with accurate 5’ ends. Nature 2017, 543, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Jordan, R.; Wai, Y.C.; Saumya, A.; Chang, J.-C.; Ciani, Y.; Kulakovskiy, I.V.; Mendez, M.; Li, J.; Ooi, C.; Ouyang, J.F.; et al. Functional Annotation of Human Long Non-Coding RNAs via Molecular Phenotyping. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Simon, M.D. Insight into lncRNA biology using hybridization capture analyses. Biochim. Biophys. Acta 2016, 1859, 121–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, C.; Qu, K.; Zhong, F.L.; Artandi, S.E.; Chang, H.Y. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol. Cell 2011, 44, 667–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, M.D.; Wang, C.I.; Kharchenko, P.V.; West, J.A.; Chapman, B.A.; Alekseyenko, A.A.; Borowsky, M.L.; Kuroda, M.I.; Kingston, R.E. The genomic binding sites of a noncoding RNA. Proc. Natl. Acad. Sci. USA 2011, 108, 20497–20502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sridhar, B.; Rivas-Astroza, M.; Nguyen, T.C.; Chen, W.; Yan, Z.; Cao, X.; Hebert, L.; Zhong, S. Systematic Mapping of RNA-Chromatin Interactions In Vivo. Curr. Biol. 2017, 27, 610–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhou, B.; Chen, L.; Gou, L.T.; Li, H.; Fu, X.D. GRID-seq reveals the global RNA-chromatin interactome. Nat. Biotechnol. 2017, 35, 940–950. [Google Scholar] [CrossRef]
- Bell, J.C.; Jukam, D.; Teran, N.A.; Risca, V.I.; Smith, O.K.; Johnson, W.L.; Skotheim, J.M.; Greenleaf, W.J.; Straight, A.F. Chromatin-associated RNA sequencing (ChAR-seq) maps genome-wide RNA-to-DNA contacts. Elife 2018, 7. [Google Scholar] [CrossRef]
- Bonetti, A.; Agostini, F.; Suzuki, A.M.; Hashimoto, K.; Pascarella, G.; Gimenez, J.; Roos, L.; Nash, A.J.; Ghilotti, M.; Cameron, C.J.F.; et al. RADICL-seq identifies general and cell type-specific principles of genome-wide RNA-chromatin interactions. Nat. Commun. 2020, 11, 1018. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Huang, N.; Wu, W.; Chen, W.; Jiang, Y.; Chen, J.; Huang, X.; Wen, X.; Xu, J.; Jin, Q.; et al. Genome-wide colocalization of RNA-DNA interactions and fusion RNA pairs. Proc. Natl. Acad. Sci. USA 2019, 116, 3328–3337. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Li, X.; Luo, D.; Lim, D.H.; Zhou, Y.; Fu, X.D. GRID-seq for comprehensive analysis of global RNA-chromatin interactions. Nat. Protoc. 2019, 14, 2036–2068. [Google Scholar] [CrossRef]
- Jukam, D.; Limouse, C.; Smith, O.K.; Risca, V.I.; Bell, J.C.; Straight, A.F. Chromatin-Associated RNA Sequencing (ChAR-seq). Curr. Protoc. Mol. Biol. 2019, 126, e87. [Google Scholar] [CrossRef]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef]
- Chujo, T.; Yamazaki, T.; Kawaguchi, T.; Kurosaka, S.; Takumi, T.; Nakagawa, S.; Hirose, T. Unusual semi-extractability as a hallmark of nuclear body-associated architectural noncoding RNAs. EMBO J. 2017, 36, 1447–1462. [Google Scholar] [CrossRef] [PubMed]
- Abdurashitov, M.A.; Tomilov, V.N.; Chernukhin, V.A.; Degtyarev, S.K.h. A physical map of human Alu repeats cleavage by restriction endonucleases. BMC Genom. 2008, 9, 305. [Google Scholar] [CrossRef] [Green Version]
- He, H.H.; Meyer, C.A.; Hu, S.S.; Chen, M.W.; Zang, C.; Liu, Y.; Rao, P.K.; Fei, T.; Xu, H.; Long, H.; et al. Refined DNase-seq protocol and data analysis reveals intrinsic bias in transcription factor footprint identification. Nat. Methods 2014, 11, 73–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Yan, Z.; Nguyen, T.C.; Bouman Chen, Z.; Chien, S.; Zhong, S. Mapping RNA-chromatin interactions by sequencing with iMARGI. Nat. Protoc. 2019, 14, 3243–3272. [Google Scholar] [CrossRef]
- Yaffe, E.; Tanay, A. Probabilistic modeling of Hi-C contact maps eliminates systematic biases to characterize global chromosomal architecture. Nat. Genet. 2011, 43, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Yan, Z.; Li, S.; Huang, N.; Huang, X.; Zhang, J.; Zhong, S. RNAs as Proximity-Labeling Media for Identifying Nuclear Speckle Positions Relative to the Genome. iScience 2018, 4, 204–215. [Google Scholar] [CrossRef]
- Engreitz, J.M.; Sirokman, K.; McDonel, P.; Shishkin, A.A.; Surka, C.; Russell, P.; Grossman, S.R.; Chow, A.Y.; Guttman, M.; Lander, E.S. RNA-RNA interactions enable specific targeting of noncoding RNAs to nascent Pre-mRNAs and chromatin sites. Cell 2014, 159, 188–199. [Google Scholar] [CrossRef] [Green Version]
- Flynn, R.A.; Do, B.T.; Rubin, A.J.; Calo, E.; Lee, B.; Kuchelmeister, H.; Rale, M.; Chu, C.; Kool, E.T.; Wysocka, J.; et al. 7SK-BAF axis controls pervasive transcription at enhancers. Nat. Struct. Mol. Biol. 2016, 23, 231–238. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Cao, C.; Ji, L.; Ye, R.; Wang, D.; Xia, C.; Wang, S.; Du, Z.; Hu, N.; Yu, X.; et al. RIC-seq for global in situ profiling of RNA–RNA spatial interactions. Nature 2020. [Google Scholar] [CrossRef]
Figure 1.
Overview of one-to-all methods to capture RNA-interacting genomic loci. Chromatin Isolation by RNA Purification (ChIRP), Capture Hybridization Analysis of RNA Targets (CHART), and RAP-DNA (RNA antisense purification) use biotinylated probes for the target RNA. After the chromatin fraction is pulled down by the use of streptavidin beads, RNase is used to elute genomic DNA. The resulting library is prepared for high-through put sequencing.
Figure 1.
Overview of one-to-all methods to capture RNA-interacting genomic loci. Chromatin Isolation by RNA Purification (ChIRP), Capture Hybridization Analysis of RNA Targets (CHART), and RAP-DNA (RNA antisense purification) use biotinylated probes for the target RNA. After the chromatin fraction is pulled down by the use of streptavidin beads, RNase is used to elute genomic DNA. The resulting library is prepared for high-through put sequencing.

Figure 2.
Overview of all-to-all methods to capture RNA-interacting genomic loci. All four methods (MARGI (mapping RNA–genome interactions), GRID-seq (global RNA interaction with DNA sequencing), RADICL-seq (RNA and DNA interacting complexes ligated and sequenced), and ChAR-seq (chromatin-associated RNA sequencing)) use fixed cells and biotinylated 5′-adenylated linkers. The steps above the line are performed in nuclei and those below the line are performed in solution.
Figure 2.
Overview of all-to-all methods to capture RNA-interacting genomic loci. All four methods (MARGI (mapping RNA–genome interactions), GRID-seq (global RNA interaction with DNA sequencing), RADICL-seq (RNA and DNA interacting complexes ligated and sequenced), and ChAR-seq (chromatin-associated RNA sequencing)) use fixed cells and biotinylated 5′-adenylated linkers. The steps above the line are performed in nuclei and those below the line are performed in solution.


{kind=link}
{kind=link}
Table 1.
Comparison of conditions for MARGI, GRID-seq, ChAR-seq, and RADICL-seq.
| Technical Aspect | MARGI (iMARGI) * | GRID-Seq | ChAR-Seq | RADICL-Seq |
|---|---|---|---|---|
| Crosslinking condition | 1% formaldehyde | DSG + 3% formaldehyde | 3% formaldehyde | 1% formaldehyde |
| Genomic digestion | HaeIII (AluI) * | AluI | DpnII | DNase I |
| Reduction of nascent transcripts | none | GridTools | none | RNase H Actinomycin D One-sided cumulative binomial test |
| Carrier to pellet nuclei and wash between enzymatic steps | Streptavidin beads | none | none | SPRI beads |
| Length of RNA and DNA tags | long | 18-20 bp | long | 25–25 bp |
| Cell types tested | human HEK293 H9 (hESC) (Human foreskin fibroblast [HFF]) * | human MDA-MB-231 (breast cancer cells) MM.1S (multiple myeloma cells) mouse mESC Drosophila S2 cells | Drosophila CME-W1-cl8+ cells (male) Kc167 (female) | moues mESC OPC |
| Cell number | 4 × 108 cells (5 × 106 cells) * | 2 × 106 cells (mammalian) 1 × 107 cells (Drosophila) | 1–1.5 × 108 cells (Drosophila) 1–1.5 × 107 cells (human) | 2 × 106 cells |
* Items marked with asterisks pertain to iMARGI.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kato, M.; Carninci, P. Genome-Wide Technologies to Study RNA–Chromatin Interactions. Non-Coding RNA 2020, 6, 20. https://0-doi-org.brum.beds.ac.uk/10.3390/ncrna6020020
AMA Style
Kato M, Carninci P. Genome-Wide Technologies to Study RNA–Chromatin Interactions. Non-Coding RNA. 2020; 6(2):20. https://0-doi-org.brum.beds.ac.uk/10.3390/ncrna6020020
Chicago/Turabian StyleKato, Masaki, and Piero Carninci. 2020. "Genome-Wide Technologies to Study RNA–Chromatin Interactions" Non-Coding RNA 6, no. 2: 20. https://0-doi-org.brum.beds.ac.uk/10.3390/ncrna6020020
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.