Indole-3-Acetic Acid Alleviates Nonalcoholic Fatty Liver Disease in Mice via Attenuation of Hepatic Lipogenesis, and Oxidative and Inflammatory Stress


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Animals and Treatments
2.3. Biochemical Analysis
2.4. Histopathological Examination
2.5. Quantitative Real-time Polymerase Chain Reaction (RT-qPCR)
2.6. Analysis of Oxidative Stress Indexes
2.7. Immunofluorescence Histochemistry
2.8. Statistical Analysis
3. Results
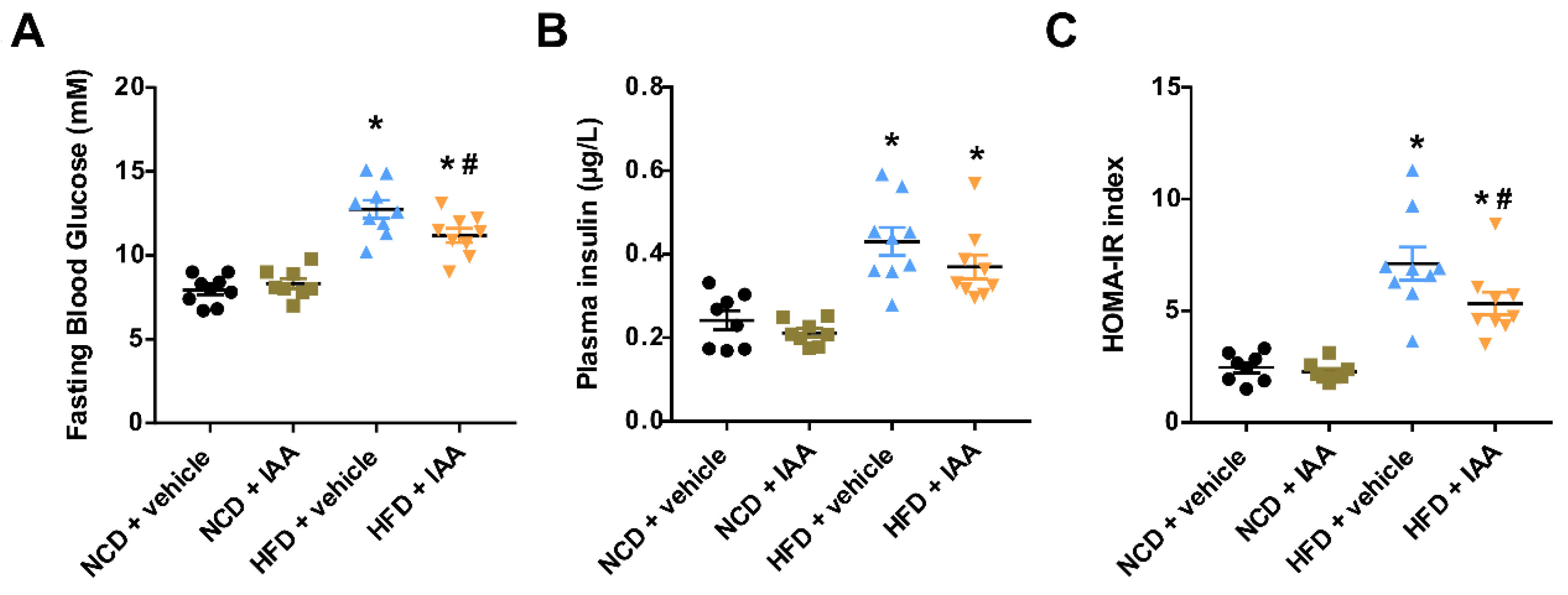
3.1. Indole-3-Acetic Acid (IAA) Ameliorates High-Fat Diet-Induced Systemic Insulin Resistance
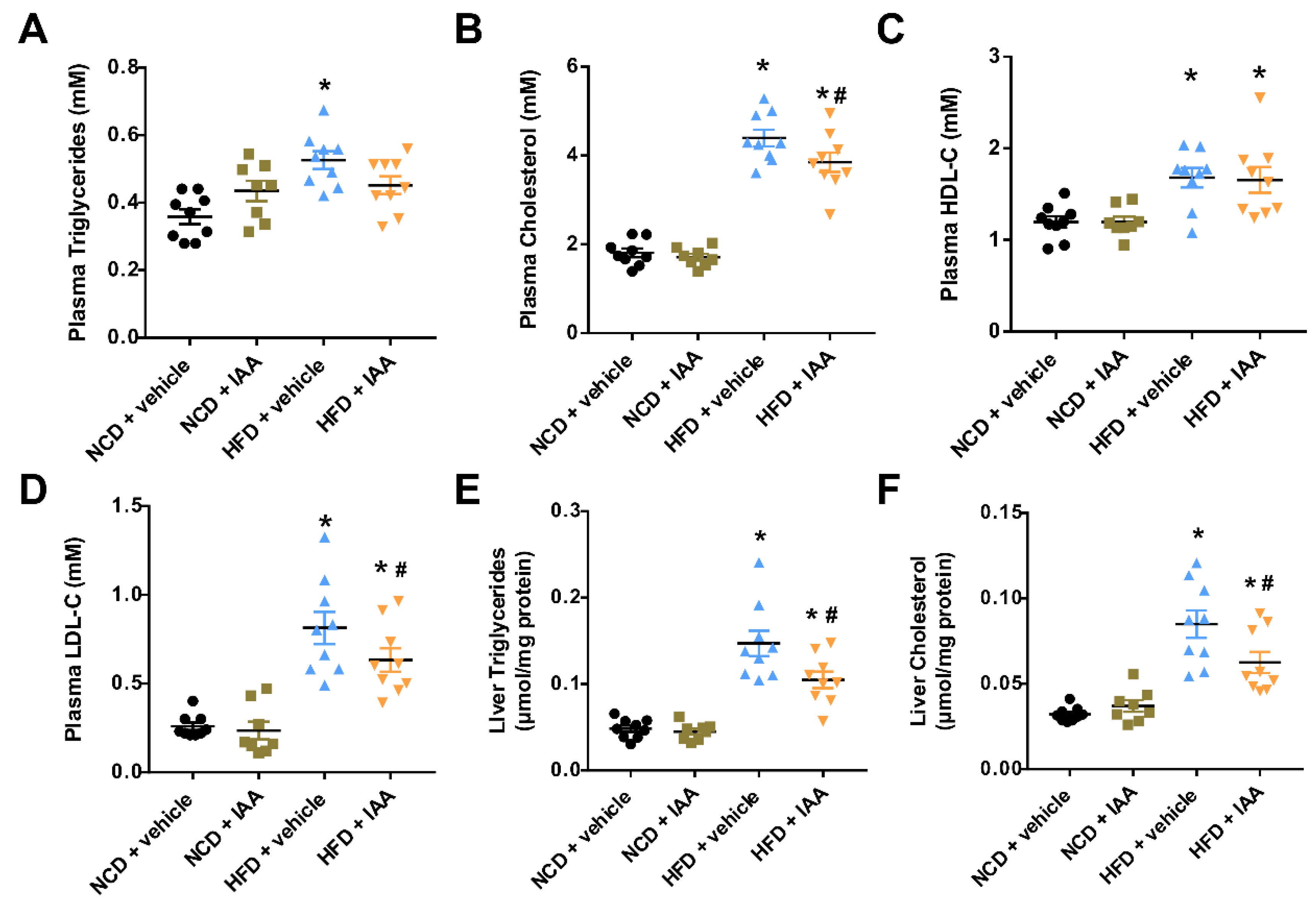
3.2. The Plasma and Hepatic Lipids Content of Mice Exposed to a High-Fat-Diet Feeding and Indole-3-Acetic Acid Treatment
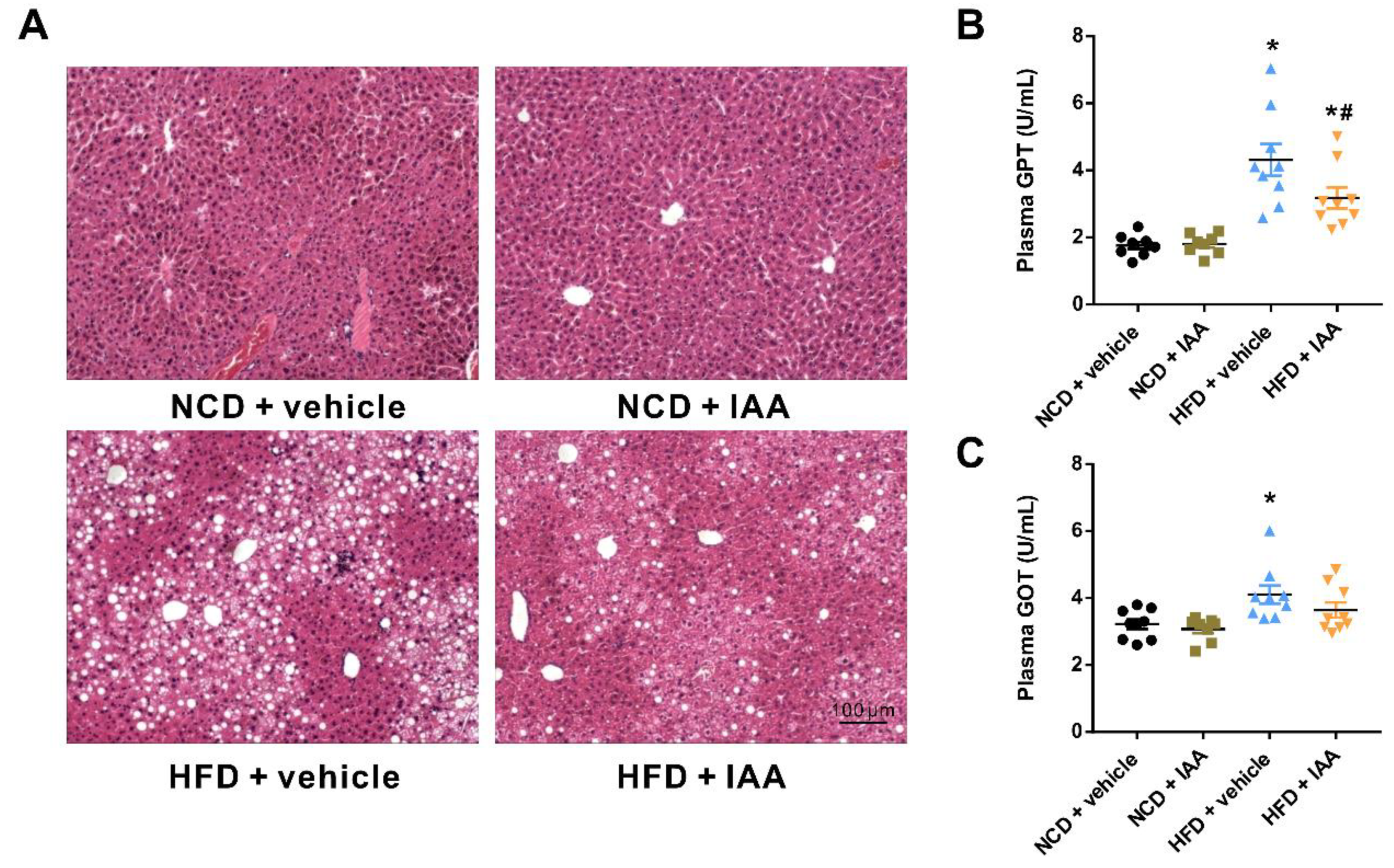
3.3. Indole-3-Acetic Acid Alleviates Liver Injury in Mice Fed with a High Fat Diet
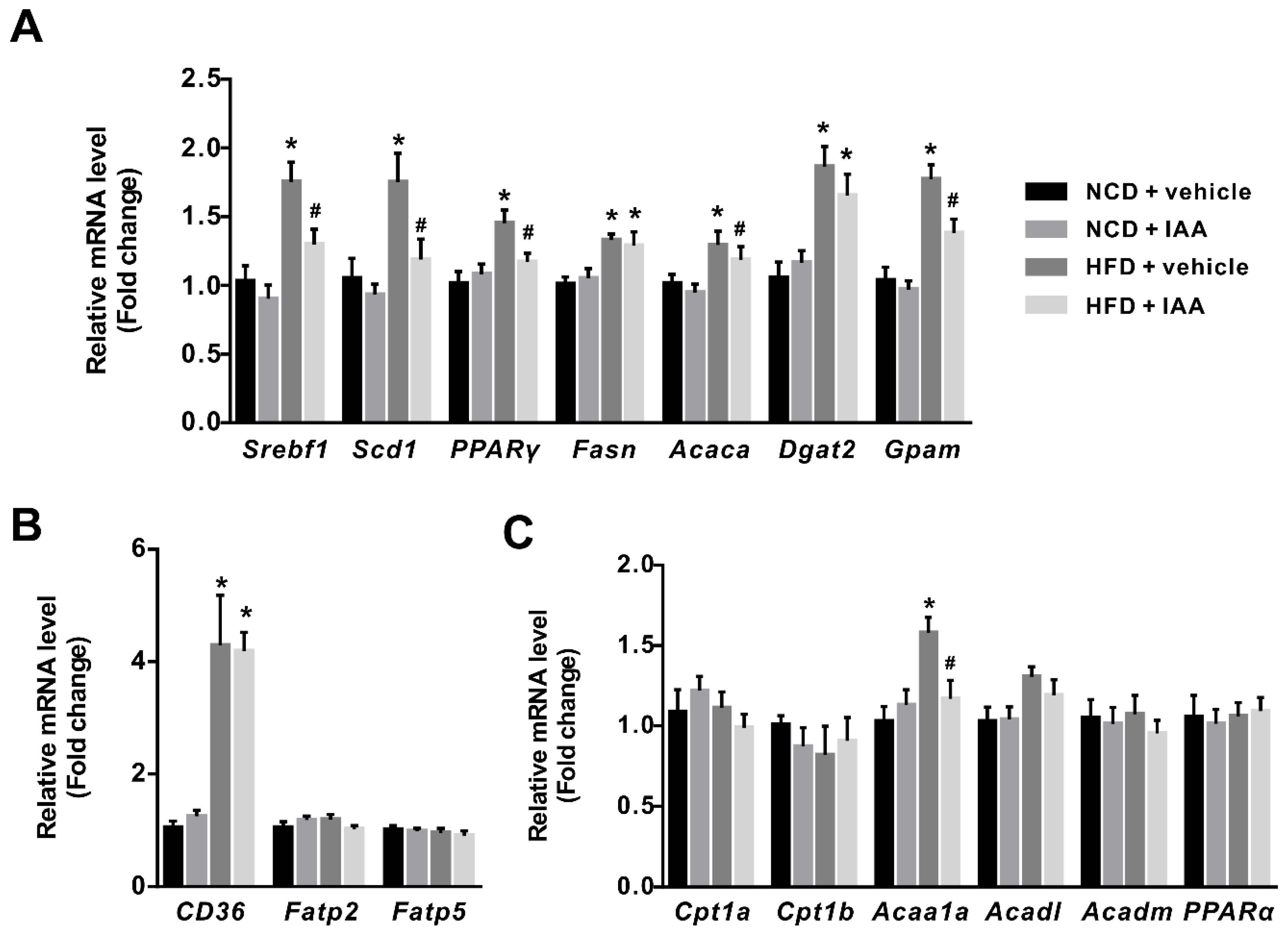
3.4. The Regulation of Indole-3-Acetic Acid on the Expression of Genes Linked with Lipid Metabolism in Liver
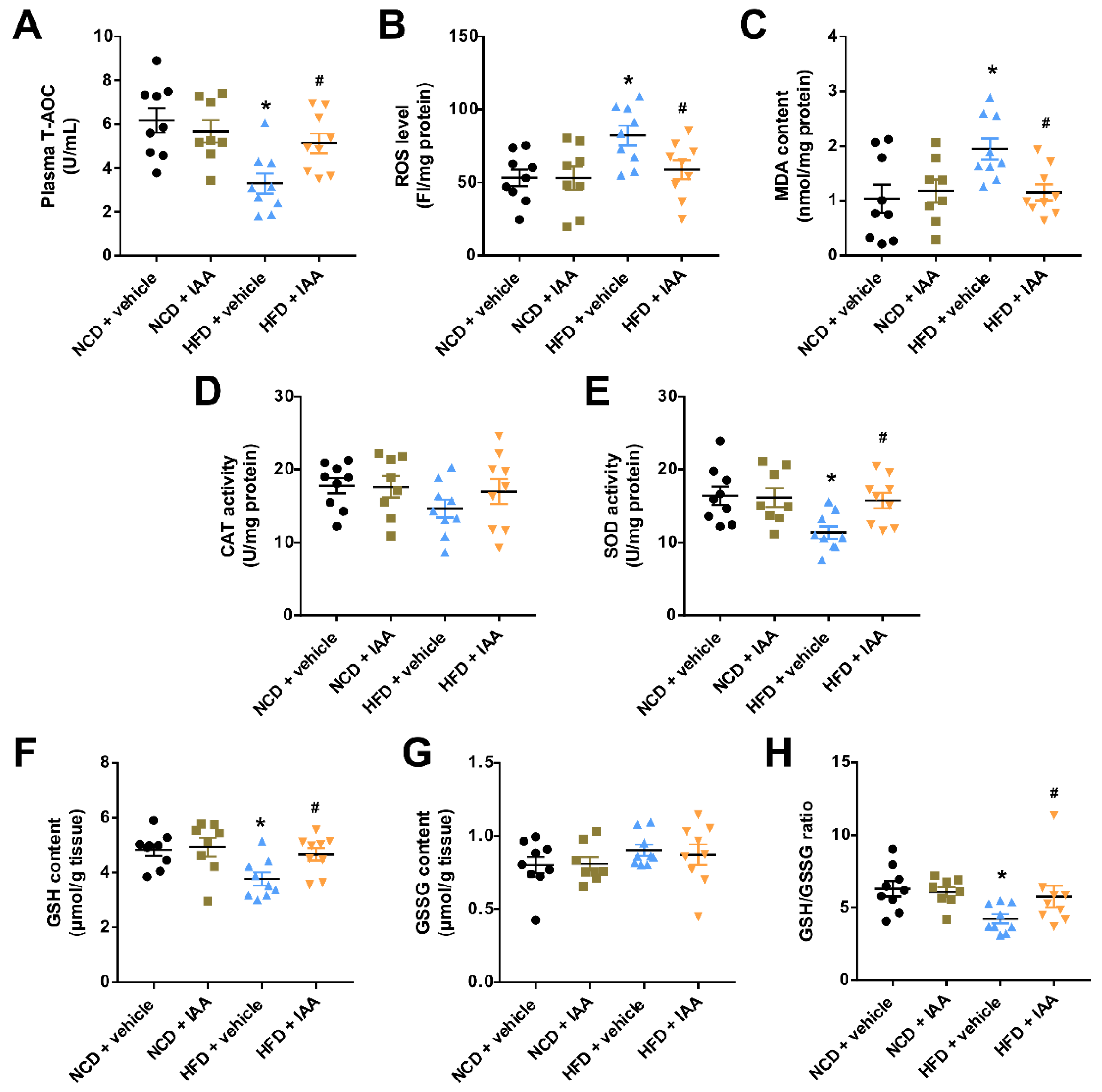
3.5. Indole-3-Acetic Acid Attenuates High Fat Diet-Induced Oxidative Stress in Hepatic Tissue
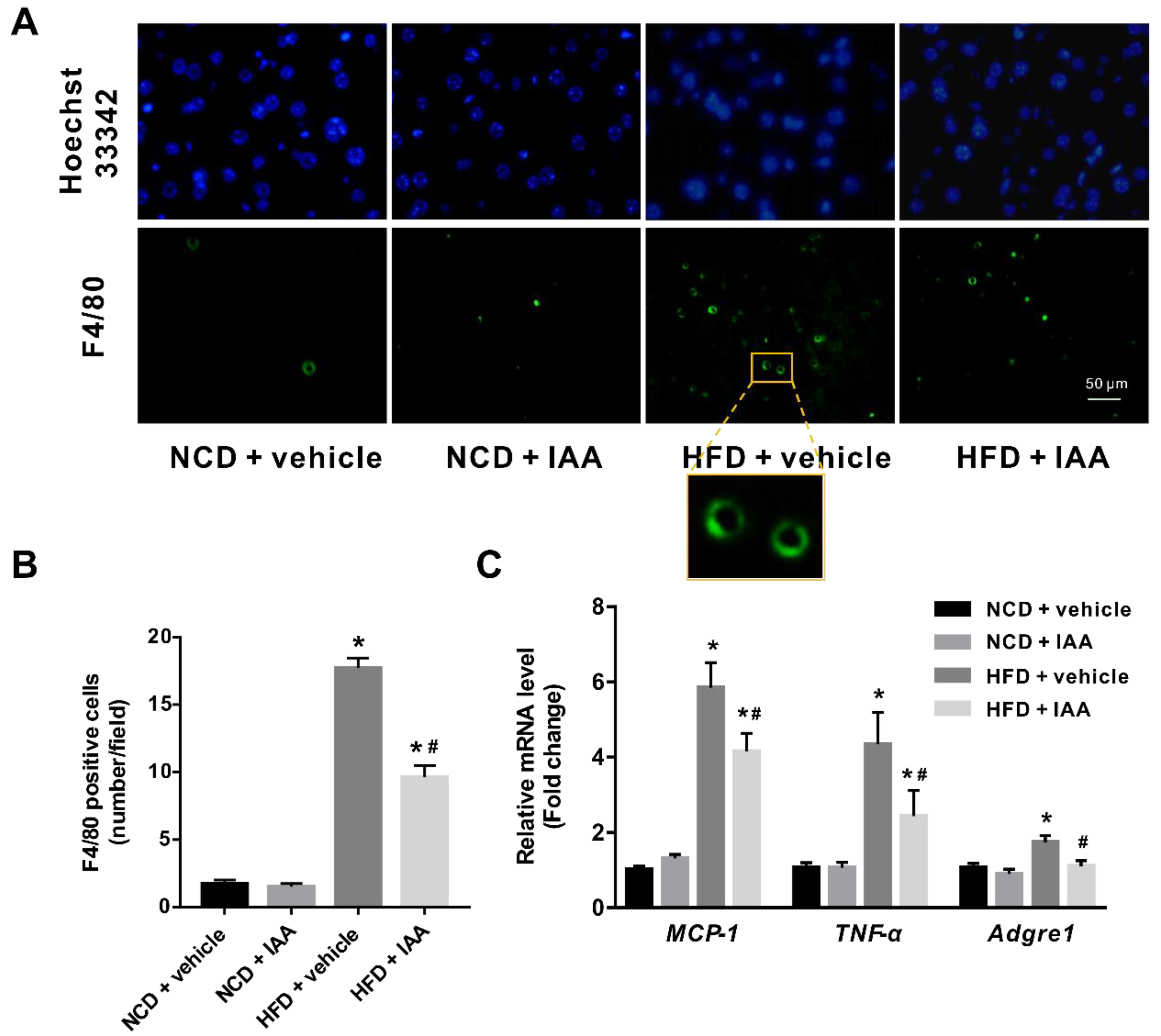
3.6. Indole-3-Acetic Acid Mitigates Hepatic Inflammatory Stress Induced by a High-Fat Diet
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Norheim, F.; Hui, S.T.; Kulahcioglu, E.; Mehrabian, M.; Cantor, R.M.; Pan, C.; Parks, B.W.; Lusis, A.J. Genetic and hormonal control of hepatic steatosis in female and male mice. J. Lipid Res. 2017, 58, 178–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abd El-Kader, S.M.; El-Den Ashmawy, E.M. Non-alcoholic fatty liver disease: The diagnosis and management. World J. Hepatol. 2015, 7, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Rau, M.; Schilling, A.K.; Meertens, J.; Hering, I.; Weiss, J.; Jurowich, C.; Kudlich, T.; Hermanns, H.M.; Bantel, H.; Beyersdorf, N.; et al. Progression from Nonalcoholic Fatty Liver to Nonalcoholic Steatohepatitis Is Marked by a Higher Frequency of Th17 Cells in the Liver and an Increased Th17/Resting Regulatory T Cell Ratio in Peripheral Blood and in the Liver. J. Immunol. 2016, 196, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Fukusato, T. Histopathology of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J. Gastroenterol. 2014, 20, 15539–15548. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.L.; Chen, H.; Wang, C.L.; Liang, L. Pathogenesis of non-alcoholic fatty liver disease in children and adolescence: From “two hit theory” to “multiple hit model”. World J. Gastroenterol. 2018, 24, 2974–2983. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.Z.; Berk, M.; McIntyre, T.M.; Feldstein, A.E. Hepatic lipid partitioning and liver damage in nonalcoholic fatty liver disease: Role of stearoyl-CoA desaturase. J. Biol. Chem. 2009, 284, 5637–5644. [Google Scholar] [CrossRef]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid. Med. Cell. Longev. 2018, 2018, 9547613. [Google Scholar] [CrossRef]
- Li, S.; Hong, M.; Tan, H.Y.; Wang, N.; Feng, Y. Insights into the Role and Interdependence of Oxidative Stress and Inflammation in Liver Diseases. Oxid. Med. Cell. Longev. 2016, 2016, 4234061. [Google Scholar] [CrossRef] [PubMed]
- Juarez-Hernandez, E.; Chavez-Tapia, N.C.; Uribe, M.; Barbero-Becerra, V.J. Role of bioactive fatty acids in nonalcoholic fatty liver disease. Nutr. J. 2016, 15, 72. [Google Scholar] [CrossRef] [PubMed]
- Roager, H.M.; Licht, T.R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 2018, 9, 3294. [Google Scholar] [CrossRef] [PubMed]
- Rath, S.; Heidrich, B.; Pieper, D.H.; Vital, M. Uncovering the trimethylamine-producing bacteria of the human gut microbiota. Microbiome 2017, 5, 54. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, H.; Zhang, M.; Guo, G.L. Fatty liver diseases, bile acids, and FXR. Acta Pharm. Sin. B 2016, 6, 409–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Y.; Yin, Y.; Li, Z.; Zhang, W. Gut Microbiota-Derived Components and Metabolites in the Progression of Non-Alcoholic Fatty Liver Disease (NAFLD). Nutrients 2019, 11, 1712. [Google Scholar] [CrossRef]
- Hubbard, T.D.; Murray, I.A.; Perdew, G.H. Indole and Tryptophan Metabolism: Endogenous and Dietary Routes to Ah Receptor Activation. Drug Metab. Dispos. 2015, 43, 1522–1535. [Google Scholar] [CrossRef] [Green Version]
- Arnao, M.B.; Sanchez-Bravo, J.; Acosta, M. Indole-3-carbinol as a scavenger of free radicals. Biochem. Mol. Biol. Int. 1996, 39, 1125–1134. [Google Scholar] [CrossRef]
- Kim, D.; Kim, H.; Kim, K.; Roh, S. The Protective Effect of Indole-3-Acetic Acid (IAA) on H2O2-Damaged Human Dental Pulp Stem Cells Is Mediated by the AKT Pathway and Involves Increased Expression of the Transcription Factor Nuclear Factor-Erythroid 2-Related Factor 2 (Nrf2) and Its Downstream Target Heme Oxygenase 1 (HO-1). Oxid. Med. Cell. Longev. 2017, 2017, 8639485. [Google Scholar]
- Krishnan, S.; Ding, Y.; Saedi, N.; Choi, M.; Sridharan, G.V.; Sherr, D.H.; Yarmush, M.L.; Alaniz, R.C.; Jayaraman, A.; Lee, K. Gut Microbiota-Derived Tryptophan Metabolites Modulate Inflammatory Response in Hepatocytes and Macrophages. Cell Rep. 2018, 23, 1099–1111. [Google Scholar] [CrossRef]
- Folkes, L.K.; Wardman, P. Enhancing the efficacy of photodynamic cancer therapy by radicals from plant auxin (indole-3-acetic acid). Cancer Res. 2003, 63, 776–779. [Google Scholar] [PubMed]
- Mather, K. Surrogate measures of insulin resistance: Of rats, mice, and men. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E398–E399. [Google Scholar] [CrossRef] [PubMed]
- Natividad, J.M.; Agus, A.; Planchais, J.; Lamas, B.; Jarry, A.C.; Martin, R.; Michel, M.L.; Chong-Nguyen, C.; Roussel, R.; Straube, M.; et al. Impaired Aryl Hydrocarbon Receptor Ligand Production by the Gut Microbiota Is a Key Factor in Metabolic Syndrome. Cell Metab. 2018, 28, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.R.; Flamm, S.L.; Di Bisceglie, A.M.; Bodenheimer, H.C. Public Policy Committee of the American Association for the Study of Liver, D. Serum activity of alanine aminotransferase (ALT) as an indicator of health and disease. Hepatology 2008, 47, 1363–1370. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yoo, W.; Park, H.M.; Lim, S.Y.; Shin, D.H.; Kim, S.; Park, H.Y.; Jeong, T.S. Arazyme Suppresses Hepatic Steatosis and Steatohepatitis in Diet-Induced Non-Alcoholic Fatty Liver Disease-Like Mouse Model. Int. J. Mol. Sci. 2019, 20, 2325. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.F.; Wang, X.N.; Huang, C.C.; Hu, L.; Xiao, Y.F.; Guan, X.H.; Qian, Y.S.; Deng, K.Y.; Xin, H.B. Inhibition of NAMPT aggravates high fat diet-induced hepatic steatosis in mice through regulating Sirt1/AMPKalpha/SREBP1 signaling pathway. Lipids Health Dis. 2017, 16, 82. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Park, S.Y.; Lee, M.H.; Rho, J.H.; Oh, Y.J.; Jung, H.U.; Yoo, S.H.; Jeong, N.Y.; Lee, H.J.; Suh, S.; et al. Hepatic STAMP2 alleviates high fat diet-induced hepatic steatosis and insulin resistance. J. Hepatol. 2015, 63, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Hendrikx, T.; Duan, Y.; Wang, Y.; Oh, J.H.; Alexander, L.M.; Huang, W.; Starkel, P.; Ho, S.B.; Gao, B.; Fiehn, O.; et al. Bacteria engineered to produce IL-22 in intestine induce expression of REG3G to reduce ethanol-induced liver disease in mice. Gut 2018. [Google Scholar] [CrossRef] [PubMed]
- Tanos, R.; Murray, I.A.; Smith, P.B.; Patterson, A.; Perdew, G.H. Role of the Ah receptor in homeostatic control of fatty acid synthesis in the liver. Toxicol. Sci. 2012, 129, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Spahis, S.; Delvin, E.; Borys, J.M.; Levy, E. Oxidative Stress as a Critical Factor in Nonalcoholic Fatty Liver Disease Pathogenesis. Antioxid. Redox Signal. 2017, 26, 519–541. [Google Scholar] [CrossRef]
- Polimeni, L.; Del Ben, M.; Baratta, F.; Perri, L.; Albanese, F.; Pastori, D.; Violi, F.; Angelico, F. Oxidative stress: New insights on the association of non-alcoholic fatty liver disease and atherosclerosis. World J. Hepatol. 2015, 7, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, R.M.P.; Romero, R.V. Effects of bixin in high-fat diet-fed-induced fatty liver in C57BL/6J mice. Asian Pac. J. Trop. Biomed. 2016, 6, 1015–1021. [Google Scholar] [CrossRef] [Green Version]
- Echeverria, F.; Valenzuela, R.; Bustamante, A.; Alvarez, D.; Ortiz, M.; Soto-Alarcon, S.A.; Munoz, P.; Corbari, A.; Videla, L.A. Attenuation of High-Fat Diet-Induced Rat Liver Oxidative Stress and Steatosis by Combined Hydroxytyrosol- (HT-) Eicosapentaenoic Acid Supplementation Mainly Relies on HT. Oxid. Med. Cell. Longev. 2018, 2018, 5109503. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.Y.; Jiang, N.; Yang, J.; Tu, J.; Zhou, Y.; Xiao, X.; Dong, Y. Silybum marianum oil attenuates hepatic steatosis and oxidative stress in high fat diet-fed mice. Biomed. Pharmacother. 2018, 100, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Mourao, L.R.; Santana, R.S.; Paulo, L.M.; Pugine, S.M.; Chaible, L.M.; Fukumasu, H.; Dagli, M.L.; de Melo, M.P. Protective action of indole-3-acetic acid on induced hepatocarcinoma in mice. Cell Biochem. Funct. 2009, 27, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Del Campo, J.A.; Gallego, P.; Grande, L. Role of inflammatory response in liver diseases: Therapeutic strategies. World J. Hepatol. 2018, 10, 1–7. [Google Scholar] [CrossRef]
- Cha, J.Y.; Kim, D.H.; Chun, K.H. The role of hepatic macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Lab. Anim. Res. 2018, 34, 133–139. [Google Scholar] [CrossRef]
- Kazankov, K.; Jorgensen, S.M.D.; Thomsen, K.L.; Moller, H.J.; Vilstrup, H.; George, J.; Schuppan, D.; Gronbaek, H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 145–159. [Google Scholar] [CrossRef]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef]
- Lee, H.; Ahn, J.; Shin, S.S.; Yoon, M. Ascorbic acid inhibits visceral obesity and nonalcoholic fatty liver disease by activating peroxisome proliferator-activated receptor alpha in high-fat-diet-fed C57BL/6J mice. Int. J. Obes. 2018. [Google Scholar] [CrossRef]
- Lu, H.J.; Tzeng, T.F.; Liou, S.S.; Chang, C.J.; Yang, C.; Wu, M.C.; Liu, I.M. Ruscogenin ameliorates experimental nonalcoholic steatohepatitis via suppressing lipogenesis and inflammatory pathway. Biomed. Res. Int. 2014, 2014, 652680. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, S.; Kishina, M.; Koda, M.; Yamamoto, Y.; Tanaka, K.; Harada, Y.; Yoshida, A.; Hisatome, I. Nimesulide, a cyclooxygenase-2 selective inhibitor, suppresses obesity-related non-alcoholic fatty liver disease and hepatic insulin resistance through the regulation of peroxisome proliferator-activated receptor gamma. Int. J. Mol. Med. 2016, 38, 721–728. [Google Scholar] [CrossRef] [PubMed]
- Kirovski, G.; Dorn, C.; Huber, H.; Moleda, L.; Niessen, C.; Wobser, H.; Schacherer, D.; Buechler, C.; Wiest, R.; Hellerbrand, C. Elevated systemic monocyte chemoattractrant protein-1 in hepatic steatosis without significant hepatic inflammation. Exp. Mol. Pathol. 2011, 91, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Kakino, S.; Ohki, T.; Nakayama, H.; Yuan, X.; Otabe, S.; Hashinaga, T.; Wada, N.; Kurita, Y.; Tanaka, K.; Hara, K.; et al. Pivotal Role of TNF-alpha in the Development and Progression of Nonalcoholic Fatty Liver Disease in a Murine Model. Horm. Metab. Res. 2018, 50, 80–87. [Google Scholar] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, Y.; Gao, Y.; Chen, H.; Yin, Y.; Zhang, W. Indole-3-Acetic Acid Alleviates Nonalcoholic Fatty Liver Disease in Mice via Attenuation of Hepatic Lipogenesis, and Oxidative and Inflammatory Stress. Nutrients 2019, 11, 2062. https://0-doi-org.brum.beds.ac.uk/10.3390/nu11092062
Ji Y, Gao Y, Chen H, Yin Y, Zhang W. Indole-3-Acetic Acid Alleviates Nonalcoholic Fatty Liver Disease in Mice via Attenuation of Hepatic Lipogenesis, and Oxidative and Inflammatory Stress. Nutrients. 2019; 11(9):2062. https://0-doi-org.brum.beds.ac.uk/10.3390/nu11092062
Chicago/Turabian StyleJi, Yun, Yuan Gao, Hong Chen, Yue Yin, and Weizhen Zhang. 2019. "Indole-3-Acetic Acid Alleviates Nonalcoholic Fatty Liver Disease in Mice via Attenuation of Hepatic Lipogenesis, and Oxidative and Inflammatory Stress" Nutrients 11, no. 9: 2062. https://0-doi-org.brum.beds.ac.uk/10.3390/nu11092062