Early Hepatic Oxidative Stress and Mitochondrial Changes Following Western Diet in Middle Aged Rats

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Design
2.2. Metabolic Analyses
2.3. Hepatic Mitochondrial Function
2.4. Hepatic Oxidative Status and Inflammation
2.5. Lipogenesis in the Liver
2.6. Western Blotting of Hepatic Proteins
2.7. Statistical Analysis
3. Results
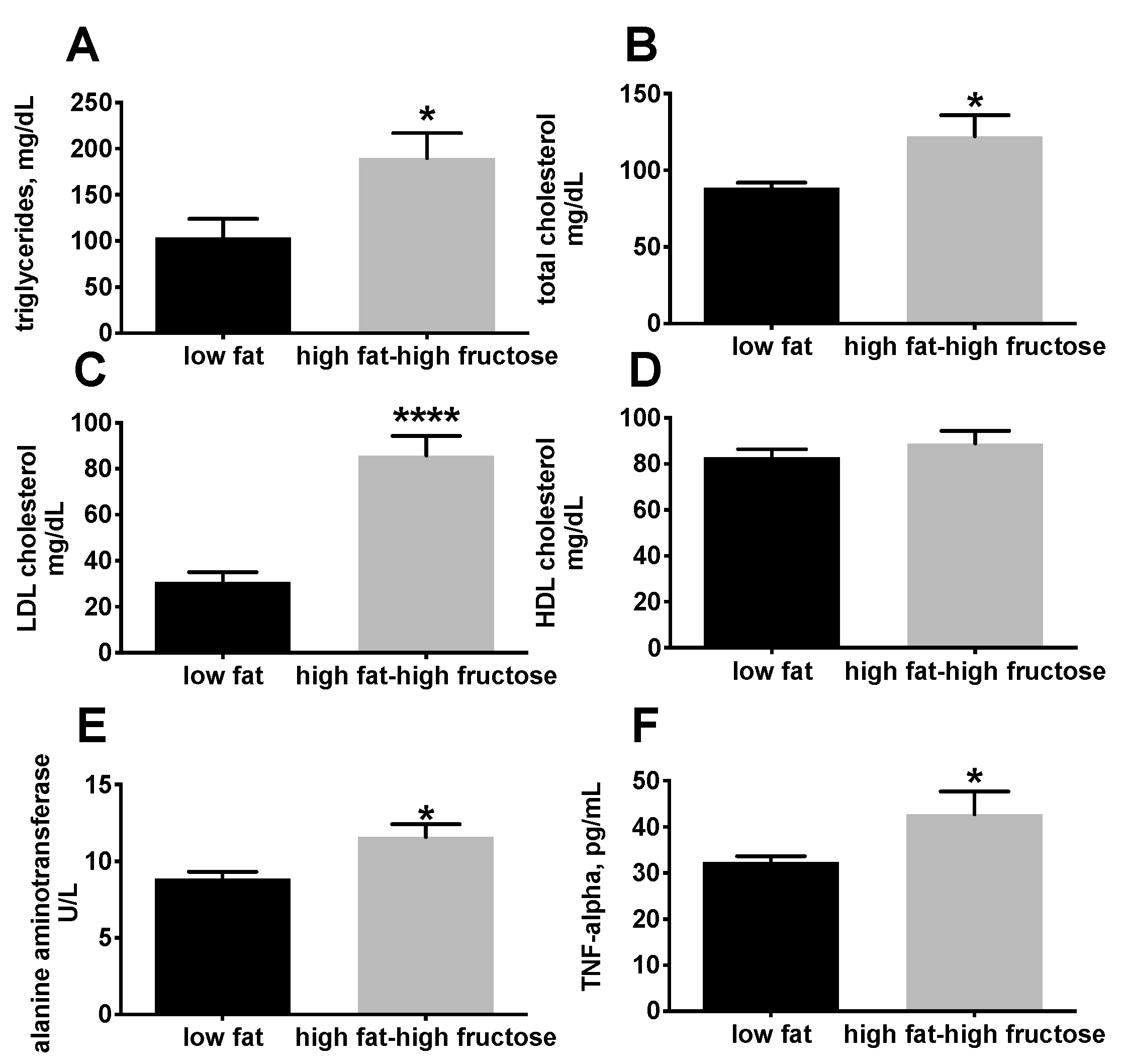
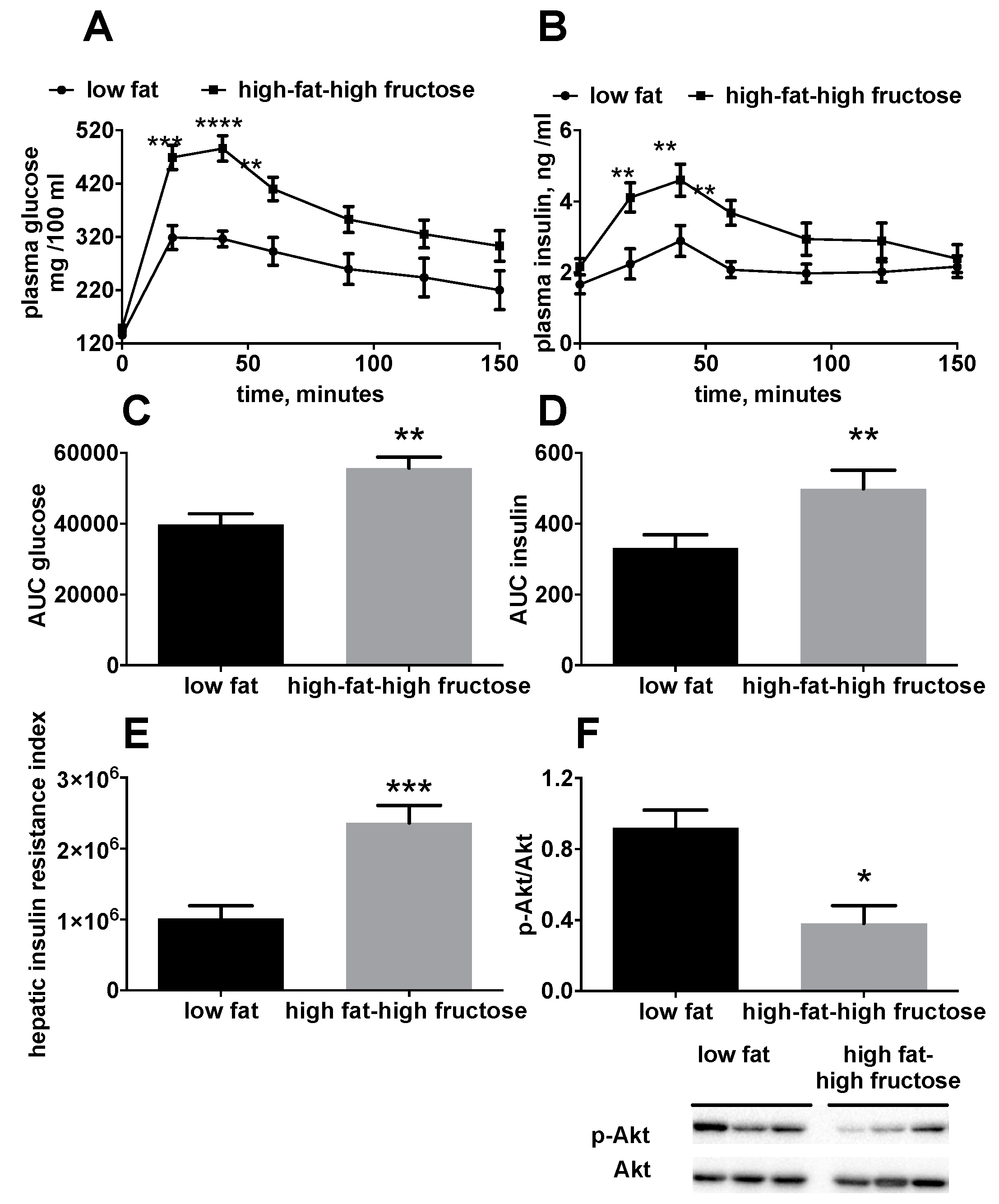
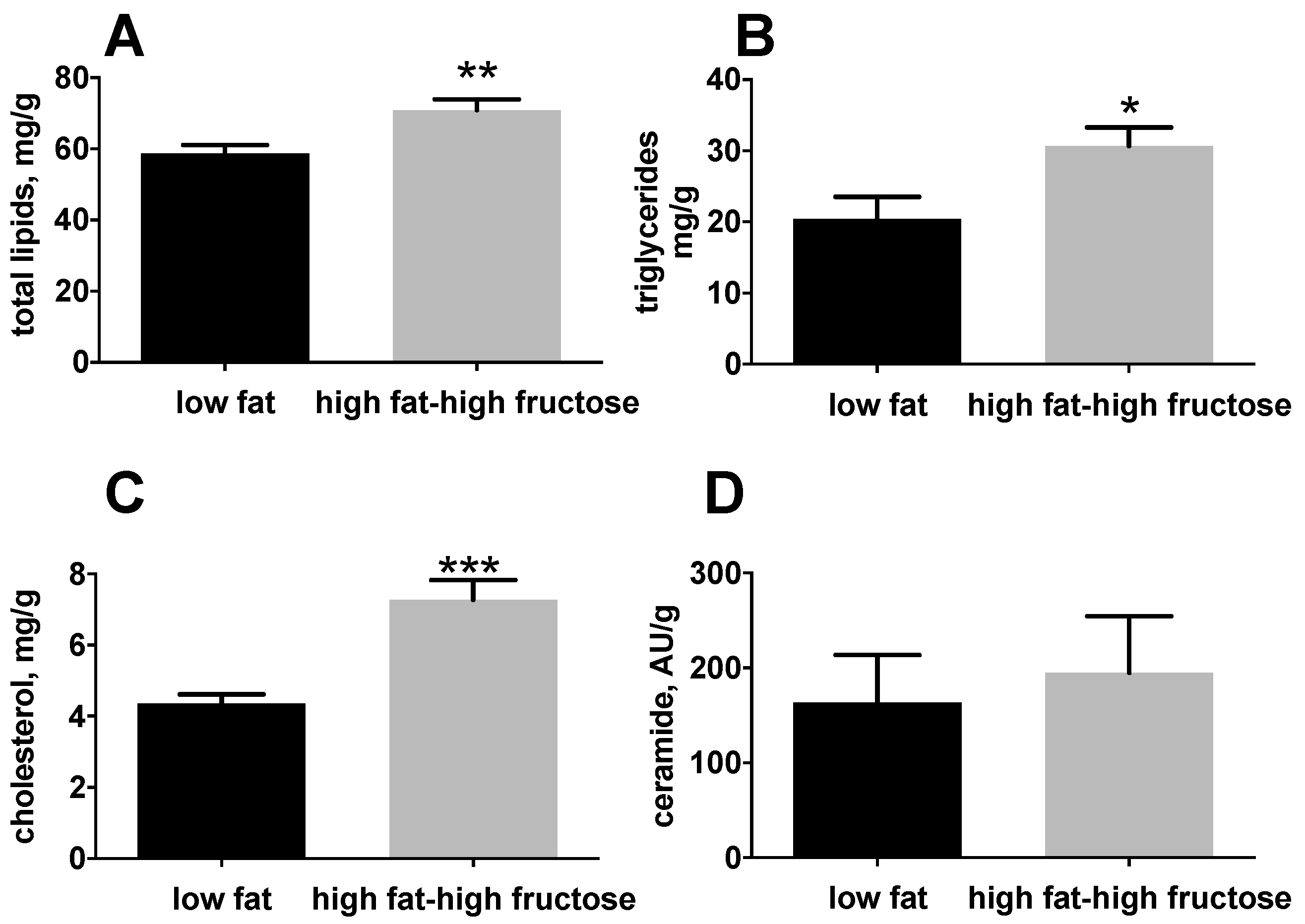
3.1. Body and Liver Composition and Plasma Parameters
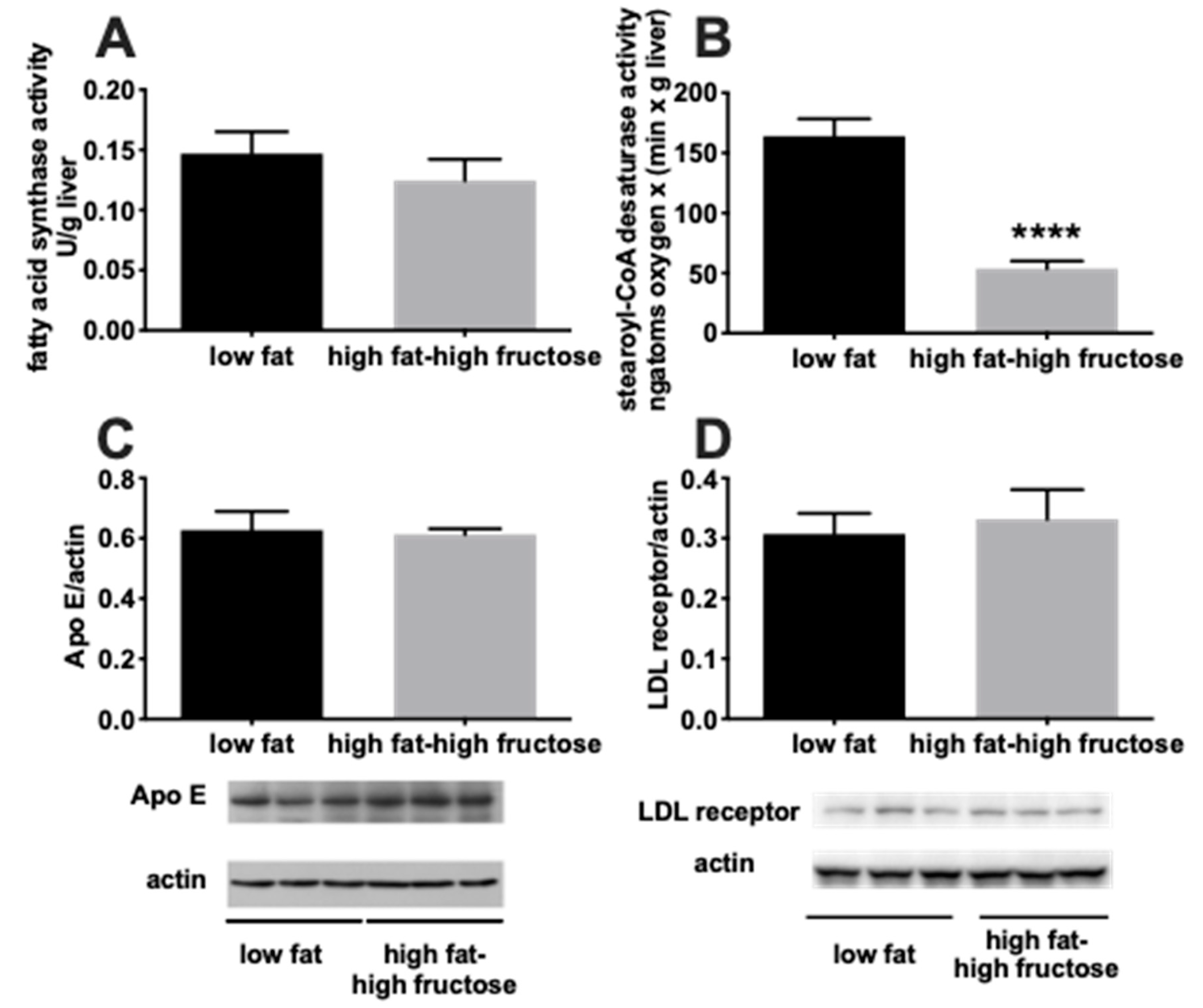
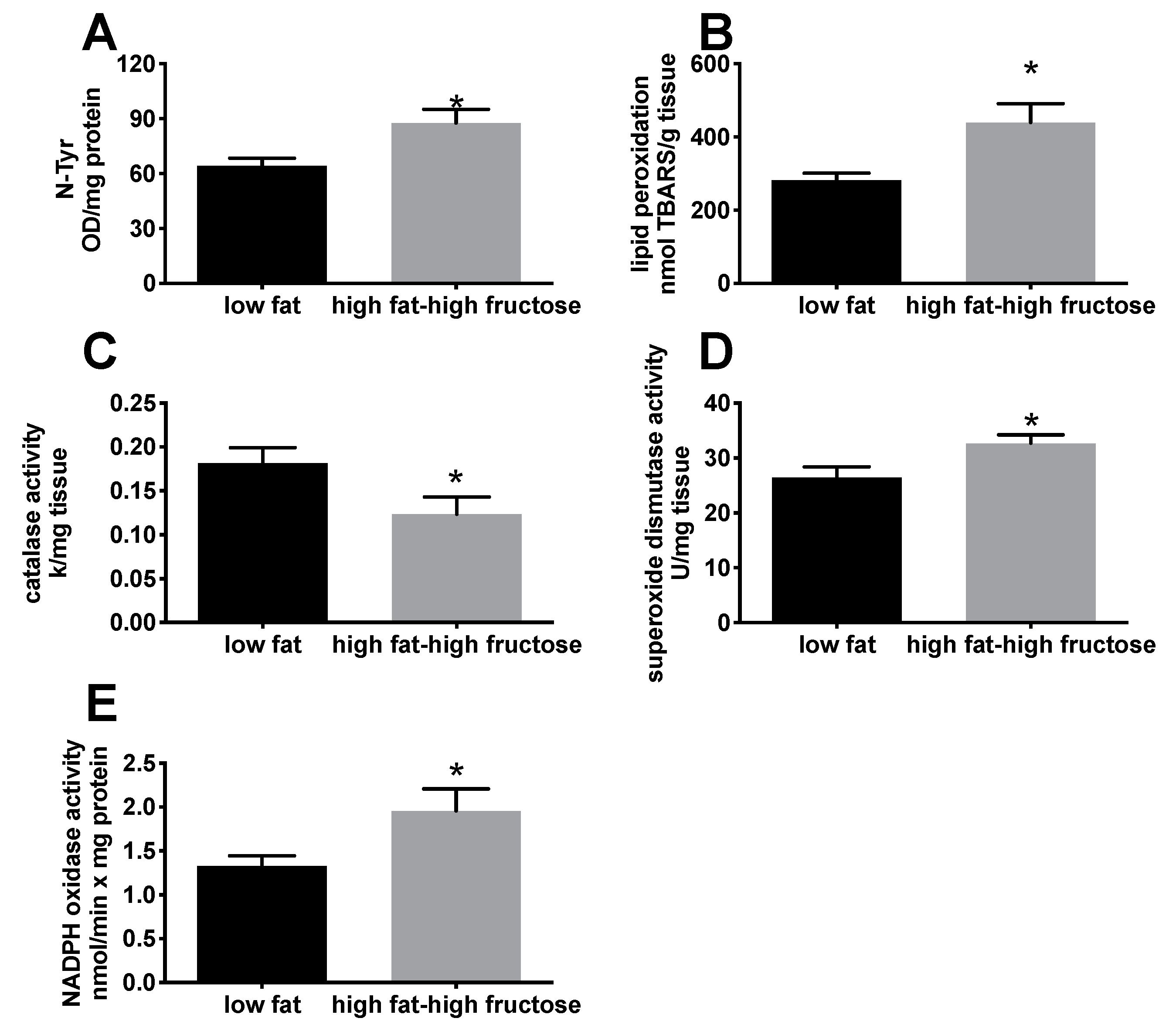
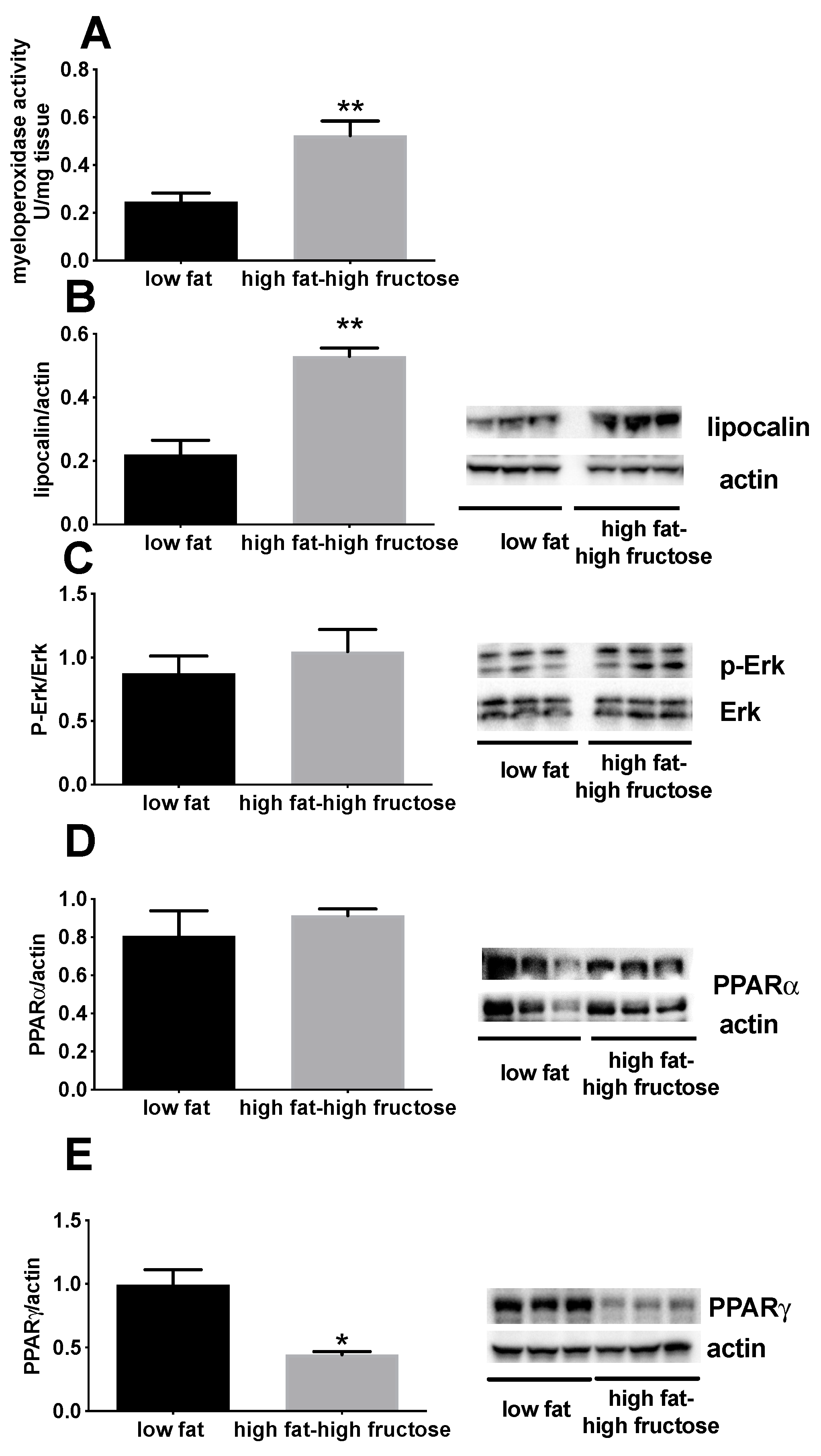
3.2. Liver Oxidative Status and Inflammation Markers
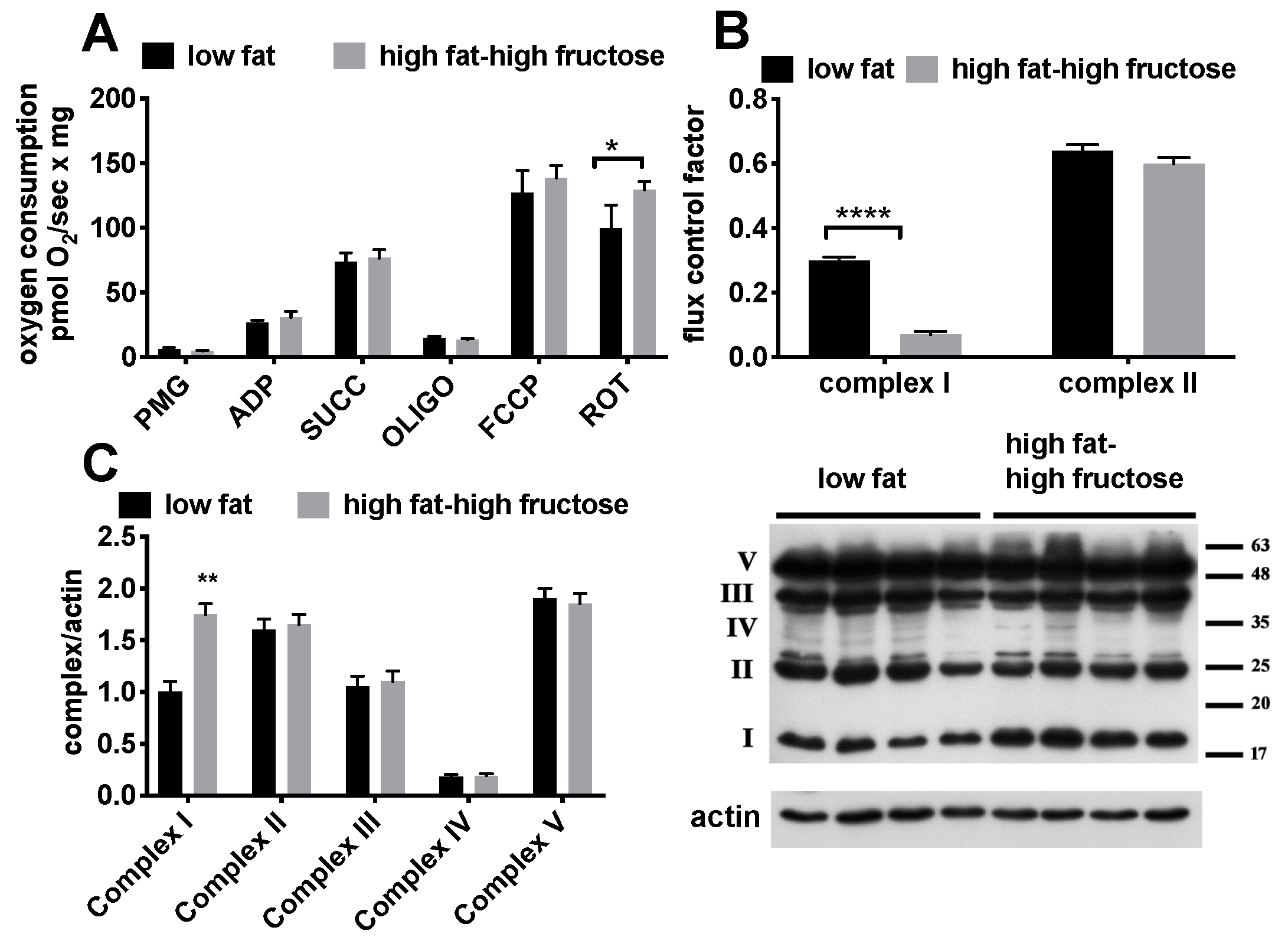
3.3. Intrinsic Mitochondrial Function
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Piper, M.D.W.; Bartke, A. Diet and aging. Cell Metab. 2008, 8, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Bohr, V.A.; de Cabo, R. Nutrition and aging. Mech. Ageing Dev. 2010, 131, 223–224. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Galluzzi, L.; Freije, J.M.P.; Madeo, F.; Kroemer, G. Metabolic Control of Longevity. Cell 2016, 166, 802–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crescenzo, R.; Bianco, F.; Mazzoli, A.; Giacco, A.; Liverini, G.; Iossa, S. A possible link between hepatic mitochondrial dysfunction and diet-induced insulin resistance. Eur. J. Nutr. 2016, 55, 1–6. [Google Scholar] [CrossRef]
- Lozano, I.; Van der Werf, R.; Bietiger, W.; Seyfritz, E.; Peronet, C.; Pinget, M.; Jeandidier, N.; Maillard, E.; Marchioni, E.; Sigrist, E.; et al. High-fructose and high-fat diet-induced disorders in rats: Impact on diabetes risk, hepatic and vascular complications. Nutr. Metab. 2016, 13, 15. [Google Scholar] [CrossRef]
- Lohr, K.; Pachl, F.; Moghaddas Gholami, A.; Geillinger, K.E.; Daniel, H.; Kuster, B.; Klingenspor, M. Reduced mitochondrial mass and function add to age-related susceptibility toward diet-induced fatty liver in C57BL/6J mice. Physiol. Rep. 2016, 4, e12988. [Google Scholar] [CrossRef] [Green Version]
- Sheedfar, F.; Sung, M.M.; Aparicio-Vergara, M.; Kloosterhuis, N.J.; Miquilena-Colina, M.E.; Vargas-Castrillón, J.; Febbraio, M.; Jacobs, R.L.; de Bruin, A.; Vinciguerra, M.; et al. Increased hepatic CD36 expression with age is associated with enhanced susceptibility to nonalcoholic fatty liver disease. Aging 2014, 6, 281–295. [Google Scholar] [CrossRef] [Green Version]
- Roberts, M.D.; Mobley, C.B.; Toedebush, R.G.; Heese, A.J.; Zhu, C.; Krieger, A.E.; Cruthirds, C.L.; Lockwood, C.M.; Hofheins, J.C.; Wiedmeyer, C.E.; et al. Western diet-induced hepatic steatosis and alterations in the liver transcriptome in adult Brown-Norway rats. BMC Gastroenterol. 2015, 15, 151. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, F.; Wu, O.; Li, M.; Zhu, Y.; Song, S.; Zhu, J.; Ma, Y.; Li, H.; Shi, X.; et al. Fish oil diet may reduce inflammatory levels in the liver of middle-aged rats. Sci. Rep. 2017, 7, 6241. [Google Scholar] [CrossRef] [Green Version]
- Ogden, C.L.; Carroll, M.D.; Fryar, C.D.; Flegal, K.M. Prevalence of obesity among adults and youth: United States, 2011–2014. NCHS Data Brief 2015, 219, 1–8. [Google Scholar]
- Bonomini, F.; Rodella, L.F.; Rezzani, R. Metabolic Syndrome, Aging and Involvement of Oxidative Stress. Aging Dis. 2015, 6, 109–120. [Google Scholar] [CrossRef]
- den Boer, M.; Voshol, P.J.; Kuipers, F.; Havekes, L.M.; Romijn, J.A. Hepatic steatosis: A mediator of the metabolic syndrome. Lessons from animal models. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 644–649. [Google Scholar] [CrossRef]
- Qureshiand, K.; Abrams, G.A. Metabolic liver disease of obesity and role of adipose tissue in the pathogenesis of nonalcoholic fatty liver disease. World J. Gastroenterol. 2007, 13, 3540–3553. [Google Scholar] [CrossRef] [PubMed]
- Meng, R.; Zhu, D.L.; Bi, Y.; Yang, D.H.; Wang, Y.P. Anti- oxidative effect of apocynin on insulin resistance in high-fat diet mice. Ann. Clin. Lab. Sci. 2011, 41, 236–243. [Google Scholar]
- Fouret, G.; Gaillet, S.; Lecomte, J.; Bonafos, B.; Djohan, F.; Barea, B.; Badia, E.; Coudray, C.; Feillet-Coudray, C. 20-week follow-up of hepatic steatosis installation and liver mitochondrial structure and activity and their interrelation in rats fed a high-fat–high-fructose diet. Br. J. Nutr. 2018, 119, 368–380. [Google Scholar] [CrossRef]
- Abdul-Ghani, M.A.; Matsuda, M.; Balas, B.; Defronzo, R.A. Muscle and liver insulin resistance indexes derived from the oral glucose tolerance test. Diabetes Care 2007, 30, 89–94. [Google Scholar] [CrossRef]
- Crescenzo, R.; Bianco, F.; Falcone, I.; Coppola, P.; Dulloo, A.G.; Liverini, G.; Iossa, S. Mitochondrial energetics in liver and skeletal muscle after energy restriction in young rats. Br. J. Nutr. 2012, 108, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Stanley, G.H.S. A simple method for the isolation and purification of total lipids from animal tissues. J. Biol. Chem. 1957, 226, 497–510. [Google Scholar] [PubMed]
- Crescenzo, R.; Bianco, F.; Coppola, P.; Mazzoli, A.; Cigliano, L.; Liverini, G.; Iossa, S. Increased skeletal muscle mitochondrial efficiency in rats with fructose-induced alteration in glucose tolerance. Br. J. Nutr. 2013, 110, 1996–2003. [Google Scholar] [CrossRef] [Green Version]
- Burtscher, J.; Zangrandi, L.; Schwarzer, C.; Gnaiger, E. Differences in mitochondrial function in homogenated samples from healthy and epileptic specific brain tissues revealed by high-resolution respirometry. Mitochondrion 2015, 25, 104–112. [Google Scholar] [CrossRef]
- Ernster, L.; Nordenbrand, K. Microsomal lipid peroxidation. In Methods in Enzymology; Colowick, S.P., Kaplan, N.O., Eds.; Academic Press: New York, NY, USA, 1967; pp. 574–580. [Google Scholar]
- Cigliano, L.; Spagnuolo, M.S.; Crescenzo, R.; Cancelliere, R.; Iannotta, L.; Mazzoli, A.; Liverini, G.; Iossa, S. Short-Term Fructose Feeding Induces Inflammation and Oxidative Stress in the Hippocampus of Young and Adult Rats. Mol. Neurobiol. 2018, 55, 2869–2883. [Google Scholar] [CrossRef] [PubMed]
- Maehly, A.C.; Chance, B. The assay of catalases and peroxidases. Methods Biochem. Anal. 1954, 1, 357–424. [Google Scholar]
- Flohè, L.; Otting, F. Superoxide dismutase assay. Methods Enzymol. 1984, 105, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Bettaieb, A.; Vazquez Prieto, M.A.; Rodriguez Lanzi, C.; Miatello, R.M.; Haj, F.G.; Fraga, C.G.; Oteiza, P.I. Epicatechin mitigates high fructose-associated insulin resistance by modulating redox signaling and endoplasmic reticulum stress. Free Radic. Biol. Med. 2014, 72, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Shajib, M.S.; Manocha, M.M.; Khan, W.I. Investigating intestinal inflammation in DSS-induced model of IBD. J. Vis. Exp. 2012, 60, 3678. [Google Scholar] [CrossRef]
- Strittmatter, P.; Spatz, L.; Corcoran, D.; Rogers, M.J.; Setlow, B.; Redline, R. Purification and properties of rat liver microsomal stearyl coenzyme A desaturase. Proc. Natl. Acad. Sci. USA 1974, 71, 4565–4569. [Google Scholar] [CrossRef]
- Penicaud, L.; Ferre, P.; Assimacopoulos-Jeannet, F.; Perdereau, D.; Leturque, A.; Jeanrenaud, B.; Picon, L.; Girard, J. Increased gene expression of lipogenic enzymes and glucose transporter in white adipose tissue of suckling and weaned obese Zucker rats. Biochem. J. 1991, 279, 303–308. [Google Scholar] [CrossRef] [Green Version]
- Ozera, J.; Ratnerb, M.; Shaw, M.; Baileya, W.; Schomaker, S. The current state of serum biomarkers of hepatotoxicity. Toxicology 2008, 245, 194–205. [Google Scholar] [CrossRef]
- Krawisz, J.E.; Sharon, P.; Stenson, W.F. Quantitative assay for acute intestinal inflammation based on myeloperoxidase activity. Assessment of inflammation in rat and hamster models. Gastroenterology 1984, 87, 1344–1350. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, Y.; Zhang, Y.; LeRoith, D.; Bernlohr, D.A.; Chen, X. The Role of Lipocalin 2 in the Regulation of Inflammation in Adipocytes and Macrophages. Mol. Endocrinol. 2008, 22, 1416–1426. [Google Scholar] [CrossRef] [Green Version]
- Drake, I.; Sonestedt, E.; Ericson, U.; Wallström, P.; Orho-Melander, M. A Western dietary pattern is prospectively associated with cardio-metabolic traits and incidence of the metabolic syndrome. Br. J. Nutr. 2018, 119, 1168–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dardevet, D.; Sornet, C.; Balage, M.; Grizard, J. Stimulation of in vitro rat muscle protein synthesis by leucine decreases with age. J. Nutr. 2000, 130, 2630–2635. [Google Scholar] [CrossRef] [PubMed]
- Crescenzo, R.; Bianco, F.; Coppola, P.; Mazzoli, A.; Tussellino, M.; Carotenuto, R.; Liverini, G.; Iossa, S. Fructose supplementation worsens the deleterious effects of short-term high-fat feeding on hepatic steatosis and lipid metabolism in adult rats. Exp. Physiol. 2014, 99, 1203–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crescenzo, R.; Bianco, F.; Coppola, P.; Mazzoli, A.; Cigliano, L.; Liverini, G.; Iossa, S. The effect of high-fat--high-fructose diet on skeletal muscle mitochondrial energetics in adult rats. Eur. J. Nutr. 2015, 54, 183–192. [Google Scholar] [CrossRef]
- Crescenzo, R.; Bianco, F.; Falcone, I.; Tsalouhidou, S.; Yepuri, G.; Mougios, V.; Dulloo, A.G.; Liverini, G.; Iossa, S. Hepatic mitochondrial energetics during catch-up fat with high-fat diets rich in lard or safflower oil. Obesity 2012, 20, 1763–1772. [Google Scholar] [CrossRef] [PubMed]
- Fernández Gianotti, T.; Burgueño, A.; Gonzales Mansilla, N.; Pirola, C.J.; Sookoian, S. Fatty liver is associated with transcriptional downregulation of stearoyl-CoA desaturase and impaired protein dimerization. PLoS ONE 2013, 8, e76912. [Google Scholar] [CrossRef] [PubMed]
- Stefan, N.; Peter, A.; Cegan, A.; Staiger, H.; Machann, J.; Schick, F.; Claussen, C.D.; Fritsche, A.; Häring, H.U.; Schleicher, E. Low hepatic stearoyl-CoA desaturase 1 activity is associated with fatty liver and insulin resistance in obese humans. Diabetologia 2008, 51, 648–656. [Google Scholar] [CrossRef] [Green Version]
- Mensenkamp, A.R.; Havekes, L.M.; Romijn, J.A.; Kuipers, F. Hepatic steatosis and very low density lipoprotein secretion: The involvement of apolipoprotein E. J. Hepatol. 2001, 35, 816–822. [Google Scholar] [CrossRef]
- Ntambi, J.M.; Miyazaki, M. Regulation of stearoyl-CoA desaturases and role in metabolism. Prog. Lipid Res. 2004, 43, 91–104. [Google Scholar] [CrossRef]
- Iossa, S.; Lionetti, L.; Mollica, M.P.; Crescenzo, R.; Barletta, A.; Liverini, G. Effect of long-term high-fat feeding on energy balance and liver oxidative activity in rats. Br. J. Nutr. 2000, 84, 377–385. [Google Scholar] [CrossRef] [Green Version]
- Garnol, T.; Endlicher, R.; Kučera, O.; Drahota, Z.; Cervinková, Z. Impairment of mitochondrial function of rat hepatocytes by high fat diet and oxidative stress. Physiol. Res. 2014, 63, 271–274. [Google Scholar] [PubMed]
- Endlicher, R.; Kriváková, P.; Rauchová, H.; Nusková, H.; Cervinková, Z.; Drahota, Z. Peroxidative damage of mitochondrial respiration is substrate-dependent. Physiol. Res. 2009, 58, 685–692. [Google Scholar] [PubMed]
- Guichard, C.; Moreau, R.; Pessayre, D.; Epperson, T.K.; Krause, K.H. NOX family NADPH oxidases in liver and in pancreatic islets: A role in the metabolic syndrome and diabetes? Biochem. Soc. Trans. 2008, 36, 920–929. [Google Scholar] [CrossRef] [PubMed]
- De Minicis, S.; Seki, E.; Paik, Y.H.; Osterreicher, C.H.; Kodama, Y.; Kluwe, J.; Torozzi, L.; Miyai, K.; Benedetti, A.; Schwabe, R.F.; et al. Role and cellular source of nicotinamide adenine dinucleotide phosphate oxidase in hepatic fibrosis. Hepatology 2010, 52, 1420–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Ruiz, I.; Solís-Muñoz, P.; Fernández-Moreira, D.; Grau, M.; Colina, F.; Muñoz-Yagüe, T.; Solís-Herruzo, J.A. High fat diet decreases activity of the oxidative phosphorylation enzymes and causes nonalcoholic steatohepatitis in mice. Dis. Model. Mech. 2014, 7, 1287–1296. [Google Scholar] [CrossRef] [PubMed]
- Feillet-Coudray, C.; Fouret, G.; Vigor, C.; Bonafos, B.; Jover, B.; Blachnio-Zabielska, A.; Rieusset, J.; Casas, F.; Gaillet, S.; Landrier, J.F.; et al. Long-term measures of dyslipidemia.; inflammation.; and oxidative stress in rats fed a high-fat/high-fructose diet. Lipids 2019, 54, 81–97. [Google Scholar] [CrossRef]
- García-Ruiz, I.; Solís-Muñoz, P.; Fernández-Moreira, D.; Muñoz-Yagüe, T.; Solís-Herruzo, J.A. In vitro treatment of HepG2 cells with saturated fatty acids reproduces mitochondrial dysfunction found in nonalcoholic steatohepatitis. Dis. Model. Mech. 2015, 8, 183–191. [Google Scholar] [CrossRef]
- Cheranov, S.Y.; Jaggar, J.H. TNF-α dilates cerebral arteries via NAD(P)H oxidase-dependent Ca2+ spark activation. Am. J. Physiol. 2006, 290, C964–C971. [Google Scholar] [CrossRef]
- Silva, A.K.S.; Peixoto, C.A. Role of peroxisome proliferator-activated receptors in non-alcoholic fatty liver disease inflammation. Cell. Mol. Life Sci. 2018, 75, 2951–2961. [Google Scholar] [CrossRef]
- Sultan, S.; Pascucci, M.; Ahmad, S.; Malik, I.A.; Bianchi, A.; Ramadori, P.; Ahmad, G.; Ramadori, G. Lipocalin-2 is a major acute-phase protein in a rat and mouse model of sterile abscess. Shock 2012, 37, 191–196. [Google Scholar] [CrossRef]
- Wang, Y.; Lam, K.S.; Kraegen, E.W.; Sweeney, G.; Zhang, J.; Tso, A.W.; Chow, W.S.; Wat, N.M.; Xu, J.Y.; Hoo, R.L.; et al. Lipocalin-2 is an inflammatory marker closely associated with obesity, insulin resistance, and hyperglycemia in humans. Clin. Chem. 2007, 53, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Nilsen-Hamilton, M. Identification of a new acute phase protein. J. Biol. Chem. 1995, 270, 22565–22570. [Google Scholar] [CrossRef] [PubMed]
- Alwahsh, S.M.; Xu, M.; Seyhan, H.A.; Ahmad, S.; Mihm, S.; Ramadori, G.; Schultze, F.C. Diet high in fructose leads to an overexpression of lipocalin-2 in rat fatty liver. World J. Gastroenterol. 2014, 20, 1807–1821. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Low Fat | High Fat-High Fructose | |
|---|---|---|
| Component, g/1000 g | ||
| Standard Chow a | 395.3 | 231.5 |
| Sunflower oil | 19.3 | 19.3 |
| Casein | 59.7 | 133.3 |
| Water | 175.7 | 175.4 |
| AIN-93 Mineral mix | 11.4 | 11.4 |
| AIN-93 Vitamin mix | 3.2 | 3.2 |
| Choline | 0.7 | 0.7 |
| Methionine | 0.9 | 0.9 |
| Cornstarch | 333.8 | ------- |
| Butter | ------- | 129.8 |
| Fructose | ------- | 294.6 |
| Energy content and composition | ||
| ME content, kJ/g b | 11.2 | 14.9 |
| Lipids, J/100 J | 10.5 | 39.3 |
| Proteins, J/100J | 19.9 | 19.8 |
| Complex carbohydrates, J/100 J | 63.9 | 7.5 |
| Simple sugars, J/100 J | 5.7 | 33.4 |
| Initial Values | Low Fat | High Fat-High Fructose | |
|---|---|---|---|
| Body Composition | |||
| Initial body weight, g | 642.0 ± 40.0 | 643.0 ± 40.0 | |
| Final body weight, g | 654.0 ± 40.0 | 678.0 ± 40.0 | |
| Body weight gain, g | 12.0 ± 1.0 | 35.0 ± 2.0# | |
| Body energy, kJ | 15.0 ± 0.6 | 15.5 ± 0.3 | 15.2 ± 0.7 |
| Body lipids, g/100 g b.w. | 21.6 ± 1.8 | 25.3 ± 1.6 * | 26.7 ± 3.0 * |
| Body proteins, g/100 g b.w. | 27.6 ± 1.8 | 23.6 ± 2.0 * | 20.0 ± 2.0 * |
| Body water, g/100 g b.w. | 58.5 ± 1.2 | 56.7 ± 0.9 | 55.5 ± 2.4 |
| Energy Balance | |||
| Food intake, g | 868.0 ± 31.0 | 719.0 ± 29.0 | |
| Metabolizable energy intake, kJ | 9524.0 ± 450.0 | 10,715.0 ± 864.0 | |
| Body energy gain, kJ | 477.0 ± 40.0 | 598.0 ± 30.0 # | |
| Body lipid gain, kJ | 1091.0 ± 100.0 | 1697 ± 100.0 # | |
| Body protein gain, kJ | −584.0 ± 30.0 | −1068.0 ± 100.0 # | |
| Energy expenditure, kJ | 9048.0 ± 400.0 | 10,117.0 ± 1000.0 | |
| Energetic efficiency, % | 5.0 ± 0.2 | 5.5 ± 0.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazzoli, A.; Crescenzo, R.; Cigliano, L.; Spagnuolo, M.S.; Cancelliere, R.; Gatto, C.; Iossa, S. Early Hepatic Oxidative Stress and Mitochondrial Changes Following Western Diet in Middle Aged Rats. Nutrients 2019, 11, 2670. https://0-doi-org.brum.beds.ac.uk/10.3390/nu11112670
Mazzoli A, Crescenzo R, Cigliano L, Spagnuolo MS, Cancelliere R, Gatto C, Iossa S. Early Hepatic Oxidative Stress and Mitochondrial Changes Following Western Diet in Middle Aged Rats. Nutrients. 2019; 11(11):2670. https://0-doi-org.brum.beds.ac.uk/10.3390/nu11112670
Chicago/Turabian StyleMazzoli, Arianna, Raffaella Crescenzo, Luisa Cigliano, Maria Stefania Spagnuolo, Rosa Cancelliere, Cristina Gatto, and Susanna Iossa. 2019. "Early Hepatic Oxidative Stress and Mitochondrial Changes Following Western Diet in Middle Aged Rats" Nutrients 11, no. 11: 2670. https://0-doi-org.brum.beds.ac.uk/10.3390/nu11112670