Oxidative Stress in Chronic Liver Disease and Portal Hypertension: Potential of DHA as Nutraceutical

 , , ,
, , , {kind=link}
{kind=link}
Abstract
:1. The Liver: Function and Cell Components
1.1. The Hepatic Lobule
1.2. Sinusoidal Cell Components
2. Liver Disease: Relevance of ACLD and Portal Hypertension
3. Major Contributors of ACLD Pathophysiology
3.1. Inflammation
3.2. Oxidative Stress
3.3. Fibrosis
3.4. Portal Hypertension and Other Complications
4. TG-DHA as a Novel Nutraceutical Approach
4.1. TG-DHA’s Mechanism of Action and Demonstrated Benefits for the Human Health
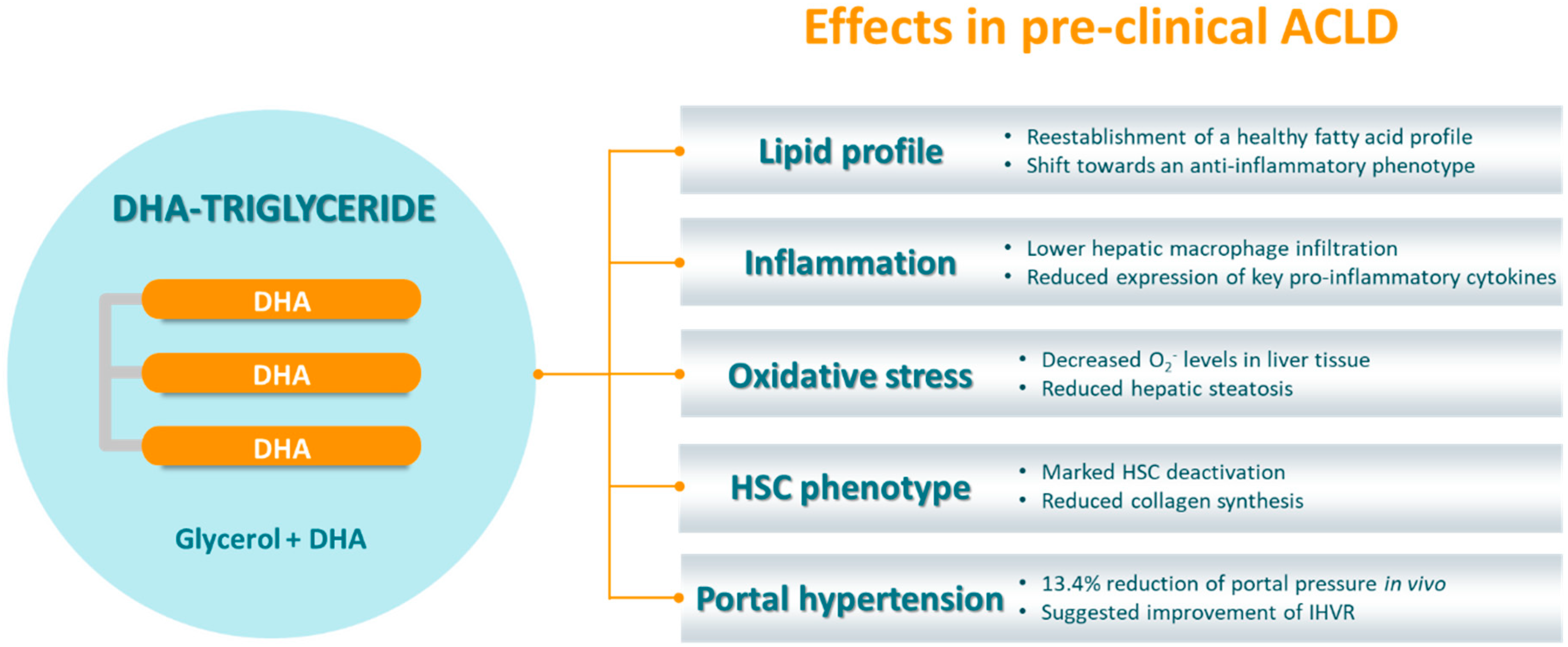
4.2. TG-DHA in ACLD
5. Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Prin, M.; Bakker, J.; Wagener, G. Hepatosplanchnic circulation in cirrhosis and sepsis. World J. Gastroenterol. 2015, 21, 2582–2592. [Google Scholar] [CrossRef]
- Trefts, E.; Gannon, M.; Wasserman, D.H. The liver. Curr. Biol. 2017, 27, R1147–R1151. [Google Scholar] [CrossRef]
- Sasse, D.; Spornitz, U.M.; Maly, I.P. Liver architecture. Enzyme 1992, 46, 8–32. [Google Scholar] [CrossRef] [PubMed]
- Hijmans, B.S.; Grefhorst, A.; Oosterveer, M.H.; Groen, A.K. Zonation of glucose and fatty acid metabolism in the liver: Mechanism and metabolic consequences. Biochimie 2014, 96, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Berndt, N.; Kolbe, E.; Gajowski, R.; Eckstein, J.; Ott, F.; Meierhofer, D.; Holzhütter, H.G.; Matz-Soja, M. Functional consequences of metabolic zonation in murine livers: New insights for an old story. Hepatology 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marrone, G.; Gracia-Sancho, J. Hepatic microcirculation in chronic liver disease. In Liver Fail; Nova Science Publishers: Hauppauge, NY, USA, 2013; pp. 90–98. [Google Scholar]
- Braet, F.; Wisse, E. Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: A review. Comp. Hepatol. 2002, 12, 51–56. [Google Scholar]
- DeLeve, L.D. The hepatic sinusoidal endothelial cell: Morphology, function, and pathobiology. In The Liver: Biology and Pathobiology; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2009; pp. 371–388. [Google Scholar]
- Pinzani, M.; Gentilini, P. Biology of hepatic stellate cells and their possible relevance in the pathogenesis of portal hypertension in cirrhosis. Semin. Liver Dis. 1999, 19, 397–410. [Google Scholar] [CrossRef]
- Sato, M.; Suzuki, S.; Senoo, H. Hepatic stellate cells: Unique characteristics in cell biology and phenotype. Cell Struct. Funct. 2003, 28, 105–112. [Google Scholar] [CrossRef] [Green Version]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef]
- Ju, C.; Tacke, F. Hepatic macrophages in homeostasis and liver diseases: From pathogenesis to novel therapeutic strategies. Cell Mol. Immunol. 2016, 13, 316–327. [Google Scholar] [CrossRef] [Green Version]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Alcoholic liver disease. Nat. Rev. Dis. Prim. 2018, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Petta, S.; Grave, D.R. Diet, weight loss, and liver health in nonalcoholic fatty liver disease: Pathophysiology, evidence, and practice. Hepatology 2016, 63, 2032–2043. [Google Scholar] [CrossRef]
- Al-Dayyat, H.M.; Rayyan, Y.M.; Tayyem, R.F. Non-alcoholic fatty liver disease and associated dietary and lifestyle risk factors. Diabetes Metab. Syndr. Clin. Res. Rev. 2018, 12, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Ringelhan, M.; McKeating, J.A.; Protzer, U. Viral hepatitis and liver cancer. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 1732. [Google Scholar] [CrossRef] [Green Version]
- Schuppan, D.; Afdhal, N.H. Liver cirrhosis. Lancet 2008, 371, 838–851. [Google Scholar] [CrossRef]
- Rosselli, M.; MacNaughtan, J.; Jalan, R.; Pinzani, M. Beyond scoring: A modern interpretation of disease progression in chronic liver disease. Gut 2013, 62, 1234–1241. [Google Scholar] [CrossRef]
- Tsochatzis, E.A.; Bosch, J.; Burroughs, A.K. Liver cirrhosis. Lancet 2014, 383, 1749–1761. [Google Scholar] [CrossRef]
- Bosch, J.; Abraldes, J.G.; Berzigotti, A.; García-Pagan, J.C. The clinical use of HVPG measurements in chronic liver disease. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 573–582. [Google Scholar] [CrossRef]
- D’Amico, G.; Garcia-Tsao, G.; Pagliaro, L. Natural history and prognostic indicators of survival in cirrhosis: A systematic review of 118 studies. J. Hepatol. 2006, 44, 217–231. [Google Scholar] [CrossRef]
- Garcia-Tsao, G.; Bosch, J. Management of varices and variceal hemorrhage in cirrhosis. N. Engl. J. Med. 2010, 362, 823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Nagalli, S. Chronic liver disease. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar] [CrossRef]
- Berzigotti, A.; Bosch, J. Pharmacologic management of portal hypertension. Clin. Liver. Dis. 2014, 18, 303–317. [Google Scholar] [CrossRef] [PubMed]
- Vilaseca, M.; Guixé-Muntet, S.; Fernández-Iglesias, A.; Gracia-Sancho, J. Advances in therapeutic options for portal hypertension. Therap. Adv. Gastroenterol. 2018, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Guixé-Muntet, S.; Zhu, C.P.; Xie, W.F.; Gracia-Sancho, J. Novel therapeutics for portal hypertension and fibrosis in chronic liver disease. Pharmacol. Ther. 2020, 215, 107626. [Google Scholar] [CrossRef]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Milić, S.; Lulić, D.; Štimac, D. Non-alcoholic fatty liver disease and obesity: Biochemical, metabolic and clinical presentations. World J. Gastroenterol. 2014, 20, 9330–9337. [Google Scholar] [CrossRef]
- Hardy, T.; Oakley, F.; Anstee, Q.M.; Day, C.P. Nonalcoholic fatty liver disease: Pathogenesis and disease spectrum. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 451–496. [Google Scholar] [CrossRef]
- Luedde, T.; Schwabe, R.F. NF-κB in the liver-linking injury, fibrosis and hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 108–118. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Iglesias, A.; Gracia-Sancho, J. How to face chronic liver disease: The sinusoidal perspective. Front. Med. 2017, 4, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Baeck, C.; Wehr, A.; Karlmark, K.R.; Heymann, F.; Vucur, M.; Gassler, N.; Huss, S.; Klussmann, S.; Eulberg, D.; Luedde, T.; et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut 2012, 61, 416–426. [Google Scholar] [CrossRef]
- Roh, Y.S.; Zhang, B.; Loomba, R.; Seki, E. TLR2 and TLR9 contribute to alcohol-mediated liver injury through induction of CXCL1 and neutrophil infiltration. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, 30–41. [Google Scholar] [CrossRef] [Green Version]
- Tacke, F.; Zimmermann, H.W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 2014, 60, 1090–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marra, F.; Tacke, F. Roles for chemokines in liver disease. Gastroenterology 2014, 147, 577–594.e1. [Google Scholar] [CrossRef] [PubMed]
- Koyama, Y.; Brenner, D.A. Liver inflammation and fibrosis. J. Clin. Investig. 2017, 127, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Guillot, A.; Tacke, F. Liver macrophages: Old dogmas and new insights. Hepatol. Commun. 2019, 3, 730–743. [Google Scholar] [CrossRef] [Green Version]
- Liaskou, E.; Wilson, D.V.; Oo, Y.H. Innate immune cells in liver inflammation. Mediat. Inflamm. 2012, 2012, 949157. [Google Scholar] [CrossRef] [Green Version]
- Hilscher, M.B.; Sehrawat, T.; Arab, J.P.; Zeng, Z.; Gao, J.; Liu, M.; Kostallari, E.; Gao, Y.; Simonetto, D.A.; Yaqoob, U.; et al. Mechanical stretch increases expression of CXCL1 in liver sinusoidal endothelial cells to recruit neutrophils, generate sinusoidal microthombi, and promote portal hypertension. Gastroenterology 2019, 157, 193–209.e9. [Google Scholar] [CrossRef]
- Gao, B.; Tsukamoto, H. Inflammation in alcoholic and nonalcoholic fatty liver disease: Friend or foe? Gastroenterology 2016, 150, 1704–1709. [Google Scholar] [CrossRef] [Green Version]
- Brieger, K.; Schiavone, S.; Miller, F.J.; Krause, K.H. Reactive oxygen species: From health to disease. Swiss Med. Wkly. 2012, 142, w13659. [Google Scholar] [CrossRef]
- Li, S.; Tan, H.Y.; Wang, N.; Zhang, Z.J.; Lao, L.; Wong, C.W.; Feng, Y. The role of oxidative stress and antioxidants in liver diseases. Int. J. Mol. Sci. 2015, 16, 26087–26124. [Google Scholar] [CrossRef] [Green Version]
- Gracia-Sancho, J.; Laviña, B.; Rodríguez-Vilarrupla, A.; García-Calderó, H.; Fernández, M.; Bosch, J.; García-Pagán, J.-C. Increased oxidative stress in cirrhotic rat livers: A potential mechanism contributing to reduced nitric oxide bioavailability. Hepatology 2008, 47, 1248–1256. [Google Scholar] [CrossRef] [PubMed]
- Abdelmegeed, M.A.; Song, B.J. Functional roles of protein nitration in acute and chronic liver diseases. Oxid. Med. Cell. Longev. 2014, 2014, 149627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Ruiz, C.; Fernández-Checa, J.C. Mitochondrial oxidative stress and antioxidants balance in fatty liver disease. Hepatol. Commun. 2018, 2, 1425–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [Green Version]
- Urtasun, R.; Nieto, N. Hepatic stellate cells and oxidative stress. Rev. Esp. Enferm. Dig. 2007, 99, 223–230. [Google Scholar]
- Moreno-Alvarez, P.; Sosa-Garrocho, M.; Briones-Orta, M.A.; González-Espinosa, C.; Medina-Tamayo, J.; Molina-Jijón, E.; Pedraza-Chaverri, J.; Macías-Silva, M. Angiotensin II increases mRNA levels of all TGF-β isoforms in quiescent and activated rat hepatic stellate cells. Cell. Biol. Int. 2010, 34, 969–978. [Google Scholar] [CrossRef]
- Zhang, F.; Ni, C.; Kong, D.; Zhang, X.; Zhu, X.; Chen, L.; Lu, Y.; Zheng, S. Ligustrazine attenuates oxidative stress-induced activation of hepatic stellate cells by interrupting platelet-derived growth factor-β receptor-mediated ERK and p38 pathways. Toxicol. Appl. Pharmacol. 2012, 265, 51–60. [Google Scholar] [CrossRef]
- Gandhi, C.R. Oxidative stress and hepatic stellate cells: A paradoxical relationship. Trends Cell. Mol. Biol. 2012, 7, 1–10. [Google Scholar]
- Cogger, V.C.; Muller, M.; Fraser, R.; McLean, A.J.; Khan, J.; Le Couteur, D.G. The effects of oxidative stress on the liver sieve. J. Hepatol. 2004, 41, 370–376. [Google Scholar] [CrossRef]
- Förstermann, U. Nitric oxide and oxidative stress in vascular disease. Pflugers Arch. Eur. J. Physiol. 2010, 459, 923–939. [Google Scholar] [CrossRef]
- Puche, J.E.; Saiman, Y.; Friedman, S.L. Hepatic stellate cells and liver fibrosis. Compr. Physiol. 2013, 3, 1473–1492. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Sancho, J.; Marrone, G.; Fernández-Iglesias, A. Hepatic microcirculation and mechanisms of portal hypertension. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Iredale, J.P.; Benyon, R.C.; Pickering, J.; McCullen, M.; Northrop, M.; Pawley, S.; Hovell, C.; Arthur, M.J. Mechanisms of spontaneous resolution of rat liver fibrosis: Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J. Clin. Investig. 1998, 102, 538–549. [Google Scholar] [CrossRef]
- Chang, T.T.; Liaw, Y.F.; Wu, S.S.; Schiff, E.; Han, K.H.; Lai, C.L.; Safadi, R.; Lee, S.S.; Halota, W.; Goodman, Z. Long-term entecavir therapy results in the reversal of fibrosis/cirrhosis and continued histological improvement in patients with chronic hepatitis B. Hepatology 2010, 52, 886–893. [Google Scholar] [CrossRef]
- Ellis, E.L.; Mann, D.A. Clinical evidence for the regression of liver fibrosis. J. Hepatol. 2012, 56, 1171–1180. [Google Scholar] [CrossRef] [Green Version]
- Marcellin, P.; Gane, E.; Buti, M.; Afdhal, N.; Sievert, W.; Jacobson, I.M.; Washington, M.K.; Germanidis, G.; Flaherty, J.F.; Schall, R.A. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: A 5-year open-label follow-up study. Lancet 2013, 381, 468–475. [Google Scholar] [CrossRef]
- Gracia-Sancho, J.; Laleman, W. Mechanisms of portal hypertension: Bench to bedside. Clin. Liver Dis. 2016, 8, 160–166. [Google Scholar] [CrossRef]
- Bhathal, P.S.; Grossman, H.J. Reduction of the increased portal vascular resistance of the isolated perfused cirrhotic rat liver by vasodilators. J. Hepatol. 1985, 1, 325–337. [Google Scholar] [CrossRef]
- Vorobioff, J.; Bredfeldt, J.E.; Groszmann, R.J. Increased blood flow through the portal system in cirrhotic rats. Gastroenterology 1984, 87, 1120–1126. [Google Scholar] [CrossRef]
- Iwakiri, Y.; Groszmann, R.J. The hyperdynamic circulation of chronic liver diseases: From the patient to the molecule. Hepatology 2006, 43, S121–S131. [Google Scholar] [CrossRef] [PubMed]
- Cales, P.; Zabotto, B.; Meskens, C.; Caucanas, J.-P.; Vinel, J.-P.; Desmorat, H.; Fermanian, J.; Pascal, J.P. Gastroesophageal endoscopic features in cirrhosis: Observer variability, interassociations, and relationship to hepatic dysfunction. Gastroenterology 1990, 98, 156–162. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Bosch, J.; Blei, A.; Arroyo, V. Portal hypertension and its complications. Gastroenterology 2008, 134, 1715–1728. [Google Scholar] [CrossRef] [Green Version]
- Plourde, M.; Cunnane, S.C. Extremely limited synthesis of long chain polyunsaturates in adults: Implications for their dietary essentiality and use as supplements. Appl. Physiol. Nutr. Metab. 2007, 32, 619–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyerberg, J.; Madsen, P.; Møller, J.M.; Aardestrup, I.; Schmidt, E.B. Bioavailability of marine n-3 fatty acid formulations. Prostaglandins Leukot. Essent. Fat. Acids 2010, 83, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Burdge, G.C.; Calder, P.C. Dietary α-linolenic acid and health-related outcomes: A metabolic perspective. Nutr. Res. Rev. 2006, 19, 26–52. [Google Scholar] [CrossRef] [PubMed]
- Arterburn, L.M.; Hall, E.B.; Oken, H. Distribution, interconversion, and dose response of n-3 fatty acids in humans. Am. J. Clin. Nutr. 2006, 83, 1467S–1476S. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Mechanisms of action of (n-3) fatty acids. J. Nutr. 2012, 142, 592S–599S. [Google Scholar] [CrossRef] [Green Version]
- Calder, P.C. Omega-3 fatty acids and inflammatory processes. Nutrients 2010, 2, 355–374. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Cruz, M.; Serna, D.S. Nutrigenomics of ω-3 fatty acids: Regulators of the master transcription factors. Nutrition 2017, 41, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Saini, R.K.; Keum, Y.S. Omega-3 and omega-6 polyunsaturated fatty acids: Dietary sources, metabolism, and significance—A review. Life Sci. 2018, 203, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Pedrol, J.C.D.; Garcia, J.A.V. Use of DHA for Treating a Pathology Associated with Cellular Oxidative Damage. European Patent EP1962825B1, 2 April 2014. [Google Scholar]
- Gómez-Soler, M.; Cordobilla, B.; Morató, X.; Fernández-Dueñas, V.; Domingo, J.C.; Ciruela, F. Triglyceride form of docosahexaenoic acid mediates neuroprotection in experimental parkinsonism. Front. Neurosci. 2018, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mancera, P.; Wappenhans, B.; Cordobilla, B.; Virgili, N.; Pugliese, M.; Rueda, F.; Espinosa-Parrilla, J.F.; Domingo, J.C. Natural docosahexaenoic acid in the triglyceride form attenuates in vitro microglial activation and ameliorates autoimmune encephalomyelitis in mice. Nutrients 2017, 9, 681. [Google Scholar] [CrossRef]
- González-Herrero, M.E.R.; Ruiz, M.; Román, F.J.L.; Sánchez, J.M.M.; Domingo, J.C. Supplementation with a highly concentrated docosahexaenoic acid plus xanthophyll carotenoid multivitamin in nonproliferative diabetic retinopathy: Prospective controlled study of macular function by fundus microperimetry. Clin. Ophthalmol. 2018, 12, 1011–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, C.; García, T.; Areces, D.; Fernández, E.; García-Noriega, M.; Domingo, J.C. Supplementation with high-content docosahexaenoic acid triglyceride in attentiondeficit hyperactivity disorder: A randomized double-blind placebo-controlled trial. Neuropsychiatr. Dis. Treat. 2019, 15, 1193–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Soto, J.C.; Domingo, J.C.; Cordobilla, B.; Nicolás, M.; Fernández, L.; Albero, P.; Gadea, J.; Landeras, J. Dietary supplementation with docosahexaenoic acid (DHA) improves seminal antioxidant status and decreases sperm DNA fragmentation. Syst. Biol. Reprod. Med. 2016, 62, 387–395. [Google Scholar] [CrossRef] [Green Version]
- Boyer-Diaz, Z.; Domingo, J.C.; de Gregorio, E.; Manicardi, N.; Aristu-Zabalza, P.; Cordobilla, B.; Abad-Jordà, L.; Ortega-Ribera, M.; Fernández-Iglesias, A.; Marí, M.; et al. A nutraceutical rich in docosahexaenoic acid improves portal hypertension in a preclinical model of advanced chronic liver disease. Nutrients 2019, 11, 2358. [Google Scholar] [CrossRef] [Green Version]
- Bosch, J.; Berzigotti, A.; Garcia-Pagan, J.C.; Abraldes, J.G. The management of portal hypertension: Rational basis, available treatments and future options. J. Hepatol. 2008, 48, 68–92. [Google Scholar] [CrossRef] [Green Version]
- Valenzuela, R.; Espinosa, A.; González-Mañán, D.; D’Espessailles, A.; Fernández, V.; Videla, L.A.; Tapia, G. N-3 long-chain polyunsaturated fatty acid supplementation significantly reduces liver oxidative stress in high fat induced steatosis. PLoS ONE 2012, 7, e46400. [Google Scholar] [CrossRef] [Green Version]
- El-Badry, A.M.; Moritz, W.; Contaldo, C.; Tian, Y.; Graf, R.; Clavien, P.A. Prevention of reperfusion injury and microcirculatory failure in macrosteatotic mouse liver by omega-3 fatty acids. Hepatology 2007, 45, 855–863. [Google Scholar] [CrossRef] [PubMed]
- El-Badry, A.M.; Graf, R.; Clavien, P.A. Omega 3-Omega 6: What is right for the liver? J. Hepatol. 2007, 47, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Dueñas, V.; Azuaje, J.; Morató, X.; Cordobilla, B.; Domingo, J.C.; Sotelo, E.; Ciruela, F. Synthesis and characterization of a new bivalent ligand combining caffeine and docosahexaenoic acid. Molecules 2017, 22, 366. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boyer-Diaz, Z.; Morata, P.; Aristu-Zabalza, P.; Gibert-Ramos, A.; Bosch, J.; Gracia-Sancho, J. Oxidative Stress in Chronic Liver Disease and Portal Hypertension: Potential of DHA as Nutraceutical. Nutrients 2020, 12, 2627. https://0-doi-org.brum.beds.ac.uk/10.3390/nu12092627
Boyer-Diaz Z, Morata P, Aristu-Zabalza P, Gibert-Ramos A, Bosch J, Gracia-Sancho J. Oxidative Stress in Chronic Liver Disease and Portal Hypertension: Potential of DHA as Nutraceutical. Nutrients. 2020; 12(9):2627. https://0-doi-org.brum.beds.ac.uk/10.3390/nu12092627
Chicago/Turabian StyleBoyer-Diaz, Zoe, Paloma Morata, Peio Aristu-Zabalza, Albert Gibert-Ramos, Jaime Bosch, and Jordi Gracia-Sancho. 2020. "Oxidative Stress in Chronic Liver Disease and Portal Hypertension: Potential of DHA as Nutraceutical" Nutrients 12, no. 9: 2627. https://0-doi-org.brum.beds.ac.uk/10.3390/nu12092627