
Berry-Derived Polyphenols in Cardiovascular Pathologies: Mechanisms of Disease and the Role of Diet and Sex



1
Department of Nutrition, Georgia State University, Atlanta, GA 30302, USA
2
Department of Kinesiology and Health, Georgia State University, Atlanta, GA 30302, USA
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Nutrients 2021, 13(2), 387; https://doi.org/10.3390/nu13020387
Submission received: 15 December 2020
/
Revised: 21 January 2021
/
Accepted: 22 January 2021
/
Published: 27 January 2021
(This article belongs to the Special Issue Gender and Diet Interaction in Cardiovascular Disease)
Abstract
:Cardiovascular disease (CVD) prevalence, pathogenesis, and manifestation is differentially influenced by biological sex. Berry polyphenols target several signaling pathways pertinent to CVD development, including inflammation, oxidative stress, and cardiac and vascular remodeling, and there are innate differences in these pathways that also vary by sex. There is limited research systematically investigating sex differences in berry polyphenol effects on these pathways, but there are fundamental findings at this time that suggest a sex-specific effect. This review will detail mechanisms within these pathological pathways, how they differ by sex, and how they may be individually targeted by berry polyphenols in a sex-specific manner. Because of the substantial polyphenolic profile of berries, berry consumption represents a promising interventional tool in the treatment and prevention of CVD in both sexes, but the mechanisms in which they function within each sex may vary.
{kind=link}
{kind=link}
{kind=link}
1. Introduction
There are sex differences in the prevalence and pathogenesis of cardiovascular diseases (CVD). These differences are likely related to endogenous estrogen effects, as epidemiological data suggest the menopausal transition, which occurs at approximately 50 years of age, is associated with significant changes in cardiovascular health and CVD diagnoses in females. For example, between ages 20–34 and ages 35–44, 13% and 32% of females are diagnosed with hypertension compared to 26% and 43% of males, respectively; however, 65% of females and 66% of males are diagnosed with hypertension between ages 55–64, and diagnoses in females exceed that of age-matched males in higher age brackets [1]. A similar trend is seen with the prevalence of total CVD, including hypertension, coronary artery disease (CAD), heart failure (HF), and stroke, where there is a 17% prevalence in females and 30% prevalence in males between ages 20–39, but prevalence increases to 52% and 57% from ages 40–59 and further to 78% and 77% from ages 60–79, respectively [1]. Regarding pathogenesis, approximately two-thirds of ischemic CVD cases in males are obstructive in nature, whereas two-thirds of ischemic CVD cases in females are non-obstructive [2]. This illustrates a greater atherosclerotic burden in males, especially in larger vessels, but a greater impact of dysregulated vascular function in females, and possibly a larger role for microvascular dysfunction in particular [3,4,5]. Divergent mechanisms of pathogenesis persist into HF as well, where females are more likely to develop HF with preserved ejection fraction (HFpEF) compared to males, who are more likely to develop HF with reduced ejection fraction (HFrEF) [6]. Collectively, this indicates the presence of sex-specific pathways in CVD pathogenesis, which likely includes a specific role for sex steroids and corresponding receptors, given the association with menopause in females. Further, the sex-specific pathogenesis of CVD indicates a potential for sex-specific mechanisms of intervention.
Inflammation, oxidative stress, and pathological remodeling of the cardiovascular system are consistent across a number of CVD-related pathologies and are major drivers of disease [7,8,9]. Biological sex (e.g., female and male) is a non-modifiable CVD risk factor and can influence these pathways through physiological mechanisms [10]. Whether there are sex-specific effects of lifestyle modifications (i.e., changes in diet) in CVD pathogenesis is not well understood. Dietary approaches that emphasize plants are associated with a reduced risk of CVD events [11,12], and consumption of such diets may reduce systemic inflammation [13], attenuate angina, improve endothelial function, reduce atherosclerotic lesions, and improve cardiac function and morphology in humans with CVD [14,15,16]. A major bioactive component of these diets are polyphenols, which likely play a key role in mediating these clinical effects at the cellular level [17]. Berries are an exceptionally rich source of polyphenols, particularly flavonoids and phenolic acids [18]. The dietary inclusion of berries can improve CVD risk factors, including a reduction of blood pressure and serum inflammatory cytokines [19,20], and is associated with reduced CVD mortality [21,22]. Thus, consumption of berries presents a major therapeutic strategy for CVD.
Polyphenol intake is greater in females compared to males, especially polyphenols from fruits and vegetables [23]. In a longitudinal study, males had lower total dietary polyphenol intake and lower total urinary polyphenols than females [24]. Though it is possible that these behavioral factors can influence sex differences in berry polyphenol-related outcomes, sex-specific outcomes are likely more so related to cellular effects of berry polyphenols in cardiovascular health and disease. Because males and females portray differences in cellular mechanisms of inflammation, oxidative stress, and pathological remodeling, the cellular effects of berry polyphenols may also affect CVD pathology in a sex-specific manner. In this review, we will discuss mechanisms of CVD and how polyphenols may serve to sex-specifically mitigate these effects. While we recognize the potential influence of gender (i.e., sociological or cultural gender roles) on human physiology and CVD risk, we will use terms referring to biological sex (i.e., male and female) throughout this review for brevity. Lastly, we focus, as much as possible, on human-based studies; however, in order to explain potential mechanisms, we do make reference to cell culture and animal studies where mechanistic studies in humans are either lacking or may be difficult to perform in humans.
2. Berry-Derived Polyphenols
Polyphenols are subdivided into four major classes: flavonoids, phenolic acids, lignans, and stilbenes. In berries, lignans are relatively absent [25] and stilbenes, particularly trans-resveratrol, constitute a small portion of their total polyphenol content [26]. In contrast, flavonoids and phenolic acids comprise the majority of the polyphenolic profile of berries [18]. Flavonoids are subdivided into six major subclasses: flavanols, flavonols, anthocyanins, flavones, flavanones, and isoflavones [27]. In berries, flavanols, flavonols, and anthocyanins are the dominant subclasses. Flavonols contain ketones with a hydroxyl group at position 3 of C ring, while flavanols lack the ketone of flavonols. Further, anthocyanins contain a positively charged oxygen at position 4 of C ring.
Phenolic acids are the most diverse class of polyphenols in berries and are derived in a number of ways. While phenolic acids can be unbound and free in the fruit, they can be derived from (1) metabolic and microbial degradation of flavonoids and (2) hydrolytic liberation of phenolic acid polymers (e.g., ellagitannins) during digestion [28]. Phenolic acids can be classified into two overarching categories, hydroxycinnamic acids, which contain a saturated tail followed by a carboxylic acid, and hydroxybenzoic acids, which lack tail saturation.
2.1. Metabolism of Berry-Derived Polyphenols
Polyphenol parent compounds are rarely detected in serum following consumption [29,30], as they are hydrolyzed into metabolites in the small intestine and undergo significant structural modifications by the gut microbiota and in the liver [31,32]. For example, 29 different metabolites were identified postprandially in serum of male subjects who consumed a 500 mg oral bolus of cyanidin-3-glucoside [30]. Polyphenols in the microbiome undergo a plethora of chemical reactions, including C- and O- glycosylation, hydrolysis of esters, dehydroxylation, and demethylation, all of which increase intestinal absorption [32]. Ellagitannins, rich in strawberries, blackberries, and raspberries [33,34], are hydrolysable polymers of ellagic acid and gallic acid. However, in the presence of gastric enzymes, hydrolysis does not occur to liberate ellagic and gallic acid, and instead microbial metabolism liberates these compounds, yielding further metabolism of ellagic acid into dibenzopyranone urolithins A and B. [35]. Following absorption, polyphenols are further modified in the liver, and undergo monooxygenation by cytochrome P450, and these modified compounds are then further catalyzed by phase II enzymes, UDP-glucuronosyl transferases and sulfotransferases, to increase solubility [36]. Isoforms of these enzymes have differing activity in males and females with no clear cumulative trend [37,38]. Further, intestinal transport of polyphenols differs between sexes, as females were found to intestinally hydrolyze quercetin from rutin (quercetin 3-rutinoside) far more efficiently than males, which led to increased serum concentrations of quercetin compared to males [39]. Once in serum, polyphenol metabolites are often albumin-bound, which can lower serum polyphenol concentrations [40], but albumin also stabilizes these metabolites from oxidative modifications [41] and may allow for slower release. This evidence does not suggest that lower concentrations of polyphenols observed physiologically are not efficacious. For example, bilberry extracts, which contain a number of anthocyanins, including cyanidin, delphinidin, petunidin, peonidin, and malvidin derivatives, reduced ischemia-reperfusion (I/R) injury in isolated male Wistar rat hearts at somewhat physiologically relevant concentrations of 0.1–1 mg/L [42]. This concentration reduced cardiac arrest, ventricular fibrillations, and tachycardia compared to control animals, but higher concentrations of bilberry (5–50 mg/L) resulted in a dose-dependent worsening of cumulative arrhythmias [42].
Polyphenols almost certainly exert their effects intracellularly due to changes in gene and protein expression (discussed below). However, berry-derived polyphenols are typically hydrophilic due to numerous hydroxyl groups. As such, crossing lipid bilayers poses a challenge. It has been observed that as hydroxyl group count decreases, hydrophobicity for membrane vesicles inversely increases [43]. Polyphenol metabolites, which contain far fewer hydroxyl groups, could passively diffuse across lipid bilayers. Indeed, microbial metabolism of polyphenols can yield metabolites that have been completely dehydroxylated [44,45]. Nonetheless, membrane transporters likely facilitate the uptake of more common hydrophilic moieties. In endothelial cells, bilitranslocase, a cell membrane transporter, was responsible for the uptake of the anthocyanin cyanidin-3-glucoside and corresponded with increased cellular antioxidant activity [46]. Further, in isolated male rat hearts that underwent I/R, cyanidin-3-glucoside significantly improved coronary flow, which was completely blunted by bilitranslocase antibody, preventing the absorption of cyanidin-3-glucoside. Bilitranslocase may also shuttle other anthocyanins, quercetin, and other polyphenols [47,48,49]. ATP-binding cassette transporters and solute carrier transporters may also be of relevance in polyphenol membrane transport, but evidence is ambiguous [50]. Males and females appear to express membrane transporters differently, with mixed findings in both sexes, but the clinical significance of these sex differences is unclear [51]. Further complicating matters, cross-species differences between metabolite appearance in serum and phase II enzyme activity between humans, rats, and mice differ substantially [52]. Thus, utilizing animal models as a proxy for humans to study sex-specific polyphenol metabolism may not be appropriate, and more human studies are needed.
2.2. Sex-Specific Effects of Polyphenols on the Microbiota
The gut microbiota is associated with cardiovascular function and disease (reviewed extensively by Battson et al. [53] and Witkowsky et al. [54]). Physiologic factors, such as age, biological sex, and hormone status, influence characteristics of the microbiota, such as diversity and abundance [55,56,57,58]. Females have increased microbiota diversity [56], although mixed observations have been observed regarding the abundance of Bacteriodetes (tend to be beneficial) versus Firmicutes (tend to unfavorable) in both males and females [57,59,60]. Hormone status or age may modulate these differences [55,58], as microbiome parameters are similar between males and postmenopausal females but not similar between postmenopausal and premenopausal females [55]. However, one of the greatest determinants of microbiota diversity and abundance of bacterial taxa is diet. This change in diversity can be observed within mere days. For example, in both males and females, switching from a conventional diet to a fiber-rich plant-based diet increased beneficial genus Prevotella, while switching to a low-fiber animal-based diet reduced Prevotella within four days [61]. Increased Prevotella corresponded with increased short-chain fatty acid synthesis, potent anti-inflammatory compounds which can attenuate CVD pathogenesis [62]. Considering the significant contribution of microbial metabolism in polyphenol metabolite generation, it is likely that berries modulate microbiota diversity in a positive manner.
Indeed, a number of berry polyphenols can positively improve gut microbiota diversity in animal models. For example, in male C57BL/6J mice fed a high-fat diet, supplementation of 200 mg/kg/d of blueberry extract for twelve weeks resulted in the reduction of Clostridium, a pathogenic microbe, compared to high-fat diet control animals [63]. Peculiarly, Prevotella also significantly decreased in blueberry extract-supplemented animals. Nonetheless, cardiac hypertrophy was significantly reduced as assessed by reduced heart weight. In high-fat diet-fed Wistar rats (sex unspecified), 30 mg/kg/d of quercetin for six weeks significantly decreased the Firmicutes/Bacteroidetes ratio, which corresponded with increased intestinal short-chain fatty acid production of acetate, propionate, and butyrate [64]. In male Fischer F344 rats treated with dextran sulfate sodium, an inducer of colitis, 1 mg/kg/d of resveratrol increased intestinal Lactobacillus and Bifidobacterium microbes compared to control animals [65], and these microbes are associated with reduced CVD risk factors [66,67]. Positive changes to microbiota in animal models due to berry polyphenols reflect positive clinical changes in human studies utilizing polyphenol-rich foods. For example, in overweight and obese males and females, 450 mg of pomegranate extract increased butyrate-producing Faecalibacterium, Butyricicoccus, and Butyricimonas, representing a positive microbial shift [68]. Additionally, the consumption of 494 cocoa flavanols for four weeks in healthy males and females significantly increased beneficial Lactobacillus and Bifidobacterium populations, while Clostridium significantly decreased [69]. In a separate investigation in males with metabolic syndrome, the 30-day consumption of 272 mL/day of red wine that contains a polyphenol profile similar to that of berries (rich in flavanols, flavonols, anthocyanins, and stilbenes), significantly reduced the Firmicutes/Bacteroidetes ratio and increased Faecalibacterium, Lactobacillus, and Bifidobacterium [70]. Lastly, a pilot study indicates a trend towards an increase in microbiota diversity in older females, who were not specified as postmenopausal but ranged from 65–77 years old, following six weeks of a blueberry-enriched diet, but there was no observed difference with the intervention in younger females (21–39 years) [71]. Considering the basal differences in microbiota population between sexes, it is likely that a sex-specific polyphenol effect does exist; however, systematic investigation of sex differences in polyphenol-mediated microbiota alterations are currently limited in humans but warranted.
2.3. Berry Polyphenol and Estrogen Receptor Interaction
There are physiological differences between males and females that impact many processes, including CVD pathogenesis, in a sex-specific manner [72]. Although females and males produce estrogens and androgens and possess estrogen and androgen receptors (ER and AR, respectively), there are sex differences in circulating sex steroid concentrations and downstream receptor-mediated signaling pathways. ER and AR are expressed throughout the cardiovascular system, in vascular smooth muscle cells (VSMC), endothelial cells, cardiomyocytes, cardiac fibroblasts, monocytes, macrophages, and other immune cells [73,74,75] in both sexes. Therefore, mediation of ER and AR can contribute to cardiovascular health and disease. 17β-estradiol (E2) is a non-specific ER agonist and the primary estrogen subtype in premenopausal females [76], and E2 is implicated in cardio-protection in premenopausal females. Testosterone is converted to E2 via aromatase, so E2 concentration in males is dependent on both testosterone and aromatase concentrations. E2 elicits several protective effects in cardiac and vascular tissues, primarily through ER-dependent mechanisms. Moreover, acute fluctuations in E2 across the menstrual cycle inversely affect inflammation [77] and oxidative stress [78] and directly affect antioxidant status in females [79]. When pertinent, this review will focus on effects of endogenous E2 and will not discuss synthetic estrogens or progestins, such as in oral contraceptives.
There appears to be nearly equivalent ER concentration across the cardiovascular system of male and female rats [80] and in myocardial samples of middle-aged, human males and postmenopausal females [81]. ER expression between human males and premenopausal females is currently unknown. Estrogen status is implicated in ER expression in humans, with postmenopausal females expressing significantly less ERα than premenopausal females [82]. Further, acute fluctuations in E2 in premenopausal females alter ERα expression, where ERα content was decreased by 30% during the early follicular phase of the menstrual cycle (i.e., low E2) compared to the late follicular phase of the menstrual cycle (i.e., elevated E2) [82]. Serum E2 concentration is under-investigated in human males [83], but one investigation allows a direct comparison of serum E2 in a sample of healthy, human postmenopausal females (n = 23), premenopausal females (n = 20, 60% of samples were donated during the ‘follicular’ phase), and males (n = 18) [84]. Serum parent estradiol averaged 31.2 pM/L (range: 10.3–90.9 pM/L) in postmenopausal females, 287 pM/L (range: 55.7–1507 pM/L) in premenopausal females, and 104 pM/L (range: 58.7–245 pM/L) in males [84]. This could suggest greater ER expression in premenopausal females than males based on estrogen concentration; however, empirical evidence is currently lacking.
Sex-specific polyphenolic action is likely related to ER mediation, possibly due to dynamic competitive binding interactions with E2, though there is evidence of beneficial effects of polyphenols that are ER-independent (discussed in detail below). Non-isoflavone polyphenols, such as ellagic acid, kaempferol, myricetin, epicatechin, and quercetin, may act as phytoestrogens, as they can act as ER ligands. However, it is unclear how ER concentration between sexes may affect polyphenolic actions in humans. Due to the cardio-protective effects of ER, it is possible that polyphenols may be of particular importance in the prevention of CVD in males who tend to be at greater risk. Polyphenol binding to ER occurs due to the structure of the ER binding pocket, in which there is a requirement for a phenolic ring to facilitate binding [85]. The hydroxyl group on the phenol ring of polyphenols further enhances ER binding due to hydrogen bonding within the pocket. This indicates that non-isoflavone polyphenols may induce estrogenic or antiestrogenic effects, as they may induce cellular responses similar to that of endogenous E2 or limit endogenous E2 effects by competing for ER binding sites. Some berry polyphenols possess differing ER subtype affinity. For instance, caffeic acid and quercetin have a greater binding affinity for ERβ than ERα [86,87], yet ellagic acid can bind more strongly to ERα and antagonizes ERβ [88]. Therefore, polyphenolic ER subtype selectivity may also contribute to sex-specific outcomes and yield cellular effects in an ER-dependent manner.
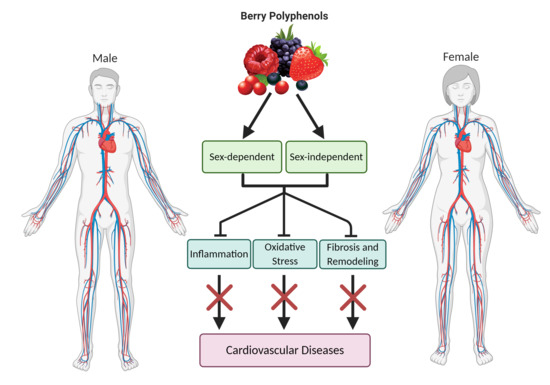
In the context of CVD, berry polyphenols target a variety of cellular pathways that drive these disease processes forward (Figure 1), including: (1) reducing inflammatory signaling, (2) reducing oxidative stress, and (3) reducing apoptosis and pathological remodeling. As suggested above, polyphenols may modulate these pathways in a sex-specific manner, facilitating targeted therapies in CVD. The majority of preclinical investigations using berry polyphenols to treat CVD have been performed with male animals, but polyphenols likely exert differing effects between sexes in a multifactorial manner, including ER-mediated effects (Figure 2). Supplementary Table S1 displays the search terms used for study compilation in this narrative review.
3. Inflammatory Signaling
In CVD, inflammation is a major driver of atherosclerosis, endothelial dysfunction, hypertension, and HF. In particular, activation of toll-like receptor (TLR)-4 and tumor necrosis factor receptor (TNFR) and their downstream signaling of mitogen activated protein kinase (MAPK) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) are primarily responsible for these deleterious effects [89,90,91,92,93,94]. NF-κB phosphorylation and nuclear translocation facilitates inflammatory cytokine and chemokine production, promoting leukocyte recruitment and infiltration into the endothelium and myocardium, as well as breakdown of the extracellular matrix and collagen deposition. In conjunction with NF-κB, MAPK phosphorylation facilitates VSMC and cardiac hypertrophy as well as cellular apoptosis [95,96,97,98,99,100,101].
3.1. Toll-Like Receptor-4
TLR-4 is found ubiquitously in the cardiovascular system and has a number of ligands. While classically activated by exogenous lipopolysaccharide (LPS), including low concentrations of LPS due to a high-fat meal [102], a variety of endogenous ligands are likely also relevant in CVD, including palmitate [103], oxidized low-density lipoprotein (LDL) [104], fibrinogen [105], and cardiac myosin [106]. Of note in TLR-4 activation is the adaptor protein myeloid differentiation factor (Myd88), which leads to recruitment of TNF receptor-associated factor-6 (TRAF6) and activation of transforming growth factor-β (TGF-β)-activated kinase 1 (TAK1) [107]. TAK1 is responsible for NF-κB and MAPK activation by phosphorylating upstream effectors IκB kinase (IKK) complex and mitogen-activated protein kinase kinase (MEK), respectively. E2 has been shown to inhibit LPS gene products by downregulating NF-κB signaling [75,108]. Sex differences in TLR-4 expression remain unclear, but it appears that E2 and testosterone may affect TLR-4 expression differently based on whether the target tissue is healthy or diseased [75].
While TLR-4 deficient animals are protected from atherosclerosis and HF [109,110], the isolated effects on endothelial function and hypertension are less clear [90,111]. Angiotensin (Ang) II is an oligopeptide and product of the renin-angiotensin-aldosterone system (RAAS), which elicits vasoconstriction classically through Ang II receptor type 1 (AT1R). Ang II may interact with glycoprotein MD2 on the TLR-4 receptor independent of AT1R, leading to its activation [112]. While TLR-4 blockade reduces blood pressure in spontaneously hypertensive rats (SHR) and deoxycorticosterone acetate-salt sensitive rats, these effects are not always consistent in Ang II-infused animals [90]. It appears that localized TLR-4 deletion in the central nervous system (CNS) is of particular importance in reducing blood pressure by mediating sympathetic and parasympathetic tone, thus reducing cardiac stress [113]. Inhibition of TLR-4 in the CNS results in a reduction of vasoconstricting properties of RAAS, namely reduced systemic norepinephrine, reduced cardiac AT1R, and reduced angiotensin converting enzyme (ACE) [113]. However, TLR-4 inhibition in the CNS also upregulated the ACE2 and Mas receptor, which act on the vasodilator side of the RAAS axis [113]. ACE2 catalyzes the conversion of Ang II to Ang (1–7) and Mas is the Ang (1–7) receptor. In humans, there are mixed results on Ang (1–7) concentration and associations with clinical outcomes between sexes [114,115].
3.2. Tumor Necrosis Factor Receptor
Tumor necrosis factor (TNF)-α is an inflammatory cytokine, which is elevated in those with CVD [116]. TNF-α can elicit cardiomyocyte apoptosis [117], endothelial dysfunction [118,119], and cardiac dysfunction. Additionally, deletion of the TNF gene from VSMCs improves microvascular function and improves blood pressure [120]. Interestingly, the detrimental effects of TNF-α appear entirely reversible upon removal [121]. While TNF-α is an inducer of NF-κB and MAPK signaling, these effects appear to be primarily mediated via increased NADPH oxidases (Nox)-derived reactive oxygen species (ROS) [122]. TNFR1 and TNFR2 are the major isoforms responsible for TNF-α signaling and have somewhat divergent roles [123]. For example, in a model of ischemic HF, TNFR1 knockout mice were protected from cardiac dysfunction, hypertrophy, and cardiomyocyte apoptosis and had reduced cardiac NF-κB and MAPK protein expression. In contrast, TNFR2 knockout significantly exacerbated these effects due to an attenuation of NF-κB signaling [124]. Increased ROS via TNFR1 appears to be the major difference between these receptors, as TNFR1 can overwhelm protective functions of TNFR2 in cardiomyocytes [123]. The effects of TNF-α and its receptors are likely influenced by sex. In a murine model of cardiomyopathy, transgenic female mice that overexpressed TNF-α had a six-month survival rate of 89% compared to 52% in transgenic male mice [125]. Further, both wild-type males and transgenic male mice had significantly greater TNFR1 expression compared to their female counterparts. These sex differences in TNFR1 may provide some explanation as to why males are at greater risk of CVD compared to females. Cardioprotective effects of TNFR2 are also sex specific. In cardiac I/R injury, TNFR2 in female mice appears cardioprotective by increasing suppressor of cytokine signaling-3, a mediator of pro-inflammatory signaling, whereas in male mice, TNFR2 operates by suppressing the MAPK Jun N-terminal kinase (JNK) [126]. E2 has also been independently associated with suppressing JNK and decreasing downstream TNF gene transcription [127].
In addition to the detrimental effects of inflammation, TNF-α has direct effects on endothelial nitric oxide synthase (eNOS), a nitric oxide (NO) producing enzyme critical to vasodilation and vascular health. TNF-α decreases eNOS mRNA in endothelial cells in a time-dependent manner [128], as well as eNOS protein expression [129], leading to reduced NO metabolite concentration. A negative feedback mechanism exists where NF-κB increases eNOS transcription in response to laminar shear stress, and endothelial NO then inhibits NF-κB, preventing sustained NF-κB activation [130]. E2 positively regulates eNOS and eNOS-derived NO in an ER-dependent manner. In endothelial cells, E2 binding to ER activates PI3K [131,132], leading to protein kinase B (Akt) phosphorylation [132,133]. This results in the phosphorylation of eNOS at Ser1177 [134] and promotes the constitutive production of NO, which is anti-inflammatory at normal physiological concentrations. E2 further prevents activation of NF-κB in endothelial cells by blocking IKK [135]. The role of androgens in anti-inflammatory signaling is less clear. Androgens have been shown to increase TNF-α-induced apoptosis and to induce vascular cell adhesion molecule-1 (VCAM-1) expression, a marker of endothelial inflammation, in endothelial cells through NF-κB signaling, but testosterone has also been shown to phosphorylate eNOS and increase NO production, all in an AR-dependent manner [136]. Normal physiological concentrations of testosterone are generally associated with cardioprotective effects in males [10,137], but females with elevated circulating androgen concentrations are more likely to portray an inflammatory profile [138]. Interestingly, co-incubation of endothelial cells with N-acetylcysteine (30 mmol/L), an antioxidant, for 24 h ameliorated detrimental effects of TNF-α (40 ng/mL) in eNOS protein expression, suggesting a major role of ROS in TNF-α signaling [129]. Additionally, TNF-α signaling inhibits argininosuccinate synthase expression, a critical enzyme in the regeneration of l-arginine, a co-substrate of eNOS [139]. NF-κB signaling also appears to play a role in regulating argininosuccinate synthase expression by decreasing argininosuccinate synthase-DNA promoter binding activity [139].
3.3. Berry Polyphenols in Inflammation
Clinically, berries tend to elicit anti-inflammatory effects. For example, Karlsen et al. [140] studied a sample of middle-aged, overweight males (n = 47) and females (n = 17) with at least one CVD risk factor (hypertension, hyperlipidemia, or smoking). They consumed water or 330 mL of bilberry juice for four weeks, and bilberry juice consumption resulted in a reduction of serum IL-6 and C-reactive protein (CRP), but not TNF-α, all of which are NF-κB transcriptional products [140]. However, in overweight females with metabolic syndrome, the consumption of bilberry extract equivalent to 100 g of fresh berries for approximately four weeks did result in a significant reduction of TNF-α [141]. This suggests possible sex-specific effects, as participants in the trial conducted by Karlsen et al. [140] were mostly male. However, due to the differing disease states within studies, a direct comparison cannot be made. Further complicating matters, due to the unique polyphenol profile of each berry, sex differences may also be apparent depending on which polyphenols are most prevalent. In contrast to Lehtonen et al. [141], which found anti-inflammatory effects with bilberry consumption in females with metabolic syndrome, the consumption of 50 g of freeze-dried strawberry for four weeks did not reduce systemic inflammation, as assessed by high-sensitivity CRP (hs-CRP), in a separate investigation of females with metabolic syndrome [142]. However, VCAM-1 did significantly decrease after eight weeks of freeze-dried strawberry consumption in adults (92% female) with metabolic syndrome [143]. Strawberries also did not appear efficacious in obese, hyperlipidemic adults (92% female) in reducing systemic inflammation [144]. Bilberries are a particularly rich source of the anthocyanin petunidin-3-glucoside [145], while strawberries are rich in pelargonidin-3-glucoside [146]; these differing polyphenolic profiles likely account for these anti-inflammatory differences. Nonetheless, in mixed-sex studies that do not include strawberries, berry consumption does appear overall anti-inflammatory. For example, 750 mg of freeze-dried black raspberry or placebo were consumed daily by middle aged males and females for twelve weeks [147]. Following the intervention, serum TNF-α and IL-6 were significantly reduced compared to control [147]. In 72 dyslipidemic participants (75% male, 25% female), consumption of 300 mL of blackberry juice with pulp every day for eight weeks reduced hs-CRP [148]. Further illustrating these differences, the consumption of 250 g frozen red raspberries every day for four weeks in diabetic males (20%) and females (80%) significantly reduced IL-6 and TNF-α [149]. Further human studies are needed to elucidate sex differences as it relates to specific polyphenolic profiles found in berries.
Berries likely exert their anti-inflammatory effects by acting as regulators of TLR-4 and TNFR signaling at multiple protein targets. For example, in a pilot study conducted by Nair et al. [150], nine males and ten females with metabolic syndrome consumed 45 g/d of freeze-dried blueberry powder for six weeks. Following the intervention, mRNA isolated from monocytes showed decreased TLR-4, IL-6, and TNF-α gene expression. These effects are mediated by the polyphenols found in berries. For example, in murine RAW 264.7 macrophages, the flavonol quercetin and flavanol catechin were used in isolation and in combination with LPS treatment, a TLR-4 ligand, for 24 h [151]. TLR-4, Myd88, TRAF6, and TAK1 mRNA expression were reduced, and the combination of quercetin and catechin was particularly effective in the reduction of Myd88 protein expression compared to their use in isolation. As expected, phosphorylation of MAPKs, including ERK1/2, p38MAPK, and JNK, were significantly reduced in a similar manner, as was the phosphorylation of p65, the subunit of NF-κB that binds to κB region of DNA encoding inflammatory cytokine transcription. Further, secretion of inflammatory cytokines TNF-α and IL-1β was reduced.
These findings highlight the multiple targets of polyphenols in inflammatory signaling, and the protective effects of polyphenols in macrophages are replicated in models of the cardiovascular system. For example, in human umbilical vein endothelial cells (HUVECs), pre-treatment with 25 μM quercetin for 1 h followed by 50 μM oxidized LDL, a TLR-4 ligand, for 24 h significantly reduced TLR-4 protein expression compared to oxidized LDL treatment alone and also decreased p65 nuclear translocation [152]. Resveratrol also decreased oxidized LDL and LPS-induced inflammation in HUVECs primarily by reducing TLR-4 protein expression [153]. In an ischemic model of anoxia-reoxygenation (A/R) injury, rat cardiomyocytes (donor sex unspecified) were treated with 5, 10, or 20 μM of resveratrol for 5 min following 3 h anoxia and 2 h reoxygenation [154]. TLR-4 protein expression was significantly upregulated due to A/R, but resveratrol decreased TLR-4 in a dose-dependent manner. Further, inhibitor of NF-κB (IκBα), the protein of the NF-κB complex that sequesters p65 into the cytoplasm, was reduced due to A/R, which coincided inversely with increased nuclear p65 translocation. Resveratrol reversed these effects, increasing cytosolic IκBα and decreasing nuclear p65.
This theme is replicated in several in vitro models, as berry polyphenols reduce p65 nuclear translocation induced by Ang II [155] and TNF-α [156] in endothelial cells. In a pressure overload-induced HF model in male mice, the flavonol myricetin (200 mg/kg/d for six weeks) significantly reduced cardiac phosphorylation of TAK1, which coincided with reduced p65 nuclear translocation and reduced phosphorylation of p38MAPK and JNK, but not ERK1/2 [157]. This resulted in significantly improved cardiac ejection fraction and reduced cardiac hypertrophy. Thus, berry polyphenols likely possess potent anti-inflammatory effects, primarily mediated by decreased NF-κB and MAPK activation, with potential sex effects. While not cardiovascular-specific tissue, in breast cancer MCF-7 cells, E2 mitigates NF-κB activity induced by TNF-α signaling via ERα, partially through inhibition of NF-κB acetylation. Resveratrol also partially mediates deacetylation of NF-κB in an ERα/Sirt1-dependent mechanism [158]. As such, the anti-inflammatory activity of polyphenols may be higher in tissues that express ERα [158]. Since ERα is expressed in endothelial cells, VSMCs, and cardiomyocytes, it is likely that resveratrol could disrupt inflammatory signaling in CVD. Because ERα expression is upregulated by higher E2 concentration, the anti-inflammatory effect of resveratrol may be greater in females compared to males, and it is possible that this relationship exists with other polyphenols as well (Figure 2). Further experimental details for some of the investigations cited in this section are displayed in Supplementary Table S2.
4. Oxidative Stress
Similar to inflammation, oxidative stress is a characteristic feature of CVD pathogenesis. A balance between ROS production and antioxidant enzyme activity is necessary to maintain normal cellular function. Oxidative stress occurs when the formation of ROS surpasses antioxidant capacity, facilitating NF-κB and MAPK signaling due to upstream redox-sensitive IKK and MEK activation, respectively [159]. Major sources of ROS include Nox and xanthine oxidase (XO). These enzymes produce superoxide (O2−) and hydrogen peroxide (H2O2), which contribute significantly to pathological processes in the cardiovascular system, including endothelial dysfunction, hypertension, atherosclerosis, and HF [160,161,162,163,164,165,166,167]. Overall, ROS leads to decreased NO production and bioavailability, increased vasoconstriction, decreased endothelium-dependent and -independent vasodilation, increased inflammation, vascular remodeling, and fibrosis [168]. In general, males have elevated systemic [169], central, and myocardial oxidative stress [170,171] under basal conditions compared to females. This is likely attributable to sex steroids, as castration in female Wistar rats resulted in a 200% increase in myocardial lipid peroxidation [170], demonstrating the protective effects of E2 in oxidative stress.
4.1. NADPH Oxidase and Xanthine Oxidase
Oxidative enzymes, such as Nox and XO, are major producers of ROS within the cardiovascular system. Nox1, Nox2, Nox4, and Nox5 are the dominant Nox isoforms in the cardiovascular system, in which all isoforms are present ubiquitously in endothelial cells, VSMCs, and cardiomyocytes [172] at differing concentrations. Nox use oxygen as an electron acceptor, abstracting electrons from NADPH, resulting in O2− generation and, in the case of Nox4, H2O2 generation [173]. Nox is bound to lipid bilayers of organelles or the outer plasma membrane via six transmembrane α-helices. While O2− cannot freely pass between lipid bilayers, H2O2 is able to do so [174]. The micro-environment within the lumen of organelles are under reducing conditions resulting in the spontaneous conversion of O2− to H2O2. Thus, cellular localization of each Nox determines whether O2− or H2O2 will be the predominant end-product. A variety of stimuli can lead to Nox transcription and over-expression, including changes in shear stress [175], Ang II [176], endothelin-1 [177], TNF-α [178], and α-adrenergic receptor agonists [179]. In cerebral arteries of Sprague Dawley rats, reduced Nox activity and lesser Nox1 and Nox4 expression was observed in females compared to males [180]. However, evidence is equivocal in different animal models [180,181,182,183], and human studies assessing sex differences in Nox isoform activity and expression are lacking.
In urea metabolism, nucleotide-derived purines are converted to hypoxanthine (derived from adenosine) and then oxidized by XO to xanthine (derived from guanosine) [184]. XO can then further catalyze the oxidation of xanthine to form uric acid. XO catalysis involves reducing molecular oxygen and the release of ROS in the form of O2− or H2O2. XO is nearly absent from the human heart [185]; however, it is found in abundance in the endothelium [186]. Males with CAD have a 200% increase in arterial XO activity compared with healthy controls [186]. Interestingly, in microvascular endothelial cells, both E2 and E2 stereoisomer (17α-estradiol) reduce XO activity despite the inability of 17α-estradiol to bind to ER, demonstrating ER-independent effects of E2, which may include post-translational modifications of XO [187]. Despite the vasodilator effect of testosterone [188], testosterone also appears to increase O2− and peroxynitrite (ONOO−) generation and upregulate XO activity through AR-dependent mechanisms [189]. Upregulation of XO can occur due to hypoxia, inflammatory signaling [190], and crosstalk with Nox [191]. Collectively, cardiovascular Nox and XO are implicated in the pathogenesis and/or can be targeted in the treatment of several CVDs [192,193,194,195,196,197], including hypertension, CAD, and HF.
4.2. Role of NOS in Oxidative Stress
NOS isoforms can also contribute to oxidative stress. NOS produce NO, which is largely cardioprotective when produced constitutively by the endothelium through eNOS [198,199]. Endothelial NO at physiologic concentrations facilitates vasodilation and decreases vascular resistance by activating VSMC soluble guanylate cyclase (sGC) to produce cyclic guanosine 3’,5’-monophosphate (cGMP) [200]. Endothelial dysfunction is characterized by decreased available NO and is an early indicator of CVD development [201]. Dysfunction of eNOS (i.e., eNOS uncoupling) can contribute to oxidative stress, where eNOS uncoupling produces O2− instead of NO [202]. eNOS uncoupling can occur when there is decreased bioavailability of eNOS cofactors, including tetrahydrobiopterin (BH4) and l-arginine. This can occur when there is reduced BH4 synthesis [203] or when BH4 is oxidized or depleted [204]. Further, l-arginine availability can be reduced via diminished argininosuccinate synthase activity [205,206,207,208], increased arginase activity [206,209], or increased asymmetric dimethylarginine (ADMA) production [210]. Argininosuccinate synthase acts in the regeneration of arginine from citrulline, arginase catalyzes the conversion of l-arginine to l-ornithine and urea in the urea cycle [206,209], and ADMA is the result of arginine proteolysis and is a competitive inhibitor of eNOS [211].
The production of NO and eNOS coupling are known to be affected by E2. The binding of E2 to ERα increases stimulatory phosphorylation sites Ser1177 and Ser635 on eNOS, and E2 binding to ERα or ERβ reduces inhibitory phosphorylation site Thr495 on eNOS [212], which aid in the constitutive production of cardioprotective NO. E2 may also prevent eNOS uncoupling and subsequent O2− production, potentially by affecting l-arginine and BH4 availability [213,214]. In peri- or postmenopausal females, but not premenopausal females, impaired brachial artery flow-mediated dilation (FMD; a measure of conduit vessel endothelial function) was associated with relative l-arginine deficiency and increased markers of l-arginine metabolism [213], and FMD improved with BH4 supplementation [214], suggesting the reduction in E2 in peri- and postmenopausal women resulted in a decrease in bioavailable precursors required for NO production via eNOS. The effect of androgens on eNOS (un)coupling are still unknown.
When NO is overproduced, such as through activation of inducible NOS (iNOS), it can directly contribute to oxidative stress. iNOS is activated through MAPK and NF-κB signaling under pathological conditions [215,216] and is upregulated in CVDs [217,218,219]. iNOS yields approximately 1000-fold the amount of NO compared to eNOS [217]. Excess NO, which is itself a free radical molecule, is then scavenged by ROS produced from Nox and XO, leading to conversion of NO to ONOO− [218,219]. Nox- and XO-derived ROS and ONOO− oxidize numerous molecules, including sGC. sGC oxidation increases VSMC intracellular Ca2+ flux and depletes BH4, which consequently results in endothelial dysfunction [220]. In addition, ONOO− can oxidize low density lipoprotein, which is linked to atherogenic processes, including foam cell formation and leukocyte adherence to endothelial cells [221]. iNOS may represent a pathway of specific interest in females, as E2 non-selectively binds to both ERα and ERβ, but they yield opposing effects on iNOS activation; ERα inhibits while ERβ stimulates iNOS [222]. Testosterone appears to affect iNOS activity differently based on concentration [223]. Macrophages treated with testosterone concentrations of 10−10 M and 10−8 M showed less iNOS protein compared to those treated with 10−12 M testosterone, but iNOS protein treated with 10−6 M testosterone was similar to that of 10−12 M [223]. This indicates that testosterone concentrations, both low or high, could elicit detrimental effects on iNOS activation and subsequent ONOO− generation.
4.3. Endogenous Antioxidant System
Antioxidant enzymes, such as superoxide dismutase (SOD), glutathione peroxidase (GPx), and catalase, also contribute to the redox balance by dissociating ROS. SOD1 (a cytoplasmic enzyme) and SOD2 (a mitochondrial enzyme) catalyze the dismutation of O2− into H2O2 and O2 [220]. GPx and catalase both reduce H2O2 to form H2O and O2. SOD, GPx, and catalase activity are all inversely associated with CVD risk [224,225,226]. SOD and GPx activity, as well as SOD expression, are also inversely related to atherosclerosis severity [225,226,227]. Both SOD1 and SOD2 upregulation in transgenic mice appear to offer cardioprotective effects in HF, including improved ejection fraction and reduced inflammatory cytokines compared to wild-type mice [228,229]. Further, there is an established relationship between NO and SOD, where SOD protects the bioavailable NO, and NO upregulates the expression of SOD [220,230]. Estrogen affects mitochondrial respiration, mitochondrial ROS production, and antioxidant capacity [231]. Estrogen appears to increase oxidative phosphorylation while simultaneously decreasing mitochondrial production of O2− and H2O2, likely related to estrogenic increases in SOD expression [170,232] and activity [170]. At this time, there is no empirical evidence that physiological doses of estrogen or testosterone influence expression of GPx or catalase [170,232]. Supraphysiological doses of testosterone decrease SOD, GPx, and catalase expression [233], but testosterone deficiency also decreases antioxidant capacity [234].
Other components of the antioxidant system include heme oxygenase (HO)-1 and NQO1. HO-1 catalyzes free heme, a pro-oxidant [235] and major component of a variety of enzymes, to produce carbon monoxide (CO), iron (Fe2+), and biliverdin [236]. CO itself has anti-inflammatory effects [237] and acts in a similar manner to NO, increasing cGMP in VSMCs, potentially improving vasodilation [238]. There is greater HO-1 expression and total HO activity in cardiac and aortic tissues of female compared to male rats [239], but sex differences in HO-1 expression or activity in humans is unknown. Additionally, NQO1, an NADPH-reductase, is a ROS scavenger, in part due to the flavin co-factor, which facilitates dismutation of O2− to H2O2 [240]. Further, NQO1 mRNA expression and activity are higher in females compared to males in rats [241], and human data are limited but suggest no sex differences [242]. The principal transcriptional regulators of these antioxidant enzymes are Nrf2 [243] and Sirt1 [240].
Nrf2 and Sirt1
Nrf2 is continually synthesized, but it is sequestered in the cytosol by the enzyme Keap1, which undergoes continuous ubiquitination and degradation when the Keap1-Nrf2 complex is formed. Under conditions of oxidative stress, such as excessive H2O2, reactive cysteine residues on KEAP1 undergo reduction, leading to conformational shifts in Keap1, preventing Nrf2 binding [244]. As such, Nrf2 is able to accumulate in the cytosol and translocate into the nucleus. Similar to NF-κB, Nrf2 binds to anchoring protein CBP, which allows it to bind to the transcriptional region of DNA encoding the antioxidant responsive element (ARE). This leads to the transcription and synthesis of antioxidant enzymes, particularly HO-1 and NQO1 [245]. Because CBP can bind to both NF-κB and Nrf2, these proteins compete for binding [246]. Under conditions of inflammation and oxidative stress, NF-κB appears to bind 10-fold greater to CBP than Nrf2 as observed in HepG2 cells treated with 12-O-tetradecanoylphorbol-13-acetate (PMA; a protein kinase C activator) [247]. Thus, under pathological conditions observed in CVD, it can be assumed that Nrf2 would be deprived of CBP binding, and under these circumstances, insufficient ARE transcription would occur not eliciting an overwhelming enough antioxidant response to counteract ROS. ER-dependent mechanisms appear to positively modulate Nrf2 activity [248]. For example, in cardiomyocytes that underwent hypoxia followed by reoxygenation, a model of I/R injury, the addition of E2 significantly increased Nrf2 nuclear translocation [249]. Conversely, ICI 182,780 (ICI), a non-specific ER antagonist, abolished these effects and reduced Nrf2 protein leading to reduced HO-1 and SOD1 mRNA.
In addition to Nrf2, Sirt1, a redox sensitive deacetylase, has multiple cellular effects, including antioxidant, anti-inflammatory, and anti-apoptotic effects. A direct transcriptional product of Sirt1 activity is SOD2, and Sirt1 increases eNOS, Nrf2, GPx, and catalase expression and inhibits NF-κB activity and apoptotic transcription factor p53 [250]. In animal models of Ang II-induced hypertension and pressure-overload-induced HF, Nrf2 and Sirt1 genetic knockdown exacerbates these conditions [251,252,253,254], while overexpression has protective effects [255,256,257]. As with Nrf2, E2 may also increase Sirt1 protein expression [258], demonstrating the multi-targeted role of E2 on oxidative stress and antioxidant targets. Increasing the activity of these enzymes is of major therapeutic importance in the treatment of CVD.
4.4. Berry Polyphenols in Oxidative Stress
Berries appear efficacious in mediating antioxidant status in humans [259]. In a number of acute studies, berry consumption increases plasma antioxidant status [260,261,262,263,264,265,266]. This increase has been shown in healthy middle-aged males after consumption of 240 g of bilberries, lingonberries, or blackcurrants [260] and 100 g freeze-dried blueberry powder with a high-fat meal [261,262]. Increased plasma antioxidant capacity has also been shown in females following ingestion of a blueberry beverage, including 200 g of blueberries, 50 g banana, and 200 mL of apple juice, but this was investigated in relation to exercise-induced muscle injury [266]. A number of mixed-sex samples confirm similar findings following consumption of 400 mL of an antioxidant-rich juice (30% white grape, 25% blackcurrant, 15% elderberry, 10% sour cherry, 10% blackberry, and 10% aronia) [263], 400 mL of elderberry juice [264], or 1 kg of strawberries [265]. Results from another mixed-sex sample indicate that acute consumption of cranberry polyphenols (456 mL cranberry leaf extract beverage or 480 mL of cranberry juice cocktail) may positively alter endogenous antioxidant systems, including increases in GPx and SOD activity [267]. This collectively indicates the efficacy of berry polyphenols in acutely improving antioxidant status in both sexes, but additional investigation of the effect of berry polyphenols in females and on endogenous antioxidant systems is warranted.
Berry polyphenols tend to reduce vascular oxidative stress in a number of chronic studies. For example, in an eight-week trial, 48 postmenopausal females with pre- and stage 1-hypertension were assigned to consume 22 g/d of freeze-dried blueberry powder or placebo [268]. Serum concentrations of NO metabolites increased in subjects consuming the blueberry powder by 68%, which was significantly greater than control [268]. Further, systolic and diastolic blood pressure decreased by 7 and 5 mmHg, respectively, whereas blood pressure did not change in control subjects [268]. In a separate investigation, 44 obese females and four obese males with metabolic syndrome consumed 50 g/d freeze-dried blueberries or placebo for eight weeks [269]. Following the intervention, markers of oxidative stress, including serum oxidized LDL and combined malondialdehyde and hydroxynonenal (indicators of fatty acid oxidation) significantly decreased by 28% and 17%, respectively, which was significantly greater than control [269]. In the previously described investigation by Nair et al. [150] in which males and females consumed 45 g/d of freeze-dried blueberry for six weeks, both serum and isolated monocytes expressed decreased total ROS and serum O2− compared to placebo. Further, in pre- and stage 1-hypertensive postmenopausal females, 50 g/d of freeze-dried strawberry for eight weeks significantly increased NO metabolites compared to baseline, although these did not correspond with changes in blood pressure at this dose [270].
Sex differences in the efficacy of berry polyphenols likely exist with regard to reductions in oxidative stress. For example, 26 hyperlipidemic males and females consumed 10 g of freeze-dried strawberry for six weeks [271]. Oxidized LDL decreased to a much greater extent in females compared with males, suggesting greater efficacy of strawberry polyphenols in females. In a 12-week study including 44 subjects with prediabetes (70% female), black raspberry consumption (900 or 1800 mg/day) decreased oxidized LDL in a dose-dependent manner [272]. However, it is likely that these suggestive sex differences are polyphenol specific, although further human investigations are needed to assess these differences.
4.4.1. Berry Polyphenols in Sirt1 and Nrf2 Regulation
Berry polyphenols exert cellular antioxidant effects, not merely due to exogenous ROS scavenging, but also by exerting cytoprotective effects, as polyphenols act as effectors for multiple proteins. For example, under basal conditions, quercetin increases Sirt1 mRNA in a dose-dependent manner in HUVECs [273]. Additionally, in HUVECs, the treatment of 10 μM quercetin for 24 h followed by exposure to 150 μg/mL oxidized LDL for another 24 h resulted in reduced Nox2 and Nox4 expression as well as increased eNOS expression compared to oxidized LDL alone [273]. However, Sirt1 siRNA abolished these protective effects. In vivo, similar findings were observed in a diabetic cardiomyopathy murine model (gender not specified), as resveratrol (25 mg/kg/d for five days) significantly increased Sirt1, reduced cardiac hypertrophy, and improved ejection fraction, but this was abrogated by Sirt1-specific cardiac deletion. Nrf2 activity can also be regulated by polyphenols [274]. This effect was attributed to potential binding to Keap1, causing conformational shifts that prevent Nrf2 from binding to Keap1, thus leading to increased cytosolic concentration of Nrf2 and subsequent nuclear translocation. Computational studies revealed that kaempferol may directly bind to Keap1 in this fashion and prevent Nrf2 binding [275]. However, in both human and rat-specific proteins, it was revealed that gallic and ellagic acid were incapable of binding to Keap1 in the domain associated with Nrf2 binding regulation. Although direct interactions of polyphenols with Keap1 may alter its binding affinity to Nrf2, other interacting proteins that regulate Keap1-Nrf2 complex affinity are also of relevance [276].
For example, p62, a scaffolding protein involved in a variety of cellular processes, such as autophagy, can directly bind to the binding pocket of Keap1 that associates with Nrf2, and thus prevent Nrf2 from binding to Keap1 [277]. In an I/R model of HF in male rats, urolithin B, a downstream microbiome-derived metabolite of ellagic acid metabolism, was injected into the intraperitoneal cavity 0, 24, and 48 h prior to surgery in rats at a concentration of 0.7 mg/kg [278]. Concentrations of p62 in cardiac tissue were significantly decreased in I/R rats that did not receive treatment; however, urolithin B injection significantly abrogated reductions in p62, which was not different than p62 in sham control. This preservation of p62 corresponded with a significant reduction in myocardial infarct size and improved cardiac hemodynamics compared to I/R controls. Further, although nuclear Nrf2 was increased in I/R control compared to sham, urolithin B treatment elicited roughly three-fold greater nuclear Nrf2 compared to I/R control. Increased NQO1 and HO-1 expression was also observed.
4.4.2. Berry Polyphenols in Endothelial Function
Berry polyphenols may also improve endothelial function mediated by reduced oxidative stress. In endothelial cells, exposure to the anthocyanin cyanidin-3-glucoside (5, 10, or 20 μM) for 2 h followed by 1 μM Ang II for 24 h resulted in a significant attenuation of ROS, reduction of iNOS, and increase in nuclear Nrf2 translocation, which coincided with increased HO-1 and SOD in a dose-dependent manner compared to Ang II alone [155]. These positive in vitro effects in endothelial cells reflect in vivo findings. For example, in male mice fed a high-cholesterol, high-fat diet with or without cyanidin-3-O-β-glucoside (2 g/kg) for eight weeks, aortic O2− was significantly reduced and cGMP was significantly increased due to anthocyanin supplementation [279]. The anthocyanin-supplemented diet also resulted in improved endothelium-dependent vasodilation and increased phosphorylated eNOS. In a separate investigation, fourteen weeks of supplementation with the phenolic acid ellagic acid (30 mg/kg/d) reduced atherosclerotic lesion area in the aortic arch of high-fat diet-fed male mice compared to mice with no phenolic acid supplementation. Additionally, aortic Nrf2 and HO-1 were significantly increased, and aortic NOS activity was significantly higher with ellagic acid supplementation [280]. Preservation of eNOS due to berry polyphenols under pathological stimulus is a common theme. Cyanidin, for example, increased eNOS protein under basal conditions and also abrogated TNF-α-induced transcriptional downregulation of eNOS in endothelial cells [281]. In male SHR, gallic acid supplementation (1% of drinking water) for sixteen weeks significantly reduced aortic components of RAAS, including ACE and AT1R expression, compared to control SHRs [282]. Additionally, blood pressure and left ventricle (LV) Nox2 protein expression were significantly reduced, and this coincided with reduced LV hypertrophy. Gallic acid also appeared protective in Ang-II infused male mice (490 ng/kg/min) [283]. After two weeks of infusion, gallic acid dose-dependently (5 and 20 mg/kg/d) improved blood pressure and increased aortic total eNOS and phosphorylated eNOS protein expression. In excised aortas from male SHR that consumed an 8% blueberry diet for eight weeks, blueberry supplementation dramatically reduced phenylephrine-induced vasoconstriction compared to SHR controls, which was completely abolished by eNOS inhibitor, l-NG-monomethyl-arginine [284].
A number of berries have clinically demonstrated improved vascular function in humans. In healthy males, the consumption of wild blueberries increased postprandial FMD [285]. Interestingly, a biphasic response was observed, with peak FMD detected at 1–2 h and 6 h, suggesting microbial metabolism of parent flavonoids to yield secondary metabolites at the later time point. Plasma phenolic acids, ferulic acid and caffeic acid, peaked at 1–2 h, while hippuric acid and homovanillic acid peaked at 6 h. These changes in plasma polyphenol concentrations also reflected decreased neutrophil Nox activity, which significantly decreased at 1–2 h and 6 h. Also, in young males, 200 and 400 g of red raspberries significantly improved FMD at both 2 and 24 h compared to control [286]. Ellagic acid serum concentrations peaked at 2 h, while urolithin metabolites peaked at 24 h, which may explain these vascular effects. Similarly, 50 g of freeze-dried strawberry powder improved serum nitrate/nitrite concentrations, which positively influenced reactive hyperemia, a measure of blood flow, in young obese males after one week [287]. These results reflected positive changes in reactive hyperemia in pre-hypertensive postmenopausal females, in which 25 g/d of freeze-dried strawberry was efficacious after eight weeks [270]. In mixed-sex studies, black raspberry appears particularly efficacious in improving vascular function in both hypertensive subjects and subjects with metabolic syndrome [147,288,289]. Studies investigating the effect of berry polyphenols on vascular function variables in premenopausal females alone or systematically comparing outcomes of female and male participants are scant; however, there is strong evidence to suggest berries and their polyphenols have beneficial effects in reducing oxidative stress and improving vascular function in humans.
Although sex differences in vascular function in response to berry polyphenols have not been fully elucidated, it is likely that sex differences exist. For example, red wine polyphenols (which include gallic acid, caffeic acid, and anthocyanins) induced endothelium-dependent vasodilation in aortas from female and male rats, but relaxation was more pronounced in female aortic rings than those from males [290]. This effect was shown to be mediated exclusively by NO, via inclusion of a NOS inhibitor [290]. The addition of non-specific ER-antagonist ICI in this study did not affect red wine polyphenol-induced aortic relaxation in either sex, but it reduced E2-induced aortic relaxation [290]. It is important to note that the effect of ICI on E2-induced relaxation was only tested in female aortas in this study, and that the non-specific antagonistic nature of ICI does not allow for the assessment of the isolated impact of ERα or ERβ. This suggests red wine polyphenols impact endothelium-dependent vasodilation and endothelial NO production in an ER-independent manner (Figure 2). In this investigation, red wine polyphenols appear to elicit this effect via mediation of antioxidant enzymes, including SOD and catalase (evidenced by addition of SOD and catalase analogues) and by increasing phosphorylation of eNOS through the PI3K/Akt pathway [290]. Therefore, some sex-specific effects of polyphenols may be due to mediation of ROS. Considering decreased FMD in females at risk for CVD compared to male counterparts [291], these sex-specific effects of polyphenols in vascular function warrant further investigation in humans.
4.4.3. Potential ROS-Independent Effects of Berry Polyphenols
While ROS can independently mediate inflammatory signaling (MAPKs and NF-κB), such as by modifying redox sensitive IKK and MEK [292], polyphenols likely have a multi-targeted role, inhibiting ROS and inflammation independent of each other. For example, in a pressure-overload model of HF in male mice, myricetin (200 mg/kg/d for six weeks) administration resulted in significantly reduced MAPK and NF-κB phosphorylation and increased Nrf2 and HO-1 in cardiac tissue compared to sham animals [157]. However, Nrf2 silencing still resulted in reduced NF-κB and MAPK phosphorylation, which corresponded with comparable inhibition of cardiac hypertrophy with and without Nrf2 silencing. In vitro findings suggest that this anti-inflammatory effect occurred despite significant cellular oxidative stress in neonatal rat cardiomyocytes co-treated with H2O2, myricetin, and Nrf2 siRNA as indicated by increased 4-hydroxynonenal expression, a metabolite of lipid peroxidation, compared to control. It is likely that myricetin prevented MAPK and NF-κB activation at multiple upstream sites, namely reduced TRAF6 degradation, preventing TAK1 activation. Nonetheless, these results do not preclude oxidative modification of IKK and MEK, suggesting that this polyphenol may also inhibit these enzymes independent of ROS. Further experimental details for some of the investigations cited in this section are displayed in Supplementary Table S3.
5. Cardiac and Vascular Remodeling
Cardiac and vessel architecture and plasticity largely reflect function; thus, pathological remodeling of the vasculature can result in CVDs, including HF, hypertension, and atherosclerosis [9,293,294]. Apoptotic cells must be replaced to maintain cardiac and vessel structure. In the heart, remodeling of the myocardium begins with apoptosis, leading to subsequent fibrosis, and causing impaired LV function. Similarly, in arteries, hypertrophic remodeling is characterized by VSMC apoptosis and fibrosis, impairing vessel dilation and promoting cardiac ischemia. Thus, cardiac and vascular remodeling induced by apoptosis and fibrosis are key features of CVDs and represent a therapeutic target.
5.1. Apoptosis
Apoptosis involves either the extrinsic pathway due to cytokine signaling (e.g., TNFR1) or the intrinsic pathway due to intracellular damage, such as DNA or mitochondrial damage [295]. Under pathological conditions observed in CVD, it is likely that both are of relevance and significant overlap and convergence exists between pathways. In the inner mitochondrial membrane, cytochrome c (Cyt c) is a phospholipid-bound heme protein that facilitates electron transfer between complex III and IV of the electron transport chain. Under conditions of oxidative stress, mitochondrial membrane permeability increases, allowing release of Cyt c into the cytosol [296]. Upon cytosolic translocation, Cyt c in conjunction with ATP (to a greater extent dATP) allosterically activate apoptosis-protease activating factor 1, facilitating caspase-9 activation due to proteolytic cleavage [297]. Subsequently, caspase-9 facilities caspase-3 activation via cleavage, leading to cell death [298]. Illustrating the role of caspase, caspase inhibition following myocardial infarct in rats reduced LV cardiomyocyte apoptosis, reduced fibrosis, and preserved LV function [299]. A regulator of these apoptotic effects is the cytosolic translocation and oligomerization of both Bax and Bcl-2 homologous antagonist killer (Bak) on the mitochondria, in which mitochondrial membrane transition pore opening occurs, facilitating Cyt c release [295]. The transcriptional regulation of Bax, and Bak to a lesser extent, is regulated by p53 [300,301], which can be activated by oxidative stress [302]. Indeed, patients with HF have significantly greater p53 protein expression in their hearts compared with healthy controls [303]. Following transcription and synthesis, Bax and Bak can be activated along the intrinsic pathway due to DNA damage and can also be activated extrinsically by caspase-8 or JNK due to inflammatory cytokine signaling via TNFR [295,304]. To counteract these effects, anti-apoptotic protein Bcl-2 can inhibit caspase-8 activity, and Bcl extra-large (Bcl-xL) can prevent the oligomerization of Bax and Bak. Thus, Bcl-2 and Bcl-xL antagonize Bax/Bak apoptotic effects.
There are marked sex differences in cell survival and cell death, where males show significantly more age-related [305] and HF-related [306] cardiomyocyte loss, post-infarct apoptosis [307], and I/R induced apoptosis [308,309,310] than females. One mechanism that contributes to differences in cell survival between males and females is the PI3K/Akt pathway. Akt activation inhibits cardiomyocyte apoptosis [311] and reduces I/R injury [312] in in vitro and in vivo models of HF. Akt interferes with pro-apoptotic pathways by inhibiting p38MAPK (a pro-apoptotic inducer [304]), p53, and caspases [313]. Importantly, premenopausal females have elevated levels of phosphorylated Akt localized in cardiomyocyte nuclei compared to age-matched males and postmenopausal females [314]. Akt phosphorylates murine double minute 2 (MDM2), causing MDM2 nuclear translocation and consequent p53 ubiquitination and degradation [315,316,317,318]. MDM2 and p53 form a negative-feedback loop, as p53 increases the expression of MDM2, which leads to p53 degradation [319]. Akt also phosphorylates caspase-9, which inhibits caspase-9 cleavage [320]. E2 is an activator of the PI3K/Akt pathway [314,321] and induces nuclear translocation of Akt in cardiomyocytes [319]. Testosterone has also been indicated to increase Akt phosphorylation [322], but some models show refuting results, where acute testosterone infusion has exhibited a downregulation of Akt [323] and exacerbated cardiac damage [324,325] in I/R models. E2 also modulates p38 isoforms [326] and inhibits p53 activity [327]. In ovariectomized female rats, there is increased activated caspase-8, -9, and -3, and cytosolic Cyt c compared to female controls [328], which is reversed by E2 treatment [329,330]. Further, ovariectomized female SHR showed increased activated caspase-9 and activated caspase-3 compared to wild-type and sham-operated female SHRs [331]. Pro- and anti-apoptotic protein content also appears to differ by sex. For instance, under physiologic conditions, male rats show greater expression of anti-apoptotic Bcl-2 protein than female rats, but Bax and phosphorylated p38MAPK content was similar between sexes [332]. Additionally, following I/R, Bcl-2 protein decreased in male rats, Bax decreased in female rats, and phosphorylated p38 increased in male rats [332].
5.2. Fibrosis
In an inflammatory state, the endothelium and heart release chemoattractant molecules, which guide leukocytes, such as monocytes and neutrophils, to infiltrate these tissues [333,334]. Monocytes differentiate into macrophages to ingest apoptotic and necrotic cells and release the cytokine transforming growth factor (TGF)-β [335]. Cardiomyocytes and VSMCs also harbor the inactive form of TGF-β, which can be activated by ROS [336,337,338]. TGF-β is a key initiator of the fibrotic process by acting on fibroblasts that reside interstitially between cardiomyocytes, while arterial fibroblasts migrate inwards from the tunica adventitia [339]. TGF-β causes fibroblast differentiation into myofibroblasts, which have muscle cell phenotype characterized by α-smooth muscle actin expression [340]. In the heart, myofibroblasts disrupt the delicate architecture of the extracellular matrix between cardiomyocytes due to excessive collagen synthesis and deposition of matrix metalloproteinases (MMPs), disrupting coordinated contraction between cells and severely diminishing the functional capacity of the heart and hampering vessel plasticity [339,341].
ERs and ARs sex-specifically regulate fibrosis, collagen and MMP synthesis [342]. Sex differences in cardiac fibrosis have been reported in a number of cardiovascular conditions, including hypertension and atherosclerosis [343,344,345]. Male cardiac fibroblasts portray greater activation of collagen synthesis, greater collagen deposition, and result in a higher degree of fibrosis than that of females [342]. In response to E2 treatment, female rats showed downregulation of cardiac fibroblast collagen I and III expression, but both were upregulated in male rats [345,346]. This was similarly shown in human cardiac fibroblasts [345]. Mediation appears to be ER subtype specific as well, where an ERα-agonist is suppressive and an ERβ-agonist is stimulating in female versus male rat cardiac fibroblasts, respectively [345]. E2 also phosphorylates ER subtypes differentially in rat cardiac fibroblasts, where it phosphorylates ERα in females and ERβ in males [345]. Further, E2 inhibits MMP-2 via an ERα-mediated reduction in gene transcription in rat cardiac and human fibroblasts [347].
5.3. Berry Polyphenols in Cardiac and Vascular Remodeling
Berries are likely efficacious in the reduction of CVD-associated pathologic remodeling. For example, in a 2% blueberry diet in male Fischer-344 rats, ischemic HF was induced by coronary artery ligation [348]. The necrotic area of the myocardial infarction was significantly reduced in blueberry-supplemented animals compared to control. Further, blueberry supplementation significantly decreased apoptotic cells in myocardial sections, with 2% apoptotic cardiomyocytes detected compared to 9% detected in control. Berry polyphenols likely exert protective effects at nearly every stage of remodeling and act on a number of cellular pathways involved in these processes. In H9c2 cardiomyocytes, 24 h treatment of 10 ng/mL of anthocyanidins, including cyanidin-3-galactoside, cyanidin-3-glucoside, and cyanidin-3-arabinoside, preceded 600 μM/L H2O2 for 2 h [349]. Single anthocyanidins decreased H2O2-induced apoptosis, although their combination was more effective. Berry polyphenols likely target p53, upstream from Bax, to reduce apoptosis. For example, in rat VSMCs (sex unspecified), 200 μg/mL of blackberry, raspberry, and black raspberry polyphenolic extract for 24 h followed by 100 nM of Ang II for 72 h resulted in a significant reduction in p53 protein expression, O2− and H2O2 production, and p38MAPK and ERK1/2 phosphorylation [350]. As such, we would expect decreased Bax, and indeed, HUVECs pretreated with 50 μmol/L of quercetin for 30 min followed by 250 μmol/L H2O2 displayed significant decreases in DNA fragmentation, Bax, and cleaved caspase-3 compared to treatment with H2O2 alone [351]. Further, H2O2 decreased Bcl-2 and was restored with quercetin. In neonatal rat cardiomyocytes (donor sex unspecified), 3 h of anoxia (95% N2 and 5% CO2) significantly increased caspase-3 activity and apoptosis but was markedly reduced by resveratrol in a dose-dependent manner [154]. Thus, in multiple cells of the cardiovascular system, apoptotic signaling is reduced with berry polyphenols. These anti-apoptotic effects in vitro are reproducible in vivo. For example, in gallic acid-treated male SHR (1% of drinking water), cardiac Bax and cleaved caspase-3 expression was significantly reduced compared to control animals [352]. Additionally, in urolithin B pretreated H9c2 cells that underwent 3 h of hypoxia and 3 h of reoxygenation, urolithin B significantly reduced cleaved caspase-3 and apoptosis; however, Nrf2 silencing neutralized these effects [278]. This suggests a clear role for ROS in mediating apoptosis [278], but polyphenols may also act in ROS-independent manners [157].
In addition to apoptotic signaling, berry polyphenols also mediate fibrotic processes. For example, in male mice that underwent trans-aortic constriction to induce HF (pressure-overload model), 100 mg/kg/day of gallic acid or medications used in the treatment of HF, including 3 mg/kg/day of losartan (AT1R blocker), 1 mg/kg/day carvedilol (β-blocker), or 3 mg/kg/day furosemide (3 mg/kg/day), for two weeks following confirmed HF resulted in a reduction of cardiac collagen I and α-smooth muscle actin to a large extent with gallic acid treatment but minimally with medications [353]. In accordance with these findings, cardiac perivascular fibrosis was dramatically reduced with gallic acid treatment but not with medications. Rutin and quercetin appear to have TGF-β reducing effects in an isoproterenol model of HF in male Wistar rats [354]; however, polyphenols likely directly prevent differentiation of fibroblasts to myofibroblasts even with pathological stimulus. For example, resveratrol pretreatment (25 μM) for 30 min significantly reduced α-smooth muscle actin in Ang II- (100 nM) and TGF-β-treated (200 pM) cardiac fibroblasts derived from male rats [355]. Interestingly, despite 3 h pretreatment with 5 ng/mL TGF-β, gallic acid was still able to halt differentiation of fibroblasts as evidenced by decreased α-smooth muscle actin and decreased collagen I protein expression [353]. While human studies that assess berries or berry polyphenols in pathological vascular and cardiac remodeling are lacking, the consumption of cranberry juice for twelve weeks (increasing in quantity from 125 mL at baseline to 500 mL at twelve weeks) resulted in a significant reduction of serum MMP-9 concentrations in normotensive and pre-hypertensive males [356]. Based on promising preclinical findings, it is likely that berries and their polyphenols can mitigate the pathological processes of vascular and cardiac remodeling. Further experimental details for some of the investigations cited in this section are displayed in Supplementary Table S4.
6. Conclusions
Based on promising preclinical studies and limited human data, berry polyphenols likely target many of the underlying cellular drivers of CVD, including inflammation, oxidative stress, and pathological remodeling. Systematic investigation of sex differences on the effects of berry polyphenols on cardiovascular health is currently limited. Additional studies are needed to elucidate mechanistic differences in the effects of polyphenols on the pathological pathways of CVD between and within sexes. Despite the paucity of data explicitly investigating sex differences, there is a substantial amount of research regarding two factors that support the hypothesis that polyphenols exert sex-specific effects, including (1) the effects of estrogen and estrogen-signaling in the human cardiovascular system and (2) the effects of berry-derived polyphenols and their cellular targets, which appear to mimic that of endogenous estrogen. Considering the increased risk of CVD in males versus females, increased berry consumption in males should be encouraged for cardio-protection. However, the complex nature of androgens and cardiovascular health remains incompletely understood, yielding this as an unresolved question. Continued research into the sex-specific effects of sex hormones, ERs, and ARs in cardiovascular health is warranted alone and in conjunction with polyphenol interventions. Regardless of a sex-specific effect, berry consumption, due to their rich polyphenolic profile, represents a promising interventional tool in the treatment and prevention of CVD.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2072-6643/13/2/387/s1: Table S1: Search terms used for study compilation, Table S2: Berry Polyphenols and Inflammation, Table S3: Berry Polyphenols and Oxidative Stress, Table S4: Berry Polyphenols and Pathological Remodeling.
Author Contributions
Conceptualization: R.G.F. and B.J.W.; investigation: R.S.N. and C.G.T.; resources: R.G.F. and B.J.W.; writing—original draft preparation: R.S.N. and C.G.T.; writing—review and editing: R.S.N., C.G.T., R.G.F., and B.J.W.; supervision: R.G.F. and B.J.W.; funding acquisition: R.G.F. All authors have read and agreed to the published version of the manuscript.
Funding
This work is supported in part by the Agriculture and Food Research Initiative [grant no. 2019-67017-29257/project accession no. 1018642] from the USDA National Institute of Food and Agriculture.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Virani, S.; Alonso, A.; Benjamin, E.; Bittencourt, M.; Callaway, C.; Carson, A.; Chamberlain, A.; Chang, A.; Cheng, S.; Delling, F.; et al. Heart Disease and Stroke Statistics—2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef] [PubMed]
- Waheed, N.; Elias-Smale, S.; Malas, W.; Maas, A.; Sedlak, T.; Tremmel, J.; Mehta, P. Sex differences in non-obstructive coronary artery disease. Cardiovasc. Res. 2020, 116, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.; Rosengren, A.; Ekman, I. Symptoms in acute coronary syndromes: Does sex make a difference? Am. Heart J. 2004, 148, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Muller-Delp, J. The coronary microvasculation in health and disease. Physiology 2013, 2013, 1–24. [Google Scholar]
- Schwertz, D.; Penckofer, S. Sex differences and the effects of sex hormones on hemostasis and vascular reactivity. Heart Lung J. Crit. Care 2001, 30, 401–426. [Google Scholar] [CrossRef]
- Lam, C.; Arnott, C.; Beale, A.; Chandramouli, C.; Hilfiker-Kleiner, D.; Kaye, D.; Ky, B.; Santema, B.; Silwa, K.; Voors, A. Sex differences in heart failure. Eur. Heart J. 2019, 40, 3859c–3868c. [Google Scholar] [CrossRef]
- Azevedo, P.S.; Polegato, B.F.; Minicucci, M.F.; Paiva, S.A.; Zornoff, L.A. Cardiac Remodeling: Concepts, Clinical Impact, Pathophysiological Mechanisms and Pharmacologic Treatment. Arq. Bras. Cardiol. 2016, 106, 62–69. [Google Scholar] [CrossRef]
- Steven, S.; Frenis, K.; Oelze, M.; Kalinovic, S.; Kuntic, M.; Bayo Jimenez, M.T.; Vujacic-Mirski, K.; Helmstadter, J.; Kroller-Schon, S.; Munzel, T.; et al. Vascular Inflammation and Oxidative Stress: Major Triggers for Cardiovascular Disease. Oxid. Med. Cell. Longev. 2019, 2019, 7092151. [Google Scholar] [CrossRef] [Green Version]
- Renna, N.F.; de Las Heras, N.; Miatello, R.M. Pathophysiology of vascular remodeling in hypertension. Int. J. Hypertens. 2013, 2013, 808353. [Google Scholar] [CrossRef] [Green Version]
- Boese, A.; Kim, S.; Yin, K.-J.; Lee, J.-P.; Hamblin, M. Sex differences in vascular physiology and pathophysiology: Estrogen and androgen signaling in health and disease. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H524–H545. [Google Scholar] [CrossRef]
- Dinu, M.; Abbate, R.; Gensini, G.F.; Casini, A.; Sofi, F. Vegetarian, vegan diets and multiple health outcomes: A systematic review with meta-analysis of observational studies. Crit. Rev. Food Sci. Nutr. 2017, 57, 3640–3649. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Caulfield, L.E.; Garcia-Larsen, V.; Steffen, L.M.; Coresh, J.; Rebholz, C.M. Plant-Based Diets Are Associated With a Lower Risk of Incident Cardiovascular Disease, Cardiovascular Disease Mortality, and All-Cause Mortality in a General Population of Middle-Aged Adults. J. Am. Heart Assoc. 2019, 8, e012865. [Google Scholar] [CrossRef] [PubMed]
- Najjar, R.S.; Moore, C.E.; Montgomery, B.D. Consumption of a defined, plant-based diet reduces lipoprotein(a), inflammation, and other atherogenic lipoproteins and particles within 4 weeks. Clin. Cardiol. 2018, 41, 1062–1068. [Google Scholar] [CrossRef] [PubMed]
- Najjar, R.S.; Montgomery, B.D. A defined, plant-based diet as a potential therapeutic approach in the treatment of heart failure: A clinical case series. Complement. Ther. Med. 2019, 45, 211–214. [Google Scholar] [CrossRef]
- Esselstyn, C.B., Jr.; Gendy, G.; Doyle, J.; Golubic, M.; Roizen, M.F. A way to reverse CAD? J. Fam. Pract. 2014, 63, 356b–364b. [Google Scholar]
- Lin, P.; Leslie, D.; Levine, M.; Davis, G.; Esselstyn, C. Plant-Based Diet Reverses Vascular Endothelial Dysfunction in Patients with Peripheral Arterial Disease. Int. J. Dis. Reversal Prev. 2020, 2, 15. [Google Scholar]
- Upadhyay, S.; Dixit, M. Role of Polyphenols and Other Phytochemicals on Molecular Signaling. Oxid. Med. Cell. Longev. 2015, 2015, 504253. [Google Scholar] [CrossRef]
- Olas, B. Berry Phenolic Antioxidant-Implications for Human Health? Front. Pharmacol. 2018, 9, 78. [Google Scholar] [CrossRef]
- Kimble, R.; Keane, K.M.; Lodge, J.K.; Howatson, G. Dietary intake of anthocyanins and risk of cardiovascular disease: A systematic review and meta-analysis of prospective cohort studies. Crit. Rev. Food Sci. Nutr. 2019, 59, 3032–3043. [Google Scholar] [CrossRef]
- McCullough, M.L.; Peterson, J.J.; Patel, R.; Jacques, P.F.; Shah, R.; Dwyer, J.T. Flavonoid intake and cardiovascular disease mortality in a prospective cohort of US adults. Am. J. Clin. Nutr. 2012, 95, 454–464. [Google Scholar] [CrossRef]
- Huang, H.; Chen, G.; Liao, D.; Zhu, Y.; Xue, X. Effects of Berries Consumption on Cardiovascular Risk Factors: A Meta-analysis with Trial Sequential Analysis of Randomized Controlled Trials. Sci. Rep. 2016, 6, 23625. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gallegos, J.L.; Haskell-Ramsay, C.; Lodge, J.K. Effects of chronic consumption of specific fruit (berries, citrus and cherries) on CVD risk factors: A systematic review and meta-analysis of randomised controlled trials. Eur. J. Nutr. 2020. [Google Scholar] [CrossRef] [PubMed]
- Del Bo’, C.; Bernardi, S.; Marino, M.; Porrini, M.; Tucci, M.; Guglielmetti, S.; Cherubini, A.; Carrieri, B.; Kirkup, B.; Kroon, P.; et al. Systematic Review on Polyphenol Intake and Health Outcomes: Is there Sufficient Evidence to Define a Health-Promoting Polyphenol-Rich Dietary Pattern? Nutrients 2019, 11, 1355. [Google Scholar]
- Zamora-Ros, R.; Rabassa, M.; Cherubini, A.; Urpí-Sardà, M.; Bandinelli, S.; Ferrucci, L.; Andres-Lacueva, C. High Concentrations of a Urinary Biomarker of Polyphenol Intake Are Associated with Decreased Mortality in Older Adults. J. Nutr. 2013, 143, 1445–1450. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Garcia, C.; Sanchez-Quesada, C.; Toledo, E.; Delgado-Rodriguez, M.; Gaforio, J.J. Naturally Lignan-Rich Foods: A Dietary Tool for Health Promotion? Molecules 2019, 24, 917. [Google Scholar] [CrossRef] [Green Version]
- Blaszczyk, A.; Sady, S.; Sielicka, M. The stilbene profile in edible berries. Phytochem. Rev. 2019, 18, 37–67. [Google Scholar] [CrossRef] [Green Version]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef] [Green Version]
- Pathak, S.B.A.; Banerjee, A.; Celep, G.S.; Bissi, L.; Marotta, F. Metabolism of Dietary Polyphenols by Human Gut Microbiota and Their Health Benefits; Academic Press: New York, NY, USA, 2018. [Google Scholar]
- Sandhu, A.K.; Miller, M.G.; Thangthaeng, N.; Scott, T.M.; Shukitt-Hale, B.; Edirisinghe, I.; Burton-Freeman, B. Metabolic fate of strawberry polyphenols after chronic intake in healthy older adults. Food Funct. 2018, 9, 96–106. [Google Scholar] [CrossRef]
- de Ferrars, R.M.; Czank, C.; Zhang, Q.; Botting, N.P.; Kroon, P.A.; Cassidy, A.; Kay, C.D. The pharmacokinetics of anthocyanins and their metabolites in humans. Br. J. Pharmacol. 2014, 171, 3268–3282. [Google Scholar] [CrossRef] [Green Version]
- Manach, C.; Scalbert, A.; Morand, C.; Remesy, C.; Jimenez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr. 2004, 79, 727–747. [Google Scholar] [CrossRef] [Green Version]
- Williamson, G.; Clifford, M.N. Colonic metabolites of berry polyphenols: The missing link to biological activity? Br. J. Nutr. 2010, 104 (Suppl. 3), S48–S66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clifford, M.N.; Scalbert, A. Ellagitannins-nature, occurrence and dietary burden. J. Sci. Food Agric. 2000, 80, 1118–1125. [Google Scholar] [CrossRef]
- Koponen, J.M.; Happonen, A.M.; Mattila, P.H.; Torronen, A.R. Contents of anthocyanins and ellagitannins in selected foods consumed in Finland. J. Agric. Food Chem. 2007, 55, 1612–1619. [Google Scholar] [CrossRef] [PubMed]
- Lipinska, L.; Klewicka, E.; Sojka, M. The structure, occurrence and biological activity of ellagitannins: A general review. Acta Sci. Pol. Technol. Aliment. 2014, 13, 289–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basheer, L.; Kerem, Z. Interactions between CYP3A4 and Dietary Polyphenols. Oxid. Med. Cell. Longev. 2015, 2015, 854015. [Google Scholar] [CrossRef] [Green Version]
- Campesi, I.; Marino, M.; Cipolletti, M.; Romani, A.; Franconi, F. Put “gender glasses” on the effects of phenolic compounds on cardiovascular function and diseases. Eur. J. Nutr. 2018, 57, 2677–2691. [Google Scholar] [CrossRef]
- Ostlund, J.; Zlabek, V.; Zamaratskaia, G. In vitro inhibition of human CYP2E1 and CYP3A by quercetin and myricetin in hepatic microsomes is not gender dependent. Toxicology 2017, 381, 10–18. [Google Scholar] [CrossRef]
- Erlund, I.; Kosonen, T.; Alfthan, G.; Mäenpää, J.; Perttunen, K.; Kenraali, J.; Parantainen, J.; Aro, A. Pharmacokinetics of quercetin from quercetin aglycone and rutin in healthy volunteers. Pharmacokinet. Dispos. 2000, 56, 545–553. [Google Scholar] [CrossRef]
- Dufour, C.; Dangles, O. Flavonoid-serum albumin complexation: Determination of binding constants and binding sites by fluorescence spectroscopy. Biochim. Biophys. Acta 2005, 1721, 164–173. [Google Scholar] [CrossRef]
- Kitson, T.M. Spectrophotometric and kinetic studies on the binding of the bioflavonoid quercetin to bovine serum albumin. Biosci. Biotechnol. Biochem. 2004, 68, 2165–2170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziberna, L.; Lunder, M.; Moze, S.; Vanzo, A.; Tramer, F.; Passamonti, S.; Drevensek, G. Acute cardioprotective and cardiotoxic effects of bilberry anthocyanins in ischemia-reperfusion injury: Beyond concentration-dependent antioxidant activity. Cardiovasc. Toxicol. 2010, 10, 283–294. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, C.; Driessen, A.J.; Recourt, K. The uncoupling efficiency and affinity of flavonoids for vesicles. Biochem. Pharmacol. 2000, 60, 1593–1600. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, B.L.; Ruthven, C.R.; Sandler, M. Gut flora and the origin of some urinary aromatic phenolic compounds. Biochem. Pharmacol. 1994, 47, 2294–2297. [Google Scholar] [CrossRef]
- Phipps, A.N.; Stewart, J.; Wright, B.; Wilson, I.D. Effect of diet on the urinary excretion of hippuric acid and other dietary-derived aromatics in rat. A complex interaction between diet, gut microflora and substrate specificity. Xenobiotica 1998, 28, 527–537. [Google Scholar] [CrossRef]
- Ziberna, L.; Tramer, F.; Moze, S.; Vrhovsek, U.; Mattivi, F.; Passamonti, S. Transport and bioactivity of cyanidin 3-glucoside into the vascular endothelium. Free Radic. Biol. Med. 2012, 52, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Maestro, A.; Terdoslavich, M.; Vanzo, A.; Kuku, A.; Tramer, F.; Nicolin, V.; Micali, F.; Decorti, G.; Passamonti, S. Expression of bilitranslocase in the vascular endothelium and its function as a flavonoid transporter. Cardiovasc. Res. 2010, 85, 175–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziberna, L.; Kim, J.H.; Auger, C.; Passamonti, S.; Schini-Kerth, V. Role of endothelial cell membrane transport in red wine polyphenols-induced coronary vasorelaxation: Involvement of bilitranslocase. Food Funct. 2013, 4, 1452–1456. [Google Scholar] [CrossRef]
- Ziberna, L.; Lunder, M.; Tramer, F.; Drevensek, G.; Passamonti, S. The endothelial plasma membrane transporter bilitranslocase mediates rat aortic vasodilation induced by anthocyanins. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 68–74. [Google Scholar] [CrossRef]
- Ziberna, L.; Fornasaro, S.; Čvorović, J.; Tramer, F.; Passamonti, S. Bioavailability of Flavonoids: The Role of Cell Membrane Transporters. In Polyphenols in Human Health and Disease; Academic Press: New York, NY, USA, 2014; pp. 489–511. [Google Scholar] [CrossRef]
- Morris, M.E.; Lee, H.J.; Predko, L.M. Gender differences in the membrane transport of endogenous and exogenous compounds. Pharmacol. Rev. 2003, 55, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Soukup, S.T.; Helppi, J.; Muller, D.R.; Zierau, O.; Watzl, B.; Vollmer, G.; Diel, P.; Bub, A.; Kulling, S.E. Phase II metabolism of the soy isoflavones genistein and daidzein in humans, rats and mice: A cross-species and sex comparison. Arch. Toxicol. 2016, 90, 1335–1347. [Google Scholar] [CrossRef]
- Battson, M.L.; Lee, D.M.; Weir, T.L.; Gentile, C.L. The gut microbiota as a novel regulator of cardiovascular function and disease. J. Nutr. Biochem. 2018, 56, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, M.; Weeks, T.L.; Hazen, S.L. Gut Microbiota and Cardiovascular Disease. Circ. Res. 2020, 127, 553–570. [Google Scholar] [CrossRef] [PubMed]
- Santos-Marcos, J.; Rangel-Zuñiga, O.; Jimenez-Lucena, R.; Quintana-Navarro, G.; Garcia-Carpintero, S.; Malagon, M.; Landa, B.; Tena-Sempere, M.; Perez-Martinez, P.; Lopez-Miranda, J.; et al. Influence of gender and menopausal status on gut microbiota. Maturitas 2018, 116, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Edogawa, S.; Peters, S.; Jenkins, G.; Gurunathan, S.; Sundt, W.; Johnson, S.; Lennon, R.; Dyer, R.; Camilleri, M.; Kashyap, P.; et al. Sex differences in NSAID-induced perturbation of human intestinal barrier function and microbiota. FASEB J. 2018, 32. [Google Scholar] [CrossRef] [PubMed]
- Haro, C.; Rangel-Zuniga, O.A.; Alcala-Diaz, J.F.; Gomez-Delgado, F.; Perez-Martinez, P.; Delgado-Lista, J.; Quintana-Navarro, G.M.; Landa, B.B.; Navas-Cortes, J.A.; Tena-Sempere, M.; et al. Intestinal Microbiota Is Influenced by Gender and Body Mass Index. PLoS ONE 2016, 11, e0154090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, T.; Vila, A.; Garmaeva, S.; Jankipersadsing, S.; Imhann, F.; Collij, V.; Bonder, M.; Jiang, X.; Gurry, T.; Alm, E.; et al. Analysis of 1135 gut metagenomes identifies sex-specific resistome profiles. Gut Microbes 2018, 10, 358–366. [Google Scholar] [CrossRef] [Green Version]
- Santos-Marcos, J.A.; Haro, C.; Vega-Rojas, A.; Alcala-Diaz, J.F.; Molina-Abril, H.; Leon-Acuna, A.; Lopez-Moreno, J.; Landa, B.B.; Tena-Sempere, M.; Perez-Martinez, P.; et al. Sex Differences in the Gut Microbiota as Potential Determinants of Gender Predisposition to Disease. Mol. Nutr. Food Res. 2019, 63, e1800870. [Google Scholar] [CrossRef]
- Dominianni, C.; Sinha, R.; Goedert, J.J.; Pei, Z.; Yang, L.; Hayes, R.B.; Ahn, J. Sex, body mass index, and dietary fiber intake influence the human gut microbiome. PLoS ONE 2015, 10, e0124599. [Google Scholar] [CrossRef] [Green Version]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [Green Version]
- Richards, L.B.; Li, M.; van Esch, B.C.; Garssen, J.; Folkerts, G. The effects of short-chain fatty acids on the cardiovascular system. PharmaNutrition 2016, 4, 68–111. [Google Scholar] [CrossRef]
- Jiao, X.; Wang, Y.; Lin, Y.; Lang, Y.; Li, E.; Zhang, X.; Zhang, Q.; Feng, Y.; Meng, X.; Li, B. Blueberry polyphenols extract as a potential prebiotic with anti-obesity effects on C57BL/6 J mice by modulating the gut microbiota. J. Nutr. Biochem. 2019, 64, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Etxeberria, U.; Arias, N.; Boque, N.; Macarulla, M.T.; Portillo, M.P.; Martinez, J.A.; Milagro, F.I. Reshaping faecal gut microbiota composition by the intake of trans-resveratrol and quercetin in high-fat sucrose diet-fed rats. J. Nutr. Biochem. 2015, 26, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Larrosa, M.; Yanez-Gascon, M.J.; Selma, M.V.; Gonzalez-Sarrias, A.; Toti, S.; Ceron, J.J.; Tomas-Barberan, F.; Dolara, P.; Espin, J.C. Effect of a low dose of dietary resveratrol on colon microbiota, inflammation and tissue damage in a DSS-induced colitis rat model. J. Agric. Food Chem. 2009, 57, 2211–2220. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; Bose, S.; Seo, J.G.; Chung, W.S.; Lim, C.Y.; Kim, H. The effects of co-administration of probiotics with herbal medicine on obesity, metabolic endotoxemia and dysbiosis: A randomized double-blind controlled clinical trial. Clin. Nutr. 2014, 33, 973–981. [Google Scholar] [CrossRef] [PubMed]
- DiRienzo, D.B. Effect of probiotics on biomarkers of cardiovascular disease: Implications for heart-healthy diets. Nutr. Rev. 2014, 72, 18–29. [Google Scholar] [CrossRef]
- Gonzalez-Sarrias, A.; Romo-Vaquero, M.; Garcia-Villalba, R.; Cortes-Martin, A.; Selma, M.V.; Espin, J.C. The Endotoxemia Marker Lipopolysaccharide-Binding Protein is Reduced in Overweight-Obese Subjects Consuming Pomegranate Extract by Modulating the Gut Microbiota: A Randomized Clinical Trial. Mol. Nutr. Food Res. 2018, 62, e1800160. [Google Scholar] [CrossRef]
- Tzounis, X.; Rodriguez-Mateos, A.; Vulevic, J.; Gibson, G.R.; Kwik-Uribe, C.; Spencer, J.P. Prebiotic evaluation of cocoa-derived flavanols in healthy humans by using a randomized, controlled, double-blind, crossover intervention study. Am. J. Clin. Nutr. 2011, 93, 62–72. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Indias, I.; Sanchez-Alcoholado, L.; Perez-Martinez, P.; Andres-Lacueva, C.; Cardona, F.; Tinahones, F.; Queipo-Ortuno, M.I. Red wine polyphenols modulate fecal microbiota and reduce markers of the metabolic syndrome in obese patients. Food Funct. 2016, 7, 1775–1787. [Google Scholar] [CrossRef] [Green Version]
- Ntemiri, A.; Ghosh, T.; Gheller, M.; Tran, T.; Blum, J.; Pellanda, P.; Vlckova, K.; Neto, M.; Howell, A.; Thalacker-Mercer, A.; et al. Whole Blueberry and Isolated Polyphenol-Rich Fractions Modulate Specific Gut Microbes in an In Vitro Colon Model and in a Pilot Study in Human Consumers. Nutrients 2020, 12, 2800. [Google Scholar] [CrossRef]
- Stanhewicz, A.; Wenner, M.; Stachenfeld, N. Sex differences in endothelial function important to vascular health and overall cardiovascular disease risk across the lifespan. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1569–H1588. [Google Scholar] [CrossRef]
- Villablanca, A.; Jayachandran, M.; Banka, C. Atherosclerosis and sex hormones: Current concepts. Clin. Sci. 2010, 119, 493–513. [Google Scholar] [CrossRef] [PubMed]
- Gilliver, S. Sex steroids as inflammatory regulators. J. Steroid Biochem. Mol. Biol. 2010, 120, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Fairweather, D. Sex Differences in Inflammation During Atherosclerosis. Clin. Med. Insights Cardiol. 2014, 8, 49–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, J.; Shen, Y.; Li, R. Estrogen synthesis and signaling pathways during ageing: From periphery to brain. Trends Mol. Med. 2013, 19, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Gaskins, A.; Wilchesky, M.; Mumford, S.; Whitcomb, B.; Browne, R.; Wactawski-Wende, J.; Perkins, N.; Schisterman, E. Endogenous Reproductive Hormones and C-reactive Protein Across the Menstrual Cycle: The BioCycle Study. Am. J. Epidemiol. 2012, 175, 423–431. [Google Scholar] [CrossRef]
- Karowicz-Bilinska, A.; Plodzidym, M.; Krol, J.; Lewinska, A.; Bartosz, G. Changes of markers of oxidative stress during menstrual cycle. Redox Rep. 2008, 13, 237–240. [Google Scholar] [CrossRef]
- Palan, P.; Magneson, A.; Castillo, M.; Dunne, J.; Mikhail, M. Effects of menstrual cycle and oral contraceptive use on serum levels of lipid-soluble antioxidants. Am. J. Obstet. Gynecol. 2006, 194, e35–e38. [Google Scholar] [CrossRef]
- Hutson, D.D.; Gurrala, R.; Ogola, B.O.; Zimmerman, M.A.; Mostany, R.; Satou, R.; Lindsey, S.H. Estrogen receptor profiles across tissues from male and female Rattus norvegicus. Biol. Sex Differ. 2019, 10, 4. [Google Scholar] [CrossRef] [Green Version]
- Nordmeyer, J.; Eder, S.; Mahmoodzadeh, S.; Martus, P.; Fielitz, J.; Bass, J.; Bethke, N.; Zurbrügg, H.; Pregla, R.; Hetzer, R.; et al. Upregulation of Myocardial Estrogen Receptors in Human Aortic Stenosis. Circulation 2004, 110, 3270–3275. [Google Scholar] [CrossRef] [Green Version]
- Gavin, K.; Seals, D.; Silver, A.; Moreau, K. Vascular Endothelial Estrogen Receptor Alpha Is Modulated by Estrogen Status and Related to Endothelial Function and Endothelial Nitric Oxide Synthase in Healthy Women. J. Clin. Endocrinol. Metab. 2009, 94, 3513–3520. [Google Scholar] [CrossRef] [Green Version]
- Cooke, P.; Nanjappa, M.; Ko, C.; Prins, G.; Hess, R. Estrogen in Male Physiology. Physiol. Rev. 2017, 97, 995–1043. [Google Scholar] [CrossRef] [PubMed]
- Coburn, S.; Stanczyk, F.; Falk, R.; McGlynn, K.; Brinton, L.; Sampson, J.; Bradwin, G.; Xu, X.; Trabert, B. Comparability of serum, plasma, and urinary estrogen and estrogen metabolite measurements by sex and menopausal status. Cancer Causes Control 2019, 30, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Brzozowski, A.M.; Pike, A.C.; Dauter, Z.; Hubbard, R.E.; Bonn, T.; Engstrom, O.; Ohman, L.; Greene, G.L.; Gustafsson, J.A.; Carlquist, M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997, 389, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Jung, B.; Kim, M.; Kim, H.; Kim, D.; Yang, J.; Her, S.; Song, Y. Caffeic acid phenethyl ester, a component of beehive propolis, is a novel selective estrogen receptor modulator. Phytother. Res. 2010, 24, 295–300. [Google Scholar] [CrossRef] [PubMed]
- van der Woude, H.; Ter Veld, M.G.; Jacobs, N.; van der Saag, P.T.; Murk, A.J.; Rietjens, I.M. The stimulation of cell proliferation by quercetin is mediated by the estrogen receptor. Mol. Nutr. Food Res. 2005, 49, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Papoutsi, Z.; Kassi, E.; Tsiapara, A.; Fokialakis, N.; Chrousos, G.P.; Moutsatsou, P. Evaluation of estrogenic/antiestrogenic activity of ellagic acid via the estrogen receptor subtypes ERalpha and ERbeta. J. Agric. Food Chem. 2005, 53, 7715–7720. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Sun, B. Toll-like receptor 4 in atherosclerosis. J. Cell. Mol. Med. 2007, 11, 88–95. [Google Scholar] [CrossRef] [Green Version]
- Nunes, K.P.; de Oliveira, A.A.; Lima, V.V.; Webb, R.C. Toll-Like Receptor 4 and Blood Pressure: Lessons From Animal Studies. Front. Physiol. 2019, 10, 655. [Google Scholar] [CrossRef]
- Yu, L.; Feng, Z. The Role of Toll-Like Receptor Signaling in the Progression of Heart Failure. Mediat. Inflamm. 2018, 2018, 9874109. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Peppel, K.; Sivashanmugam, P.; Orman, E.S.; Brian, L.; Exum, S.T.; Freedman, N.J. Expression of tumor necrosis factor receptor-1 in arterial wall cells promotes atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1087–1094. [Google Scholar] [CrossRef] [Green Version]
- Mehaffey, E.; Majid, D.S.A. Tumor necrosis factor-alpha, kidney function, and hypertension. Am. J. Physiol. Renal Physiol. 2017, 313, F1005–F1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duerrschmid, C.; Trial, J.; Wang, Y.; Entman, M.L.; Haudek, S.B. Tumor necrosis factor: A mechanistic link between angiotensin-II-induced cardiac inflammation and fibrosis. Circ. Heart Fail. 2015, 8, 352–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, T.; Ascher, E.; Alapat, D.; Olevskaia, Y.; Hingorani, A. Activation of p38MAPK signaling cascade in a VSMC injury model: Role of p38MAPK inhibitors in limiting VSMC proliferation. Eur. J. Vasc. Endovasc. Surg. 2005, 29, 470–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ushio-Fukai, M.; Alexander, R.W.; Akers, M.; Griendling, K.K. p38 Mitogen-activated protein kinase is a critical component of the redox-sensitive signaling pathways activated by angiotensin II. Role in vascular smooth muscle cell hypertrophy. J. Biol. Chem. 1998, 273, 15022–15029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshizumi, M.; Tsuchiya, K.; Kirima, K.; Kyaw, M.; Suzaki, Y.; Tamaki, T. Quercetin inhibits Shc- and phosphatidylinositol 3-kinase-mediated c-Jun N-terminal kinase activation by angiotensin II in cultured rat aortic smooth muscle cells. Mol. Pharmacol. 2001, 60, 656–665. [Google Scholar]
- Liu, Q.; Chen, Y.; Auger-Messier, M.; Molkentin, J.D. Interaction between NFkappaB and NFAT coordinates cardiac hypertrophy and pathological remodeling. Circ. Res. 2012, 110, 1077–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaub, M.C.; Hefti, M.A.; Harder, B.A.; Eppenberger, H.M. Various hypertrophic stimuli induce distinct phenotypes in cardiomyocytes. J. Mol. Med. 1997, 75, 901–920. [Google Scholar] [CrossRef]
- Liao, P.; Georgakopoulos, D.; Kovacs, A.; Zheng, M.; Lerner, D.; Pu, H.; Saffitz, J.; Chien, K.; Xiao, R.P.; Kass, D.A.; et al. The in vivo role of p38 MAP kinases in cardiac remodeling and restrictive cardiomyopathy. Proc. Natl. Acad. Sci. USA 2001, 98, 12283–12288. [Google Scholar] [CrossRef] [Green Version]
- Petrich, B.G.; Molkentin, J.D.; Wang, Y. Temporal activation of c-Jun N-terminal kinase in adult transgenic heart via cre-loxP-mediated DNA recombination. FASEB J. 2003, 17, 749–751. [Google Scholar] [CrossRef]
- Deopurkar, R.; Ghanim, H.; Friedman, J.; Abuaysheh, S.; Sia, C.L.; Mohanty, P.; Viswanathan, P.; Chaudhuri, A.; Dandona, P. Differential effects of cream, glucose, and orange juice on inflammation, endotoxin, and the expression of Toll-like receptor-4 and suppressor of cytokine signaling-3. Diabetes Care 2010, 33, 991–997. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xia, G.; Zhang, Y.; Liu, J.; Liu, X.; Li, W.; Lv, Y.; Wei, S.; Liu, J.; Quan, J. Palmitate induces VSMC apoptosis via toll like receptor (TLR)4/ROS/p53 pathway. Atherosclerosis 2017, 263, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Sanchez, L.; Madrid-Miller, A.; Chavez-Rueda, K.; Legorreta-Haquet, M.V.; Tesoro-Cruz, E.; Blanco-Favela, F. Activation of TLR2 and TLR4 by minimally modified low-density lipoprotein in human macrophages and monocytes triggers the inflammatory response. Hum. Immunol. 2010, 71, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Smiley, S.T.; King, J.A.; Hancock, W.W. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J. Immunol. 2001, 167, 2887–2894. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Cox, C.J.; Alvarez, K.M.; Cunningham, M.W. Cutting edge: Cardiac myosin activates innate immune responses through TLRs. J. Immunol. 2009, 183, 27–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dauphinee, S.M.; Karsan, A. Lipopolysaccharide signaling in endothelial cells. Lab. Investig. 2006, 86, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Deshpande, R.; Khalili, H.; Pergolizzi, R.; Michael, S.; Change, M. Estradiol down-regulates LPS-induced cytokine production and NFκB activation in murine macrophages. Am. J. Reprod. Immunol. 1997, 38, 46–64. [Google Scholar] [CrossRef]
- Michelsen, K.S.; Wong, M.H.; Shah, P.K.; Zhang, W.; Yano, J.; Doherty, T.M.; Akira, S.; Rajavashisth, T.B.; Arditi, M. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc. Natl. Acad. Sci. USA 2004, 101, 10679–10684. [Google Scholar] [CrossRef] [Green Version]
- Riad, A.; Jager, S.; Sobirey, M.; Escher, F.; Yaulema-Riss, A.; Westermann, D.; Karatas, A.; Heimesaat, M.M.; Bereswill, S.; Dragun, D.; et al. Toll-like receptor-4 modulates survival by induction of left ventricular remodeling after myocardial infarction in mice. J. Immunol. 2008, 180, 6954–6961. [Google Scholar] [CrossRef] [Green Version]
- Menghini, R.; Campia, U.; Tesauro, M.; Marino, A.; Rovella, V.; Rodia, G.; Schinzari, F.; Tolusso, B.; di Daniele, N.; Federici, M.; et al. Toll-like receptor 4 mediates endothelial cell activation through NF-kappaB but is not associated with endothelial dysfunction in patients with rheumatoid arthritis. PLoS ONE 2014, 9, e99053. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Zou, C.; Mei, L.; Zhang, Y.; Qian, Y.; You, S.; Pan, Y.; Xu, Z.; Bai, B.; Huang, W.; et al. MD2 mediates angiotensin II-induced cardiac inflammation and remodeling via directly binding to Ang II and activating TLR4/NF-kappaB signaling pathway. Basic Res. Cardiol. 2017, 112, 9. [Google Scholar] [CrossRef]
- Dange, R.B.; Agarwal, D.; Masson, G.S.; Vila, J.; Wilson, B.; Nair, A.; Francis, J. Central blockade of TLR4 improves cardiac function and attenuates myocardial inflammation in angiotensin II-induced hypertension. Cardiovasc. Res. 2014, 103, 17–27. [Google Scholar] [CrossRef]
- Sullivan, J.; Rodriguez-Miguelez, P.; Zimmerman, M.; Harris, R. Differences in angiotensin (1–7) between men and women. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H1171–H1176. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Engel, A.; Morcillo, L.; Aranda, F.; Ruiz, M.; Gaitan, M.; Mayor-Olea, A.; Aranda, P.; Ferrario, C. Influence of gender and genetic variability on plasma angiotensin peptides. J. Renin Angiotensin Aldosterone Syst. 2006, 7, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Rifai, N.; Pfeffer, M.; Sacks, F.; Lepage, S.; Braunwald, E. Elevation of tumor necrosis factor-alpha and increased risk of recurrent coronary events after myocardial infarction. Circulation 2000, 101, 2149–2153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haudek, S.B.; Taffet, G.E.; Schneider, M.D.; Mann, D.L. TNF provokes cardiomyocyte apoptosis and cardiac remodeling through activation of multiple cell death pathways. J. Clin. Investig. 2007, 117, 2692–2701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Xu, X.; Potter, B.J.; Wang, W.; Kuo, L.; Michael, L.; Bagby, G.J.; Chilian, W.M. TNF-alpha contributes to endothelial dysfunction in ischemia/reperfusion injury. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 475–480. [Google Scholar] [CrossRef]
- Picchi, A.; Gao, X.; Belmadani, S.; Potter, B.J.; Focardi, M.; Chilian, W.M.; Zhang, C. Tumor necrosis factor-alpha induces endothelial dysfunction in the prediabetic metabolic syndrome. Circ. Res. 2006, 99, 69–77. [Google Scholar] [CrossRef] [Green Version]
- Kroetsch, J.T.; Levy, A.S.; Zhang, H.; Aschar-Sobbi, R.; Lidington, D.; Offermanns, S.; Nedospasov, S.A.; Backx, P.H.; Heximer, S.P.; Bolz, S.S. Constitutive smooth muscle tumour necrosis factor regulates microvascular myogenic responsiveness and systemic blood pressure. Nat. Commun. 2017, 8, 14805. [Google Scholar] [CrossRef] [Green Version]
- Bozkurt, B.; Kribbs, S.B.; Clubb, F.J., Jr.; Michael, L.H.; Didenko, V.V.; Hornsby, P.J.; Seta, Y.; Oral, H.; Spinale, F.G.; Mann, D.L. Pathophysiologically relevant concentrations of tumor necrosis factor-alpha promote progressive left ventricular dysfunction and remodeling in rats. Circulation 1998, 97, 1382–1391. [Google Scholar] [CrossRef] [Green Version]
- Moe, K.T.; Khairunnisa, K.; Yin, N.O.; Chin-Dusting, J.; Wong, P.; Wong, M.C. Tumor necrosis factor-alpha-induced nuclear factor-kappaB activation in human cardiomyocytes is mediated by NADPH oxidase. J. Physiol. Biochem. 2014, 70, 769–779. [Google Scholar] [CrossRef]
- Defer, N.; Azroyan, A.; Pecker, F.; Pavoine, C. TNFR1 and TNFR2 signaling interplay in cardiac myocytes. J. Biol. Chem. 2007, 282, 35564–35573. [Google Scholar] [CrossRef] [Green Version]
- Hamid, T.; Gu, Y.; Ortines, R.V.; Bhattacharya, C.; Wang, G.; Xuan, Y.T.; Prabhu, S.D. Divergent tumor necrosis factor receptor-related remodeling responses in heart failure: Role of nuclear factor-kappaB and inflammatory activation. Circulation 2009, 119, 1386–1397. [Google Scholar] [CrossRef] [PubMed]
- Kadokami, T.; McTiernan, C.F.; Kubota, T.; Frye, C.S.; Feldman, A.M. Sex-related survival differences in murine cardiomyopathy are associated with differences in TNF-receptor expression. J. Clin. Investig. 2000, 106, 589–597. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Crisostomo, P.R.; Markel, T.A.; Wang, Y.; Meldrum, D.R. Mechanisms of sex differences in TNFR2-mediated cardioprotection. Circulation 2008, 118, S38–S45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S.; Weitzmann, M.; Cenci, S.; Ross, F.; Adler, S.; Pacifici, R. Estrogen decreases TNF gene expression by blocking JNK activity and the resulting production of c-Jun and JunD. J. Clin. Investig. 1999, 104, 503–513. [Google Scholar] [CrossRef] [Green Version]
- Yoshizumi, M.; Perrella, M.A.; Burnett, J.C., Jr.; Lee, M.E. Tumor necrosis factor downregulates an endothelial nitric oxide synthase mRNA by shortening its half-life. Circ. Res. 1993, 73, 205–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Z.; Liu, M.; Wu, Y.; Sharma, V.; Luo, T.; Ouyang, J.; McNeill, J.H. N-acetylcysteine attenuates TNF-alpha-induced human vascular endothelial cell apoptosis and restores eNOS expression. Eur. J. Pharmacol. 2006, 550, 134–142. [Google Scholar] [CrossRef]
- Grumbach, I.; Chen, W.; Mertens, S.; Harrison, D. A negative feedback mechanism involving nitric oxide and nuclear factor kappa-B modulates endothelial nitric oxide synthase transcription. J. Mol. Cell. Cardiol. 2005, 39, 595–603. [Google Scholar] [CrossRef]
- Revankar, C.; Cimino, D.; Sklar, L.; Arterburn, J.; Prossnitz, E. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 2005, 307, 1625–1630. [Google Scholar] [CrossRef] [Green Version]
- Stirone, C.; Boroujerdi, A.; Duckles, S.; Krause, D. Estrogen receptor activation of phosphoinositide-3 kinase, akt, and nitric oxide signaling in cerebral blood vessels: Rapid and long-term effects. Mol. Pharmacol. 2005, 67, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Sharma, G.; Prossnitz, E. Mechanisms of estradiol-induced insulin secretion by the G protein-coupled estrogen receptor GPR30/GPER in pancreatic β-cells. Endocrinology 2011, 152, 3030–3039. [Google Scholar] [CrossRef] [PubMed]
- Peluso, A.A.; Bertelsen, J.B.; Andersen, K.; Mortsensen, T.P.; Hansen, P.B.; Sumners, C.; Bader, M.; Santos, R.A.; Steckelings, U.M. Identification of protein phosphatase involvement in the AT2 receptor-induced activation of endothelial nitric oxide synthase. Clin. Sci. 2018, 132, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Simoncini, T.; Maffei, S.; Basta, G.; Barsacchi, G.; Genazzani, A.; Liao, J.; De Caterina, R. Estrogens and glucocorticoids inhibit endothelial vascular cell adhesion molecule-1 expression by different transcriptional mechanisms. Circ. Res. 2000, 87, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Torres-Estay, V.; Carreño, D.; Francisco, I.S.; Sotomayor, P.; Godoy, A.; Smith, G. Androgen receptor in human endothelial cells. J. Endocrinol. 2015, 224, R131–R137. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Death, A.; Handelsman, D. Androgens and cardiovascular disease. Endocr. Rev. 2003, 24, 313–340. [Google Scholar] [CrossRef]
- Diamanti-Kandarakis, E. Polycystic ovarian syndrome: Pathophysiology, molecular aspects and clinical implications. Expert Rev. Mol. Med. 2008, 10, e3. [Google Scholar] [CrossRef]
- Goodwin, B.L.; Pendleton, L.C.; Levy, M.M.; Solomonson, L.P.; Eichler, D.C. Tumor necrosis factor-alpha reduces argininosuccinate synthase expression and nitric oxide production in aortic endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1115–H1121. [Google Scholar] [CrossRef]
- Karlsen, A.; Paur, I.; Bohn, S.K.; Sakhi, A.K.; Borge, G.I.; Serafini, M.; Erlund, I.; Laake, P.; Tonstad, S.; Blomhoff, R. Bilberry juice modulates plasma concentration of NF-kappaB related inflammatory markers in subjects at increased risk of CVD. Eur. J. Nutr. 2010, 49, 345–355. [Google Scholar] [CrossRef]
- Lehtonen, H.M.; Suomela, J.P.; Tahvonen, R.; Yang, B.; Venojarvi, M.; Viikari, J.; Kallio, H. Different berries and berry fractions have various but slightly positive effects on the associated variables of metabolic diseases on overweight and obese women. Eur. J. Clin. Nutr. 2011, 65, 394–401. [Google Scholar] [CrossRef] [Green Version]
- Basu, A.; Wilkinson, M.; Penugonda, K.; Simmons, B.; Betts, N.M.; Lyons, T.J. Freeze-dried strawberry powder improves lipid profile and lipid peroxidation in women with metabolic syndrome: Baseline and post intervention effects. Nutr. J. 2009, 8, 43. [Google Scholar] [CrossRef]
- Basu, A.; Fu, D.X.; Wilkinson, M.; Simmons, B.; Wu, M.; Betts, N.M.; Du, M.; Lyons, T.J. Strawberries decrease atherosclerotic markers in subjects with metabolic syndrome. Nutr. Res. 2010, 30, 462–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, A.; Betts, N.M.; Nguyen, A.; Newman, E.D.; Fu, D.; Lyons, T.J. Freeze-dried strawberries lower serum cholesterol and lipid peroxidation in adults with abdominal adiposity and elevated serum lipids. J. Nutr. 2014, 144, 830–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanoeva, J.P.; Stefova, M.; Andonovska, K.B.; Vankova, A.; Stafilov, T. Phenolics and mineral content in bilberry and bog bilberry from Macedonia. Int. J. Food Prop. 2017, 20, S863–S883. [Google Scholar] [CrossRef]
- Aaby, K.; Wrolstad, R.E.; Ekeberg, D.; Skrede, G. Polyphenol composition and antioxidant activity in strawberry purees; impact of achene level and storage. J. Agric. Food Chem. 2007, 55, 5156–5166. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.S.; Kim, S.; Hong, S.J.; Choi, S.C.; Choi, J.H.; Kim, J.H.; Park, C.Y.; Cho, J.Y.; Lee, T.B.; Kwon, J.W.; et al. Black Raspberry Extract Increased Circulating Endothelial Progenitor Cells and Improved Arterial Stiffness in Patients with Metabolic Syndrome: A Randomized Controlled Trial. J. Med. Food. 2016, 19, 346–352. [Google Scholar] [CrossRef]
- Aghababaee, S.K.; Vafa, M.; Shidfar, F.; Tahavorgar, A.; Gohari, M.; Katebi, D.; Mohammadi, V. Effects of blackberry (Morus nigra L.) consumption on serum concentration of lipoproteins, apo A-I, apo B, and high-sensitivity-C-reactive protein and blood pressure in dyslipidemic patients. J. Res. Med. Sci. 2015, 20, 684–691. [Google Scholar] [CrossRef]
- Schell, J.; Betts, N.M.; Lyons, T.J.; Basu, A. Raspberries Improve Postprandial Glucose and Acute and Chronic Inflammation in Adults with Type 2 Diabetes. Ann. Nutr. Metab. 2019, 74, 165–174. [Google Scholar] [CrossRef]
- Nair, A.R.; Mariappan, N.; Stull, A.J.; Francis, J. Blueberry supplementation attenuates oxidative stress within monocytes and modulates immune cell levels in adults with metabolic syndrome: A randomized, double-blind, placebo-controlled trial. Food Funct. 2017, 8, 4118–4128. [Google Scholar] [CrossRef]
- Li, T.; Li, F.; Liu, X.; Liu, J.; Li, D. Synergistic anti-inflammatory effects of quercetin and catechin via inhibiting activation of TLR4-MyD88-mediated NF-kappaB and MAPK signaling pathways. Phytother. Res. 2019, 33, 756–767. [Google Scholar] [CrossRef]
- Bhaskar, S.; Sudhakaran, P.R.; Helen, A. Quercetin attenuates atherosclerotic inflammation and adhesion molecule expression by modulating TLR-NF-kappaB signaling pathway. Cell. Immunol. 2016, 310, 131–140. [Google Scholar] [CrossRef]
- Zhang, M.; Xue, Y.; Chen, H.; Meng, L.; Chen, B.; Gong, H.; Zhao, Y.; Qi, R. Resveratrol Inhibits MMP3 and MMP9 Expression and Secretion by Suppressing TLR4/NF-kappaB/STAT3 Activation in Ox-LDL-Treated HUVECs. Oxid. Med. Cell. Longev. 2019, 2019, 9013169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Lin, G.; Wan, W.; Li, X.; Zeng, B.; Yang, B.; Huang, C. Resveratrol, a polyphenol phytoalexin, protects cardiomyocytes against anoxia/reoxygenation injury via the TLR4/NF-kappaB signaling pathway. Int. J. Mol. Med. 2012, 29, 557–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivasinprasasn, S.; Pantan, R.; Thummayot, S.; Tocharus, J.; Suksamrarn, A.; Tocharus, C. Cyanidin-3-glucoside attenuates angiotensin II-induced oxidative stress and inflammation in vascular endothelial cells. Chem. Biol. Interact. 2016, 260, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.Y.; Liu, Y.M.; Wang, J.; Wang, X.N.; Li, C.Y. Anti-inflammatory effect of the blueberry anthocyanins malvidin-3-glucoside and malvidin-3-galactoside in endothelial cells. Molecules 2014, 19, 12827–12841. [Google Scholar] [CrossRef] [Green Version]
- Liao, H.H.; Zhang, N.; Meng, Y.Y.; Feng, H.; Yang, J.J.; Li, W.J.; Chen, S.; Wu, H.M.; Deng, W.; Tang, Q.Z. Myricetin Alleviates Pathological Cardiac Hypertrophy via TRAF6/TAK1/MAPK and Nrf2 Signaling Pathway. Oxid. Med. Cell. Longev. 2019, 2019, 6304058. [Google Scholar] [CrossRef]
- Nwachukwu, J.; Srinivasan, S.; Bruno, N.; Parent, A.; Hughes, T.; Pollock, J.; Gjyshi, O.; Cavett, V.; Nowak, J.; Garcia-Ordonez, R.; et al. Resveratrol modulates the inflammatory response via an estrogen receptor-signal integration network. eLife 2014, 3. [Google Scholar] [CrossRef]
- Han, D.; Ybanez, M.D.; Ahmadi, S.; Yeh, K.; Kaplowitz, N. Redox regulation of tumor necrosis factor signaling. Antioxid. Redox Signal. 2009, 11, 2245–2263. [Google Scholar] [CrossRef] [Green Version]
- Dikalov, S.I.; Ungvari, Z. Role of mitochondrial oxidative stress in hypertension. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1417–H1427. [Google Scholar] [CrossRef] [Green Version]
- Dey, S.; DeMazumder, D.; Sidor, A.; Foster, D.B.; O’Rourke, B. Mitochondrial ROS Drive Sudden Cardiac Death and Chronic Proteome Remodeling in Heart Failure. Circ. Res. 2018, 123, 356–371. [Google Scholar] [CrossRef]
- Kornfeld, O.S.; Hwang, S.; Disatnik, M.H.; Chen, C.H.; Qvit, N.; Mochly-Rosen, D. Mitochondrial reactive oxygen species at the heart of the matter: New therapeutic approaches for cardiovascular diseases. Circ. Res. 2015, 116, 1783–1799. [Google Scholar] [CrossRef]
- Hajjar, R.J.; Leopold, J.A. Xanthine oxidase inhibition and heart failure: Novel therapeutic strategy for ventricular dysfunction? Circ. Res. 2006, 98, 169–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, H.; DeLano, F.A.; Parks, D.A.; Jamshidi, N.; Granger, D.N.; Ishii, H.; Suematsu, M.; Zweifach, B.W.; Schmid-Schonbein, G.W. Xanthine oxidase activity associated with arterial blood pressure in spontaneously hypertensive rats. Proc. Natl. Acad. Sci. USA 1998, 95, 4754–4759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murdoch, C.E.; Alom-Ruiz, S.P.; Wang, M.; Zhang, M.; Walker, S.; Yu, B.; Brewer, A.; Shah, A.M. Role of endothelial Nox2 NADPH oxidase in angiotensin II-induced hypertension and vasomotor dysfunction. Basic Res. Cardiol. 2011, 106, 527–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouabout, G.; Ayme-Dietrich, E.; Jacob, H.; Champy, M.F.; Birling, M.C.; Pavlovic, G.; Madeira, L.; Fertak, L.E.; Petit-Demouliere, B.; Sorg, T.; et al. Nox4 genetic inhibition in experimental hypertension and metabolic syndrome. Arch. Cardiovasc. Dis. 2018, 111, 41–52. [Google Scholar] [CrossRef]
- Langbein, H.; Brunssen, C.; Hofmann, A.; Cimalla, P.; Brux, M.; Bornstein, S.R.; Deussen, A.; Koch, E.; Morawietz, H. NADPH oxidase 4 protects against development of endothelial dysfunction and atherosclerosis in LDL receptor deficient mice. Eur. Heart J. 2016, 37, 1753–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Oria, R.; Schipani, R.; Leonardini, A.; Natalicchio, A.; Perrini, S.; Cignarelli, A.; Laviola, L.; Giorgino, F. The Role of Oxidative Stress in Cardiac Disease: From Physiological Response to Injury Factor. Oxid. Med. Cell. Longev. 2020, 2020, 5732956. [Google Scholar] [CrossRef]
- Ide, T.; Tsutsui, H.; Ohashi, N.; Hayashidani, S.; Suematsu, N.; Tsuchihashi, M.; Tamai, H.; Takeshita, A. Greater oxidative stress in healthy young men compared with premenopausal women. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 438–442. [Google Scholar] [CrossRef] [Green Version]
- Barp, J.; Araújo, A.; Fernandes, T.G.; Rigatto, K.; Llesuy, S.; Belló-Klein, A.; Singal, P. Myocardial antioxidant and oxidative stress changes due to sex hormones. Braz. J. Med. Biol. Res. 2002, 35, 1075–1081. [Google Scholar] [CrossRef] [Green Version]
- Khalifa, A.R.; Abdel-Rahman, E.A.; Mahmoud, A.M.; Ali, M.H.; Noureldin, M.; Saber, S.H.; Mohsen, M.; Ali, S.S. Sex-specific differences in mitochondria biogenesis, morphology, respiratory function, and ROS homeostasis in young mouse heart and brain. Physiol. Rep. 2017, 5. [Google Scholar] [CrossRef]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH oxidases in vascular pathology. Antioxid. Redox Signal. 2014, 20, 2794–2814. [Google Scholar] [CrossRef] [Green Version]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, R.M. Reactive oxygen species at phospholipid bilayers: Distribution, mobility and permeation. Biochim. Biophys. Acta 2014, 1838, 438–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, J.; Ing, M.H.; Salazar, A.; Lassegue, B.; Griendling, K.; Navab, M.; Sevanian, A.; Hsiai, T.K. Pulsatile versus oscillatory shear stress regulates NADPH oxidase subunit expression: Implication for native LDL oxidation. Circ. Res. 2003, 93, 1225–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.M.; Shah, A.M. Mechanism of endothelial cell NADPH oxidase activation by angiotensin II. Role of the p47phox subunit. J. Biol. Chem. 2003, 278, 12094–12100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duerrschmidt, N.; Wippich, N.; Goettsch, W.; Broemme, H.J.; Morawietz, H. Endothelin-1 induces NAD(P)H oxidase in human endothelial cells. Biochem. Biophys. Res. Commun. 2000, 269, 713–717. [Google Scholar] [CrossRef]
- Frey, R.S.; Rahman, A.; Kefer, J.C.; Minshall, R.D.; Malik, A.B. PKCzeta regulates TNF-alpha-induced activation of NADPH oxidase in endothelial cells. Circ. Res. 2002, 90, 1012–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, L.; Pimentel, D.R.; Wang, J.; Singh, K.; Colucci, W.S.; Sawyer, D.B. Role of reactive oxygen species and NAD(P)H oxidase in alpha(1)-adrenoceptor signaling in adult rat cardiac myocytes. Am. J. Physiol. Cell Physiol. 2002, 282, C926–C934. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.A.; Drummond, G.R.; Mast, A.E.; Schmidt, H.H.; Sobey, C.G. Effect of gender on NADPH-oxidase activity, expression, and function in the cerebral circulation: Role of estrogen. Stroke 2007, 38, 2142–2149. [Google Scholar] [CrossRef] [Green Version]
- Wong, P.S.; Randall, M.D.; Roberts, R.E. Sex differences in the role of NADPH oxidases in endothelium-dependent vasorelaxation in porcine isolated coronary arteries. Vasc. Pharmacol. 2015, 72, 83–92. [Google Scholar] [CrossRef]
- De Silva, T.M.; Broughton, B.R.; Drummond, G.R.; Sobey, C.G.; Miller, A.A. Gender influences cerebral vascular responses to angiotensin II through Nox2-derived reactive oxygen species. Stroke 2009, 40, 1091–1097. [Google Scholar] [CrossRef]
- Zhang, R.; Thor, D.; Han, X.; Anderson, L.; Rahimian, R. Sex differences in mesenteric endothelial function of streptozotocin-induced diabetic rats: A shift in the relative importance of EDRFs. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H1183–H1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacher, P.; Nivorozhkin, A.; Szabo, C. Therapeutic effects of xanthine oxidase inhibitors: Renaissance half a century after the discovery of allopurinol. Pharmacol. Rev. 2006, 58, 87–114. [Google Scholar] [CrossRef] [PubMed]
- Linder, N.; Rapola, J.; Raivio, K.O. Cellular expression of xanthine oxidoreductase protein in normal human tissues. Lab. Investig. 1999, 79, 967–974. [Google Scholar] [PubMed]
- Spiekermann, S.; Landmesser, U.; Dikalov, S.; Bredt, M.; Gamez, G.; Tatge, H.; Reepschlager, N.; Hornig, B.; Drexler, H.; Harrison, D.G. Electron spin resonance characterization of vascular xanthine and NAD(P)H oxidase activity in patients with coronary artery disease: Relation to endothelium-dependent vasodilation. Circulation 2003, 107, 1383–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budhiraja, R.; Kayyali, U.; Karamsetty, M.; Fogel, M.; Hill, N.; Chalkley, R.; Finlay, G.; Hassoun, P. Estrogen modulates xanthine dehydrogenase/xanthine oxidase activity by a receptor-independent mechanism. Antioxid. Redox Signal. 2003, 5, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Seyrek, M.; Yildiz, O.; Ulusoy, H.B.; Yildirim, V. Testosterone relaxes isolated human radial artery by potassium channel opening action. J. Pharmacol. Sci. 2007, 103, 309–316. [Google Scholar] [CrossRef] [Green Version]
- Puttabyatappa, Y.; Stallone, J.; Ergul, A.; El-Remessy, A.; Kumar, S.; Black, S.; Johnson, M.; Owen, M.; White, R. Peroxynitrite mediates testosterone-induced vasodilation of microvascular resistance vessels. J. Pharmacol. Exp. Ther. 2013, 345, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Berry, C.E.; Hare, J.M. Xanthine oxidoreductase and cardiovascular disease: Molecular mechanisms and pathophysiological implications. J. Physiol. 2004, 555, 589–606. [Google Scholar] [CrossRef]
- McNally, J.; Davis, M.; Giddens, D.; Saha, A.; Hwang, J.; Dikalov, S.; Jo, H.; Harrison, D. Role of xanthine oxidoreductase and NAD(P)H oxidase in endothelial superoxide production in response to oscillatory shear stress. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2290–H2297. [Google Scholar] [CrossRef] [Green Version]
- Bredemeier, M.; Lopes, L.; Eisenreich, M.; Hickmann, S.; Bongiorno, G.; d’Avila, R.; Morsch, A.; da Silva Stein, F.; Campos, G. Xanthine oxidase inhibitors for prevention of cardiovascular events: A systematic review and meta-analysis of randomized controlled trials. BMC Cardiovasc. Disord. 2018, 18, 24. [Google Scholar] [CrossRef]
- Guzik, T.; Sadowski, J.; Guzik, B.; Jopek, A.; Kapelak, B.; Przybylowski, P.; Wierzbicki, K.; Korbut, R.; Harrison, D.; Channon, K. Coronary artery superoxide production and nox isoform expression in human coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cicero, A.; Cosentino, E.; Kuwabara, M.; Esposti, D.; Borghi, C. Effects of allopurinol and febuxostat on cardiovascular mortality in elderly heart failure patients. Intern. Emerg. Med. 2019, 14, 949–956. [Google Scholar] [CrossRef]
- Zhang, Y.; Murugesan, P.; Huang, K.; Cai, H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: Novel therapeutic targets. Nat. Rev. Cardiol. 2020, 17, 170–194. [Google Scholar] [CrossRef] [PubMed]
- Bryk, D.; Olejarz, W.; Zapolska-Downar, D. The role of oxidative stress and NADPH oxidase in the pathogenesis of atherosclerosis. Postepy Hig. Med. Dosw. (Online) 2017, 71, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef] [Green Version]
- Vallance, P.; Chan, N. Endothelial function and nitric oxide: Clinical relevance. Heart 2001, 85, 342–350. [Google Scholar] [CrossRef] [Green Version]
- Tousoulis, D.; Kampoli, A.-M.; Papageorgiou, C.; Stefanadis, C. The Role of Nitric Oxide on Endothelial Function. Curr. Vasc. Pharmacol. 2012, 10, 4–18. [Google Scholar] [CrossRef]
- Jones, S.P.; Greer, J.J.; van Haperen, R.; Duncker, D.J.; de Crom, R.; Lefer, D.J. Endothelial nitric oxide synthase overexpression attenuates congestive heart failure in mice. Proc. Natl. Acad. Sci. USA 2003, 100, 4891–4896. [Google Scholar] [CrossRef] [Green Version]
- Widmer, R.; Lerman, A. Endothelial dysfunction and cardiovascular disease. Glob. Cardiol. Sci. Pract. 2014, 2014, 291–308. [Google Scholar] [CrossRef]
- Li, Q.; Yon, J.-Y.; Cai, H. Mechanisms and Consequences of eNOS Dysfunction in Hypertension. J. Hypertens. 2015, 33, 1128–1136. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.-M.; Huang, A.; Kaley, G.; Sun, D. eNOS uncoupling and endothelial dysfunction in aged vessels. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1829–H1836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landmesser, U.; Dikalov, S.; PRice, S.; McCann, L.; Fukai, T.; Holland, S.; Mitch, W.; Harrison, D. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Investig. 2003, 111, 1201–1209. [Google Scholar] [CrossRef] [PubMed]
- White, A.; Ryoo, S.; Li, D.; Champion, H.; Steppan, J.; Wang, D.; Nyhan, D.; Shoukas, A.; Hare, J.; Berkowitz, D. Knockdown of arginase I restores NO signaling in the vasculature of old rats. Hypertension 2006, 47, 245–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holowatz, L.; Thompson, C.; Kenney, W. l-Arginine supplementation or arginase inhibition augments reflex cutaneous vasodilatation in aged human skin. J. Physiol. 2006, 574, 573–581. [Google Scholar] [CrossRef]
- Katusic, Z. Vascular endothelial dysfunction: Does tetrahydrobiopterin play a role? Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H981–H986. [Google Scholar] [CrossRef] [Green Version]
- Stroes, E.; Hijmering, M.; van Zandvoost, M.; Wever, R.; Rabelink, T.; van Faassen, E. Origin of superoxide production by endothelial nitric oxide synthase. FEBS Lett. 1998, 438, 161–164. [Google Scholar] [CrossRef] [Green Version]
- Holowatz, L.; Kenney, W. Up-regulation of arginase activity contributes to attenuated reflex cutaneous vasodilatation in hypertensive humans. J. Physiol. 2007, 581, 863–872. [Google Scholar] [CrossRef]
- Antoniades, C.; Shirodaria, C.; Leeson, P.; Antonopoulos, A.; Warrick, N.; Van-Assche, T.; Cunnington, C.; Tousoulis, D.; Pillai, R.; Ratnatunga, C.; et al. Association of plasma asymmetrical dimethylarginine (ADMA) with elevated vascular superoxide production and endothelial nitric oxide synthase uncoupling: Implications for endothelial function in human atherosclerosis. Eur. Heart J. 2009, 30, 1142–1150. [Google Scholar] [CrossRef] [Green Version]
- Böger, R. Asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase, explains the “l-arginine paradox” and acts as a novel cardiovascular risk factor. J Nutr. 2004, 134, 2842S–2847S. [Google Scholar] [CrossRef]
- Pastore, M.; Talwar, S.; Conley, M.; Magness, R. Identification of Differential ER-Alpha Versus ER-Beta Mediated Activation of eNOS in Ovine Uterine Artery Endothelial Cells. Biol. Reprod. 2016, 94, 1–9. [Google Scholar] [CrossRef]
- Klawitter, J.; Hildreth, K.; Christians, U.; Kohrt, W.; Moreau, K. A relative l-arginine deficiency contributes to endothelial dysfunction across the stages of the menopausal transition. Physiol. Rep. 2017, 5, e13409. [Google Scholar] [CrossRef] [PubMed]
- Moreau, K.; Meditz, A.; Deane, K.; Kohrt, W. Tetrahydrobiopterin improves endothelial function and decreases arterial stiffness in estrogen-deficient postmenopausal women. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1211–H1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandoo, A.; Zanten, J.V.V.; Metsios, G.; Carroll, D.; Kitas, G. The Endothelium and Its Role in Regulating Vascular Tone. Open Cardiovasc. Med. J. 2010, 4, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Tyml, K.; Wilson, J.X. iNOS expression requires NADPH oxidase-dependent redox signaling in microvascular endothelial cells. J. Cell. Physiol. 2008, 217, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Kuo, P.; Schroeder, R. The emerging multifaceted roles of nitric oxide. Ann. Surg. 1995, 221, 220–235. [Google Scholar] [CrossRef]
- Hong, H.-J.; Loh, S.-H.; Yen, M.-H. Suppression of the development of hypertension by the inhibition of inducible nitric oxide synthase. Br. J. Pharmacol. 2000, 131, 631–637. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.; Santhanam, L.; Bruning, R.; Stanhewicz, A.; Berkowitz, D.; Holowatz, L. Upregulation of inducible nitric oxide synthase contributes to attenuated cutaneous vasodilation in essential hypertensive humans. Hypertension 2011, 58, 935–942. [Google Scholar] [CrossRef] [Green Version]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [Green Version]
- Carr, A.; McCall, M.; Frei, B. Oxidation of LDL by Myeloperoxidase and Reactive Nitrogen Species: Reaction Pathways and Antioxidant Protection. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1716–1723. [Google Scholar] [CrossRef] [Green Version]
- Cignarella, A.; Bolego, C.; Pelosi, V.; Meda, C.; Krust, A.; Pinna, C.; Gaion, R.; Vegeto, E.; Maggi, A. Distinct Roles of Estrogen Receptor-a and B in the Modulation of Vascular Inducible Nitric-Oxide Synthase in Diabetes. J. Pharmacol. Exp. Ther. 2009, 328, 174–182. [Google Scholar] [CrossRef] [Green Version]
- Juliet, P.; Hayashi, T.; Daigo, S.; Matsui-Hirai, H.; Miyazaki, A.; Fukatsu, A.; Funami, J.; Iguchi, A.; Ignarro, L. Combined effect of testosterone and apocynin on nitric oxide and superoxide production in PMA-differentiated THP-1 cells. Biochim. Biophys. Acta 2004, 1693, 185–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores-Mateo, G.; Carrillo-Santisteve, P.; Elosua, R.; Guallar, E.; Marrugat, J.; Bleys, J.; Covas, M.-I. Antioxidant Enzyme Activity and Coronary Heart Disease: Meta-analyses of Observational Studies. Am. J. Epidemiol. 2009, 170, 135–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinola-Klein, C.; Rupprecht, H.; Bickel, C.; Schnabel, R.; Genth-Zotz, S.; Torzewski, M.; Lackner, K.; Munzel, T.; Blankenberg, S. Glutathione Peroxidase-1 Activity, Atherosclerotic Burden, and Cardiovascular Prognosis. Am. J. Cardiol. 2007, 99, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Buijsse, B.; Lee, D.-H.; Erickson, R.; Luepker, R.; Jacobs, D.; Holtzman, J. Low Serum Glutathione Peroxidase Activity Is Associated with Increased Cardiovascular Mortality in Individuals with Low HDLc’s. PLoS ONE 2012, 7, e38901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukai, T.; Galis, Z.; Meng, X.; Parthasarathy, S.; Harrison, D. Vascular expression of extracellular superoxide dismutase in atherosclerosis. J. Clin. Investig. 1998, 101, 2101–2111. [Google Scholar] [CrossRef]
- Tanaka, M.; Mokhtari, G.K.; Terry, R.D.; Balsam, L.B.; Lee, K.H.; Kofidis, T.; Tsao, P.S.; Robbins, R.C. Overexpression of human copper/zinc superoxide dismutase (SOD1) suppresses ischemia-reperfusion injury and subsequent development of graft coronary artery disease in murine cardiac grafts. Circulation 2004, 110, II200–II206. [Google Scholar] [CrossRef] [Green Version]
- Kang, P.T.; Chen, C.L.; Ohanyan, V.; Luther, D.J.; Meszaros, J.G.; Chilian, W.M.; Chen, Y.R. Overexpressing superoxide dismutase 2 induces a supernormal cardiac function by enhancing redox-dependent mitochondrial function and metabolic dilation. J. Mol. Cell. Cardiol. 2015, 88, 14–28. [Google Scholar] [CrossRef] [Green Version]
- Fukai, T.; Siegfried, M.; Ushio-Fukai, M.; Cheng, Y.; Kojda, G.; Harrison, D. Regulation of the vascular extracellular superoxide dismutase by nitric oxide and exercise training. J. Clin. Investig. 2000, 105, 1631–1639. [Google Scholar] [CrossRef] [Green Version]
- Duckles, S.; Krause, D.; Stirone, C.; Procaccio, V. Estrogen and Mitochondria: A New Paradigm for Vascular Protection? Mol. Interv. 2006, 6, 26–35. [Google Scholar] [CrossRef]
- Stirone, C.; Duckles, S.; Krause, D.; Procaccio, V. Estrogen increases mitochondrial efficiency and reduces oxidative stress in cerebral blood vessels. Mol. Pharmacol. 2005, 68, 959–965. [Google Scholar] [CrossRef] [Green Version]
- Skogastierna, C.; Hotzen, M.; Rane, A.; Ekstrom, L. A supraphysiological dose of testosterone induces nitric oxide production and oxidative stress. Eur. J. Prev. Cardiol. 2014, 21, 1049–1054. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wu, S.; Ruan, Y.; Hong, L.; Xing, X.; Lai, W. Testosterone suppresses oxidative stress via androgen receptor-independent pathway in murine cardiomyocytes. Mol. Med. Rep. 2011, 4, 1183–1188. [Google Scholar] [PubMed]
- Jeney, V.; Balla, J.; Yachie, A.; Varga, Z.; Vercellotti, G.M.; Eaton, J.W.; Balla, G. Pro-oxidant and cytotoxic effects of circulating heme. Blood 2002, 100, 879–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredenburgh, L.E.; Merz, A.A.; Cheng, S. Haeme oxygenase signalling pathway: Implications for cardiovascular disease. Eur. Heart J. 2015, 36, 1512–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otterbein, L.E.; Bach, F.H.; Alam, J.; Soares, M.; Tao Lu, H.; Wysk, M.; Davis, R.J.; Flavell, R.A.; Choi, A.M. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat. Med. 2000, 6, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Christova, T.; Diankova, Z.; Setchenska, M. Heme oxygenase--carbon monoxide signalling pathway as a physiological regulator of vascular smooth muscle cells. Acta Physiol. Pharmacol. Bulg. 2000, 25, 9–17. [Google Scholar] [PubMed]
- Pósa, A.; Kupai, K.; Ménesi, R.; Szalai, Z.; Szabó, R.; Pintér, Z.; Pálfi, G.; Gyöngyösi, M.; Berkó, A.; Pávó, I.; et al. Sexual Dimorphism of Cardiovascular Ischemia Susceptibility Is Mediated by Heme Oxygenase. Oxid. Med. Cell. Longev. 2013, 2013, 521563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, Z.Z.; Wang, S.; Shang, Y.C.; Maiese, K. Targeting cardiovascular disease with novel SIRT1 pathways. Future Cardiol. 2012, 8, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Augustine, L.; Fisher, C.; Lickteig, A.; Aleksunes, L.; Slitt, A.; Cherrington, N. Gender divergent expression of Nqo1 in Sprague Dawley and August Copenhagen x Irish rats. J. Biochem. Mol. Toxicol. 2008, 22, 93–100. [Google Scholar] [CrossRef]
- Rougée, L.; Riches, A.; Berman, J.; Collier, A. The Ontogeny and Population Variability of Human Hepatic NADPH Dehydrogenase Quinone Oxido-Reductase 1 (NQO1). Drug Metab. Dispos. 2016, 44, 967–974. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.M.; Maltagliati, A.J. Nrf2 at the heart of oxidative stress and cardiac protection. Physiol. Genom. 2018, 50, 77–97. [Google Scholar] [CrossRef] [PubMed]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Canning, P. Keap1, the cysteine-based mammalian intracellular sensor for electrophiles and oxidants. Arch. Biochem. Biophys. 2017, 617, 84–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 2001, 6, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-kappaB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.H.; Qu, J.; Shen, X. NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. Biophys. Acta 2008, 1783, 713–727. [Google Scholar] [CrossRef] [Green Version]
- Kerns, M.; Hakim, K.; Zieman, A.; Lu, R.; Coulombe, P. Sexual dimorphism in response to a NRF2 inducer in a model for pachyonychia congenita. J. Investig. Dermatol. 2018, 138, 1094–1100. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Zhao, Y.; Li, B.; Sun, L.; Huo, H. 17beta-estradiol regulates the expression of antioxidant enzymes in myocardial cells by increasing Nrf2 translocation. J. Biochem. Mol. Toxicol. 2012, 26, 264–269. [Google Scholar] [CrossRef]
- D’Onofrio, N.; Servillo, L.; Balestrieri, M.L. SIRT1 and SIRT6 Signaling Pathways in Cardiovascular Disease Protection. Antioxid. Redox Signal. 2018, 28, 711–732. [Google Scholar] [CrossRef]
- Li, J.; Ichikawa, T.; Villacorta, L.; Janicki, J.S.; Brower, G.L.; Yamamoto, M.; Cui, T. Nrf2 protects against maladaptive cardiac responses to hemodynamic stress. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1843–1850. [Google Scholar] [CrossRef]
- Wang, C.; Luo, Z.; Carter, G.; Wellstein, A.; Jose, P.A.; Tomlinson, J.; Leiper, J.; Welch, W.J.; Wilcox, C.S.; Wang, D. NRF2 prevents hypertension, increased ADMA, microvascular oxidative stress, and dysfunction in mice with two weeks of ANG II infusion. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 314, R399–R406. [Google Scholar] [CrossRef]
- Sanz, M.N.; Grimbert, L.; Moulin, M.; Gressette, M.; Rucker-Martin, C.; Lemaire, C.; Mericskay, M.; Veksler, V.; Ventura-Clapier, R.; Garnier, A.; et al. Inducible Cardiac-Specific Deletion of Sirt1 in Male Mice Reveals Progressive Cardiac Dysfunction and Sensitization of the Heart to Pressure Overload. Int. J. Mol. Sci. 2019, 20, 5. [Google Scholar] [CrossRef] [Green Version]
- Fry, J.L.; Shiraishi, Y.; Turcotte, R.; Yu, X.; Gao, Y.Z.; Akiki, R.; Bachschmid, M.; Zhang, Y.; Morgan, K.G.; Cohen, R.A.; et al. Vascular Smooth Muscle Sirtuin-1 Protects Against Aortic Dissection During Angiotensin II-Induced Hypertension. J. Am. Heart Assoc. 2015, 4, e002384. [Google Scholar] [CrossRef] [Green Version]
- Alcendor, R.R.; Gao, S.; Zhai, P.; Zablocki, D.; Holle, E.; Yu, X.; Tian, B.; Wagner, T.; Vatner, S.F.; Sadoshima, J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ. Res. 2007, 100, 1512–1521. [Google Scholar] [CrossRef]
- Gao, P.; Xu, T.T.; Lu, J.; Li, L.; Xu, J.; Hao, D.L.; Chen, H.Z.; Liu, D.P. Overexpression of SIRT1 in vascular smooth muscle cells attenuates angiotensin II-induced vascular remodeling and hypertension in mice. J. Mol. Med. 2014, 92, 347–357. [Google Scholar] [CrossRef]
- Wang, W.J.; Li, S.Y.; Xing, Y.F.; Horvath, G.; Wang, Q.; Cui, T.X. Cardiac Specific Overexpression of Nrf2 Protects Against Pressure Overload-Induced Heart Failure Via Enhancing Autophagic Clearance of Ubiquitinated Proteins. J. Cardiac. Fail. 2012, 18, S1. [Google Scholar] [CrossRef]
- Shimabukuro, M. SIRT1 and Gender Differences in Atherosclerotic Cardiovascular Disease. J. Atheroscler. 2020, 27, 8–10. [Google Scholar] [CrossRef] [Green Version]
- Del Bo’, C.; Martini, D.; Porrini, M.; Klimis-Zacas, D.; Riso, P. Berries and oxidative stress markers: An overview of human intervention studies. Food Funct. 2015, 6. [Google Scholar] [CrossRef]
- Marniemi, J.; Hakala, P.; Mäki, J.; Ahotupa, M. Partial resistance of low-density lipoprotein to oxidation in vivo after increased intake of berries. Nutr. Metab. Cardiovasc. Dis. 2000, 10, 331–337. [Google Scholar]
- Mazza, G.; Kay, C.; Cottrell, T.; Holub, B. Absorption of anthocyanins from blueberries and serum antioxidant status in human subjects. J. Agric. Food Chem. 2002, 50, 7731–7737. [Google Scholar] [CrossRef]
- Kay, C.; Holub, B. The effect of wild blueberry (Vaccinium angustifolium) consumption on postprandial serum antioxidant status in human subjects. Br. J. Nutr. 2002, 88, 389–397. [Google Scholar] [CrossRef] [Green Version]
- Netzel, M.; Strass, G.; Kaul, C.; Bitsch, I.; Dietrich, H.; Bitsch, R. In vivo antioxidative capacity of a composite berry juice. Food Res. Int. 2002, 35, 213–216. [Google Scholar] [CrossRef]
- Netzel, M.; Strass, G.; Herbst, M.; Dietrich, H.; Bitsch, R.; Bitsch, I.; Frank, T. The excretion and biological antioxidant activity of elderberry antioxidants in healthy humans. Food Res. Int. 2005, 38, 905–910. [Google Scholar] [CrossRef]
- Tulipani, S.; Romandini, S.; Busco, F.; Bompadre, S.; Mezzetti, B.; Battino, M. Ascorbate, not urate, modulates the plasma antioxidant capacity after strawberry intake. Food Chem. 2009, 117, 181–188. [Google Scholar] [CrossRef]
- McLeay, Y.; Barnes, M.; Mundel, T.; Hurst, S.; Hurst, R.; Stannard, S. Effect of New Zealand blueberry consumption on recovery from eccentric exercise-induced muscle damage. J. Int. Soc. Sports Nutr. 2012, 9. [Google Scholar] [CrossRef] [Green Version]
- Mathison, B.; Kimble, L.; Kaspar, K.; Khoo, C.; Chew, B. Consumption of cranberry beverage improved endogenous antioxidant status and protected against bacteria adhesion in healthy humans: A randomized controlled trial. Nutr. Res. 2014, 34, 420–427. [Google Scholar] [CrossRef]
- Johnson, S.A.; Figueroa, A.; Navaei, N.; Wong, A.; Kalfon, R.; Ormsbee, L.T.; Feresin, R.G.; Elam, M.L.; Hooshmand, S.; Payton, M.E.; et al. Daily blueberry consumption improves blood pressure and arterial stiffness in postmenopausal women with pre- and stage 1-hypertension: A randomized, double-blind, placebo-controlled clinical trial. J. Acad. Nutr. Diet 2015, 115, 369–377. [Google Scholar] [CrossRef]
- Basu, A.; Du, M.; Leyva, M.J.; Sanchez, K.; Betts, N.M.; Wu, M.; Aston, C.E.; Lyons, T.J. Blueberries decrease cardiovascular risk factors in obese men and women with metabolic syndrome. J. Nutr. 2010, 140, 1582–1587. [Google Scholar] [CrossRef] [Green Version]
- Feresin, R.G.; Johnson, S.A.; Pourafshar, S.; Campbell, J.C.; Jaime, S.J.; Navaei, N.; Elam, M.L.; Akhavan, N.S.; Alvarez-Alvarado, S.; Tenenbaum, G.; et al. Impact of daily strawberry consumption on blood pressure and arterial stiffness in pre- and stage 1-hypertensive postmenopausal women: A randomized controlled trial. Food Funct. 2017, 8, 4139–4149. [Google Scholar] [CrossRef]
- Burton-Freeman, B.; Linares, A.; Hyson, D.; Kappagoda, T. Strawberry modulates LDL oxidation and postprandial lipemia in response to high-fat meal in overweight hyperlipidemic men and women. J. Am. Coll. Nutr. 2010, 29, 46–54. [Google Scholar] [CrossRef]
- An, J.H.; Kim, D.L.; Lee, T.B.; Kim, K.J.; Kim, S.H.; Kim, N.H.; Kim, H.Y.; Choi, D.S.; Kim, S.G. Effect of Rubus Occidentalis Extract on Metabolic Parameters in Subjects with Prediabetes: A Proof-of-concept, Randomized, Double-blind, Placebo-controlled Clinical Trial. Phytother. Res. 2016, 30, 1634–1640. [Google Scholar] [CrossRef]
- Hung, C.H.; Chan, S.H.; Chu, P.M.; Tsai, K.L. Quercetin is a potent anti-atherosclerotic compound by activation of SIRT1 signaling under oxLDL stimulation. Mol. Nutr. Food Res. 2015, 59, 1905–1917. [Google Scholar] [CrossRef]
- Zhou, Y.; Jiang, Z.; Lu, H.; Xu, Z.; Tong, R.; Shi, J.; Jia, G. Recent Advances of Natural Polyphenols Activators for Keap1-Nrf2 Signaling Pathway. Chem. Biodivers. 2019, 16, e1900400. [Google Scholar] [CrossRef]
- Bello, M.; Morales-Gonzalez, J.A. Molecular recognition between potential natural inhibitors of the Keap1-Nrf2 complex. Int. J. Biol. Macromol. 2017, 105, 981–992. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef]
- Silva-Islas, C.A.; Maldonado, P.D. Canonical and non-canonical mechanisms of Nrf2 activation. Pharmacol. Res. 2018, 134, 92–99. [Google Scholar] [CrossRef]
- Zheng, D.; Liu, Z.; Zhou, Y.; Hou, N.; Yan, W.; Qin, Y.; Ye, Q.; Cheng, X.; Xiao, Q.; Bao, Y.; et al. Urolithin B, a gut microbiota metabolite, protects against myocardial ischemia/reperfusion injury via p62/Keap1/Nrf2 signaling pathway. Pharmacol. Res. 2020, 153, 104655. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, Y.; Wang, X.; Liu, Y.; Xia, M. Supplementation with cyanidin-3-O-beta-glucoside protects against hypercholesterolemia-mediated endothelial dysfunction and attenuates atherosclerosis in apolipoprotein E-deficient mice. J. Nutr. 2012, 142, 1033–1037. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Zhang, B.; Zhou, K.; Chen, M.; Wang, M.; Jia, Y.; Song, Y.; Li, Y.; Wen, A. Dietary ellagic acid improves oxidant-induced endothelial dysfunction and atherosclerosis: Role of Nrf2 activation. Int. J. Cardiol. 2014, 175, 508–514. [Google Scholar] [CrossRef]
- Xu, J.W.; Ikeda, K.; Yamori, Y. Inhibitory effect of polyphenol cyanidin on TNF-alpha-induced apoptosis through multiple signaling pathways in endothelial cells. Atherosclerosis 2007, 193, 299–308. [Google Scholar] [CrossRef]
- Jin, L.; Piao, Z.H.; Sun, S.; Liu, B.; Kim, G.R.; Seok, Y.M.; Lin, M.Q.; Ryu, Y.; Choi, S.Y.; Kee, H.J.; et al. Gallic Acid Reduces Blood Pressure and Attenuates Oxidative Stress and Cardiac Hypertrophy in Spontaneously Hypertensive Rats. Sci. Rep. 2017, 7, 15607. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Zhang, Q.Y.; Zhang, Y.L.; Han, X.; Guo, S.B.; Li, H.H. Gallic Acid Attenuates Angiotensin II-Induced Hypertension and Vascular Dysfunction by Inhibiting the Degradation of Endothelial Nitric Oxide Synthase. Front. Pharmacol. 2020, 11, 1121. [Google Scholar] [CrossRef]
- Kalea, A.Z.; Clark, K.; Schuschke, D.A.; Kristo, A.S.; Klimis-Zacas, D.J. Dietary enrichment with wild blueberries (Vaccinium angustifolium) affects the vascular reactivity in the aorta of young spontaneously hypertensive rats. J. Nutr. Biochem. 2010, 21, 14–22. [Google Scholar] [CrossRef]
- Rodriguez-Mateos, A.; Rendeiro, C.; Bergillos-Meca, T.; Tabatabaee, S.; George, T.W.; Heiss, C.; Spencer, J.P. Intake and time dependence of blueberry flavonoid-induced improvements in vascular function: A randomized, controlled, double-blind, crossover intervention study with mechanistic insights into biological activity. Am. J. Clin. Nutr. 2013, 98, 1179–1191. [Google Scholar] [CrossRef] [Green Version]
- Istas, G.; Feliciano, R.P.; Weber, T.; Garcia-Villalba, R.; Tomas-Barberan, F.; Heiss, C.; Rodriguez-Mateos, A. Plasma urolithin metabolites correlate with improvements in endothelial function after red raspberry consumption: A double-blind randomized controlled trial. Arch. Biochem. Biophys. 2018, 651, 43–51. [Google Scholar] [CrossRef]
- Djurica, D.; Holt, R.; Ren, J.; Shindel, A.; Hackman, R.; Keen, C. Effects of a dietary strawberry powder on parameters of vascular health in adolescent males. Br. J. Nutr. 2016, 116, 639–647. [Google Scholar] [CrossRef]
- Jeong, H.S.; Hong, S.J.; Lee, T.B.; Kwon, J.W.; Jeong, J.T.; Joo, H.J.; Park, J.H.; Ahn, C.M.; Yu, C.W.; Lim, D.S. Effects of black raspberry on lipid profiles and vascular endothelial function in patients with metabolic syndrome. Phytother. Res. 2014, 28, 1492–1498. [Google Scholar] [CrossRef]
- Jeong, H.S.; Hong, S.J.; Cho, J.Y.; Lee, T.B.; Kwon, J.W.; Joo, H.J.; Park, J.H.; Yu, C.W.; Lim, D.S. Effects of Rubus occidentalis extract on blood pressure in patients with prehypertension: Randomized, double-blinded, placebo-controlled clinical trial. Nutrition 2016, 32, 461–467. [Google Scholar] [CrossRef]
- Kane, M.; Anselm, E.; Rattmann, Y.; Auger, C.; Schini-Kerth, V. Role of gender and estrogen receptors in the rat aorta endothelium-dependent relaxation to red wine polyphenols. Vasc. Pharmacol. 2009, 51, 140–146. [Google Scholar] [CrossRef]
- Yao, F.; Liu, Y.; Liu, D.; Wu, S.; Lin, H.; Fan, R.; Li, C. Sex differences between vascular endothelial function and carotid intima-media thickness by Framingham Risk Score. J. Ultrasound Med. 2014, 33, 281–286. [Google Scholar] [CrossRef]
- Son, Y.; Cheong, Y.K.; Kim, N.H.; Chung, H.T.; Kang, D.G.; Pae, H.O. Mitogen-Activated Protein Kinases and Reactive Oxygen Species: How Can ROS Activate MAPK Pathways? J. Signal Transduct. 2011, 2011, 792639. [Google Scholar] [CrossRef]
- Narula, J.; Haider, N.; Arbustini, E.; Chandrashekhar, Y. Mechanisms of disease: Apoptosis in heart failure--seeing hope in death. Nat. Clin. Pract. Cardiovasc. Med. 2006, 3, 681–688. [Google Scholar] [CrossRef]
- Bennett, M.R. Apoptosis of vascular smooth muscle cells in vascular remodelling and atherosclerotic plaque rupture. Cardiovasc. Res. 1999, 41, 361–368. [Google Scholar] [CrossRef]
- Kim, R. Unknotting the roles of Bcl-2 and Bcl-xL in cell death. Biochem. Biophys. Res. Commun. 2005, 333, 336–343. [Google Scholar] [CrossRef]
- Zorov, D.B.; Filburn, C.R.; Klotz, L.O.; Zweier, J.L.; Sollott, S.J. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 2000, 192, 1001–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Porter, A.G.; Janicke, R.U. Emerging roles of caspase-3 in apoptosis. Cell. Death Differ. 1999, 6, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekhar, Y.; Sen, S.; Anway, R.; Shuros, A.; Anand, I. Long-term caspase inhibition ameliorates apoptosis, reduces myocardial troponin-I cleavage, protects left ventricular function, and attenuates remodeling in rats with myocardial infarction. J. Am. Coll. Cardiol. 2004, 43, 295–301. [Google Scholar] [CrossRef] [Green Version]
- Miyashita, T.; Reed, J.C. Tumor-Suppressor P53 Is a Direct Transcriptional Activator of the Human Bax Gene. Cell 1995, 80, 293–299. [Google Scholar]
- Graupner, V.; Alexander, E.; Overkamp, T.; Rothfuss, O.; De Laurenzi, V.; Gillissen, B.F.; Daniel, P.T.; Schulze-Osthoff, K.; Essmann, F. Differential regulation of the proapoptotic multidomain protein Bak by p53 and p73 at the promoter level. Cell Death Differ. 2011, 18, 1130–1139. [Google Scholar] [CrossRef] [Green Version]
- Cesselli, D.; Jakoniuk, I.; Barlucchi, L.; Beltrami, A.P.; Hintze, T.H.; Nadal-Ginard, B.; Kajstura, J.; Leri, A.; Anversa, P. Oxidative stress-mediated cardiac cell death is a major determinant of ventricular dysfunction and failure in dog dilated cardiomyopathy. Circ. Res. 2001, 89, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Conte, J.V., Jr.; Foster, A.H.; McLaughlin, J.S.; Wei, C. Increased p53 protein expression in human failing myocardium. J. Heart Lung Transplant. 1999, 18, 744–749. [Google Scholar] [CrossRef]
- Fan, M.; Goodwin, M.; Vu, T.; Brantley-Finley, C.; Gaarde, W.A.; Chambers, T.C. Vinblastine-induced phosphorylation of Bcl-2 and Bcl-XL is mediated by JNK and occurs in parallel with inactivation of the Raf-1/MEK/ERK cascade. J. Biol. Chem. 2000, 275, 29980–29985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivetti, G.; Giordano, G.; Corradi, D.; Melissari, M.; Lagrasta, C.; Gambert, S.; Anversa, P. Gender differences and aging: Effects on the human heart. J. Am. Coll. Cardiol. 1995, 26, 1068–1079. [Google Scholar] [CrossRef] [Green Version]
- Guerra, S.; Leri, A.; Wang, X.; Finato, N.; Di Loreto, C.; Beltrami, C.; Kajstura, J.; Anversa, P. Myocyte death in the failing human heart is gender dependent. Circ. Res. 1999, 85, 856–866. [Google Scholar] [CrossRef] [PubMed]
- Biondi-Zoccai, C.; Abate, A.; Bussani, R.; Camilot, D.; Giorgio, F.; Marino, M.; Silvestri, F.; Baldi, F.; Biasucci, L.; Baldi, A. Reduced post-infarction myocardial apoptosis in women: A clue to their different clinical course? Heart 2005, 91, 99–101. [Google Scholar] [CrossRef]
- Bouma, W.; Noma, M.; Kanemoto, S.; Matsubara, M.; Leshnower, B.; Hinmon, R.; Gorman, J.H., 3rd; Gorman, R. Sex-related resistance to myocardial ischemia-reperfusion injury is associated with high constitutive ARC expression. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1510–H1517. [Google Scholar] [CrossRef] [Green Version]
- Gabel, S.; Walker, V.; London, R.; Steenbergen, C.; Korach, K.; Murphy, E. Estrogen receptor beta mediates gender differences in ischemia/reperfusion injury. JMCC 2005, 38, 289–297. [Google Scholar] [CrossRef]
- Wang, M.; Tsai, B.; Reiger, K.; Brown, J.; Meldrum, D. 17-beta-Estradiol decreases p38 MAPK-mediated myocardial inflammation and dysfunction following acute ischemia. J. Mol. Cell. Cardiol. 2006, 40, 205–212. [Google Scholar] [CrossRef]
- Matsui, T.; Li, L.; del Monte, F.; Fukui, Y.; Franke, T.; Hajjar, R.; Rosenzweig, A. Adenoviral gene transfer of activated phosphatidylinositol 3′-kinase and Akt inhibits apoptosis of hypoxic cardiomyocytes in vitro. Circulation 1999, 100, 2373–2379. [Google Scholar] [CrossRef] [Green Version]
- Fujio, Y.; Nguyen, T.; Wencker, D.; Kitsis, R.; Walsh, K. Akt promotes survival of cardiomyocytes in vitro and protects against ischemia-reperfusion injury in mouse heart. Circulation 2000, 101, 660–667. [Google Scholar] [CrossRef]
- Abeyrathna, P.; Su, Y. The Critical Role of Akt in Cardiovascular Function. Vasc. Pharmacol. 2015, 74, 38–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camper-Kirby, D.; Welch, S.; Walker, A.; Shiraishi, I.; Schaefer, K.; Kajstura, J.; Anversa, P.; Sussman, M. Myocardial Akt activation and gender: Increased nuclear activity in females versus males. Circ. Res. 2001, 88, 1020–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawara, Y.; Kishishita, S.; Obata, T.; Isazawa, Y.; Suzuki, T.; Tanaka, K.; Masuyama, N.; Gotoh, Y. Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J. Biol. Chem. 2002, 277, 21843–21850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlieb, T.; Leal, J.; Seger, R.; Taya, Y.; Oren, M. Cross-talk between Akt, p53 and Mdm2: Possible implications for the regulation of apoptosis. Oncogene 2002, 21, 1299–1303. [Google Scholar] [CrossRef] [PubMed]
- Mayo, L.; Donner, D. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc. Natl. Acad. Sci. USA 2001, 98, 11598–11603. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B.; Liao, Y.; Xia, W.; Zou, Y.; Spohn, B.; Hung, M. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat. Cell Biol. 2001, 3, 973–982. [Google Scholar] [CrossRef]
- Moll, U.M.; Petrenko, O. The MDM2-p53 interaction. Mol. Cancer Res. 2003, 1, 1001–1008. [Google Scholar]
- Cardone, M.; Roy, N.; Stennicke, H.; Salvesen, G.; Franke, T.; Stanbridge, E.; Frisch, S.; Reed, J. Regulation of cell death protease caspase-9 by phosphorylation. Science 1998, 282, 1318–1321. [Google Scholar] [CrossRef]
- Patten, R.; Pourati, I.; Aronovitz, M.; Baur, J.; Celestin, F.; Chen, X.; Michael, A.; Haq, S.; Nuedling, S.; Grohe, C.; et al. 17beta-estradiol reduces cardiomyocyte apoptosis in vivo and in vitro via activation of phospho-inositide-3 kinase/Akt signaling. Circ. Res. 2004, 95, 692–699. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, Y.; Aihara, K.; Akaike, M.; Sato, T.; Ishikawa, K.; Ise, T.; Yagi, S.; Iwase, T.; Ueda, Y.; Yoshida, S.; et al. Androgen receptor counteracts Doxorubicin-induced cardiotoxicity in male mice. Mol. Endocrinol. 2010, 24, 1338–1348. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Gu, H.; Zhang, W.; Herrmann, J.; Wang, M. Testosterone-down-regulated Akt pathway during cardiac ischemia/reperfusion: A mechanism involving BAD, Bcl-2 and FOXO3a. J. Surg. Res. 2010, 164, e1–e11. [Google Scholar] [CrossRef] [Green Version]
- Le, T.; Ashton, A.; Mardini, M.; Stanton, P.; Funder, J.; Handelsman, D.; Mihailidou, A. Role of androgens in sex differences in cardiac damage during myocardial infarction. Endocrinology 2014, 155, 568–575. [Google Scholar] [CrossRef]
- Wang, M.; Tsai, B.; Kher, A.; Baker, L.; Wairiuko, G.; Meldrum, D. Role of endogenous testosterone in myocardial proinflammatory and proapoptotic signaling after acute ischemia-reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H221–H226. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Pedram, A.; Razandi, M.; Levin, E. Estrogen prevents cardiomyocyte apoptosis through inhibition of reactive oxygen species and differential regulation of p38 kinase isoforms. J. Biol. Chem. 2006, 281, 6760–6767. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Pedram, A.; Kim, J. Oestrogen prevents cardiomyocyte apoptosis by suppressing p38α-mediated activation of p53 and by down-regulating p53 inhibition on p38β. Cardiovasc. Res. 2011, 89, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-D.; Kuo, W.-W.; Ho, Y.-J.; Lin, A.-C.; Tsai, C.-H.; Wang, H.-F.; Kuo, C.-H.; Yang, A.-L.; Huang, C.-Y.; Hwang, J.-M. Cardiac Fas-dependent and mitochondria-dependent apoptosis in ovariectomized rats. Maturitas 2008, 61, 268–277. [Google Scholar] [CrossRef]
- Liou, C.-M.; Yang, A.-L.; Kuo, C.-H.; Tin, H.; Huang, C.-Y.; Lee, S.-D. Effects of 17beta-estradiol on cardiac apoptosis in ovariectomized rats. Cell Biochem. Funct. 2010, 28, 521–528. [Google Scholar] [CrossRef]
- Lin, Y.-Y.; Chen, J.-S.; Wu, X.-B.; Shyu, W.-C.; Chaunchaiyakul, R.; Zhao, X.-L.; Kuo, C.-H.; Cheng, Y.-J.; Yang, A.-L.; Lee, S.-D. Combined effects of 17β-estradiol and exercise training on cardiac apoptosis in ovariectomized rats. PLoS ONE 2018, 13. [Google Scholar] [CrossRef]
- Lin, Y.-Y.; Hong, Y.; Yu, S.-H.; Wu, X.-B.; Shyu, W.-C.; Chen, J.-S.; Ting, H.; Yang, A.-L.; Lee, S.-D. Antiapoptotic and mitochondrial biogenetic effects of exercise training on ovariectomized hypertensive rat hearts. J. Appl. Physiol. 2019, 126, 1661–1672. [Google Scholar] [CrossRef]
- Chen, C.; Hu, L.; Dong, T.; Wang, G.; Wang, L.; Zhou, X.; Jiang, Y.; Murao, K.; Lu, S.; Chen, J.; et al. Apoptosis and autophagy contribute to gender difference in cardiac ischemia-reperfusion induced injury in rats. Life Sci. 2013, 93, 265–270. [Google Scholar] [CrossRef]
- Gordon, J.W.; Shaw, J.A.; Kirshenbaum, L.A. Multiple facets of NF-kappaB in the heart: To be or not to NF-kappaB. Circ. Res. 2011, 108, 1122–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bujak, M.; Dobaczewski, M.; Chatila, K.; Mendoza, L.H.; Li, N.; Reddy, A.; Frangogiannis, N.G. Interleukin-1 receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am. J. Pathol. 2008, 173, 57–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huynh, M.L.; Fadok, V.A.; Henson, P.M. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J. Clin. Investig. 2002, 109, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Thompson, N.L.; Flanders, K.C.; Smith, J.M.; Ellingsworth, L.R.; Roberts, A.B.; Sporn, M.B. Expression of transforming growth factor-beta 1 in specific cells and tissues of adult and neonatal mice. J. Cell Biol. 1989, 108, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Barcellos-Hoff, M.H.; Dix, T.A. Redox-mediated activation of latent transforming growth factor-beta 1. Mol. Endocrinol. 1996, 10, 1077–1083. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, R.; Cao, Y.; Afroz, R.; Xu, S.; Ta, H.T.; Barras, M.; Zheng, W.; Little, P.J.; Kamato, D. ROS directly activates transforming growth factor beta type 1 receptor signalling in human vascular smooth muscle cells. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129463. [Google Scholar] [CrossRef] [PubMed]
- Sartore, S.; Chiavegato, A.; Faggin, E.; Franch, R.; Puato, M.; Ausoni, S.; Pauletto, P. Contribution of adventitial fibroblasts to neointima formation and vascular remodeling: From innocent bystander to active participant. Circ. Res. 2001, 89, 1111–1121. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Aspects Med. 2019, 65, 70–99. [Google Scholar] [CrossRef]
- Iwanaga, Y.; Aoyama, T.; Kihara, Y.; Onozawa, Y.; Yoneda, T.; Sasayama, S. Excessive activation of matrix metalloproteinases coincides with left ventricular remodeling during transition from hypertrophy to heart failure in hypertensive rats. J. Am. Coll. Cardiol. 2002, 39, 1384–1391. [Google Scholar] [CrossRef] [Green Version]
- Regitz-Zagrosek, V.; Kararigas, G. Mechanistic Pathways of Sex Differences in Cardiovascular Disease. Physiol. Rev. 2016, 97, 1–37. [Google Scholar] [CrossRef] [Green Version]
- Venkatesh, B.; Volpe, G.; Donekal, S.; Mewton, N.; Liu, C.; Shea, S.; Liu, K.; Burke, G.; Wu, C.; Bluemke, D.; et al. Association of longitudinal changes in left ventricular structure and function with myocardial fibrosis: The Multi-Ethnic Study of Atherosclerosis study. Hypertension 2014, 64, 508–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, D.; Somaratne, J.; Jenkins, A.; Prior, D.; Yii, M.; Kenny, J.; Newcomb, A.; Kelly, D.; Black, M. Differences in myocardial structure and coronary microvasculature between men and women with coronary artery disease. Hypertension 2011, 57, 186–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dworatzek, E.; Mahmoodzadeh, S.; Schriever, C.; Kusumoto, K.; Kramer, L.; Santos, G.; Fliegner, D.; Leung, Y.-K.; Ho, S.-M.; Zimmermann, W.-H.; et al. Sex-specific regulation of collagen I and III expression by 17β-Estradiol in cardiac fibroblasts: Role of estrogen receptors. Cardiovasc. Res. 2019, 115, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Petrov, G.; Regitz-Zagrosek, V.; Lehmkuhl, E.; Krabatsch, T.; Dunkel, A.; Dandel, M.; Dworatzek, E.; Mahmoodzadeh, S.; Schubert, C.; Becher, E.; et al. Regression of myocardial hypertrophy after aortic valve replacement: Faster in women? Circulation 2010, 122, S23–S28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmoodzadeh, S.; Dworatzek, E.; Fritschka, S.; Pham, T.; Regitz-Zagrosek, V. 17β-Estradiol inhibits matrix metalloproteinase-2 transcription via MAP kinase in fibroblasts. Cardiovasc. Res. 2010, 85, 719–728. [Google Scholar] [CrossRef] [Green Version]
- Ahmet, I.; Spangler, E.; Shukitt-Hale, B.; Juhaszova, M.; Sollott, S.J.; Joseph, J.A.; Ingram, D.K.; Talan, M. Blueberry-enriched diet protects rat heart from ischemic damage. PLoS ONE 2009, 4, e5954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isaak, C.K.; Petkau, J.C.; Blewett, H.; O, K.; Siow, Y.L. Lingonberry anthocyanins protect cardiac cells from oxidative-stress-induced apoptosis. Can. J. Physiol. Pharmacol. 2017, 95, 904–910. [Google Scholar] [CrossRef] [Green Version]
- Feresin, R.G.; Huang, J.; Klarich, D.S.; Zhao, Y.; Pourafshar, S.; Arjmandi, B.H.; Salazar, G. Blackberry, raspberry and black raspberry polyphenol extracts attenuate angiotensin II-induced senescence in vascular smooth muscle cells. Food Funct. 2016, 7, 4175–4187. [Google Scholar] [CrossRef]
- Choi, Y.J.; Kang, J.S.; Park, J.H.; Lee, Y.J.; Choi, J.S.; Kang, Y.H. Polyphenolic flavonoids differ in their antiapoptotic efficacy in hydrogen peroxide-treated human vascular endothelial cells. J. Nutr. 2003, 133, 985–991. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Piao, Z.H.; Liu, C.P.; Sun, S.; Liu, B.; Kim, G.R.; Choi, S.Y.; Ryu, Y.; Kee, H.J.; Jeong, M.H. Gallic acid attenuates calcium calmodulin-dependent kinase II-induced apoptosis in spontaneously hypertensive rats. J. Cell Mol. Med. 2018, 22, 1517–1526. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Sun, S.; Ryu, Y.; Piao, Z.H.; Liu, B.; Choi, S.Y.; Kim, G.R.; Kim, H.S.; Kee, H.J.; Jeong, M.H. Gallic acid improves cardiac dysfunction and fibrosis in pressure overload-induced heart failure. Sci. Rep. 2018, 8, 9302. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Jiang, Y.; Jing, W.; Sun, B.; Miao, C.; Ren, L. Quercetin provides greater cardioprotective effect than its glycoside derivative rutin on isoproterenol-induced cardiac fibrosis in the rat. Can. J. Physiol. Pharmacol. 2013, 91, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.R.; Naugle, J.E.; Zhang, X.; Bomser, J.A.; Meszaros, J.G. Inhibition of cardiac fibroblast proliferation and myofibroblast differentiation by resveratrol. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H1131–H1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruel, G.; Pomerleau, S.; Couture, P.; Lemieux, S.; Lamarche, B.; Couillard, C. Plasma matrix metalloproteinase (MMP)-9 levels are reduced following low-calorie cranberry juice supplementation in men. J. Am. Coll. Nutr. 2009, 28, 694–701. [Google Scholar] [CrossRef]
Figure 1.
Mechanisms of Berry Polyphenols in CVD. Polyphenols found in berries can attenuate the pathological cellular processes of cardiovascular disease (CVD) in a multi-targeted manner. (A) Berry polyphenols, such as myricetin, reduce inflammation by decreasing toll-like receptor (TLR)-4 activation and phosphorylation of transforming growth factor-β (TGF-β)-activated kinase 1 (TAK1) while also mitigating tumor necrosis factor receptor 1 (TNFR1)-dependent NADPH-oxidase (Nox) activation. Thus, phosphorylation of mitogen-activated protein kinase kinase (MEK) and IκB kinase (IKK) and redox modifications are reduced, preventing downstream activity of mitogen activated protein kinase (MAPK) and nuclear factor kappa-light-chain-enhancer of activated B (NF-κB), respectively. (B) Berry polyphenols, such as resveratrol, reduce oxidative stress by increasing nuclear factor erythroid 2-related factor (Nrf2) protein expression via a number of mechanisms, of which include possible reduction of kelch-like ECH-associated protein 1 (Keap1) binding affinity to Nrf2, reducing Keap1-mediated ubiquitination. Increased Nrf2 nuclear translocation results in increased transcription of heme oxygenase (HO)-1 and NADPH quinone dehydrogenase (NQO1) due to antioxidant responsive element (ARE) transcriptional activity. Additionally, berry polyphenols increase sirtuin 1 (Sirt1) protein expression, which targets a number of proteins, reducing NF-κB and p53 activity while increasing Nrf2 and endothelial nitric oxide synthase (eNOS) activity. Further, Sirt1 increases antioxidant enzymes superoxide dismutase (SOD), glutathione peroxidase (GPx), and catalase, which neutralize superoxide O2− and H2O2. (C) In endothelial cells, berry polyphenols, such as cyanidin, increase phosphoinositide 3-kinase (PI3K) activity, resulting in increased phosphorylation of protein kinase B (Akt) and eNOS, resulting in increased nitric oxide (NO), promoting vasodilation. Additionally, berry polyphenols preserve eNOS protein expression under a number of pathological stimuli, including due to TNFR1 activation. (D) Berry polyphenols reduce apoptosis by increasing B-cell lymphoma 2 (Bcl-2), reducing caspase-8 mediated activation of Bcl-2-associated X protein (Bax) and Bcl-2 homologous antagonist killer (Bak), while Bcl extra-large (Bcl-xL) reduces Bax/Bak oligomerization. (E) Berry polyphenols, such as gallic acid, reduce fibrosis by decreasing TGF-β protein expression and prevent fibroblast differentiation to myofibroblasts, resulting in reduced collagen synthesis. Created in BioRender.com.
Figure 1.
Mechanisms of Berry Polyphenols in CVD. Polyphenols found in berries can attenuate the pathological cellular processes of cardiovascular disease (CVD) in a multi-targeted manner. (A) Berry polyphenols, such as myricetin, reduce inflammation by decreasing toll-like receptor (TLR)-4 activation and phosphorylation of transforming growth factor-β (TGF-β)-activated kinase 1 (TAK1) while also mitigating tumor necrosis factor receptor 1 (TNFR1)-dependent NADPH-oxidase (Nox) activation. Thus, phosphorylation of mitogen-activated protein kinase kinase (MEK) and IκB kinase (IKK) and redox modifications are reduced, preventing downstream activity of mitogen activated protein kinase (MAPK) and nuclear factor kappa-light-chain-enhancer of activated B (NF-κB), respectively. (B) Berry polyphenols, such as resveratrol, reduce oxidative stress by increasing nuclear factor erythroid 2-related factor (Nrf2) protein expression via a number of mechanisms, of which include possible reduction of kelch-like ECH-associated protein 1 (Keap1) binding affinity to Nrf2, reducing Keap1-mediated ubiquitination. Increased Nrf2 nuclear translocation results in increased transcription of heme oxygenase (HO)-1 and NADPH quinone dehydrogenase (NQO1) due to antioxidant responsive element (ARE) transcriptional activity. Additionally, berry polyphenols increase sirtuin 1 (Sirt1) protein expression, which targets a number of proteins, reducing NF-κB and p53 activity while increasing Nrf2 and endothelial nitric oxide synthase (eNOS) activity. Further, Sirt1 increases antioxidant enzymes superoxide dismutase (SOD), glutathione peroxidase (GPx), and catalase, which neutralize superoxide O2− and H2O2. (C) In endothelial cells, berry polyphenols, such as cyanidin, increase phosphoinositide 3-kinase (PI3K) activity, resulting in increased phosphorylation of protein kinase B (Akt) and eNOS, resulting in increased nitric oxide (NO), promoting vasodilation. Additionally, berry polyphenols preserve eNOS protein expression under a number of pathological stimuli, including due to TNFR1 activation. (D) Berry polyphenols reduce apoptosis by increasing B-cell lymphoma 2 (Bcl-2), reducing caspase-8 mediated activation of Bcl-2-associated X protein (Bax) and Bcl-2 homologous antagonist killer (Bak), while Bcl extra-large (Bcl-xL) reduces Bax/Bak oligomerization. (E) Berry polyphenols, such as gallic acid, reduce fibrosis by decreasing TGF-β protein expression and prevent fibroblast differentiation to myofibroblasts, resulting in reduced collagen synthesis. Created in BioRender.com.

Figure 2.
Potential estrogen receptor-mediated effects by berry polyphenols in CVD. Multiple cellular targets are mediated by the estrogen receptor (ER), acting in an overall protective manner. (A) Polyphenols derived from berries, such as quercetin, can enter the cell via bilitranslocase and act as ER ligands in a similar fashion to 17β-estradiol (E2). (B) Once ER is activated, it inhibits IκB kinase (IKK)-mediated IκB phosphorylation and degradation, preventing nuclear translocation of NF-κB subunits, p65 and p50. Additionally, ER recruits Sirt1 to induce deacetylation of nuclear factor kappa-light-chain-enhancer of activated B (NF-κB), further inhibiting its activity. (C) The active sulfhydryl form of xanthine oxidase (XO) undergoes posttranslational modifications by ER, hydrolyzing the sulfhydryl group and inducing phosphorylation of XO, inhibiting its activity and reducing ROS. (D) Murine double minute 2 (MDM2) activity is upregulated by ER, leading to increased p53 degradation, reducing apoptosis. (E) ER increases nuclear factor erythroid 2-related factor (Nrf2) nuclear activity, increasing endogenous detoxifying enzymes. (F) Activity of endothelial nitric oxide synthase (eNOS) is increased due to phosphorylation by phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) mediated by ER. (G) Fibrosis is reduced in myofibroblasts due to ER-mediated effects on matrix metalloproteinase (MMP) and collagen, decreasing the expression of these proteins. Thus, berry polyphenols may be protective partially due to ER-dependent activity. Created in BioRender.com.
Figure 2.
Potential estrogen receptor-mediated effects by berry polyphenols in CVD. Multiple cellular targets are mediated by the estrogen receptor (ER), acting in an overall protective manner. (A) Polyphenols derived from berries, such as quercetin, can enter the cell via bilitranslocase and act as ER ligands in a similar fashion to 17β-estradiol (E2). (B) Once ER is activated, it inhibits IκB kinase (IKK)-mediated IκB phosphorylation and degradation, preventing nuclear translocation of NF-κB subunits, p65 and p50. Additionally, ER recruits Sirt1 to induce deacetylation of nuclear factor kappa-light-chain-enhancer of activated B (NF-κB), further inhibiting its activity. (C) The active sulfhydryl form of xanthine oxidase (XO) undergoes posttranslational modifications by ER, hydrolyzing the sulfhydryl group and inducing phosphorylation of XO, inhibiting its activity and reducing ROS. (D) Murine double minute 2 (MDM2) activity is upregulated by ER, leading to increased p53 degradation, reducing apoptosis. (E) ER increases nuclear factor erythroid 2-related factor (Nrf2) nuclear activity, increasing endogenous detoxifying enzymes. (F) Activity of endothelial nitric oxide synthase (eNOS) is increased due to phosphorylation by phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) mediated by ER. (G) Fibrosis is reduced in myofibroblasts due to ER-mediated effects on matrix metalloproteinase (MMP) and collagen, decreasing the expression of these proteins. Thus, berry polyphenols may be protective partially due to ER-dependent activity. Created in BioRender.com.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Najjar, R.S.; Turner, C.G.; Wong, B.J.; Feresin, R.G. Berry-Derived Polyphenols in Cardiovascular Pathologies: Mechanisms of Disease and the Role of Diet and Sex. Nutrients 2021, 13, 387. https://0-doi-org.brum.beds.ac.uk/10.3390/nu13020387
AMA Style
Najjar RS, Turner CG, Wong BJ, Feresin RG. Berry-Derived Polyphenols in Cardiovascular Pathologies: Mechanisms of Disease and the Role of Diet and Sex. Nutrients. 2021; 13(2):387. https://0-doi-org.brum.beds.ac.uk/10.3390/nu13020387
Chicago/Turabian StyleNajjar, Rami S., Casey G. Turner, Brett J. Wong, and Rafaela G. Feresin. 2021. "Berry-Derived Polyphenols in Cardiovascular Pathologies: Mechanisms of Disease and the Role of Diet and Sex" Nutrients 13, no. 2: 387. https://0-doi-org.brum.beds.ac.uk/10.3390/nu13020387
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.