
The Role of Gut Microbiota and Metabolites in Obesity-Associated Chronic Gastrointestinal Disorders


Abstract
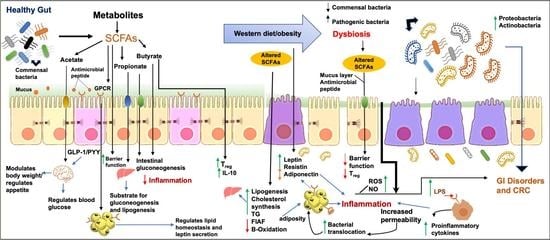
:
1. Introduction
2. Gut Microbiome
2.1. Early Colonization
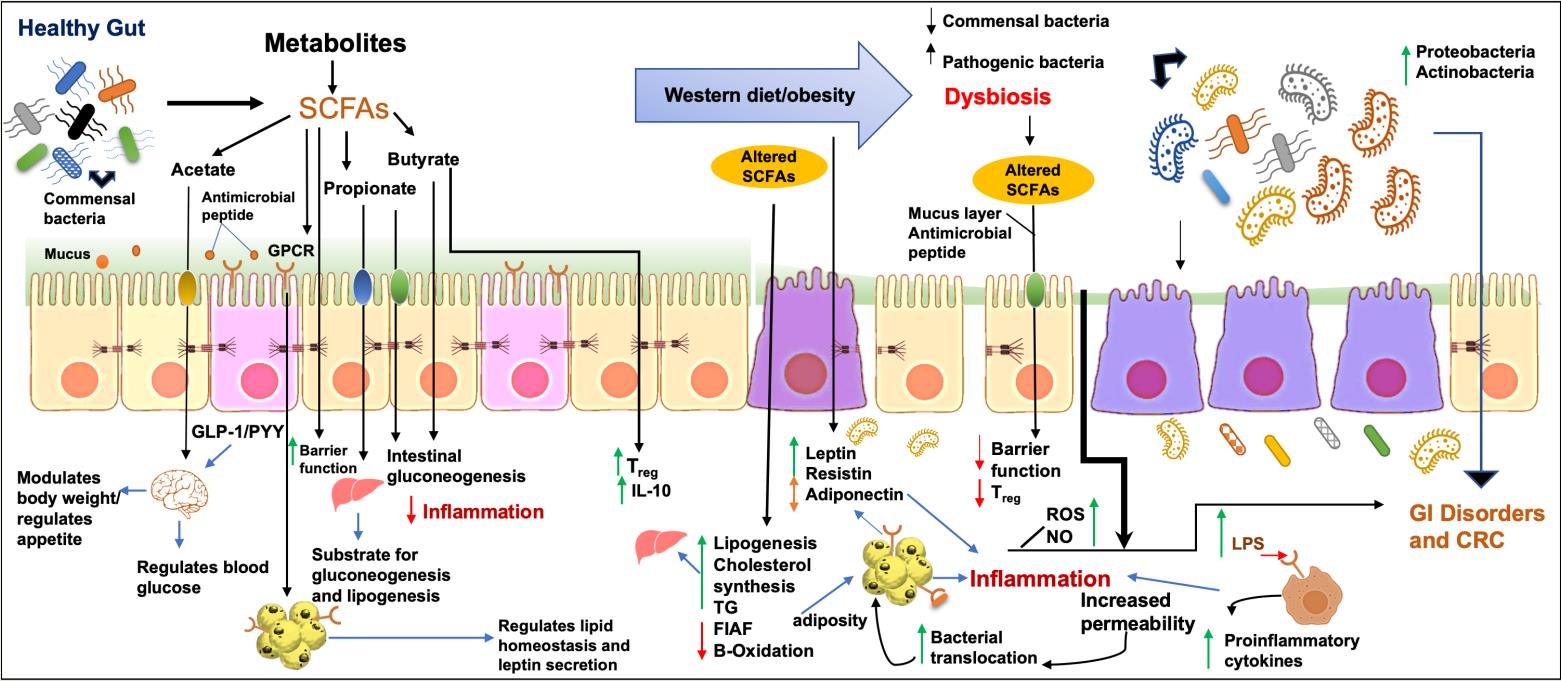
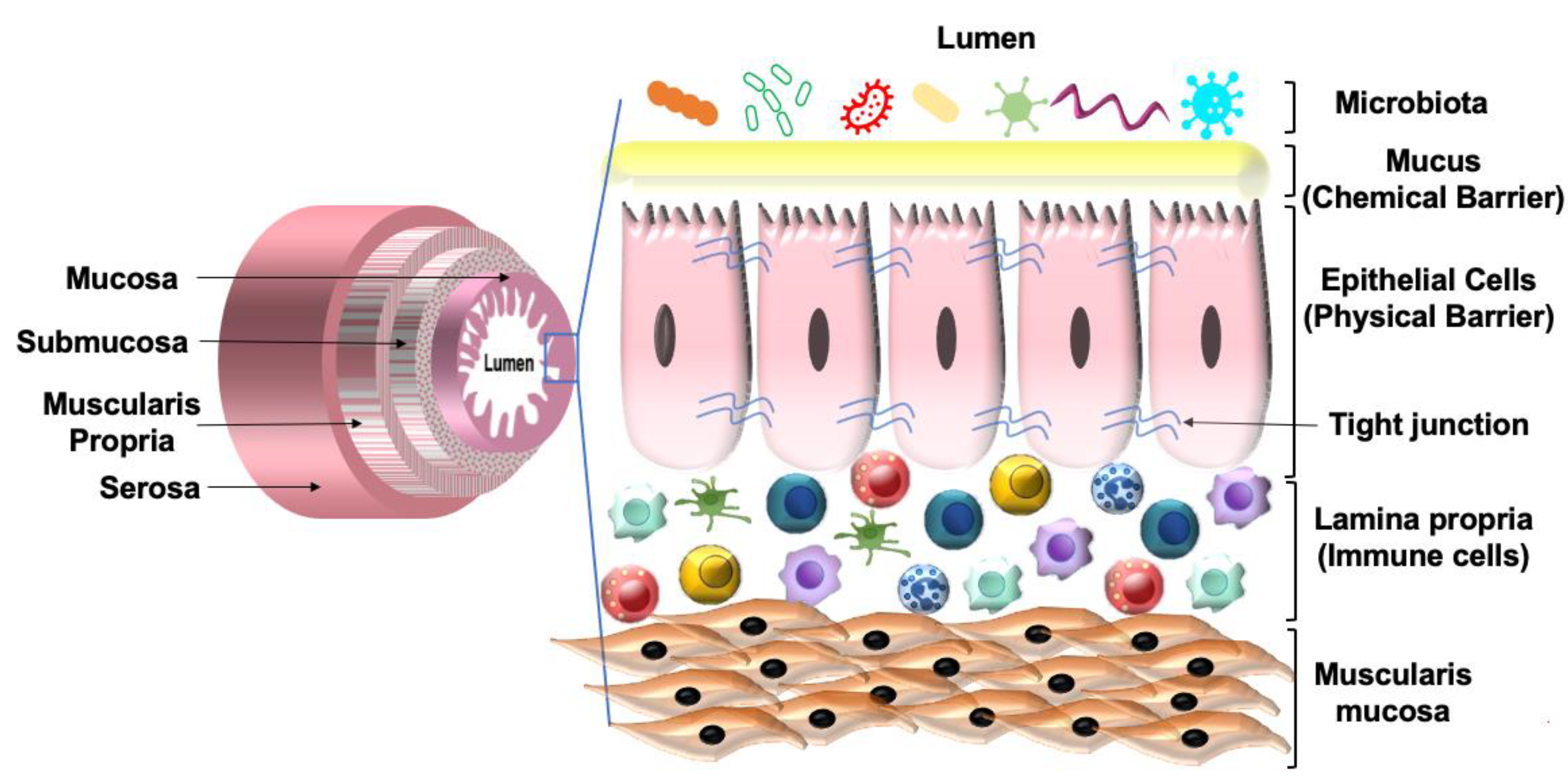
2.2. Host Defense
3. Short-Chain Fatty Acids (SCFAs)
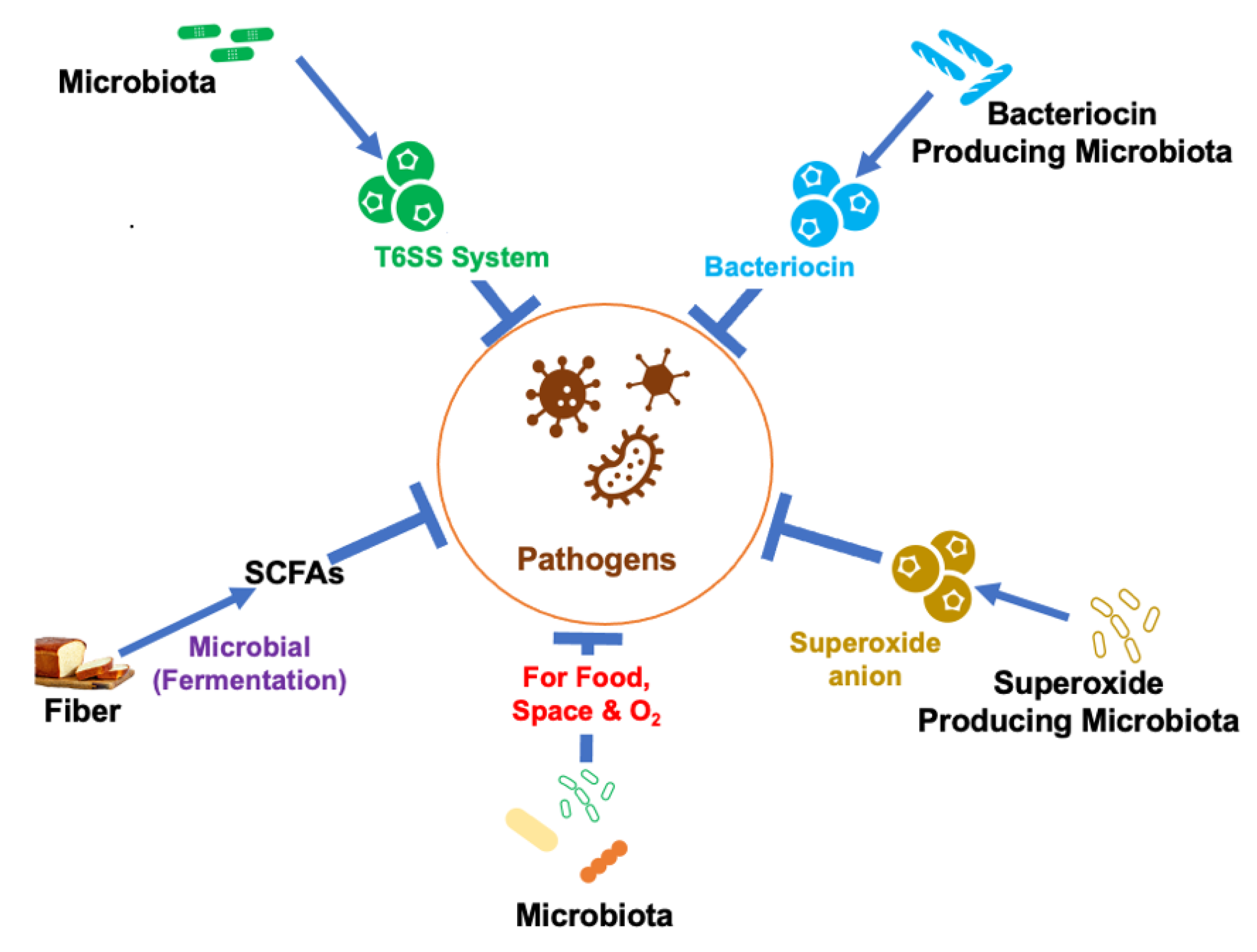
3.1. Production of SCFAs
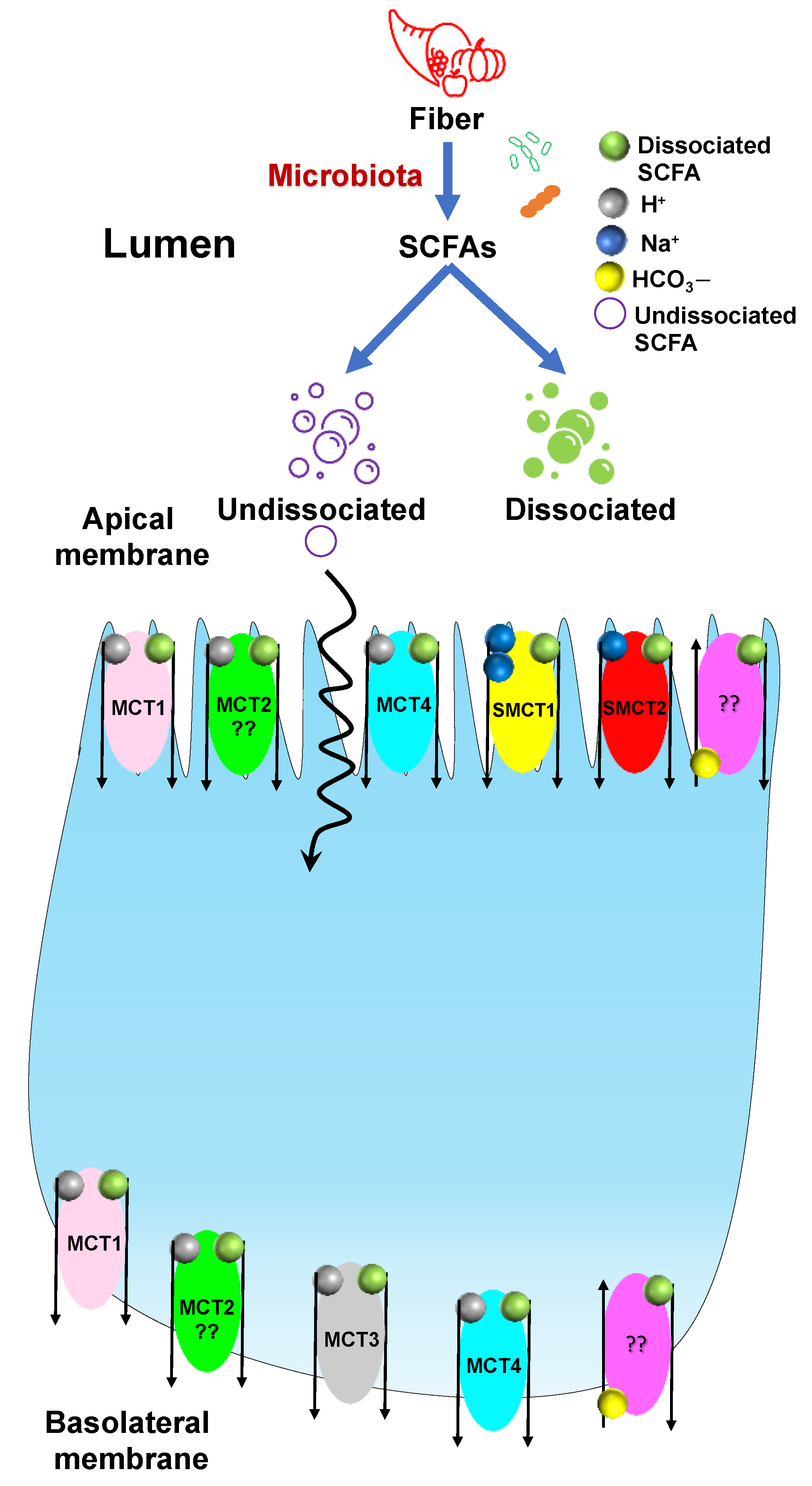
3.2. Transport of SCFAs
3.3. SCFAs as Activators of Signaling Pathways
4. Effect of SCFAs on Health
4.1. SCFAs and Gut Barrier Function
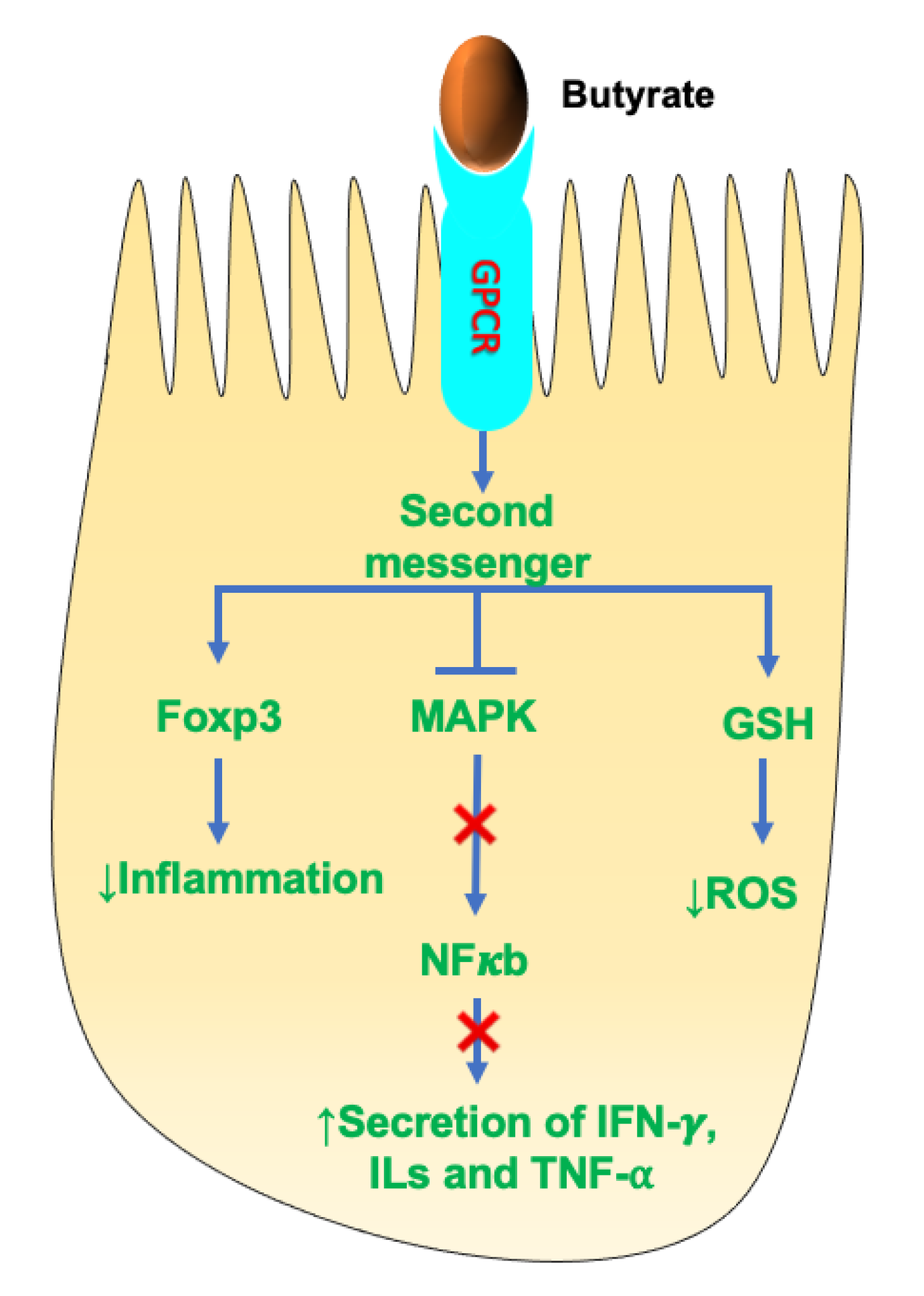
4.2. SCFAs and Anti-Inflammation
4.3. SCFAs and Energy Metabolism
5. Diet–Microbiome Interaction
5.1. Effect of Diet on Microbiota
5.2. Effect of Diet on SCFAs
6. Obesity
6.1. Obesity and Gut Microbiota
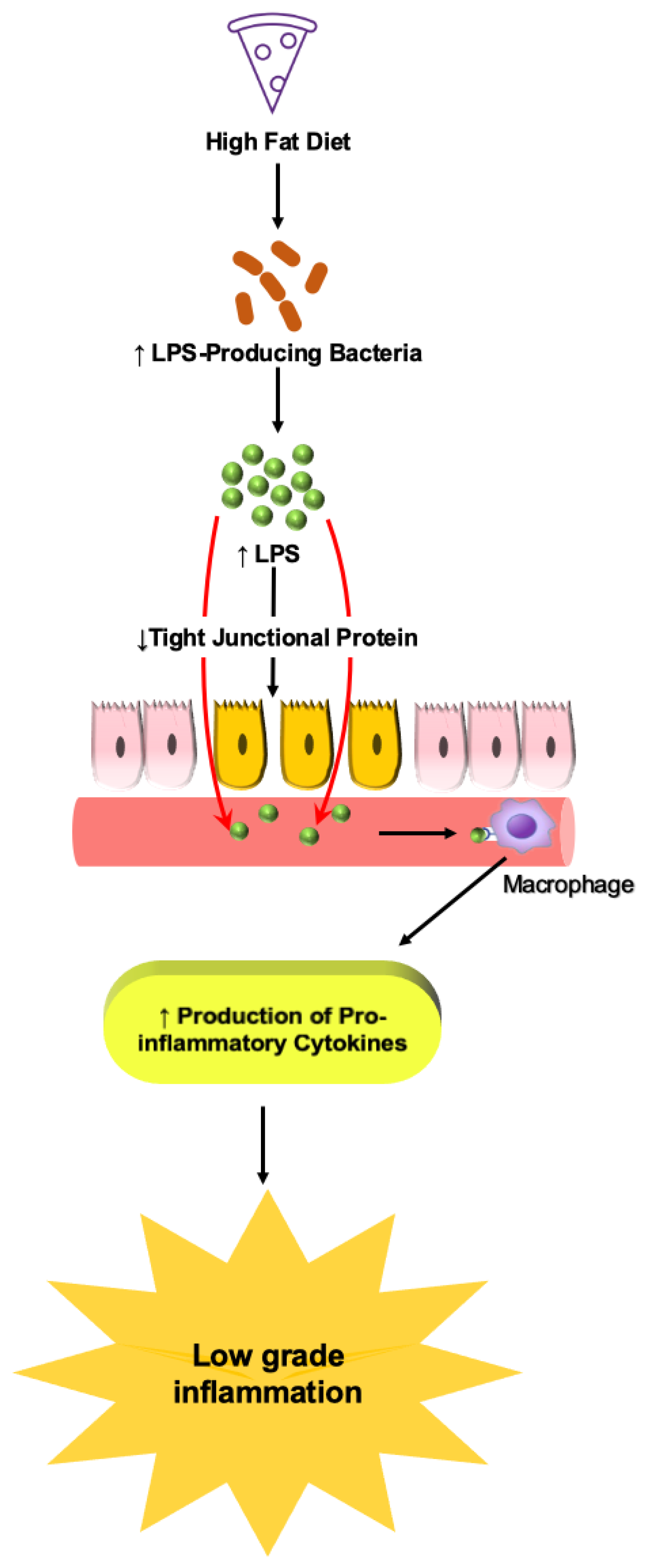
6.1.1. Role of LPS
6.1.2. Role of SCFAs
6.2. Obesity and IBD
6.2.1. Epidemiology and Risk Factor
6.2.2. Impact of Obesity on IBD
6.3. Obesity in the Pathogenesis of IBD
6.3.1. Role of Adipose Tissue
6.3.2. Role of Gut Microbiome
6.4. Creeping Fat (CF)
7. IBD and CRC
7.1. Inflammation
7.2. Gut Microbiota
8. Obesity and CRC
Mechanisms
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, Y.; Lim, H. The global childhood obesity epidemic and the association between socio-economic status and childhood obesity. Int. Rev. Psychiatry 2012, 24, 176–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emerenziani, S.; Guarino, M.P.L.; Asensio, L.M.T.; Altomare, A.; Ribolsi, M.; Balestrieri, P.; Cicala, M. Role of Overweight and Obesity in Gastrointestinal Disease. Nutrients 2019, 12, 111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, Q. A Comprehensive Review and Update on the Pathogenesis of Inflammatory Bowel Disease. J. Immunol. Res. 2019, 2019, 7247238. [Google Scholar] [CrossRef] [Green Version]
- Abraham, C.; Cho, J.H. Inflammatory Bowel Disease. N. Engl. J. Med. 2009, 361, 2066–2078. [Google Scholar] [CrossRef] [PubMed]
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011, 474, 307–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajendran, M.; Loganathan, P.; Catinella, A.P.; Hashash, J.G. A comprehensive review and update on Crohn’s disease. Dis. A Mon. 2018, 64, 20–57. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, G.G.; Ng, S.C. Understanding and Preventing the Global Increase of Inflammatory Bowel Disease. Gastroenterology 2017, 152, 313–321.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ananthakrishnan, A.N. Epidemiology and risk factors for IBD. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Rosen, M.J.; Dhawan, A.; Saeed, S.A. Inflammatory Bowel Disease in Children and Adolescents. JAMA Pediatr. 2015, 169, 1053–1060. [Google Scholar] [CrossRef] [Green Version]
- Monteleone, G.; Fina, D.; Caruso, R.; Pallone, F. New mediators of immunity and inflammation in inflammatory bowel disease. Curr. Opin. Gastroenterol. 2006, 22, 361–364. [Google Scholar] [CrossRef]
- Danese, S.; Sans, M.; Fiocchi, C. Inflammatory bowel disease: The role of environmental factors. Autoimmun. Rev. 2004, 3, 394–400. [Google Scholar] [CrossRef]
- Fiocchi, C. Inflammatory bowel disease: Etiology and pathogenesis. Gastroenterology 1998, 115, 182–205. [Google Scholar] [CrossRef]
- Bezzio, C.; Festa, S.; Saibeni, S.; Papi, C. Chemoprevention of colorectal cancer in ulcerative colitis: Digging deep in current evidence. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 339–347. [Google Scholar] [CrossRef]
- Keller, D.S.; Windsor, A.; Cohen, R.; Chand, M. Colorectal cancer in inflammatory bowel disease: Review of the evidence. Tech. Coloproctol. 2019, 23, 3–13. [Google Scholar] [CrossRef]
- Hill, M.J. Intestinal flora and endogenous vitamin synthesis. Eur. J. Cancer Prev. 1997, 6, S43–S45. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Almeida, A.; Mitchell, A.; Boland, M.; Forster, S.; Gloor, G.; Tarkowska, A.; Lawley, T.D.; Finn, R.D. A new genomic blueprint of the human gut microbiota. Nature 2019, 568, 499–504. [Google Scholar] [CrossRef] [Green Version]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef]
- Segata, N.; Haake, S.K.; Mannon, P.; Lemon, K.P.; Waldron, L.; Gevers, D.; Huttenhower, C.; Izard, J. Composition of the adult digestive tract bacterial microbiome based on seven mouth surfaces, tonsils, throat and stool samples. Genome Biol. 2012, 13, R42. [Google Scholar] [CrossRef] [Green Version]
- Paredes-Sabja, D.; Shen, A.; Sorg, J. Clostridium difficile spore biology: Sporulation, germination, and spore structural proteins. Trends Microbiol. 2014, 22, 406–416. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Li, P.; Li, J.; An, Y.; Wang, M.; Zhong, G. The Differences between Luminal Microbiota and Mucosal Microbiota in Mice. J. Microbiol. Biotechnol. 2020, 30, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Iacob, S.; Iacob, D.G.; Luminos, L.M. Intestinal Microbiota as a Host Defense Mechanism to Infectious Threats. Front. Microbiol. 2019, 9, 3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cresci, G.A.; Bawden, E. Gut Microbiome. Nutr. Clin. Pract. 2015, 30, 734–746. [Google Scholar] [CrossRef] [Green Version]
- Den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickard, J.M.; Zeng, M.Y.; Caruso, R.; Núñez, G. Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol. Rev. 2017, 279, 70–89. [Google Scholar] [CrossRef]
- Xu, J.; Gordon, J.I. Honor thy symbionts. Proc. Natl. Acad. Sci. USA 2003, 100, 10452–10459. [Google Scholar] [CrossRef] [Green Version]
- Mueller, N.; Bakacs, E.; Combellick, J.; Grigoryan, Z.; Dominguez-Bello, M.G. The infant microbiome development: Mom matters. Trends Mol. Med. 2015, 21, 109–117. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, M.; Panahi, S.; Tremblay, A. Childhood Obesity: A Role for Gut Microbiota? Int. J. Environ. Res. Public Health 2014, 12, 162–175. [Google Scholar] [CrossRef] [Green Version]
- Dreyer, J.L.; Liebl, A.L. Early colonization of the gut microbiome and its relationship with obesity. Hum. Microbiome J. 2018, 10, 1–5. [Google Scholar] [CrossRef]
- Wilson, K.H.; Perini, F. Role of competition for nutrients in suppression of Clostridium difficile by the colonic microflora. Infect. Immun. 1988, 56, 2610–2614. [Google Scholar] [CrossRef] [Green Version]
- Gantois, I.; Ducatelle, R.; Pasmans, F.; Haesebrouck, F.; Hautefort, I.; Thompson, A.; Hinton, J.C.; Van Immerseel, F. Butyrate Specifically Down-Regulates Salmonella Pathogenicity Island 1 Gene Expression. Appl. Environ. Microbiol. 2006, 72, 946–949. [Google Scholar] [CrossRef] [Green Version]
- Lawley, T.D.; Walker, A.W. Intestinal colonization resistance. Immunology 2013, 138, 1–11. [Google Scholar] [CrossRef]
- Ford, S.A.; Kao, D.; Williams, D.; King, K.C. Microbe-mediated host defence drives the evolution of reduced pathogen virulence. Nat. Commun. 2016, 7, 13430. [Google Scholar] [CrossRef] [Green Version]
- Fridman, C.M.; Keppel, K.; Gerlic, M.; Bosis, E.; Salomon, D. A comparative genomics methodology reveals a widespread family of membrane-disrupting T6SS effectors. Nat. Commun. 2020, 11, 1085. [Google Scholar] [CrossRef]
- Zhang, L.-J.; Gallo, R.L. Antimicrobial peptides. Curr. Biol. 2016, 26, R14–R19. [Google Scholar] [CrossRef]
- Medzhitov, R. Toll-like receptors and innate immunity. Nat. Rev. Immunol. 2001, 1, 135–145. [Google Scholar] [CrossRef]
- Okumura, R.; Takeda, K. Maintenance of intestinal homeostasis by mucosal barriers. Inflamm. Regen. 2018, 38, 5. [Google Scholar] [CrossRef]
- Frantz, A.L.; Rogier, E.W.; Weber, C.R.; Shen, L.; A Cohen, D.; A Fenton, L.; Bruno, M.E.C.; Kaetzel, C.S. Targeted deletion of MyD88 in intestinal epithelial cells results in compromised antibacterial immunity associated with downregulation of polymeric immunoglobulin receptor, mucin-2, and antibacterial peptides. Mucosal Immunol. 2012, 5, 501–512. [Google Scholar] [CrossRef] [Green Version]
- Bhinder, G.; Stahl, M.; Sham, H.P.; Crowley, S.M.; Morampudi, V.; Dalwadi, U.; Ma, C.; Jacobson, K.; Vallance, B.A. Intestinal Epithelium-Specific MyD88 Signaling Impacts Host Susceptibility to Infectious Colitis by Promoting Protective Goblet Cell and Antimicrobial Responses. Infect. Immun. 2014, 82, 3753–3763. [Google Scholar] [CrossRef] [Green Version]
- Lei, J.; Sun, L.; Huang, S.; Zhu, C.; Li, P.; He, J.; Mackey, V.; Coy, D.H.; He, Q. The antimicrobial peptides and their potential clinical applications. Am. J. Transl. Res. 2019, 11, 3919–3931. [Google Scholar]
- Van Der Beek, C.M.; DeJong, C.H.C.; Troost, F.J.; Masclee, A.A.M.; Lenaerts, K. Role of short-chain fatty acids in colonic inflammation, carcinogenesis, and mucosal protection and healing. Nutr. Rev. 2017, 75, 286–305. [Google Scholar] [CrossRef]
- Topping, D.L.; Clifton, P.M. Short-Chain Fatty Acids and Human Colonic Function: Roles of Resistant Starch and Nonstarch Polysaccharides. Physiol. Rev. 2001, 81, 1031–1064. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef]
- Fernández, J.; Redondo-Blanco, S.; Gutiérrez-Del-Río, I.; Miguélez, E.M.; Villar, C.J.; Lombó, F. Colon microbiota fermentation of dietary prebiotics towards short-chain fatty acids and their roles as anti-inflammatory and antitumour agents: A review. J. Funct. Foods 2016, 25, 511–522. [Google Scholar] [CrossRef]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, P.; Flint, H.J. Formation of propionate and butyrate by the human colonic microbiota. Environ. Microbiol. 2016, 19, 29–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, P.; Flint, H.J. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol. Lett. 2009, 294, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliphant, K.; Allen-Vercoe, E. Macronutrient metabolism by the human gut microbiome: Major fermentation by-products and their impact on host health. Microbiome 2019, 7, 91. [Google Scholar] [CrossRef]
- Derrien, M.; Vaughan, E.E.; Plugge, C.M.; De Vos, W.M. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int. J. Syst. Evol. Microbiol. 2004, 54, 1469–1476. [Google Scholar] [CrossRef] [Green Version]
- Louis, P.; Young, P.; Holtrop, G.; Flint, H.J. Diversity of human colonic butyrate-producing bacteria revealed by analysis of the butyryl-CoA:acetate CoA-transferase gene. Environ. Microbiol. 2010, 12, 304–314. [Google Scholar] [CrossRef]
- Rivière, A.; Selak, M.; Lantin, D.; Leroy, F.; De Vuyst, L. Bifidobacteria and Butyrate-Producing Colon Bacteria: Importance and Strategies for Their Stimulation in the Human Gut. Front. Microbiol. 2016, 7, 979. [Google Scholar] [CrossRef] [Green Version]
- Sivaprakasam, S.; Bhutia, Y.D.; Yang, S.; Ganapathy, V. Short-Chain Fatty Acid Transporters: Role in Colonic Homeostasis. In Comprehensive Physiology; Wiley: New York, NY, USA, 2017; Volume 8, pp. 299–314. [Google Scholar]
- Blaak, E.E.; Canfora, E.E.; Theis, S.; Frost, G.; Groen, A.K.; Mithieux, G.; Nauta, A.; Scott, K.; Stahl, B.; van Harsselaar, J.; et al. Short chain fatty acids in human gut and metabolic health. Benef. Microbes 2020, 11, 411–455. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Price, N.T. The proton-linked monocarboxylate transporter (MCT) family: Structure, function and regulation. Biochem. J. 1999, 343 Pt 2, 281–299. [Google Scholar] [CrossRef]
- Bonen, A.; Heynen, M.; Hatta, H. Distribution of monocarboxylate transporters MCT1-MCT8 in rat tissues and human skeletal muscle. Appl. Physiol. Nutr. Metab. 2006, 31, 31–39. [Google Scholar] [CrossRef]
- Thwaites, D.T.; Anderson, C.M. H+-coupled nutrient, micronutrient and drug transporters in the mammalian small intestine. Exp. Physiol. 2007, 92, 603–619. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.M.; Goldstein, J.L.; Brown, M.S. cDNA cloning of MEV, a mutant protein that facilitates cellular uptake of mevalonate, and identification of the point mutation responsible for its gain of function. J. Biol. Chem. 1992, 267, 23113–23121. [Google Scholar] [CrossRef]
- Garcia, C.K.; Goldstein, J.L.; Pathak, R.K.; Anderson, R.G.; Brown, M.S. Molecular characterization of a membrane transporter for lactate, pyruvate, and other monocarboxylates: Implications for the Cori cycle. Cell 1994, 76, 865–873. [Google Scholar] [CrossRef]
- Schmitt, M.G., Jr.; Soergel, K.H.; Wood, C.M.; Steff, J.J. Absorption of short-chain fatty acids from the human ileum. Am. J. Dig. Dis. 1977, 22, 340–347. [Google Scholar] [CrossRef]
- Ritzhaupt, A.; Wood, I.S.; Ellis, A.; Hosie, K.B.; Shirazi-Beechey, S.P. Identification and characterization of a monocarboxylate transporter (MCT1) in pig and human colon: Its potential to transport L-lactate as well as butyrate. J. Physiol. 1998, 513 Pt 3, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Tamai, I.; Sai, Y.; Ono, A.; Kido, Y.; Yabuuchi, H.; Takanaga, H.; Satoh, E.; Ogihara, T.; Amano, O.; Izeki, S.; et al. Immunohistochemical and functional characterization of pH-dependent intestinal absorption of weak organic acids by the monocarboxylic acid transporter MCT1. J. Pharm. Pharmacol. 1999, 51, 1113–1121. [Google Scholar] [CrossRef]
- Iwanaga, T.; Takebe, K.; Kato, I.; Karaki, S.; Kuwahara, A. Cellular expression of monocarboxylate transporters (MCT) in the digestive tract of the mouse, rat, and humans, with special reference to slc5a8. Biomed. Res. 2006, 27, 243–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, R.K.; Saksena, S.; Alrefai, W.A.; Sarwar, Z.; Goldstein, J.L.; Carroll, R.E.; Ramaswamy, K.; Dudeja, P.K. Expression and membrane localization of MCT isoforms along the length of the human intestine. Am. J. Physiol. Cell. Physiol. 2005, 289, C846–C852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kekuda, R.; Manoharan, P.; Baseler, W.; Sundaram, U. Monocarboxylate 4 mediated butyrate transport in a rat intestinal epithelial cell line. Dig. Dis. Sci. 2013, 58, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Cui, D.; Morris, M.E. The drug of abuse gamma-hydroxybutyrate is a substrate for sodium-coupled monocarboxylate transporter (SMCT) 1 (SLC5A8): Characterization of SMCT-mediated uptake and inhibition. Drug Metab. Dispos. 2009, 37, 1404–1410. [Google Scholar] [CrossRef] [Green Version]
- Schroder, O.; Opritz, J.; Stein, J. Substrate and inhibitor specificity of butyrate uptake in apical membrane vesicles of the rat distal colon. Digestion 2000, 62, 152–158. [Google Scholar] [CrossRef]
- Reynolds, D.A.; Rajendran, V.M.; Binder, H.J. Bicarbonate-stimulated [14C]butyrate uptake in basolateral membrane vesicles of rat distal colon. Gastroenterology 1993, 105, 725–732. [Google Scholar] [CrossRef]
- Tyagi, S.; Venugopalakrishnan, J.; Ramaswamy, K.; Dudeja, P.K. Mechanism of n-butyrate uptake in the human proximal colonic basolateral membranes. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G676–G682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.H.; Kang, S.G.; Park, J.H.; Yanagisawa, M.; Kim, C.H. Short-chain fatty acids activate GPR41 and GPR43 on intestinal epithelial cells to promote inflammatory responses in mice. Gastroenterology 2013, 145, 396–406. [Google Scholar] [CrossRef]
- Le Poul, E.; Loison, C.; Struyf, S.; Springael, J.Y.; Lannoy, V.; Decobecq, M.E.; Brezillon, S.; Dupriez, V.; Vassart, G.; Van Damme, J.; et al. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J. Biol. Chem. 2003, 278, 25481–25489. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Zhang, P.; Shen, L.; Niu, L.; Tan, Y.; Chen, L.; Zhao, Y.; Bai, L.; Hao, X.; Li, X.; et al. Short-Chain Fatty Acids and Their Association with Signalling Pathways in Inflammation, Glucose and Lipid Metabolism. Int. J. Mol. Sci. 2020, 21, 6356. [Google Scholar] [CrossRef]
- Husted, A.S.; Trauelsen, M.; Rudenko, O.; Hjorth, S.A.; Schwartz, T.W. GPCR-Mediated Signaling of Metabolites. Cell Metab. 2017, 25, 777–796. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 2014, 40, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Vidyasagar, S.; Ramakrishna, B.S. Effects of butyrate on active sodium and chloride transport in rat and rabbit distal colon. J. Physiol. 2002, 539, 163–173. [Google Scholar] [CrossRef]
- Tan, J.; McKenzie, C.; Potamitis, M.; Thorburn, A.N.; Mackay, C.R.; Macia, L. The role of short-chain fatty acids in health and disease. Adv. Immunol. 2014, 121, 91–119. [Google Scholar] [CrossRef]
- Konig, J.; Wells, J.; Cani, P.D.; Garcia-Rodenas, C.L.; MacDonald, T.; Mercenier, A.; Whyte, J.; Troost, F.; Brummer, R.J. Human Intestinal Barrier Function in Health and Disease. Clin. Transl. Gastroenterol. 2016, 7, e196. [Google Scholar] [CrossRef]
- Chelakkot, C.; Ghim, J.; Ryu, S.H. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp. Mol. Med. 2018, 50, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, T.M.; Leonel, A.J.; Melo, M.A.; Santos, R.R.; Cara, D.C.; Cardoso, V.N.; Correia, M.I.; Alvarez-Leite, J.I. Oral supplementation of butyrate reduces mucositis and intestinal permeability associated with 5-Fluorouracil administration. Lipids 2012, 47, 669–678. [Google Scholar] [CrossRef]
- Yan, H.; Ajuwon, K.M. Butyrate modifies intestinal barrier function in IPEC-J2 cells through a selective upregulation of tight junction proteins and activation of the Akt signaling pathway. PLoS ONE 2017, 12, e0179586. [Google Scholar] [CrossRef]
- Hung, T.V.; Suzuki, T. Dietary Fermentable Fiber Reduces Intestinal Barrier Defects and Inflammation in Colitic Mice. J. Nutr. 2016, 146, 1970–1979. [Google Scholar] [CrossRef]
- Yoo, J.Y.; Groer, M.; Dutra, S.V.O.; Sarkar, A.; McSkimming, D.I. Gut Microbiota and Immune System Interactions. Microorganisms 2020, 8, 1587. [Google Scholar] [CrossRef]
- Inan, M.S.; Rasoulpour, R.J.; Yin, L.; Hubbard, A.K.; Rosenberg, D.W.; Giardina, C. The luminal short-chain fatty acid butyrate modulates NF-kappaB activity in a human colonic epithelial cell line. Gastroenterology 2000, 118, 724–734. [Google Scholar] [CrossRef]
- Scheppach, W.; Sommer, H.; Kirchner, T.; Paganelli, G.M.; Bartram, P.; Christl, S.; Richter, F.; Dusel, G.; Kasper, H. Effect of butyrate enemas on the colonic mucosa in distal ulcerative colitis. Gastroenterology 1992, 103, 51–56. [Google Scholar] [CrossRef]
- Vernia, P.; Marcheggiano, A.; Caprilli, R.; Frieri, G.; Corrao, G.; Valpiani, D.; Di Paolo, M.C.; Paoluzi, P.; Torsoli, A. Short-chain fatty acid topical treatment in distal ulcerative colitis. Aliment. Pharmacol. Ther. 1995, 9, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, W.; Ryl, A.; Mizerski, A.; Walczakiewicz, K.; Sipak, O.; Laszczynska, M. Immunomodulatory potential of gut microbiome-derived short-chain fatty acids (SCFAs). Acta. Biochim. Pol. 2019, 66, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roediger, W.E. Utilization of nutrients by isolated epithelial cells of the rat colon. Gastroenterology 1982, 83, 424–429. [Google Scholar] [CrossRef]
- Tazoe, H.; Otomo, Y.; Kaji, I.; Tanaka, R.; Karaki, S.I.; Kuwahara, A. Roles of short-chain fatty acids receptors, GPR41 and GPR43 on colonic functions. J. Physiol. Pharmacol. 2008, 59 (Suppl. 2), 251–262. [Google Scholar]
- Bergman, E.N. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol. Rev. 1990, 70, 567–590. [Google Scholar] [CrossRef] [Green Version]
- Frost, G.; Sleeth, M.L.; Sahuri-Arisoylu, M.; Lizarbe, B.; Cerdan, S.; Brody, L.; Anastasovska, J.; Ghourab, S.; Hankir, M.; Zhang, S.; et al. The short-chain fatty acid acetate reduces appetite via a central homeostatic mechanism. Nat. Commun. 2014, 5, 3611. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Yi, C.X.; Katiraei, S.; Kooijman, S.; Zhou, E.; Chung, C.K.; Gao, Y.; van den Heuvel, J.K.; Meijer, O.C.; Berbee, J.F.P.; et al. Butyrate reduces appetite and activates brown adipose tissue via the gut-brain neural circuit. Gut 2018, 67, 1269–1279. [Google Scholar] [CrossRef] [Green Version]
- Arnoldussen, I.A.C.; Wiesmann, M.; Pelgrim, C.E.; Wielemaker, E.M.; van Duyvenvoorde, W.; Amaral-Santos, P.L.; Verschuren, L.; Keijser, B.J.F.; Heerschap, A.; Kleemann, R.; et al. Butyrate restores HFD-induced adaptations in brain function and metabolism in mid-adult obese mice. Int. J. Obes. 2017, 41, 935–944. [Google Scholar] [CrossRef]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Fan, C.; Li, P.; Lu, Y.; Chang, X.; Qi, K. Short Chain Fatty Acids Prevent High-fat-diet-induced Obesity in Mice by Regulating G Protein-coupled Receptors and Gut Microbiota. Sci. Rep. 2016, 6, 37589. [Google Scholar] [CrossRef] [Green Version]
- Batterham, R.L.; Cowley, M.A.; Small, C.J.; Herzog, H.; Cohen, M.A.; Dakin, C.L.; Wren, A.M.; Brynes, A.E.; Low, M.J.; Ghatei, M.A.; et al. Gut hormone PYY(3–36) physiologically inhibits food intake. Nature 2002, 418, 650–654. [Google Scholar] [CrossRef]
- van den Hoek, A.M.; Heijboer, A.C.; Corssmit, E.P.; Voshol, P.J.; Romijn, J.A.; Havekes, L.M.; Pijl, H. PYY3-36 reinforces insulin action on glucose disposal in mice fed a high-fat diet. Diabetes 2004, 53, 1949–1952. [Google Scholar] [CrossRef] [Green Version]
- Barrera, J.G.; Sandoval, D.A.; D’Alessio, D.A.; Seeley, R.J. GLP-1 and energy balance: An integrated model of short-term and long-term control. Nat. Rev. Endocrinol. 2011, 7, 507–516. [Google Scholar] [CrossRef]
- Sakakibara, S.; Yamauchi, T.; Oshima, Y.; Tsukamoto, Y.; Kadowaki, T. Acetic acid activates hepatic AMPK and reduces hyperglycemia in diabetic KK-A(y) mice. Biochem. Biophys. Res. Commun. 2006, 344, 597–604. [Google Scholar] [CrossRef]
- Hattori, M.; Kondo, T.; Kishi, M.; Yamagami, K. A single oral administration of acetic acid increased energy expenditure in C57BL/6J mice. Biosci. Biotechnol. Biochem. 2010, 74, 2158–2159. [Google Scholar] [CrossRef]
- Sukkar, A.H.; Lett, A.M.; Frost, G.; Chambers, E.S. Regulation of energy expenditure and substrate oxidation by short-chain fatty acids. J. Endocrinol. 2019, 242, R1–R8. [Google Scholar] [CrossRef]
- den Besten, G.; Bleeker, A.; Gerding, A.; Van Eunen, K.; Havinga, R.; Van Dijk, T.H.; Oosterveer, M.H.; Jonker, J.W.; Groen, A.K.; Reijngoud, D.-J.; et al. Short-Chain Fatty Acids Protect Against High-Fat Diet–Induced Obesity via a PPARgamma-Dependent Switch From Lipogenesis to Fat Oxidation. Diabetes 2015, 64, 2398–2408. [Google Scholar] [CrossRef] [Green Version]
- Tierney, B.; Yang, Z.; Luber, J.M.; Beaudin, M.; Wibowo, M.C.; Baek, C.; Mehlenbacher, E.; Patel, C.J.; Kostic, A.D. The Landscape of Genetic Content in the Gut and Oral Human Microbiome. Cell Host Microbe 2019, 26, 283–295.e8. [Google Scholar] [CrossRef]
- Gentile, C.L.; Weir, T.L. The gut microbiota at the intersection of diet and human health. Science 2018, 362, 776–780. [Google Scholar] [CrossRef] [Green Version]
- Fujita, K.; Sakamoto, A.; Kaneko, S.; Kotake, T.; Tsumuraya, Y.; Kitahara, K. Degradative enzymes for type II arabinogalactan side chains in Bifidobacterium longum subsp. longum. Appl. Microbiol. Biotechnol. 2018, 103, 1299–1310. [Google Scholar] [CrossRef]
- Collins, K.H.; Paul, H.A.; Hart, D.A.; Reimer, R.A.; Smith, I.C.; Rios, J.L.; Seerattan, R.A.; Herzog, W. A High-Fat High-Sucrose Diet Rapidly Alters Muscle Integrity, Inflammation and Gut Microbiota in Male Rats. Sci. Rep. 2016, 6, 37278. [Google Scholar] [CrossRef]
- LeComte, V.; Kaakoush, N.O.; Maloney, C.A.; Raipuria, M.; Huinao, K.D.; Mitchell, H.M.; Morris, M.J. Changes in Gut Microbiota in Rats Fed a High Fat Diet Correlate with Obesity-Associated Metabolic Parameters. PLoS ONE 2015, 10, e0126931. [Google Scholar] [CrossRef] [Green Version]
- Dehghan, P.; Gargari, B.P.; Jafarabadi, M.A. Oligofructose-enriched inulin improves some inflammatory markers and metabolic endotoxemia in women with type 2 diabetes mellitus: A randomized controlled clinical trial. Nutrition 2014, 30, 418–423. [Google Scholar] [CrossRef]
- Vulevic, J.; Juric, A.; Tzortzis, G.; Gibson, G.R. A Mixture of trans-Galactooligosaccharides Reduces Markers of Metabolic Syndrome and Modulates the Fecal Microbiota and Immune Function of Overweight Adults. J. Nutr. 2013, 143, 324–331. [Google Scholar] [CrossRef]
- Gibson, G.R.; Roberfroid, M.B. Dietary Modulation of the Human Colonic Microbiota: Introducing the Concept of Prebiotics. J. Nutr. 1995, 125, 1401–1412. [Google Scholar] [CrossRef]
- Jackson, K.G.; Taylor, G.R.J.; Clohessy, A.M.; Williams, C.M. The effect of the daily intake of inulin on fasting lipid, insulin and glucose concentrations in middle-aged men and women. Br. J. Nutr. 1999, 82, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-M.; Yu, Y.-N.; Wang, J.-L.; Lin, Y.-W.; Kong, X.; Yang, C.-Q.; Yang, L.; Liu, Z.-J.; Yuan, Y.-Z.; Liu, F.; et al. Decreased dietary fiber intake and structural alteration of gut microbiota in patients with advanced colorectal adenoma. Am. J. Clin. Nutr. 2013, 97, 1044–1052. [Google Scholar] [CrossRef] [Green Version]
- Russell, W.R.; Gratz, S.W.; Duncan, S.H.; Holtrop, G.; Ince, J.; Scobbie, L.; Duncan, G.; Johnstone, A.M.; Lobley, G.E.; Wallace, R.J.; et al. High-protein, reduced-carbohydrate weight-loss diets promote metabolite profiles likely to be detrimental to colonic health. Am. J. Clin. Nutr. 2011, 93, 1062–1072. [Google Scholar] [CrossRef]
- Kang, S.; Denman, S.; Morrison, M.; Yu, Z.; Dore, J.; Leclerc, M.; McSweeney, C. Dysbiosis of fecal microbiota in Crohn’s disease patients as revealed by a custom phylogenetic microarray. Inflamm. Bowel Dis. 2010, 16, 2034–2042. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- I Cook, S.; Sellin, J.H. Review article: Short chain fatty acids in health and disease. Aliment. Pharmacol. Ther. 1998, 12, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.; Su, W.; Rahat-Rozenbloom, S.; Wolever, T.M.S.; Comelli, E. Adiposity, gut microbiota and faecal short chain fatty acids are linked in adult humans. Nutr. Diabetes 2014, 4, e121. [Google Scholar] [CrossRef] [PubMed]
- Goverse, G.; Molenaar, R.; Macia, L.; Tan, J.; Erkelens, M.N.; Konijn, T.; Knippenberg, M.; Cook, E.C.L.; Hanekamp, D.; Veldhoen, M.; et al. Diet-Derived Short Chain Fatty Acids Stimulate Intestinal Epithelial Cells to Induce Mucosal Tolerogenic Dendritic Cells. J. Immunol. 2017, 198, 2172–2181. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, T.; Nanjo, F. Dietary Cycloinulooligosaccharides Enhance Intestinal Immunoglobulin A Production in Mice. Biosci. Biotechnol. Biochem. 2009, 73, 677–682. [Google Scholar] [CrossRef] [Green Version]
- Mueller, N.T.; Zhang, M.; Juraschek, S.P.; Miller, E.R., 3rd; Appel, L.J. Effects of high-fiber diets enriched with carbohydrate, protein, or unsaturated fat on circulating short chain fatty acids: Results from the OmniHeart randomized trial. Am. J. Clin. Nutr. 2020, 111, 545–554. [Google Scholar] [CrossRef]
- Cotillard, A.; Kennedy, S.P.; Kong, L.C.; Prifti, E.; Pons, N.; Le Chatelier, E.; Almeida, M.; Quinquis, B.; Levenez, F.; Galleron, N.; et al. Dietary intervention impact on gut microbial gene richness. Nature 2013, 500, 585–588. [Google Scholar] [CrossRef]
- Kopp, W. How Western Diet And Lifestyle Drive The Pandemic Of Obesity And Civilization Diseases. Diabetes Metab. Syndr. Obes. Targets Ther. 2019, 12, 2221–2236. [Google Scholar] [CrossRef] [Green Version]
- Rakhra, V.; Galappaththy, S.L.; Bulchandani, S.; Cabandugama, P.K. Obesity and the Western Diet: How We Got Here. Mo. Med. 2020, 117, 536–538. [Google Scholar]
- Hill, J.O.; Wyatt, H.R.; Peters, J.C. The Importance of Energy Balance. Eur. Endocrinol. 2010, 9, 111–115. [Google Scholar] [CrossRef] [Green Version]
- Reilly, S.M.; Saltiel, A.R. Adapting to obesity with adipose tissue inflammation. Nat. Rev. Endocrinol. 2017, 13, 633–643. [Google Scholar] [CrossRef]
- Ohlson, L.-O.; Larsson, B.; Svärdsudd, K.; Welin, L.; Eriksson, H.; Wilhelmsen, L.; Björntorp, P.; Tibblin, G. The Influence of Body Fat Distribution on the Incidence of Diabetes Mellitus: 13.5 Years of Follow-up of the Participants in the Study of Men Born in 1913. Diabetes 1985, 34, 1055–1058. [Google Scholar] [CrossRef]
- Chakraborti, C.K. New-found link between microbiota and obesity. World J. Gastrointest. Pathophysiol. 2015, 6, 110–119. [Google Scholar] [CrossRef]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.-M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef]
- Clarke, S.; Murphy, E.F.; Nilaweera, K.; Ross, R.; Shanahan, F.; O’Toole, P.W.; Cotter, P.D. The gut microbiota and its relationship to diet and obesity. Gut Microbes 2012, 3, 186–202. [Google Scholar] [CrossRef]
- Feng, J.; Tang, H.; Li, M.; Pang, X.; Wang, L.; Zhang, M.; Zhao, Y.; Zhang, X.; Shen, J. The abundance of fecal Faecalibacterium prausnitzii in relation to obesity and gender in Chinese adults. Arch. Microbiol. 2013, 196, 73–77. [Google Scholar] [CrossRef]
- Andoh, A.; Nishida, A.; Takahashi, K.; Inatomi, O.; Imaeda, H.; Bamba, S.; Kito, K.; Sugimoto, M.; Kobayashi, T. Comparison of the gut microbial community between obese and lean peoples using 16S gene sequencing in a Japanese population. J. Clin. Biochem. Nutr. 2016, 59, 65–70. [Google Scholar] [CrossRef] [Green Version]
- Ganesan, K.; Chung, S.K.; Vanamala, J.; Xu, B. Causal Relationship between Diet-Induced Gut Microbiota Changes and Diabetes: A Novel Strategy to Transplant Faecalibacterium prausnitzii in Preventing Diabetes. Int. J. Mol. Sci. 2018, 19, 3720. [Google Scholar] [CrossRef] [Green Version]
- Miquel, S.; Martin, R.; Rossi, O.; Bermudez-Humaran, L.G.; Chatel, J.M.; Sokol, H.; Thomas, M.; Wells, J.M.; Langella, P. Faecalibacterium prausnitzii and human intestinal health. Curr. Opin. Microbiol. 2013, 16, 255–261. [Google Scholar] [CrossRef]
- Balamurugan, R.; George, G.; Kabeerdoss, J.; Hepsiba, J.; Chandragunasekaran, A.M.S.; Ramakrishna, B.S. Quantitative differences in intestinal Faecalibacterium prausnitzii in obese Indian children. Br. J. Nutr. 2009, 103, 335–338. [Google Scholar] [CrossRef] [Green Version]
- Sanz, Y.; Santacruz, A.; Gauffin-Cano, P. Gut microbiota in obesity and metabolic disorders. Proc. Nutr. Soc. 2010, 69, 434–441. [Google Scholar] [CrossRef] [Green Version]
- Escobedo, G.; López-Ortiz, E.; Torres-Castro, I. Gut microbiota as a key player in triggering obesity, systemic inflammation and insulin resistance. Rev. Investig. Clin. 2015, 66, 450–459. [Google Scholar]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef]
- Ismail, N.; Ragab, S.; ElBaky, A.A.; Shoeib, A.R.; Alhosary, Y.; Fekry, D. Frequency of Firmicutes and Bacteroidetes in gut microbiota in obese and normal weight Egyptian children and adults. Arch. Med. Sci. 2011, 3, 501–507. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.W.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [Green Version]
- Graham, C.; Mullen, A.; Whelan, K. Obesity and the gastrointestinal microbiota: A review of associations and mechanisms. Nutr. Rev. 2015, 73, 376–385. [Google Scholar] [CrossRef]
- Brahe, L.K.; Astrup, A.; Larsen, L.H. Is butyrate the link between diet, intestinal microbiota and obesity-related metabolic diseases? Obes. Rev. 2013, 14, 950–959. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.V.; Frassetto, A.; Kowalik, E.J., Jr.; Nawrocki, A.R.; Lu, M.M.; Kosinski, J.R.; Hubert, J.A.; Szeto, D.; Yao, X.; Forrest, G.; et al. Butyrate and Propionate Protect against Diet-Induced Obesity and Regulate Gut Hormones via Free Fatty Acid Receptor 3-Independent Mechanisms. PLoS ONE 2012, 7, e35240. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.J.; Gerasimidis, K.; Edwards, C.A.; Shaikh, M.G. Role of Gut Microbiota in the Aetiology of Obesity: Proposed Mechanisms and Review of the Literature. J. Obes. 2016, 2016, 7353642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López, M. EJE PRIZE 2017: Hypothalamic AMPK: A golden target against obesity? Eur. J. Endocrinol. 2017, 176, R235–R246. [Google Scholar] [CrossRef]
- Kallus, S.J.; Brandt, L.J. The Intestinal Microbiota and Obesity. J. Clin. Gastroenterol. 2012, 46, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Semova, I.; Carten, J.D.; Stombaugh, J.; Mackey, L.C.; Knight, R.; Farber, S.A.; Rawls, J.F. Microbiota Regulate Intestinal Absorption and Metabolism of Fatty Acids in the Zebrafish. Cell Host Microbe 2012, 12, 277–288. [Google Scholar] [CrossRef] [Green Version]
- Landau, D.-A.; Goldberg, A.; Levi, Z.; Levy, Y.; Niv, Y.; Bar-Dayan, Y. The Prevalence of Gastrointestinal Diseases in Israeli Adolescents and its Association with Body Mass Index, Gender, and Jewish Ethnicity. J. Clin. Gastroenterol. 2008, 42, 903–909. [Google Scholar] [CrossRef]
- Anbazhagan, A.N.; Priyamvada, S.; Alrefai, W.A.; Dudeja, P.K. Pathophysiology of IBD associated diarrhea. Tissue Barriers 2018, 6, e1463897. [Google Scholar] [CrossRef]
- Moran, G.; Dubeau, M.-F.; Kaplan, G.; Panaccione, R.; Ghosh, S. The Increasing Weight of Crohn’s Disease Subjects in Clinical Trials. Inflamm. Bowel Dis. 2013, 19, 2949–2956. [Google Scholar] [CrossRef]
- Long, M.D.; Crandall, W.V.; Leibowitz, I.H.; Duffy, L.; Del Rosario, F.; Kim, S.C.; Integlia, M.J.; Berman, J.; Grunow, J.; Colletti, R.B.; et al. Prevalence and epidemiology of overweight and obesity in children with inflammatory bowel disease 12. Inflamm. Bowel Dis. 2011, 17, 2162–2168. [Google Scholar] [CrossRef] [Green Version]
- Nic Suibhne, T.; Raftery, T.C.; McMahon, O.; Walsh, C.; O’Morain, C.; O’Sullivan, M. High prevalence of overweight and obesity in adults with Crohn’s disease: Associations with disease and lifestyle factors. J. Crohns. Colitis. 2013, 7, e241–e248. [Google Scholar] [CrossRef] [Green Version]
- Steed, H.; Walsh, S.; Reynolds, N. A brief report of the epidemiology of obesity in the inflammatory bowel disease population of Tayside, Scotland. Obes. Facts. 2009, 2, 370–372. [Google Scholar] [CrossRef]
- Jensen, C.B.; Angquist, L.H.; Mendall, M.A.; Sorensen, T.I.A.; Baker, J.L.; Jess, T. Childhood body mass index and risk of inflammatory bowel disease in adulthood: A population-based cohort study. Am. J. Gastroenterol. 2018, 113, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.S.; Luben, R.; Olsen, A.; Tjonneland, A.; Kaaks, R.; Teucher, B.; Lindgren, S.; Grip, O.; Key, T.; Crowe, F.L.; et al. Body mass index and the risk for Crohn’s disease and ulcerative colitis: Data from a European Prospective Cohort Study (The IBD in EPIC Study). Am. J. Gastroenterol. 2013, 108, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Greuter, T.; Porchet, F.; Braga-Neto, M.B.; Rossel, J.B.; Biedermann, L.; Schreiner, P.; Scharl, M.; Schoepfer, A.M.; Safroneeva, E.; Straumann, A.; et al. Impact of obesity on disease activity and disease outcome in inflammatory bowel disease: Results from the Swiss inflammatory bowel disease cohort. United Eur. Gastroenterol. J. 2020, 8, 1196–1207. [Google Scholar] [CrossRef] [PubMed]
- Uko, V.; Vortia, E.; Achkar, J.P.; Karakas, P.; Fiocchi, C.; Worley, S.; Kay, M.H. Impact of abdominal visceral adipose tissue on disease outcome in pediatric Crohn’s disease. Inflamm. Bowel Dis. 2014, 20, 2286–2291. [Google Scholar] [CrossRef]
- Bertin, B.; Desreumaux, P.; Dubuquoy, L. Obesity, visceral fat and Crohn’s disease. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 574–580. [Google Scholar] [CrossRef]
- Blain, A.; Cattan, S.; Beaugerie, L.; Carbonnel, F.; Gendre, J.P.; Cosnes, J. Crohn’s disease clinical course and severity in obese patients. Clin. Nutr. 2002, 21, 51–57. [Google Scholar] [CrossRef]
- Hass, D.J.; Brensinger, C.M.; Lewis, J.D.; Lichtenstein, G.R. The impact of increased body mass index on the clinical course of Crohn’s disease. Clin. Gastroenterol. Hepatol. 2006, 4, 482–488. [Google Scholar] [CrossRef]
- Malik, T.A.; Manne, A.; Oster, R.A.; Eckhoff, A.; Inusah, S.; Gutierrez, A.M. Obesity is Associated With Poor Surgical Outcome in Crohn’s Disease. Gastroenterol. Res. 2013, 6, 85–90. [Google Scholar] [CrossRef] [Green Version]
- Egberg, M.D.; Galanko, J.A.; Banegas, M.; Roberson, M.; Strassle, P.D.; Phillips, M.; Kappelman, M.D. Obesity Increases the Risk of Hospital Readmission following Intestinal Surgery for Children with Crohn Disease. J. Pediatr. Gastroenterol. Nutr. 2021, 73, 620–625. [Google Scholar] [CrossRef]
- Erhayiem, B.; Dhingsa, R.; Hawkey, C.J.; Subramanian, V. Ratio of visceral to subcutaneous fat area is a biomarker of complicated Crohn’s disease. Clin. Gastroenterol. Hepatol. 2011, 9, 684–687.e681. [Google Scholar] [CrossRef]
- Bryant, R.V.; Schultz, C.G.; Ooi, S.; Goess, C.; Costello, S.P.; Vincent, A.D.; Schoeman, S.; Lim, A.; Bartholomeusz, F.D.; Travis, S.P.L.; et al. Visceral Adipose Tissue Is Associated With Stricturing Crohn’s Disease Behavior, Fecal Calprotectin, and Quality of Life. Inflamm. Bowel Dis. 2019, 25, 592–600. [Google Scholar] [CrossRef]
- Seminerio, J.L.; Koutroubakis, I.E.; Ramos-Rivers, C.; Hashash, J.G.; Dudekula, A.; Regueiro, M.; Baidoo, L.; Barrie, A.; Swoger, J.; Schwartz, M.; et al. Impact of Obesity on the Management and Clinical Course of Patients with Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2015, 21, 2857–2863. [Google Scholar] [CrossRef] [Green Version]
- Flores, A.; Burstein, E.; Cipher, D.J.; Feagins, L.A. Obesity in Inflammatory Bowel Disease: A Marker of Less Severe Disease. Dig. Dis. Sci. 2015, 60, 2436–2445. [Google Scholar] [CrossRef]
- Pringle, P.L.; Stewart, K.O.; Peloquin, J.M.; Sturgeon, H.C.; Nguyen, D.; Sauk, J.; Garber, J.J.; Yajnik, V.; Ananthakrishnan, A.N.; Chan, A.T.; et al. Body Mass Index, Genetic Susceptibility, and Risk of Complications Among Individuals with Crohn’s Disease. Inflamm. Bowel Dis. 2015, 21, 2304–2310. [Google Scholar] [CrossRef]
- Kim, S.K.; Lee, H.S.; Kim, B.J.; Park, J.H.; Hwang, S.W.; Yang, D.H.; Ye, B.D.; Byeon, J.S.; Myung, S.J.; Yang, S.K.; et al. The Clinical Features of Inflammatory Bowel Disease in Patients with Obesity. Can. J. Gastroenterol. Hepatol. 2021, 2021, 9981482. [Google Scholar] [CrossRef]
- Nam, S.Y. Obesity-Related Digestive Diseases and Their Pathophysiology. Gut Liver 2017, 11, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Wajchenberg, B.L. Subcutaneous and visceral adipose tissue: Their relation to the metabolic syndrome. Endocr. Rev. 2000, 21, 697–738. [Google Scholar] [CrossRef]
- Rowan, C.R.; McManus, J.; Boland, K.; O’Toole, A. Visceral adiposity and inflammatory bowel disease. Int. J. Colorectal. Dis. 2021, 36, 2305–2319. [Google Scholar] [CrossRef]
- Magro, D.O.; Barreto, M.R.L.; Cazzo, E.; Camargo, M.G.; Kotze, P.G.; Coy, C.S.R. Visceral Fat Is Increased in Individuals with Crohn’s Disease: A Comparative Analysis with Healthy Controls. Arq. Gastroenterol. 2018, 55, 142–147. [Google Scholar] [CrossRef]
- Buning, C.; von Kraft, C.; Hermsdorf, M.; Gentz, E.; Wirth, E.K.; Valentini, L.; Haas, V. Visceral Adipose Tissue in Patients with Crohn’s Disease Correlates with Disease Activity, Inflammatory Markers, and Outcome. Inflamm. Bowel Dis. 2015, 21, 2590–2597. [Google Scholar] [CrossRef] [Green Version]
- Cravo, M.L.; Velho, S.; Torres, J.; Costa Santos, M.P.; Palmela, C.; Cruz, R.; Strecht, J.; Maio, R.; Baracos, V. Lower skeletal muscle attenuation and high visceral fat index are associated with complicated disease in patients with Crohn’s disease: An exploratory study. Clin. Nutr. ESPEN 2017, 21, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Yadav, D.P.; Kedia, S.; Madhusudhan, K.S.; Bopanna, S.; Goyal, S.; Jain, S.; Vikram, N.K.; Sharma, R.; Makharia, G.K.; Ahuja, V. Body Composition in Crohn’s Disease and Ulcerative Colitis: Correlation with Disease Severity and Duration. Can. J. Gastroenterol. Hepatol. 2017, 2017, 1215035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, C.J.; Murphy, K.E.; Fernandez, M.L. Impact of Obesity and Metabolic Syndrome on Immunity. Adv. Nutr. 2016, 7, 66–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dandona, P.; Aljada, A.; Chaudhuri, A.; Mohanty, P.; Garg, R. Metabolic syndrome: A comprehensive perspective based on interactions between obesity, diabetes, and inflammation. Circulation 2005, 111, 1448–1454. [Google Scholar] [CrossRef] [Green Version]
- Baker, R.G.; Hayden, M.S.; Ghosh, S. NF-kappaB, inflammation, and metabolic disease. Cell Metab. 2011, 13, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Khalili, H.; Ananthakrishnan, A.N.; Konijeti, G.G.; Higuchi, L.M.; Fuchs, C.S.; Richter, J.M.; Chan, A.T. Measures of obesity and risk of Crohn’s disease and ulcerative colitis. Inflamm. Bowel Dis. 2015, 21, 361–368. [Google Scholar] [CrossRef] [Green Version]
- Mendall, M.A.; Gunasekera, A.V.; John, B.J.; Kumar, D. Is obesity a risk factor for Crohn’s disease? Dig. Dis. Sci. 2011, 56, 837–844. [Google Scholar] [CrossRef]
- Harpsoe, M.C.; Basit, S.; Andersson, M.; Nielsen, N.M.; Frisch, M.; Wohlfahrt, J.; Nohr, E.A.; Linneberg, A.; Jess, T. Body mass index and risk of autoimmune diseases: A study within the Danish National Birth Cohort. Int. J. Epidemiol. 2014, 43, 843–855. [Google Scholar] [CrossRef] [Green Version]
- Rahmani, J.; Kord-Varkaneh, H.; Hekmatdoost, A.; Thompson, J.; Clark, C.; Salehisahlabadi, A.; Day, A.S.; Jacobson, K. Body mass index and risk of inflammatory bowel disease: A systematic review and dose-response meta-analysis of cohort studies of over a million participants. Obes. Rev. 2019, 20, 1312–1320. [Google Scholar] [CrossRef]
- Yang, Y.K.; Chen, M.; Clements, R.H.; Abrams, G.A.; Aprahamian, C.J.; Harmon, C.M. Human mesenteric adipose tissue plays unique role versus subcutaneous and omental fat in obesity related diabetes. Cell Physiol. Biochem. 2008, 22, 531–538. [Google Scholar] [CrossRef]
- Desreumaux, P.; Ernst, O.; Geboes, K.; Gambiez, L.; Berrebi, D.; Muller-Alouf, H.; Hafraoui, S.; Emilie, D.; Ectors, N.; Peuchmaur, M.; et al. Inflammatory alterations in mesenteric adipose tissue in Crohn’s disease. Gastroenterology 1999, 117, 73–81. [Google Scholar] [CrossRef]
- Coffey, J.C.; O’Leary, D.P. The mesentery: Structure, function, and role in disease. Lancet. Gastroenterol. Hepatol. 2016, 1, 238–247. [Google Scholar] [CrossRef]
- Bilski, J.; Mazur-Bialy, A.; Wojcik, D.; Surmiak, M.; Magierowski, M.; Sliwowski, Z.; Pajdo, R.; Kwiecien, S.; Danielak, A.; Ptak-Belowska, A.; et al. Role of Obesity, Mesenteric Adipose Tissue, and Adipokines in Inflammatory Bowel Diseases. Biomolecules 2019, 9, 780. [Google Scholar] [CrossRef] [Green Version]
- Karmiris, K.; Koutroubakis, I.E.; Kouroumalis, E.A. Leptin, adiponectin, resistin, and ghrelin—Implications for inflammatory bowel disease. Mol. Nutr. Food Res. 2008, 52, 855–866. [Google Scholar] [CrossRef]
- Tuzun, A.; Uygun, A.; Yesilova, Z.; Ozel, A.M.; Erdil, A.; Yaman, H.; Bagci, S.; Gulsen, M.; Karaeren, N.; Dagalp, K. Leptin levels in the acute stage of ulcerative colitis. J. Gastroenterol. Hepatol. 2004, 19, 429–432. [Google Scholar] [CrossRef]
- Hoppin, A.G.; Kaplan, L.M.; Zurakowski, D.; Leichtner, A.M.; Bousvaros, A. Serum leptin in children and young adults with inflammatory bowel disease. J. Pediatr. Gastroenterol. Nutr. 1998, 26, 500–505. [Google Scholar] [CrossRef]
- Zhao, Q.; Liu, Y.; Tan, L.; Yan, L.; Zuo, X. Adiponectin administration alleviates DSS-induced colonic inflammation in Caco-2 cells and mice. Inflamm. Res. 2018, 67, 663–670. [Google Scholar] [CrossRef]
- Fantuzzi, G. Adiponectin and inflammation: Consensus and controversy. J. Allergy Clin. Immunol. 2008, 121, 326–330. [Google Scholar] [CrossRef]
- Konrad, A.; Lehrke, M.; Schachinger, V.; Seibold, F.; Stark, R.; Ochsenkuhn, T.; Parhofer, K.G.; Goke, B.; Broedl, U.C. Resistin is an inflammatory marker of inflammatory bowel disease in humans. Eur. J. Gastroenterol. Hepatol. 2007, 19, 1070–1074. [Google Scholar] [CrossRef]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Chassaing, B.; Koren, O.; Goodrich, J.K.; Poole, A.C.; Srinivasan, S.; Ley, R.E.; Gewirtz, A.T. Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature 2015, 519, 92–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, J.; Burke, B.; Ford, D.; Garvin, G.; Korn, C.; Sulis, C.; Bhadelia, N. Possible association between obesity and Clostridium difficile infection. Emerg. Infect. Dis. 2013, 19, 1791–1798. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Siles, M.; Enrich-Capo, N.; Aldeguer, X.; Sabat-Mir, M.; Duncan, S.H.; Garcia-Gil, L.J.; Martinez-Medina, M. Alterations in the Abundance and Co-occurrence of Akkermansia muciniphila and Faecalibacterium prausnitzii in the Colonic Mucosa of Inflammatory Bowel Disease Subjects. Front. Cell. Infect. Microbiol. 2018, 8, 281. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Medina, M.; Denizot, J.; Dreux, N.; Robin, F.; Billard, E.; Bonnet, R.; Darfeuille-Michaud, A.; Barnich, N. Western diet induces dysbiosis with increased E coli in CEABAC10 mice, alters host barrier function favouring AIEC colonisation. Gut 2014, 63, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Agus, A.; Denizot, J.; Thevenot, J.; Martinez-Medina, M.; Massier, S.; Sauvanet, P.; Bernalier-Donadille, A.; Denis, S.; Hofman, P.; Bonnet, R.; et al. Western diet induces a shift in microbiota composition enhancing susceptibility to Adherent-Invasive E. coli infection and intestinal inflammation. Sci. Rep. 2016, 6, 19032. [Google Scholar] [CrossRef] [Green Version]
- Thibault, R.; De Coppet, P.; Daly, K.; Bourreille, A.; Cuff, M.; Bonnet, C.; Mosnier, J.F.; Galmiche, J.P.; Shirazi-Beechey, S.; Segain, J.P. Down-regulation of the monocarboxylate transporter 1 is involved in butyrate deficiency during intestinal inflammation. Gastroenterology 2007, 133, 1916–1927. [Google Scholar] [CrossRef]
- De Preter, V.; Arijs, I.; Windey, K.; Vanhove, W.; Vermeire, S.; Schuit, F.; Rutgeerts, P.; Verbeke, K. Impaired butyrate oxidation in ulcerative colitis is due to decreased butyrate uptake and a defect in the oxidation pathway. Inflamm. Bowel Dis. 2012, 18, 1127–1136. [Google Scholar] [CrossRef]
- Drouet, M.; Dubuquoy, L.; Desreumaux, P.; Bertin, B. Visceral fat and gut inflammation. Nutrition 2012, 28, 113–117. [Google Scholar] [CrossRef]
- Ha, C.W.; Martin, A.; Sepich-Poore, G.D.; Shi, B.; Wang, Y.; Gouin, K.; Humphrey, G.; Sanders, K.; Ratnayake, Y.; Chan, K.S.; et al. Translocation of Viable Gut Microbiota to Mesenteric Adipose Drives Formation of Creeping Fat in Humans. Cell 2020, 183, 666–683.e17. [Google Scholar] [CrossRef]
- Amar, J.; Chabo, C.; Waget, A.; Klopp, P.; Vachoux, C.; Bermúdez-Humarán, L.G.; Smirnova, N.; Bergé, M.; Sulpice, T.; Lahtinen, S.; et al. Intestinal mucosal adherence and translocation of commensal bacteria at the early onset of type 2 diabetes: Molecular mechanisms and probiotic treatment. EMBO Mol. Med. 2011, 3, 559–572. [Google Scholar] [CrossRef]
- He, Z.; Wu, J.; Gong, J.; Ke, J.; Ding, T.; Zhao, W.; Cheng, W.M.; Luo, Z.; He, Q.; Zeng, W.; et al. Microbiota in mesenteric adipose tissue from Crohn’s disease promote colitis in mice. Microbiome 2021, 9, 228. [Google Scholar] [CrossRef]
- Gummesson, A.; Carlsson, L.M.; Storlien, L.H.; Bäckhed, F.; Lundin, P.; Löfgren, L.; Stenlöf, K.; Lam, Y.Y.; Fagerberg, B.; Carlsson, B. Intestinal Permeability Is Associated With Visceral Adiposity in Healthy Women. Obesity 2011, 19, 2280–2282. [Google Scholar] [CrossRef]
- Massier, L.; Chakaroun, R.; Tabei, S.; Crane, A.; Didt, K.D.; Fallmann, J.; Von Bergen, M.; Haange, S.-B.; Heyne, H.; Stumvoll, M.; et al. Adipose tissue derived bacteria are associated with inflammation in obesity and type 2 diabetes. Gut 2020, 69, 1796–1806. [Google Scholar] [CrossRef]
- Zulian, A.; Cancello, R.; Cesana, E.; Rizzi, E.; Consolandi, C.; Severgnini, M.; Panizzo, V.; Di Blasio, A.M.; Micheletto, G.; Invitti, C. Adipose tissue microbiota in humans: An open issue. Int. J. Obes. 2016, 40, 1643–1648. [Google Scholar] [CrossRef]
- Munkholm, P. Review article: The incidence and prevalence of colorectal cancer in inflammatory bowel disease. Aliment. Pharmacol. Ther. 2003, 18, 1–5. [Google Scholar] [CrossRef]
- Long, A.G.; Lundsmith, E.T.; Hamilton, K.E. Inflammation and Colorectal Cancer. Curr. Color. Cancer Rep. 2017, 13, 341–351. [Google Scholar] [CrossRef]
- De Marzo, A.M.; Platz, E.A.; Sutcliffe, S.; Xu, J.; Grönberg, H.; Drake, C.G.; Nakai, Y.; Isaacs, W.B.; Nelson, W.G. Inflammation in prostate carcinogenesis. Nat. Cancer 2007, 7, 256–269. [Google Scholar] [CrossRef] [Green Version]
- Poullis, A.; Foster, R.; Northfield, T.C.; Mendall, M.A. Faecal markers in the assessment of activity in inflammatory bowel disease. Aliment. Pharmacol. Ther. 2002, 16, 675–681. [Google Scholar] [CrossRef]
- Røseth, A.G.; Aadland, E.; Jahnsen, J.; Raknerud, N. Assessment of Disease Activity in Ulcerative Colitis by Faecal Calprotectin, a Novel Granulocyte Marker Protein. Digestion 1997, 58, 176–180. [Google Scholar] [CrossRef]
- Klampfer, L. Cytokines, Inflammation and Colon Cancer. Curr. Cancer Drug Targets 2011, 11, 451–464. [Google Scholar] [CrossRef]
- Kojima, M.; Morisaki, T.; Sasaki, N.; Nakano, K.; Mibu, R.; Tanaka, M.; Katano, M. Increased nuclear factor-kB activation in human colorectal carcinoma and its correlation with tumor progression. Anticancer. Res. 2004, 24, 675–681. [Google Scholar] [PubMed]
- Ullman, T.A.; Itzkowitz, S.H. Intestinal Inflammation and Cancer. Gastroenterology 2011, 140, 1807–1816.e1. [Google Scholar] [CrossRef] [PubMed]
- Klaunig, E.J.; Xu, Y.; Isenberg, J.S.; Bachowski, S.; Kolaja, K.L.; Jiang, J.; Stevenson, E.D.; Walborg, E.F. The role of oxidative stress in chemical carcinogenesis. Environ. Health Perspect. 1998, 106, 289–295. [Google Scholar] [CrossRef]
- Kimura, H.; Hokari, R.; Miura, S.; Shigematsu, T.; Hirokawa, M.; Akiba, Y.; Kurose, I.; Higuchi, H.; Fujimori, H.; Tsuzuki, Y.; et al. Increased expression of an inducible isoform of nitric oxide synthase and the formation of peroxynitrite in colonic mucosa of patients with active ulcerative colitis. Gut 1998, 42, 180–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rachmilewitz, D.; Stamler, J.S.; Bachwich, D.; Karmeli, F.; Ackerman, Z.; Podolsky, D.K. Enhanced colonic nitric oxide generation and nitric oxide synthase activity in ulcerative colitis and Crohn’s disease. Gut 1995, 36, 718–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Q.; Cang, S.; Jiao, J.; Rong, W.; Xu, H.; Bi, K.; Li, Q.; Liu, R. Integrated study of metabolomics and gut metabolic activity from ulcerative colitis to colorectal cancer: The combined action of disordered gut microbiota and linoleic acid metabolic pathway might fuel cancer. J. Chromatogr. A 2020, 1629, 461503. [Google Scholar] [CrossRef]
- Phinney, S.D. Metabolism of Exogenous and Endogenous Arachidonic Acid in Cancer. Adv. Exp. Med. Biol. 1996, 399, 87–94. [Google Scholar] [CrossRef]
- Ahmed, I.; Umar, S. Microbiome and Colorectal Cancer. Curr. Color. Cancer Rep. 2018, 14, 217–225. [Google Scholar] [CrossRef]
- Kang, M.; Martin, A. Microbiome and colorectal cancer: Unraveling host-microbiota interactions in colitis-associated colorectal cancer development. Semin. Immunol. 2017, 32, 3–13. [Google Scholar] [CrossRef]
- McCoy, A.N.; Araújo-Pérez, F.; Azcarate-Peril, M.A.; Yeh, J.J.; Sandler, R.S.; Keku, T.O. Fusobacterium Is Associated with Colorectal Adenomas. PLoS ONE 2013, 8, e53653. [Google Scholar] [CrossRef]
- Arthur, J.C.; Perez-Chanona, E.; Mühlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.-J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal Inflammation Targets Cancer-Inducing Activity of the Microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef] [Green Version]
- Dai, Z. Obesity and colorectal cancer risk: A meta-analysis of cohort studies. World J. Gastroenterol. 2007, 13, 4199–4206. [Google Scholar] [CrossRef]
- Wunderlich, C.M.; Ackermann, P.J.; Ostermann, A.L.; Adams-Quack, P.; Vogt, M.C.; Tran, M.-L.; Nikolajev, A.; Waisman, A.; Garbers, C.; Theurich, S.; et al. Obesity exacerbates colitis-associated cancer via IL-6-regulated macrophage polarisation and CCL-20/CCR-6-mediated lymphocyte recruitment. Nat. Commun. 2018, 9, 1646. [Google Scholar] [CrossRef]
- Liu, P.-H.; Wu, K.; Ng, K.; Zauber, A.G.; Nguyen, L.; Song, M.; He, X.; Fuchs, C.S.; Ogino, S.; Willett, W.C.; et al. Association of Obesity With Risk of Early-Onset Colorectal Cancer Among Women. JAMA Oncol. 2019, 5, 37–44. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Yang, Y.; Wang, F.; Zhang, P.; Shi, C.; Zou, Y.; Qin, H. Obesity and Risk of Colorectal Cancer: A Systematic Review of Prospective Studies. PLoS ONE 2013, 8, e53916. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Fan, W.; Luo, B.; Xu, Z.; Wang, P.; Tang, S.; Xu, P.; Yu, M. Circulating Resistin Levels and Risk of Colorectal Cancer: A Meta-Analysis. BioMed Res. Int. 2016, 2016, 7367485. [Google Scholar] [CrossRef] [Green Version]
- Campisciano, G.; De Manzini, N.; Delbue, S.; Cason, C.; Cosola, D.; Basile, G.; Ferrante, P.; Comar, M.; Palmisano, S. The Obesity-Related Gut Bacterial and Viral Dysbiosis Can Impact the Risk of Colon Cancer Development. Microorganisms 2020, 8, 431. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
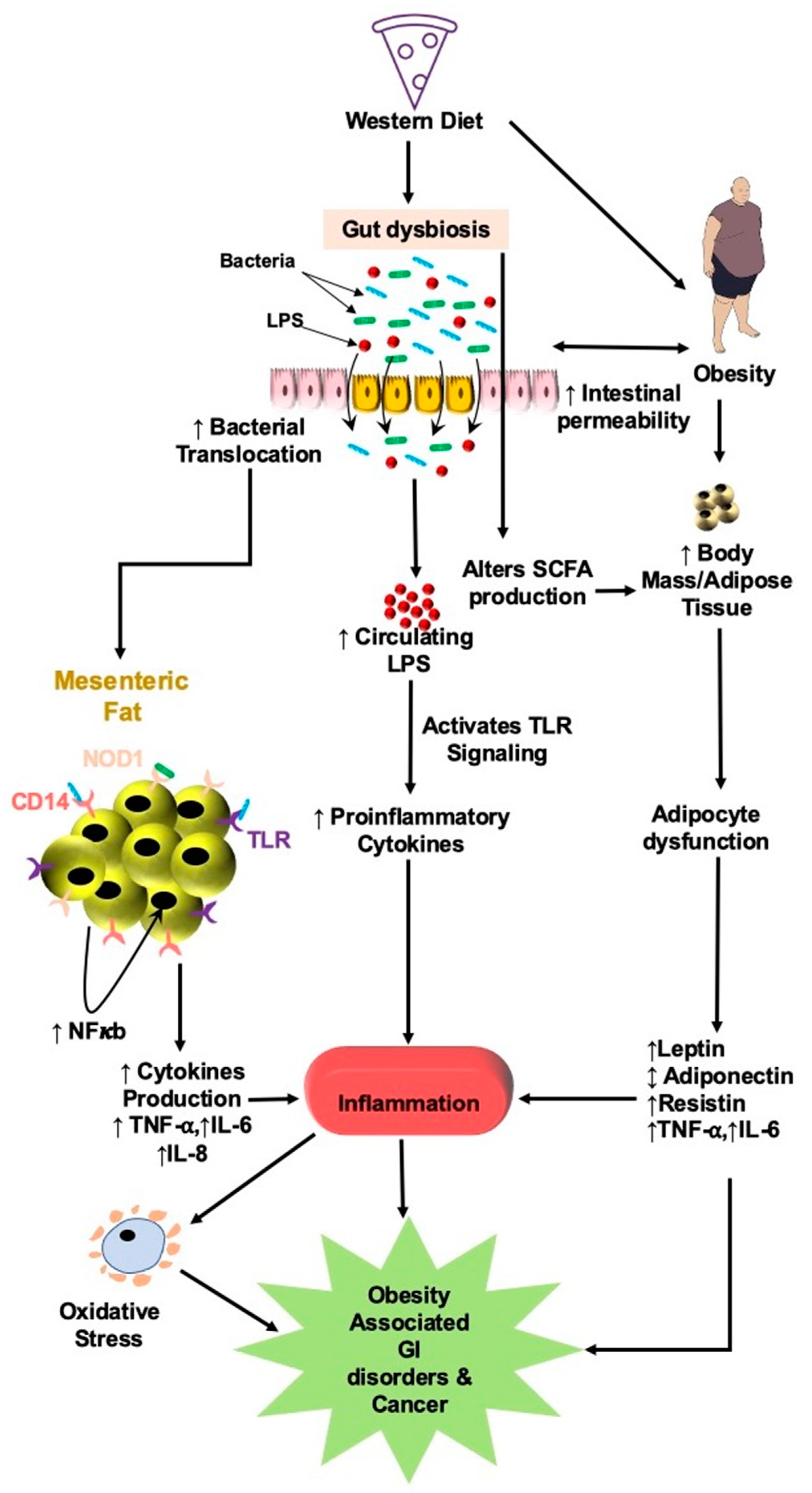{kind=link}
| Name | Lipid No. | Major Producers | References | |
|---|---|---|---|---|
| Acetate | C2:0 | Lactobacillus spp. *, Bifidobacterium spp., Akkermansia muciniphila, Bacteroides spp., Prevotella spp., Ruminococcus spp., Streptococcus spp. | [43,44] | |
| Major SCFAs | Propionate | C3:0 | Phascolarctobacterium succinatutens, Akkermansia muciniphila, Bacteroides spp., Dialister spp.,Megasphaera elsdenii, Veillonella spp., Coprococcus catus, Roseburia inulinivorans, Ruminococcus obeum, Salmonella spp. | [45,46] |
| Butyrate | C4:0 | Faecalibacterium prausnitzii, Clostridium leptum, Eubacterium rectale, Roseburia spp. | [47] | |
| Minor SCFAs | Formate | C1:0 | Bifidobacterium spp., Prevotella spp., Parabacteroides spp., Bacteroides spp., Alistipes spp., Eubacterium spp., Erysipelatoclostridium spp., Blautia (Clostridium cluster XIVa) spp., Coprococcus, Dorea, Roseburia (Clostridium cluster XIVa) spp., Lactobacillus spp., Faecalibacterium (Clostridium cluster IV) spp., Ruminococcus (Clostridium cluster IV) spp., Streptococcus spp., Veillonella spp., Escherichia spp. | [48] |
| Valerate | C5:0 | Clostridium (Clostridium cluster I) spp. | [48] |
| Transporter | Species | Cell/Tissue-Type | Localization | References |
|---|---|---|---|---|
| MCT1 | Hamster | IECs | Basolateral membrane | [58] |
| Human, Rat, Pig | IECs | Apical membrane | [59,60,61] | |
| Human, Mouse, Rat | Enterocytes | Basolateral membrane | [62] | |
| MCT2 | Hamster | Parietal Cells | Unknown | [62] |
| MCT3 | Human | Ileum, Colon | Basolateral membrane | [60,62,63] |
| MCT4 | Rat | IEC-cell line | Apical membrane | [62,64] |
| Mouse | IECs (Villus and Crypts) | Basolateral membrane | [62,64] | |
| Human | Ileum, Colon | Basolateral membrane | [62,64] |
| SCFAs | Absorption | Site of Utilization | Function | References |
|---|---|---|---|---|
| Butyrate | Colonocytes | Colon | Differentiation and proliferation of colonocytes | [86] |
| Acetate | Proximal Colon | Liver | Regulation of appetite, body weight, and cholesterol synthesis | [89] |
| Propionate | Colonocytes | Liver | Used for gluconeogenesis, lipogenesis, and protein synthesis | [41,45] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Islam, M.R.; Arthur, S.; Haynes, J.; Butts, M.R.; Nepal, N.; Sundaram, U. The Role of Gut Microbiota and Metabolites in Obesity-Associated Chronic Gastrointestinal Disorders. Nutrients 2022, 14, 624. https://0-doi-org.brum.beds.ac.uk/10.3390/nu14030624
Islam MR, Arthur S, Haynes J, Butts MR, Nepal N, Sundaram U. The Role of Gut Microbiota and Metabolites in Obesity-Associated Chronic Gastrointestinal Disorders. Nutrients. 2022; 14(3):624. https://0-doi-org.brum.beds.ac.uk/10.3390/nu14030624
Chicago/Turabian StyleIslam, Maafi R., Subha Arthur, Jennifer Haynes, Molly R. Butts, Niraj Nepal, and Uma Sundaram. 2022. "The Role of Gut Microbiota and Metabolites in Obesity-Associated Chronic Gastrointestinal Disorders" Nutrients 14, no. 3: 624. https://0-doi-org.brum.beds.ac.uk/10.3390/nu14030624