
Elucidating the Local Transmission Dynamics of Highly Pathogenic Avian Influenza H5N6 in the Republic of Korea by Integrating Phylogenetic Information

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Source
2.2. Spatiotemporal Cluster Analyses
2.3. Estimation of Spatial Transmission Risk
2.4. Identification of Factors Associated with the Local Transmissibility of Highly Pathogenic Avian Influenza (HPAI)
2.5. Simulation of Ring Culling Radii
3. Results
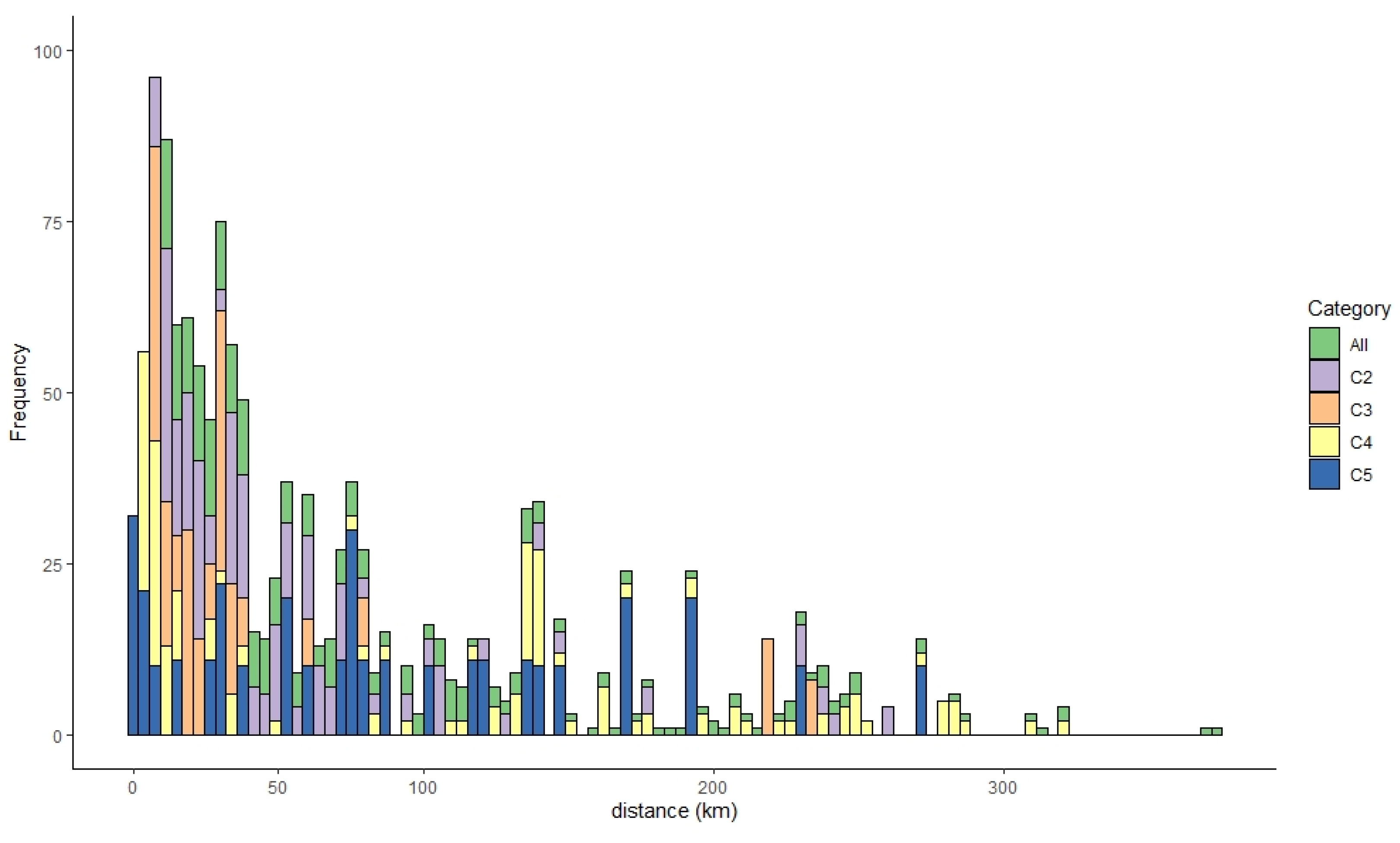
3.1. Spatiotemporal Characteristics of the HPAI H5N6 Epidemic
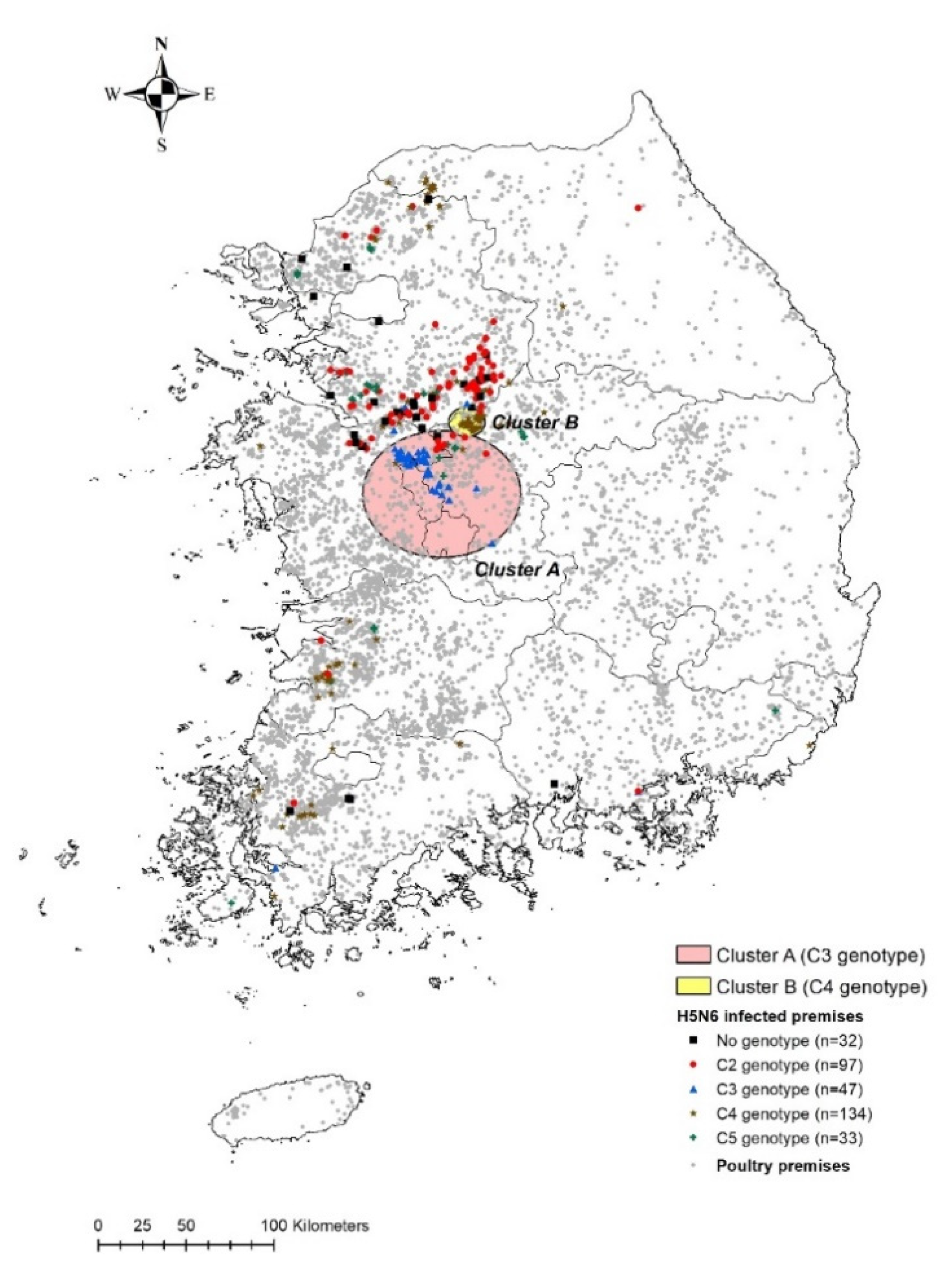
3.2. Cluster Analyses
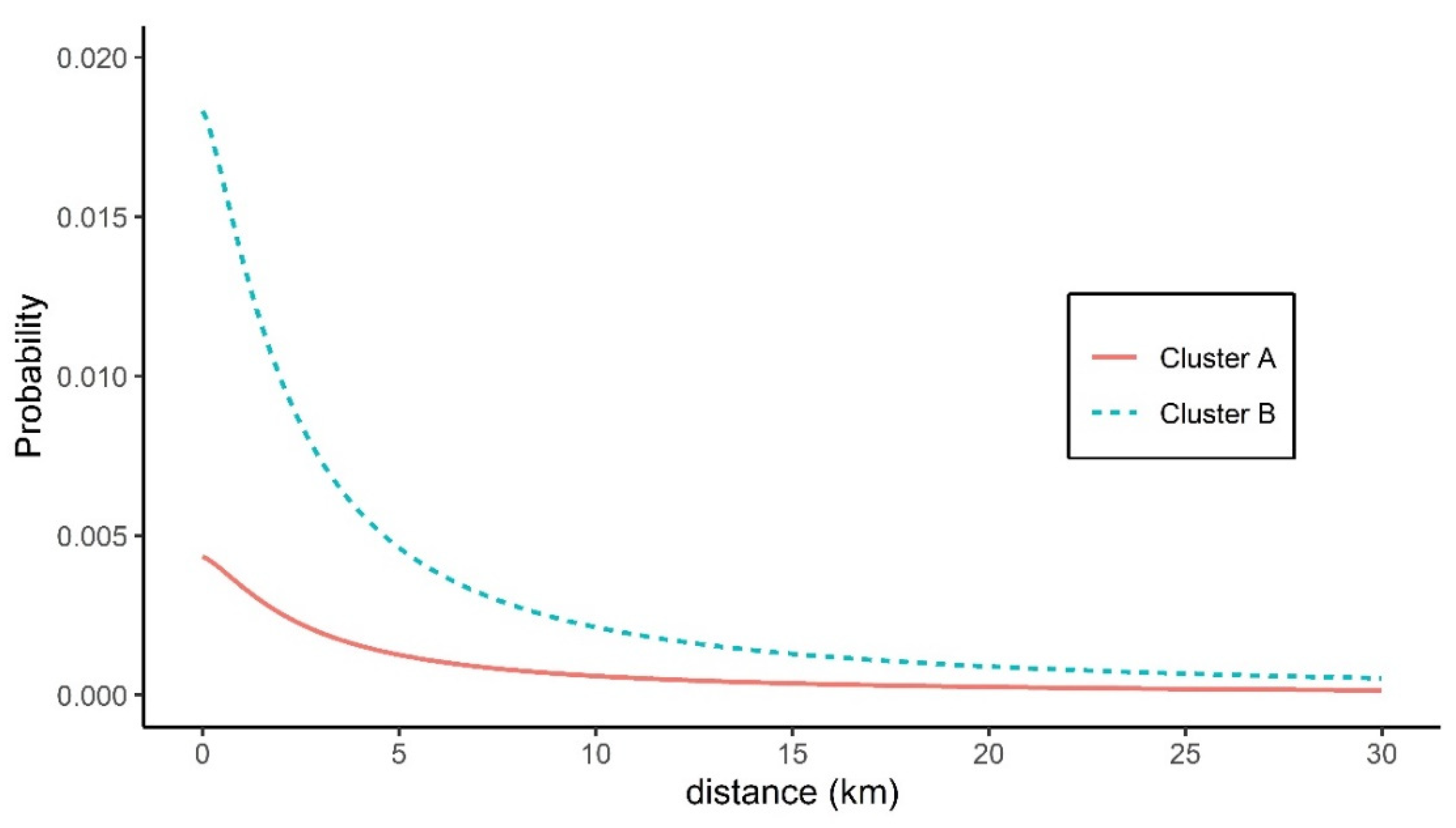
3.3. Spatial Transmission Kernel Estimates
3.4. Factors Associated with the Local Transmissibility of HPAI
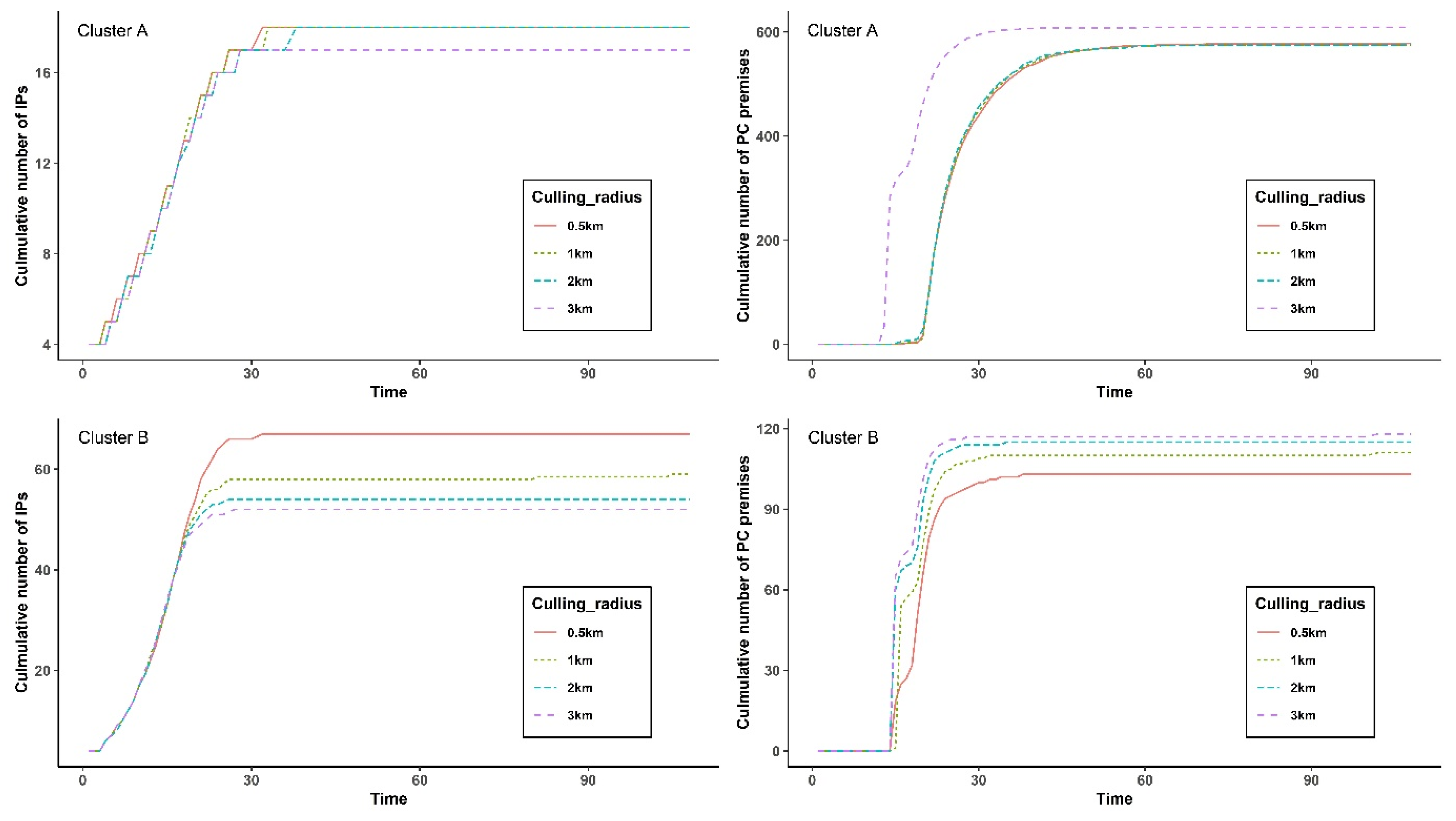
3.5. Simulations with Different Preemptive Culling Radii
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boyce, W.M.; Sandrock, C.; Kreuder-Johnson, C.; Kelly, T.; Cardona, C. Avian influenza viruses in wild birds: A moving target. Comp. Immunol. Microbiol. Infect. Dis. 2009, 32, 275–286. [Google Scholar] [CrossRef]
- Heo, D.; Woo, H.J.; Hwang, Y.J.; Lee, Y.G.; Kim, T.R. The Analysis of Response Strategy against Highly Pathogenic Avian Influenza (AI) Epidemic during the 2017–2018 in the Republic of Korea; Korea Rural Economical Institute: Naju, Korea, 2020. [Google Scholar]
- Auty, H.; Mellor, D.; Gunn, G.; Boden, L.A. The Risk of Foot and Mouth Disease Transmission Posed by Public Access to the Countryside during an Outbreak. Front. Vet. Sci. 2019, 6, 381. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Ko, Y.; Jung, E. Effective control measures considering spatial heterogeneity to mitigate the 2016–2017 avian influenza epidemic in the Republic of Korea. PLoS ONE 2019, 14, e0218202. [Google Scholar] [CrossRef]
- Nielsen, S.S.; Alvarez, J.; Bicout, D.J.; Calistri, P.; Depner, K.; Drewe, J.A.; Garin-Bastuji, B.; Gonzales Rojas, J.L.; Schmidt, C.G.; Herskin, M.; et al. Scientific Opinion on the assessment of the control measures of the category A diseases of Animal Health Law: Highly Pathogenic Avian Influenza. EFSA J. 2021, 19, e06372. [Google Scholar]
- Boender, G.J.; van Roermund, H.J.; de Jong, M.C.; Hagenaars, T.J. Transmission risks and control of foot-and-mouth disease in The Netherlands: Spatial patterns. Epidemics 2010, 2, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Takemae, N.; Tsunekuni, R.; Sharshov, K.; Tanikawa, T.; Uchida, Y.; Ito, H.; Soda, K.; Usui, T.; Sobolev, I.; Shestopalov, A.; et al. Five distinct reassortants of H5N6 highly pathogenic avian influenza A viruses affected Japan during the winter of 2016–2017. Virology 2017, 512, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Dorigatti, I.; Mulatti, P.; Rosa, R.; Pugliese, A.; Busani, L. Modelling the spatial spread of H7N1 avian influenza virus among poultry farms in Italy. Epidemics 2010, 2, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Salvador, R.; Tanquilut, N.; Na Lampang, K.; Chaisowwong, W.; Pfeiffer, D.; Punyapornwithaya, V. Identification of High-Risk Areas for the Spread of Highly Pathogenic Avian Influenza in Central Luzon, Philippines. Vet. Sci. 2020, 7, 107. [Google Scholar] [CrossRef] [PubMed]
- Bonney, P.J.; Malladi, S.; Boender, G.J.; Weaver, J.T.; Ssematimba, A.; Halvorson, D.A.; Cardona, C.J. Spatial transmission of H5N2 highly pathogenic avian influenza between Minnesota poultry premises during the 2015 outbreak. PLoS ONE 2018, 13, e0204262. [Google Scholar] [CrossRef] [Green Version]
- Firestone, S.M.; Hayama, Y.; Lau, M.S.Y.; Yamamoto, T.; Nishi, T.; Bradhurst, R.A.; Demirhan, H.; Stevenson, M.A.; Tsutsui, T. Transmission network reconstruction for foot-and-mouth disease outbreaks incorporating farm-level covariates. PLoS ONE 2020, 15, e0235660. [Google Scholar] [CrossRef] [PubMed]
- De Maio, N.; Wu, C.H.; Wilson, D.J. SCOTTI: Efficient Reconstruction of Transmission within Outbreaks with the Structured Coalescent. PLoS Comput. Biol. 2016, 12, e1005130. [Google Scholar] [CrossRef]
- Mulatti, P.; Fusaro, A.; Scolamacchia, F.; Zecchin, B.; Azzolini, A.; Zamperin, G.; Terregino, C.; Cunial, G.; Monne, I.; Marangon, S. Integration of genetic and epidemiological data to infer H5N8 HPAI virus transmission dynamics during the 2016-2017 epidemic in Italy. Sci. Rep. 2018, 8, 18037. [Google Scholar] [CrossRef] [Green Version]
- World Organisation for Animal Health Organization. OIE Terrestrial Manual; World Organisation for Animal Health Organization: Paris, France, 2018; pp. 821–843. [Google Scholar]
- Epidemiology report for 2016-2017 Outbreak of Highly Pathogen-Ic Avian Influenza (HPAI) in Republic of Korea; Division of Veterinary Epidemiology, Animal and Plant Quaranitne Agency, Ministry of Agriculture, Food and Rural Affairs: Gimcheon, Korea, 2017; Volume 1, pp. 19, 161–162.
- Lee, E.-K.; Song, B.-M.; Lee, Y.-N.; Heo, G.-B.; Bae, Y.-C.; Joh, S.-J.; Park, S.-C.; Choi, K.-S.; Lee, H.-J.; Jang, I. Multiple novel H5N6 highly pathogenic avian influenza viruses, South Korea, 2016. Infect. Genet. Evol. 2017, 51, 21–23. [Google Scholar] [CrossRef] [PubMed]
- Si, Y.J.; Lee, I.W.; Kim, E.H.; Kim, Y.I.; Kwon, H.I.; Park, S.J.; Nguyen, H.D.; Kim, S.M.; Kwon, J.J.; Choi, W.S.; et al. Genetic characterisation of novel, highly pathogenic avian influenza (HPAI) H5N6 viruses isolated in birds, South Korea, November 2016. Euro Surveill 2017, 22, 30434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, I.; Kulldorff, M.; Richard, O.J. A spatial scan statistic for multinomial data. Stat. Med. 2010, 29, 1910–1918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, H.; Jung, I. Optimizing the maximum reported cluster size for normal-based spatial scan statistics. Commun. Stat. Appl. Methods 2018, 25, 373–383. [Google Scholar] [CrossRef]
- Swayne, D.E. Animal Influenza; John Wiley & Sons: Hoboken, NJ, USA, 2016. [Google Scholar]
- Kulldorff, M. SaTScan™ User Guide; Harvard Medical School and Harvard Pilgrim Health Care Institute: Boston, MA, USA, 2018; Available online: https://www.satscan.org/ (accessed on 25 January 2020).
- Salvador, R.; Tanquilut, N.; Macmac, R.; Na Lampang, K.; Chaisowwong, W.; Pfeiffer, D.; Punyapornwithaya, V. Evaluation of strategies using simulation model to control a potential outbreak of highly pathogenic avian influenza among poultry farms in Central Luzon, Philippines. PLoS ONE 2020, 15, e0238815. [Google Scholar] [CrossRef] [PubMed]
- Bolker, B.; Team, R. bbmle: Tools for General Maximum Likelihood Estimation, R package version 0.9; University of Florida: Gainesville, FL, USA, 2010; Available online: https://github.com/bbolker/bbmle (accessed on 25 January 2020).
- Kuznetsova, A.; Brockhoff, P.B.; Christensen, R.H.B. Package ‘Lmertest’, R package version; Technical University of Denmark: Kongens Lyngby, Denmark, 2015; Available online: https://github.com/runehaubo/lmerTestR (accessed on 25 January 2020).
- Guinat, C.; Nicolas, G.; Vergne, T.; Bronner, A.; Durand, B.; Courcoul, A.; Gilbert, M.; Guerin, J.L.; Paul, M.C. Spatio-temporal patterns of highly pathogenic avian influenza virus subtype H5N8 spread, France, 2016 to 2017. Euro Surveill 2018, 23, 1700791. [Google Scholar] [CrossRef]
- Kim, W.H.; An, J.U.; Kim, J.; Moon, O.K.; Bae, S.H.; Bender, J.B.; Cho, S. Risk factors associated with highly pathogenic avian influenza subtype H5N8 outbreaks on broiler duck farms in South Korea. Transbound. Emerg. Dis. 2018, 65, 1329–1338. [Google Scholar] [CrossRef]
- Kwon, J.H.; Bahl, J.; Swayne, D.E.; Lee, Y.N.; Lee, Y.J.; Song, C.S.; Lee, D.H. Domestic ducks play a major role in the maintenance and spread of H5N8 highly pathogenic avian influenza viruses in South Korea. Transbound. Emerg. Dis. 2020, 67, 844–851. [Google Scholar] [CrossRef]
- So, H.H.; Bae, Y.; Mo, I. Risk Assessment of Highly Pathogenic Avian Influenza by Comparison of Biosecurity Level in Domestic Poultry Farms. Korean J. Poult. Sci. 2019, 46, 313–325. [Google Scholar] [CrossRef] [Green Version]
- Boender, G.J.; Hagenaars, T.J.; Bouma, A.; Nodelijk, G.; Elbers, A.R.; de Jong, M.C.; van Boven, M. Risk maps for the spread of highly pathogenic avian influenza in poultry. PLoS Comput. Biol. 2007, 3, e71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Jong, M.C.; Stegeman, A.; van der Goot, J.; Koch, G. Intra- and interspecies transmission of H7N7 highly pathogenic avian influenza virus during the avian influenza epidemic in The Netherlands in 2003. Rev. Sci. Tech. 2009, 28, 333–340. [Google Scholar] [CrossRef] [Green Version]
- Stegeman, A.; Bouma, A.; Elbers, A.R.; de Jong, M.C.; Nodelijk, G.; de Klerk, F.; Koch, G.; van Boven, M. Avian influenza A virus (H7N7) epidemic in The Netherlands in 2003: Course of the epidemic and effectiveness of control measures. J. Infect. Dis. 2004, 190, 2088–2095. [Google Scholar] [CrossRef] [PubMed]
- Marangon, S.; Capua, I.; Pozza, G.; Santucci, U. Field experiences in the control of avian influenza outbreaks in densely populated poultry areas. Dev. Biol. 2004, 119, 155–164. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | Distance between IPs (km) Mean (95% CI) | Outbreak Duration (Start Date, End Date) | No. of Cases |
|---|---|---|---|
| All | 66.71 (0.15, 272.43) | 108 days (16 November 2016, 3 March 2017) | 343 |
| C2 genotype | 50.93 (0.24, 234.88) | 64 days (29 November 2016, 21 January 2017) | 97 |
| C3 genotype | 29.36 (0.67, 218.29) | 51 days (16 November 2016, 5 January 2017) | 58 |
| C4 genotype | 75.72 (0.10, 283.46) | 86 days (16 November 2016, 9 February 2017) | 134 |
| C5 genotype | 50.93 (0.40, 240.37) | 45 days (20 November 2016, 3 February 2017) | 33 |
| Characteristics | Cluster A | Cluster B |
|---|---|---|
| Radius (km) | 36.11 | 8.32 |
| Duration (from start date to end date) | 26 November 2016–6 January 2017 | 16 November 2016–16 December 2016 |
| Total no. of IPs in the cluster | 58 | 48 |
| No. of IPs infected with the clustered genotype | 42 | 48 |
| RR associated with each genotype (C2, C3, C4, C5, N/A) | (0.44, 41.28, 0.074, 0.68, 0.33) | (0, 0, 3.43, 0, 0) |
| LLR | 82.26 | 51.45 |
| p-value | 0.001 | 0.001 |
| Cluster (Genotype) | h0 | r0 | α | AIC |
|---|---|---|---|---|
| Cluster A (C3) | 0.00062 (0.00042, 0.00082) | 2.603 (2.350, 2.857) | 1.363 (0.928, 1.797) | 633.99 |
| Cluster B (C4) | 0.00262 (0.00161, 0.00363) | 2.246 (0.555, 3.936) | 1.358 (0.351, 2.365) | 483.64 |
| Variable (Unit) | Coefficient Estimates | p-Value | |
|---|---|---|---|
| Mean | 95% CI Lower, Upper | ||
| Flock size (10,000 head) | −0.004 | −0.011, 0.004 | 0.349 |
| Density of duck farms (n/km2) | 0.237 | 0.177, 0.306 | <0.001 |
| Density of chicken farms (n/km2) | −0.146 | −0.300, −0.003 | 0.048 |
| Human population density (inhabitants/km2/100) | 0.001 | −0.016, 0.017 | 0.885 |
| Minimum distance to driveway from a farm (km) | −0.156 | −0.479, 0.174 | 0.359 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoo, D.-S.; Chun, B.; Min, K.-D.; Lim, J.-S.; Moon, O.-K.; Lee, K.-N. Elucidating the Local Transmission Dynamics of Highly Pathogenic Avian Influenza H5N6 in the Republic of Korea by Integrating Phylogenetic Information. Pathogens 2021, 10, 691. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10060691
Yoo D-S, Chun B, Min K-D, Lim J-S, Moon O-K, Lee K-N. Elucidating the Local Transmission Dynamics of Highly Pathogenic Avian Influenza H5N6 in the Republic of Korea by Integrating Phylogenetic Information. Pathogens. 2021; 10(6):691. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10060691
Chicago/Turabian StyleYoo, Dae-Sung, Byungchul Chun, Kyung-Duk Min, Jun-Sik Lim, Oun-Kyoung Moon, and Kwang-Nyeong Lee. 2021. "Elucidating the Local Transmission Dynamics of Highly Pathogenic Avian Influenza H5N6 in the Republic of Korea by Integrating Phylogenetic Information" Pathogens 10, no. 6: 691. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10060691