
Vesicular Stomatitis Virus: From Agricultural Pathogen to Vaccine Vector

 ,
,
Abstract
:
{kind=link}
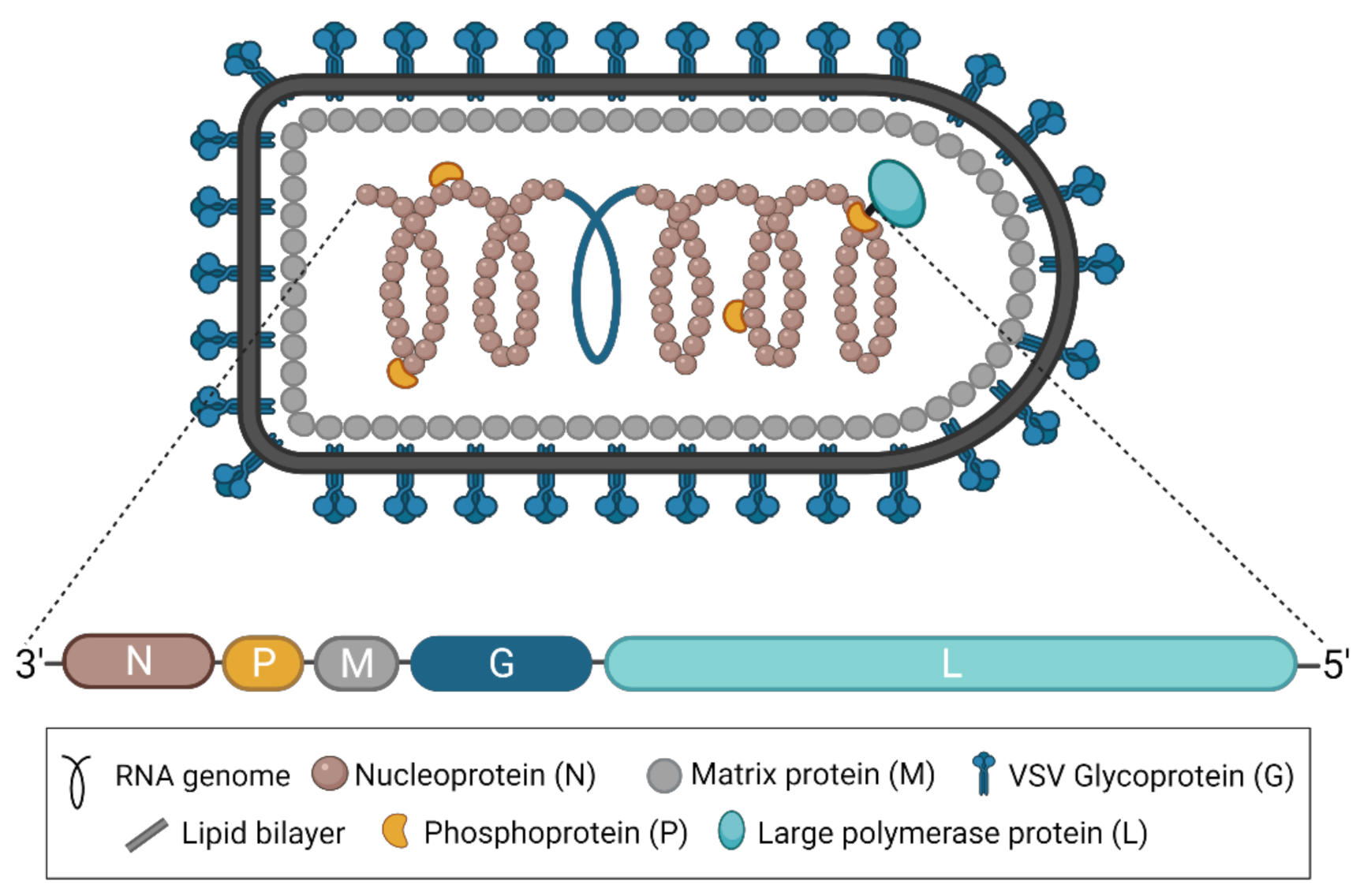{kind=link}
{kind=link}
1. Vesicular Stomatitis Virus
1.1. The Virus and Its Replication
1.2. The Disease: Vesicular Stomatitis
2. VSV and Molecular Virology
2.1. VSV Reverse Genetics
2.2. VSV as a Molecular Tool
3. VSV as a Vaccine Vector
3.1. Ebola Virus
3.1.1. Pre-Clinical Development
3.1.2. Clinical Trials and Approval
3.1.3. Mechanisms of Protection
3.1.4. Challenges
3.2. Marburg Virus and Other Filoviruses
3.3. Lassa Virus
3.4. Henipaviruses
3.5. Coronaviruses
3.6. Zika Virus
3.7. Crimean-Congo Hemorrhagic Fever Virus
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Walker, P.J.; Blasdell, K.R.; Calisher, C.H.; Dietzgen, R.G.; Kondo, H.; Kurath, G.; Longdon, B.; Stone, D.M.; Tesh, R.B.; Tordo, N.; et al. ICTV Virus Taxonomy Profile: Rhabdoviridae. J. Gen. Virol. 2018, 99, 447–448. [Google Scholar] [CrossRef] [PubMed]
- Walker, P.J.; Firth, C.; Widen, S.G.; Blasdell, K.R.; Guzman, H.; Wood, T.G.; Paradkar, P.N.; Holmes, E.C.; Tesh, R.B.; Vasilakis, N. Evolution of Genome Size and Complexity in the Rhabdoviridae. PLoS Pathog. 2015, 11, e1004664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, L.L.; Pauszek, S.J.; Bunch, T.A.; Schumann, K.R. Full-length genome analysis of natural isolates of vesicular stomatitis virus (Indiana 1 serotype) from North, Central and South America. J. Gen. Virol. 2002, 83, 2475–2483. [Google Scholar] [CrossRef] [PubMed]
- Peluso, R.W.; Richardson, J.C.; Talon, J.; Lock, M. Identification of a set of proteins (C′ and C) encoded by the bicistronic P gene of the Indiana serotype of vesicular stomatitis virus and analysis of their effect on transcription by the viral RNA polymerase. Virology 1996, 218, 335–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiropoulou, C.F.; Nichol, S.T. A small highly basic protein is encoded in overlapping frame within the P gene of vesicular stomatitis virus. J. Virol. 1993, 67, 3103–3110. [Google Scholar] [CrossRef] [Green Version]
- Ge, P.; Tsao, J.; Schein, S.; Green, T.J.; Luo, M.; Zhou, Z.H. Cryo-EM model of the bullet-shaped vesicular stomatitis virus. Science 2010, 327, 689–693. [Google Scholar] [CrossRef] [Green Version]
- Riedel, C.; Hennrich, A.A.; Conzelmann, K.K. Components and Architecture of the Rhabdovirus Ribonucleoprotein Complex. Viruses 2020, 12, 959. [Google Scholar] [CrossRef]
- Thomas, D.; Newcomb, W.W.; Brown, J.C.; Wall, J.S.; Hainfeld, J.F.; Trus, B.L.; Steven, A.C. Mass and molecular composition of vesicular stomatitis virus: A scanning transmission electron microscopy analysis. J. Virol. 1985, 54, 598–607. [Google Scholar] [CrossRef] [Green Version]
- Green, T.J.; Zhang, X.; Wartz, G.W.; Luo, M. Structure of the vesicular stomatitis virus nucleoprotein-RNA complex. Science 2006, 313, 357–360. [Google Scholar] [CrossRef]
- Liang, B.; Li, Z.; Jenni, S.; Rahmeh, A.A.; Morin, B.M.; Grant, T.; Grigorieff, N.; Harrison, S.C.; Whelan, S.P.J. Structure of the L Protein of Vesicular Stomatitis Virus from Electron Cryomicroscopy. Cell 2015, 162, 314–327. [Google Scholar] [CrossRef] [Green Version]
- Gould, J.R.; Qiu, S.; Shang, Q.; Ogino, T.; Prevelige, P.E.; Petit, C.M.; Green, T.J. The Connector Domain of Vesicular Stomatitis Virus Large Protein Interacts with the Viral Phosphoprotein. J. Virol. 2020, 94, e01729-19. [Google Scholar] [CrossRef] [PubMed]
- Leyrat, C.; Yabukarski, F.; Tarbouriech, N.; Ribeiro, E.A.; Jensen, M.R.; Blackledge, M.; Ruigrok, R.W.H.; Jamin, M. Structure of the vesicular stomatitis virus n 0-p complex. PLoS Pathog. 2011, 7, e1002248. [Google Scholar] [CrossRef]
- Ding, H.; Green, T.J.; Lu, S.; Luo, M. Crystal Structure of the Oligomerization Domain of the Phosphoprotein of Vesicular Stomatitis Virus. J. Virol. 2006, 80, 2808–2814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, T.J.; Luo, M. Structure of the vesicular stomatitis virus nucleocapsid in complex with the nucleocapsid-binding domain of the small polymerase cofactor, P. Proc. Natl. Acad. Sci. USA 2009, 106, 11713–11718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudier, M.; Gaudin, Y.; Knossow, M. Crystal structure of vesicular stomatitis virus matrix protein. EMBO J. 2002, 21, 2886–2892. [Google Scholar] [CrossRef]
- Newcomb, W.W.; Brown, J.C. Role of the vesicular stomatitis virus matrix protein in maintaining the viral nucleocapsid in the condensed form found in native virions. J. Virol. 1981, 39, 295–299. [Google Scholar] [CrossRef] [Green Version]
- Roche, S.; Rey, F.A.; Gaudin, Y.; Bressanelli, S. Structure of the prefusion form of the vesicular stomatitis virus glycoprotein G. Science 2007, 315, 843–848. [Google Scholar] [CrossRef] [Green Version]
- Roche, S.; Bressanelli, S.; Rey, F.A.; Gaudin, Y. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science 2006, 313, 187–191. [Google Scholar] [CrossRef]
- Banerjee, A.K.; Barik, S. Gene expression of vesicular stomatitis virus genome RNA. Virology 1992, 188, 417–428. [Google Scholar] [CrossRef]
- Lyles, D.S.; Kuzmin, I.V.; Rupprecht, C.E. Rhabdoviridae. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Wolters Kluwer: Philadelphia, PA, USA, 2013; pp. 885–922. [Google Scholar]
- Finkelshtein, D.; Werman, A.; Novick, D.; Barak, S.; Rubinstein, M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc. Natl. Acad. Sci. USA 2013, 110, 7306–7311. [Google Scholar] [CrossRef] [Green Version]
- Nikolic, J.; Belot, L.; Raux, H.; Legrand, P.; Gaudin, Y.; Albertini, A.A. Structural basis for the recognition of LDL-receptor family members by VSV glycoprotein. Nat. Commun. 2018, 9, 1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belot, L.; Albertini, A.; Gaudin, Y. Structural and cellular biology of rhabdovirus entry. Adv. Virus Res. 2019, 104, 147–183. [Google Scholar] [CrossRef] [PubMed]
- Mire, C.E.; White, J.M.; Whitt, M.A. A spatio-temporal analysis of matrix protein and nucleocapsid trafficking during vesicular stomatitis virus uncoating. PLoS Pathog. 2010, 6, e1000994. [Google Scholar] [CrossRef]
- Barr, J.N.; Whelan, S.P.J.; Wertz, G.W. Transcriptional control of the RNA-dependent RNA polymerase of vesicular stomatitis virus. Biochim. Biophys. Acta-Gene Struct. Expr. 2002, 1577, 337–353. [Google Scholar] [CrossRef]
- Arnheiter, H.; Davis, N.L.; Wertz, G.; Schubert, M.; Lazzarini, R.A. Role of the nucleocapsid protein in regulating vesicular stomatitis virus RNA synthesis. Cell 1985, 41, 259–267. [Google Scholar] [CrossRef]
- Lyles, D.S. Assembly and Budding of Negative-Strand RNA Viruses. Adv. Virus Res. 2013, 85, 57–90. [Google Scholar] [CrossRef]
- Fine, S.M. Vesicular Stomatitis Virus and Related Vesiculoviruses. In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases; Bennett, J.E., Dolin, R., Blaser, M.J., Eds.; Elsevier: Philadelphia, PA, USA, 2014; Volume 2, pp. 1981–1983.e1. ISBN 9996096742. [Google Scholar]
- Letchworth, G.J.; Rodriguez, L.L.; Barrera, J.D.C. Vesicular stomatitis. Vet. J. 1999, 157, 239–260. [Google Scholar] [CrossRef]
- Teidebold, T.C.; Mather, C.S.; Merrillat, L.A. Gangrenous glossitis of horses. In Proceedings of the 20th Annual Meeting of the United States Live Stock Sanitary Association, Chicago, IL, USA, 5–7 December 1916; pp. 29–42. [Google Scholar]
- Hanson, R.P. Discussion of the natural history of vesicular stomatitis. Am. J. Epidemiol. 1968, 87, 264–266. [Google Scholar] [CrossRef]
- Cotton, W.E. Vesicular stomatitis. Vet. Med. 1927, 22, 169–175. [Google Scholar]
- Rozo-Lopez, P.; Drolet, B.S.; Londoño-Renteria, B. Vesicular stomatitis virus transmission: A comparison of incriminated vectors. Insects 2018, 9, 190. [Google Scholar] [CrossRef] [Green Version]
- Corn, J.L.; Comer, J.A.; Erickson, G.A.; Nettles, V.F. Isolation of vesicular stomatitis virus New Jersey serotype from phlebotomine sand flies in Georgia. Am. J. Trop. Med. Hyg. 1990, 42, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Fields, B.N.; Hawkins, K. Human Infection with the Virus of Vesicular Stomatitis during an Epizootic. N. Engl. J. Med. 1967, 277, 989–994. [Google Scholar] [CrossRef]
- Munis, A.M.; Bentley, E.M.; Takeuchi, Y. A tool with many applications: Vesicular stomatitis virus in research and medicine. Expert Opin. Biol. Ther. 2020, 20, 1187–1201. [Google Scholar] [CrossRef] [PubMed]
- Schnell, M.J.; Mebatsion, T.; Conzelmann, K.K. Infectious rabies viruses from cloned cDNA. EMBO J. 1994, 13, 4195–4203. [Google Scholar] [CrossRef] [Green Version]
- Whelan, S.P.J.; Ball, L.A.; Barr, J.N.; Wertz, G.T.W. Efficient recovery of infectious vesicular stomatitis virus entirely from cDNA clones. Proc. Natl. Acad. Sci. USA 1995, 92, 8388–8392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawson, N.D.; Stillman, E.A.; Whitt, M.A.; Rose, J.K. Recombinant vesicular stomatitis viruses from DNA. Proc. Natl. Acad. Sci. USA 1995, 92, 4477–4481. [Google Scholar] [CrossRef] [Green Version]
- Pfaller, C.K.; Cattaneo, R.; Schnell, M.J. Reverse genetics of Mononegavirales: How they work, new vaccines, and new cancer therapeutics. Virology 2015, 479–480, 331–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Mercier, P.; Jacob, Y.; Tanner, K.; Tordo, N. A Novel Expression Cassette of Lyssavirus Shows that the Distantly Related Mokola Virus Can Rescue a Defective Rabies Virus Genome. J. Virol. 2002, 76, 2024–2027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGettigan, J.P.; Naper, K.; Orenstein, J.; Koser, M.; McKenna, P.M.; Schnell, M.J. Functional Human Immunodeficiency Virus Type 1 (HIV-1) Gag-Pol or HIV-1 Gag-Pol and Env Expressed from a Single Rhabdovirus-Based Vaccine Vector Genome. J. Virol. 2003, 77, 10889–10899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchholz, U.J.; Finke, S.; Conzelmann, K.-K. Generation of Bovine Respiratory Syncytial Virus (BRSV) from cDNA: BRSV NS2 Is Not Essential for Virus Replication in Tissue Culture, and the Human RSV Leader Region Acts as a Functional BRSV Genome Promoter. J. Virol. 1999, 73, 251–259. [Google Scholar] [CrossRef] [Green Version]
- Harty, R.N.; Brown, M.E.; Hayes, F.P.; Wright, N.T.; Schnell, M.J. Vaccinia virus-free recovery of vesicular stomatitis virus. J. Mol. Microbiol. Biotechnol. 2001, 3, 513–517. [Google Scholar] [PubMed]
- Inoue, K.I.; Shoji, Y.; Kurane, I.; Iijima, T.; Sakai, T.; Morimoto, K. An improved method for recovering rabies virus from cloned cDNA. J. Virol. Methods 2003, 107, 229–236. [Google Scholar] [CrossRef]
- Haglund, K.; Forman, J.; Kräusslich, H.G.; Rose, J.K. Expression of human immunodeficiency virus type 1 gag protein precursor and envelope proteins from a vesicular stomatitis virus recombinant: High-level production of virus-like particles containing HIV envelope. Virology 2000, 268, 112–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitt, M.A. Generation of VSV pseudotypes using recombinant ΔG-VSV for studies on virus entry, identification of entry inhibitors, and immune responses to vaccines. J. Virol. Methods 2010, 169, 365–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez-Guerrero, A.; Cosset, F.L.; Verhoeyen, E. Lentiviral Vector Pseudotypes: Precious Tools to Improve Gene Modification of Hematopoietic Cells for Research and Gene Therapy. Viruses 2020, 12, 1016. [Google Scholar] [CrossRef] [PubMed]
- Garbutt, M.; Liebscher, R.; Wahl-Jensen, V.; Jones, S.; Möller, P.; Wagner, R.; Volchkov, V.; Klenk, H.-D.; Feldmann, H.; Ströher, U. Properties of Replication-Competent Vesicular Stomatitis Virus Vectors Expressing Glycoproteins of Filoviruses and Arenaviruses. J. Virol. 2004, 78, 5458–5465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konduru, K.; Shurtleff, A.C.; Bradfute, S.B.; Nakamura, S.; Bavari, S.; Kaplan, G. Ebolavirus Glycoprotein Fc Fusion Protein Protects Guinea Pigs against Lethal Challenge. PLoS ONE 2016, 11, e0162446. [Google Scholar] [CrossRef]
- DeBuysscher, B.L.; Scott, D.; Marzi, A.; Prescott, J.; Feldmann, H. Single-dose live-attenuated Nipah virus vaccines confer complete protection by eliciting antibodies directed against surface glycoproteins. Vaccine 2014, 32, 2637–2644. [Google Scholar] [CrossRef] [Green Version]
- Takada, A.; Robison, C.; Goto, H.; Sanchez, A.; Murti, K.G.; Whitt, M.A.; Kawaoka, Y. A system for functional analysis of Ebola virus glycoprotein. Proc. Natl. Acad. Sci. USA 1997, 94, 14764–14769. [Google Scholar] [CrossRef] [Green Version]
- Chattopadhyay, A.; Rose, J.K. Complementing Defective Viruses That Express Separate Paramyxovirus Glycoproteins Provide a New Vaccine Vector Approach. J. Virol. 2011, 85, 2004–2011. [Google Scholar] [CrossRef] [Green Version]
- Ito, H.; Watanabe, S.; Sanchez, A.; Whitt, M.A.; Kawaoka, Y. Mutational Analysis of the Putative Fusion Domain of Ebola Virus Glycoprotein. J. Virol. 1999, 73, 8907–8912. [Google Scholar] [CrossRef] [Green Version]
- Ito, H.; Watanabe, S.; Takada, A.; Kawaoka, Y. Ebola Virus Glycoprotein: Proteolytic Processing, Acylation, Cell Tropism, and Detection of Neutralizing Antibodies. J. Virol. 2001, 75, 1576–1580. [Google Scholar] [CrossRef] [Green Version]
- Schnell, M.J.; Buonocore, L.; Whitt, M.A.; Rose, J.K. The minimal conserved transcription stop-start signal promotes stable expression of a foreign gene in vesicular stomatitis virus. J. Virol. 1996, 70, 2318–2323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.M.; Feldmann, H.; Ströher, U.; Geisbert, J.B.; Fernando, L.; Grolla, A.; Klenk, H.D.; Sullivan, N.J.; Volchkov, V.E.; Fritz, E.A.; et al. Live attenuated recombinant vaccine protects nonhuman primates against Ebola and Marburg viruses. Nat. Med. 2005, 11, 786–790. [Google Scholar] [CrossRef]
- Jones, S.M.; Ströher, U.; Fernando, L.; Qiu, X.; Alimonti, J.; Melito, P.; Bray, M.; Klenk, H.D.; Feldmann, H. Assessment of a vesicular stomatitis virus-based vaccine by use of the mouse model of Ebola virus hemorrhagic fever. J. Infect. Dis. 2007, 196, S404–S412. [Google Scholar] [CrossRef]
- Qiu, X.; Fernando, L.; Alimonti, J.B.; Melito, P.L.; Feldmann, F.; Dick, D.; Ströher, U.; Feldmann, H.; Jones, S.M. Mucosal immunization of cynomolgus macaques with the VSVΔG/ZEBOVGP vaccine stimulates strong ebola GP-specific immune responses. PLoS ONE 2009, 4, e5547. [Google Scholar] [CrossRef]
- Feldmann, H.; Jones, S.M.; Daddario-DiCaprio, K.M.; Geisbert, J.B.; Ströher, U.; Grolla, A.; Bray, M.; Fritz, E.A.; Fernando, L.; Feldmann, F.; et al. Effective post-exposure treatment of ebola infection. PLoS Pathog. 2007, 3, e2. [Google Scholar] [CrossRef] [Green Version]
- Tsuda, Y.; Safronetz, D.; Brown, K.; Lacasse, R.; Marzi, A.; Ebihara, H.; Feldmann, H. Protective efficacy of a bivalent recombinant vesicular stomatitis virus vaccine in the syrian hamster model of lethal Ebola virus infection. J. Infect. Dis. 2011, 204 (Suppl. 3), S1090–S1097. [Google Scholar] [CrossRef] [Green Version]
- Marzi, A.; Hanley, P.W.; Haddock, E.; Martellaro, C.; Kobinger, G.; Feldmann, H. Efficacy of Vesicular Stomatitis Virus-Ebola Virus Postexposure Treatment in Rhesus Macaques Infected with Ebola Virus Makona. J. Infect. Dis. 2016, 214, S360–S366. [Google Scholar] [CrossRef] [PubMed]
- Wong, G.; Audet, J.; Fernando, L.; Fausther-Bovendo, H.; Alimonti, J.B.; Kobinger, G.P.; Qiu, X. Immunization with vesicular stomatitis virus vaccine expressing the Ebola glycoprotein provides sustained long-term protection in rodents. Vaccine 2014, 32, 5722–5729. [Google Scholar] [CrossRef]
- Suder, E.; Furuyama, W.; Feldmann, H.; Marzi, A.; de Wit, E. The vesicular stomatitis virus-based Ebola virus vaccine: From concept to clinical trials. Hum. Vaccines Immunother. 2018, 14, 2107–2113. [Google Scholar] [CrossRef] [Green Version]
- Wolf, J.; Jannat, R.; Dubey, S.; Troth, S.; Onorato, M.T.; Coller, B.A.; Hanson, M.E.; Simon, J.K. Development of pandemic vaccines: ERVEBO case study. Vaccines 2021, 9, 190. [Google Scholar] [CrossRef]
- Huttner, A.; Dayer, J.A.; Yerly, S.; Combescure, C.; Auderset, F.; Desmeules, J.; Eickmann, M.; Finckh, A.; Goncalves, A.R.; Hooper, J.W.; et al. The effect of dose on the safety and immunogenicity of the VSV Ebola candidate vaccine: A randomised double-blind, placebo-controlled phase 1/2 trial. Lancet Infect. Dis. 2015, 15, 1156–1166. [Google Scholar] [CrossRef]
- Agnandji, S.T.; Huttner, A.; Zinser, M.E.; Njuguna, P.; Dahlke, C.; Fernandes, J.F.; Yerly, S.; Dayer, J.-A.; Kraehling, V.; Kasonta, R.; et al. Phase 1 Trials of rVSV Ebola Vaccine in Africa and Europe. N. Engl. J. Med. 2016, 374, 1647–1660. [Google Scholar] [CrossRef] [PubMed]
- Regules, J.A.; Beigel, J.H.; Paolino, K.M.; Voell, J.; Castellano, A.R.; Hu, Z.; Muñoz, P.; Moon, J.E.; Ruck, R.C.; Bennett, J.W.; et al. A Recombinant Vesicular Stomatitis Virus Ebola Vaccine. N. Engl. J. Med. 2017, 376, 330–341. [Google Scholar] [CrossRef]
- ElSherif, M.S.; Brown, C.; Mackinnon-Cameron, D.; Li, L.; Racine, T.; Alimonti, J.; Rudge, T.L.; Sabourin, C.; Silvera, P.; Hooper, J.W.; et al. Assessing the safety and immunogenicity of recombinant vesicular stomatitis virus Ebola vaccine in healthy adults: A randomized clinical trial. CMAJ 2017, 189, E819–E827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlke, C.; Kasonta, R.; Lunemann, S.; Krähling, V.; Zinser, M.E.; Biedenkopf, N.; Fehling, S.K.; Ly, M.L.; Rechtien, A.; Stubbe, H.C.; et al. Dose-dependent T-cell Dynamics and Cytokine Cascade Following rVSV-ZEBOV Immunization. EBioMedicine 2017, 19, 107–118. [Google Scholar] [CrossRef] [Green Version]
- Heppner, D.G.; Kemp, T.L.; Martin, B.K.; Ramsey, W.J.; Nichols, R.; Dasen, E.J.; Link, C.; Das, R.; Xu, Z.J.; Sheldon, E.A.; et al. Safety and immunogenicity of the rVSV∆G-ZEBOV-GP Ebola virus vaccine candidate in healthy adults: A phase 1b randomised, multicentre, double-blind, placebo-controlled, dose-response study. Lancet Infect. Dis. 2017, 17, 854–866. [Google Scholar] [CrossRef] [Green Version]
- Juan-Giner, A.; Tchaton, M.; Jemmy, J.P.; Soumah, A.; Boum, Y.; Faga, E.M.; Cisse, M.; Grais, R.F. Safety of the rVSV ZEBOV vaccine against Ebola Zaire among frontline workers in Guinea. Vaccine 2019, 37, 7171–7177. [Google Scholar] [CrossRef]
- Agnandji, S.T.; Fernandes, J.F.; Bache, E.B.; Obiang Mba, R.M.; Brosnahan, J.S.; Kabwende, L.; Pitzinger, P.; Staarink, P.; Massinga-Loembe, M.; Krähling, V.; et al. Safety and immunogenicity of rVSVΔG-ZEBOV-GP Ebola vaccine in adults and children in Lambaréné, Gabon: A phase I randomised trial. PLoS Med. 2017, 14, e1002402. [Google Scholar] [CrossRef] [Green Version]
- Poetsch, J.H.; Dahlke, C.; Zinser, M.E.; Kasonta, R.; Lunemann, S.; Rechtien, A.; Ly, M.L.; Stubbe, H.C.; Krähling, V.; Biedenkopf, N.; et al. Detectable Vesicular Stomatitis Virus (VSV)-Specific Humoral and Cellular Immune Responses Following VSV-Ebola Virus Vaccination in Humans. J. Infect. Dis. 2019, 219, 556–561. [Google Scholar] [CrossRef]
- Henao-Restrepo, A.M.; Camacho, A.; Longini, I.M.; Watson, C.H.; Edmunds, W.J.; Egger, M.; Carroll, M.W.; Dean, N.E.; Diatta, I.; Doumbia, M.; et al. Efficacy and effectiveness of an rVSV-vectored vaccine in preventing Ebola virus disease: Final results from the Guinea ring vaccination, open-label, cluster-randomised trial (Ebola Ça Suffit!). Lancet 2017, 389, 505–518. [Google Scholar] [CrossRef] [Green Version]
- Widdowson, M.A.; Schrag, S.J.; Carter, R.J.; Carr, W.; Legardy-Williams, J.; Gibson, L.; Lisk, D.R.; Jalloh, M.I.; Bash-Taqi, D.A.; Kargbo, S.A.S.; et al. Implementing an Ebola Vaccine Study—Sierra Leone. MMWR Suppl. 2016, 65, 98–106. [Google Scholar] [CrossRef]
- Halperin, S.A.; Arribas, J.R.; Rupp, R.; Andrews, C.P.; Chu, L.; Das, R.; Simon, J.K.; Onorato, M.T.; Liu, K.; Martin, J.; et al. Six-Month Safety Data of Recombinant Vesicular Stomatitis Virus-Zaire Ebola Virus Envelope Glycoprotein Vaccine in a Phase 3 Double-Blind, Placebo-Controlled Randomized Study in Healthy Adults. J. Infect. Dis. 2017, 215, 1789–1798. [Google Scholar] [CrossRef]
- Sikakulya, F.K.; Mulisya, O.; Munyambalu, D.K.; Bunduki, G.K. Ebola in the Eastern Democratic Republic of Congo: One Health approach to infectious disease control. One Health 2020, 9, 100117. [Google Scholar] [CrossRef]
- Wells, C.R.; Pandey, A.; Parpia, A.S.; Fitzpatrick, M.C.; Meyers, L.A.; Singer, B.H.; Galvani, A.P. Ebola vaccination in the Democratic Republic of the Congo. Proc. Natl. Acad. Sci. USA 2019, 116, 10178–10183. [Google Scholar] [CrossRef] [Green Version]
- Ollmann Saphire, E. A Vaccine against Ebola Virus. Cell 2020, 181, 6. [Google Scholar] [CrossRef] [PubMed]
- Khurana, S.; Fuentes, S.; Coyle, E.M.; Ravichandran, S.; Davey, R.T.; Beigel, J.H. Human antibody repertoire after VSV-Ebola vaccination identifies novel targets and virus-neutralizing IgM antibodies. Nat. Med. 2016, 22, 1439–1447. [Google Scholar] [CrossRef] [PubMed]
- Ehrhardt, S.A.; Zehner, M.; Krähling, V.; Cohen-Dvashi, H.; Kreer, C.; Elad, N.; Gruell, H.; Ercanoglu, M.S.; Schommers, P.; Gieselmann, L.; et al. Polyclonal and convergent antibody response to Ebola virus vaccine rVSV-ZEBOV. Nat. Med. 2019, 25, 1589–1600. [Google Scholar] [CrossRef] [Green Version]
- Geisbert, T.W.; Daddario-DiCaprio, K.M.; Lewis, M.G.; Geisbert, J.B.; Grolla, A.; Leung, A.; Paragas, J.; Matthias, L.; Smith, M.A.; Jones, S.M.; et al. Vesicular stomatitis virus-based Ebola vaccine is well-tolerated and protects immunocompromised nonhuman primates. PLoS Pathog. 2008, 4, e1000225. [Google Scholar] [CrossRef]
- Marzi, A.; Engelmann, F.; Feldmann, F.; Haberthur, K.; Shupert, W.L.; Brining, D.; Scott, D.P.; Geisbert, T.W.; Kawaoka, Y.; Katze, M.G.; et al. Antibodies are necessary for rVSV/ZEBOV-GP-mediated protection against lethal Ebola virus challenge in nonhuman primates. Proc. Natl. Acad. Sci. USA 2013, 110, 1893–1898. [Google Scholar] [CrossRef] [Green Version]
- Farooq, F.; Beck, K.; Paolino, K.M.; Phillips, R.; Waters, N.C.; Regules, J.A.; Bergmann-Leitner, E.S. Circulating follicular T helper cells and cytokine profile in humans following vaccination with the rVSV-ZEBOV Ebola vaccine. Sci. Rep. 2016, 6, 27944. [Google Scholar] [CrossRef] [Green Version]
- Sakabe, S.; Sullivan, B.M.; Hartnett, J.N.; Robles-Sikisaka, R.; Gangavarapu, K.; Cubitt, B.; Ware, B.C.; Kotliar, D.; Branco, L.M.; Goba, A.; et al. Analysis of CD8+ T cell response during the 2013–2016 Ebola epidemic in West Africa. Proc. Natl. Acad. Sci. USA 2018, 115, E7578–E7586. [Google Scholar] [CrossRef] [Green Version]
- Warfield, K.L.; Perkins, J.G.; Swenson, D.L.; Deal, E.M.; Bosio, C.M.; Aman, M.J.; Yokoyama, W.M.; Young, H.A.; Bavari, S. Role of natural killer cells innate protection against lethal Ebola virus infection. J. Exp. Med. 2004, 200, 169–179. [Google Scholar] [CrossRef] [Green Version]
- Marzi, A.; Robertson, S.J.; Haddock, E.; Feldmann, F.; Hanley, P.W.; Scott, D.P.; Strong, J.E.; Kobinger, G.; Best, S.M.; Feldmann, H. VSV-EBOV rapidly protects macaques against infection with the 2014/15 Ebola virus outbreak strain. Science 2015, 349, 739–742. [Google Scholar] [CrossRef] [Green Version]
- Menicucci, A.R.; Jankeel, A.; Feldmann, H.; Marzi, A.; Messaoudi, I. Antiviral innate responses induced by VSV-EBOV vaccination contribute to rapid protection. mBio 2019, 10, e00597-19. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.; Buonocore, L.; Price, R.; Forman, J.; Rose, J.K. Attenuated Vesicular Stomatitis Viruses as Vaccine Vectors. J. Virol. 1999, 73, 3723–3732. [Google Scholar] [CrossRef] [Green Version]
- Marzi, A.; Feldmann, F.; Geisbert, T.W.; Feldmann, H.; Safronetz, D. Vesicular stomatitis virus–based vaccines against lassa and ebola viruses. Emerg. Infect. Dis. 2015, 21, 305–307. [Google Scholar] [CrossRef]
- Hastie, E.; Cataldi, M.; Marriott, I.; Grdzelishvili, V.Z. Understanding and altering cell tropism of vesicular stomatitis virus. Virus Res. 2013, 176, 16–32. [Google Scholar] [CrossRef] [Green Version]
- Mire, C.E.; Miller, A.D.; Carville, A.; Westmoreland, S.V.; Geisbert, J.B.; Mansfield, K.G.; Feldmann, H.; Hensley, L.E.; Geisbert, T.W. Recombinant vesicular stomatitis virus vaccine vectors expressing filovirus glycoproteins lack neurovirulence in nonhuman primates. PLoS Negl. Trop. Dis. 2012, 6, e1567. [Google Scholar] [CrossRef]
- McWilliams, I.L.; Kielczewski, J.L.; Ireland, D.D.C.; Sykes, J.S.; Lewkowicz, A.P.; Konduru, K.; Xu, B.C.; Chan, C.C.; Caspi, R.R.; Manangeeswaran, M.; et al. Pseudovirus rVSVΔG-ZEBOV-GP Infects Neurons in Retina and CNS, Causing Apoptosis and Neurodegeneration in Neonatal Mice. Cell Rep. 2019, 26, 1718–1726.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Wit, E.; Marzi, A.; Bushmaker, T.; Brining, D.; Scott, D.; Richt, J.A.; Geisbert, T.W.; Feldmann, H. Safety of recombinant VSV-Ebola virus vaccine vector in pigs. Emerg. Infect. Dis. 2015, 21, 702–704. [Google Scholar] [CrossRef]
- Morozov, I.; Monath, T.P.; Meekins, D.A.; Trujillo, J.D.; Sunwoo, S.Y.; Urbaniak, K.; Kim, I.J.; Narayanan, S.K.; Indran, S.V.; Ma, W.; et al. High dose of vesicular stomatitis virus-vectored Ebola virus vaccine causes vesicular disease in swine without horizontal transmission. Emerg. Microbes Infect. 2021, 10, 651–663. [Google Scholar] [CrossRef] [PubMed]
- Daddario-DiCaprio, K.M.; Geisbert, T.W.; Geisbert, J.B.; Ströher, U.; Hensley, L.E.; Grolla, A.; Fritz, E.A.; Feldmann, F.; Feldmann, H.; Jones, S.M. Cross-Protection against Marburg Virus Strains by Using a Live, Attenuated Recombinant Vaccine. J. Virol. 2006, 80, 9659–9666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mire, C.E.; Geisbert, J.B.; Agans, K.N.; Satterfield, B.A.; Versteeg, K.M.; Fritz, E.A.; Feldmann, H.; Hensley, L.E.; Geisbert, T.W. Durability of a vesicular stomatitis virus-based marburg virus vaccine in nonhuman primates. PLoS ONE 2014, 9, e94355. [Google Scholar] [CrossRef]
- Daddario-DiCaprio, K.M.; Geisbert, T.W.; Ströher, U.; Geisbert, J.B.; Grolla, A.; Fritz, E.A.; Fernando, L.; Kagan, E.; Jahrling, P.B.; Hensley, L.E.; et al. Postexposure protection against Marburg haemorrhagic fever with recombinant vesicular stomatitis virus vectors in non-human primates: An efficacy assessment. Lancet 2006, 367, 1399–1404. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Hensley, L.E.; Geisbert, J.B.; Leung, A.; Johnson, J.C.; Grolla, A.; Feldmann, H. Postexposure treatment of marburg virus infection. Emerg. Infect. Dis. 2010, 16, 1119–1122. [Google Scholar] [CrossRef]
- Woolsey, C.; Geisbert, J.B.; Matassov, D.; Agans, K.N.; Borisevich, V.; Cross, R.W.; Deer, D.J.; Fenton, K.A.; Eldridge, J.H.; Mire, C.E.; et al. Postexposure Efficacy of Recombinant Vesicular Stomatitis Virus Vectors Against High and Low Doses of Marburg Virus Variant Angola in Nonhuman Primates. J. Infect. Dis. 2018, 218, S582–S587. [Google Scholar] [CrossRef] [Green Version]
- Marzi, A.; Menicucci, A.R.; Engelmann, F.; Callison, J.; Horne, E.J.; Feldmann, F.; Jankeel, A.; Feldmann, H.; Messaoudi, I. Protection against marburg virus using a recombinant VSV-vaccine depends on T and B cell activation. Front. Immunol. 2019, 9, 3071. [Google Scholar] [CrossRef] [Green Version]
- Woolsey, C.; Jankeel, A.; Matassov, D.; Geisbert, J.B.; Agans, K.N.; Borisevich, V.; Cross, R.W.; Deer, D.J.; Fenton, K.A.; Latham, T.E.; et al. Immune correlates of postexposure vaccine protection against Marburg virus. Sci. Rep. 2020, 10, 3071. [Google Scholar] [CrossRef] [Green Version]
- Mire, C.E.; Geisbert, J.B.; Marzi, A.; Agans, K.N.; Feldmann, H.; Geisbert, T.W. Vesicular Stomatitis Virus-Based Vaccines Protect Nonhuman Primates against Bundibugyo ebolavirus. PLoS Negl. Trop. Dis. 2013, 7, e2600. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Daddario-DiCaprio, K.M.; Williams, K.J.N.; Geisbert, J.B.; Leung, A.; Feldmann, F.; Hensley, L.E.; Feldmann, H.; Jones, S.M. Recombinant Vesicular Stomatitis Virus Vector Mediates Postexposure Protection against Sudan Ebola Hemorrhagic Fever in Nonhuman Primates. J. Virol. 2008, 82, 5664–5668. [Google Scholar] [CrossRef] [Green Version]
- Falzarano, D.; Feldmann, F.; Grolla, A.; Leung, A.; Ebihara, H.; Strong, J.E.; Marzi, A.; Takada, A.; Jones, S.; Gren, J.; et al. Single immunization with a monovalent vesicular stomatitis virus-based vaccine protects nonhuman primates against heterologous challenge with Bundibugyo ebolavirus. J. Infect. Dis. 2011, 204 (Suppl. 3), S1082–S1089. [Google Scholar] [CrossRef]
- Marzi, A.; Ebihara, H.; Callison, J.; Groseth, A.; Williams, K.J.; Geisbert, T.W.; Feldmann, H. Vesicular stomatitis virus-based Ebola vaccines with improved cross-protective efficacy. J. Infect. Dis. 2011, 204 (Suppl. 3), S1066–S1074. [Google Scholar] [CrossRef] [PubMed]
- Geisbert, T.W.; Geisbert, J.B.; Leung, A.; Daddario-DiCaprio, K.M.; Hensley, L.E.; Grolla, A.; Feldmann, H. Single-Injection Vaccine Protects Nonhuman Primates against Infection with Marburg Virus and Three Species of Ebola Virus. J. Virol. 2009, 83, 7296–7304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cross, R.W.; Xu, R.; Matassov, D.; Hamm, S.; Latham, T.E.; Gerardi, C.S.; Nowak, R.M.; Geisbert, J.B.; Ota-Setlik, A.; Agans, K.N.; et al. Quadrivalent VesiculoVax vaccine protects nonhuman primates from viral-induced hemorrhagic fever and death. J. Clin. Investig. 2020, 130, 539–551. [Google Scholar] [CrossRef]
- Matassov, D.; Mire, C.E.; Latham, T.; Geisbert, J.B.; Xu, R.; Ota-Setlik, A.; Agans, K.N.; Kobs, D.J.; Wendling, M.Q.S.; Burnaugh, A.; et al. Single-Dose Trivalent VesiculoVax Vaccine Protects Macaques from Lethal Ebolavirus and Marburgvirus Challenge. J. Virol. 2018, 92, e01190-17. [Google Scholar] [CrossRef] [Green Version]
- Mire, C.E.; Geisbert, J.B.; Versteeg, K.M.; Mamaeva, N.; Agans, K.N.; Geisbert, T.W.; Connor, J.H. A Single-Vector, Single-Injection Trivalent Filovirus Vaccine: Proof of Concept Study in Outbred Guinea Pigs. J. Infect. Dis. 2015, 212, S384–S388. [Google Scholar] [CrossRef] [Green Version]
- Geisbert, T.W.; Jones, S.; Fritz, E.A.; Shurtleff, A.C.; Geisbert, J.B.; Liebscher, R.; Grolla, A.; Ströher, U.; Fernando, L.; Daddario, K.M.; et al. Development of a new vaccine for the prevention of Lassa fever. PLoS Med. 2005, 2, e183. [Google Scholar] [CrossRef]
- Safronetz, D.; Mire, C.; Rosenke, K.; Feldmann, F.; Haddock, E.; Geisbert, T.; Feldmann, H. A Recombinant Vesicular Stomatitis Virus-Based Lassa Fever Vaccine Protects Guinea Pigs and Macaques against Challenge with Geographically and Genetically Distinct Lassa Viruses. PLoS Negl. Trop. Dis. 2015, 9, e0003736. [Google Scholar] [CrossRef]
- Stein, D.R.; Warner, B.M.; Soule, G.; Tierney, K.; Frost, K.L.; Booth, S.; Safronetz, D. A recombinant vesicular stomatitis-based Lassa fever vaccine elicits rapid and long-term protection from lethal Lassa virus infection in guinea pigs. npj Vaccines 2019, 4, 8. [Google Scholar] [CrossRef]
- Stein, D.R.; Sroga, P.; Warner, B.M.; Deschambault, Y.; Poliquin, G.; Safronetz, D. Evaluating temperature sensitivity of vesicular stomatitis virus–based vaccines. Emerg. Infect. Dis. 2019, 25, 1563–1566. [Google Scholar] [CrossRef]
- Van den Pol, A.N.; Mao, G.; Chattopadhyay, A.; Rose, J.K.; Davis, J.N. Chikungunya, Influenza, Nipah, and Semliki Forest Chimeric Viruses with Vesicular Stomatitis Virus: Actions in the Brain. J. Virol. 2017, 91, e02154-16. [Google Scholar] [CrossRef] [Green Version]
- Lo, M.K.; Bird, B.H.; Chattopadhyay, A.; Drew, C.P.; Martin, B.E.; Coleman, J.D.; Rose, J.K.; Nichol, S.T.; Spiropoulou, C.F. Single-dose replication-defective VSV-based Nipah virus vaccines provide protection from lethal challenge in Syrian hamsters. Antivir. Res. 2014, 101, 26–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mire, C.E.; Versteeg, K.M.; Cross, R.W.; Agans, K.N.; Fenton, K.A.; Whitt, M.A.; Geisbert, T.W. Single injection recombinant vesicular stomatitis virus vaccines protect ferrets against lethal Nipah virus disease. Virol. J. 2013, 10, 353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debuysscher, B.L.; Scott, D.; Thomas, T.; Feldmann, H.; Prescott, J. Peri-exposure protection against Nipah virus disease using a single-dose recombinant vesicular stomatitis virus-based vaccine. npj Vaccines 2016, 1, 16002. [Google Scholar] [CrossRef]
- Prescott, J.; DeBuysscher, B.L.; Feldmann, F.; Gardner, D.J.; Haddock, E.; Martellaro, C.; Scott, D.; Feldmann, H. Single-dose live-attenuated vesicular stomatitis virus-based vaccine protects African green monkeys from Nipah virus disease. Vaccine 2015, 33, 2823–2829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mire, C.E.; Geisbert, J.B.; Agans, K.N.; Versteeg, K.M.; Deer, D.J.; Satterfield, B.A.; Fenton, K.A.; Geisbert, T.W. Use of single-injection recombinant vesicular stomatitis virus vaccine to protect nonhuman primates against lethal nipah virus disease. Emerg. Infect. Dis. 2019, 25, 1144–1152. [Google Scholar] [CrossRef] [Green Version]
- Kurup, D.; Wirblich, C.; Feldmann, H.; Marzi, A.; Schnell, M.J. Rhabdovirus-Based Vaccine Platforms against Henipaviruses. J. Virol. 2015, 89, 144–154. [Google Scholar] [CrossRef] [Green Version]
- Kapadia, S.U.; Rose, J.K.; Lamirande, E.; Vogel, L.; Subbarao, K.; Roberts, A. Long-term protection from SARS coronavirus infection conferred by a single immunization with an attenuated VSV-based vaccine. Virology 2005, 340, 174–182. [Google Scholar] [CrossRef] [Green Version]
- Kapadia, S.U.; Simon, I.D.; Rose, J.K. SARS vaccine based on a replication-defective recombinant vesicular stomatitis virus is more potent than one based on a replication-competent vector. Virology 2008, 376, 165–172. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Wang, J.; Shao, Y.; Wang, X.; Zhang, H.; Shuai, L.; Ge, J.; Wen, Z.; Bu, Z. A recombinant VSV-vectored MERS-CoV vaccine induces neutralizing antibody and T cell responses in rhesus monkeys after single dose immunization. Antivir. Res. 2018, 150, 30–38. [Google Scholar] [CrossRef]
- Case, J.B.; Rothlauf, P.W.; Chen, R.E.; Kafai, N.M.; Fox, J.M.; Smith, B.K.; Shrihari, S.; McCune, B.T.; Harvey, I.B.; Keeler, S.P.; et al. Replication-Competent Vesicular Stomatitis Virus Vaccine Vector Protects against SARS-CoV-2-Mediated Pathogenesis in Mice. Cell Host Microbe 2020, 28, 465–474.e4. [Google Scholar] [CrossRef]
- Hennrich, A.A.; Sawatsky, B.; Santos-Mandujano, R.; Banda, D.H.; Oberhuber, M.; Schopf, A.; Pfaffinger, V.; Wittwer, K.; Riedel, C.; Pfaller, C.K.; et al. Safe and effective two-in-one replicon-and-VLP minispike vaccine for COVID-19: Protection of mice after a single immunization. PLoS Pathog. 2021, 17, e1009064. [Google Scholar] [CrossRef]
- Yahalom-Ronen, Y.; Tamir, H.; Melamed, S.; Politi, B.; Shifman, O.; Achdout, H.; Vitner, E.B.; Israeli, O.; Milrot, E.; Stein, D.; et al. A single dose of recombinant VSV-∆G-spike vaccine provides protection against SARS-CoV-2 challenge. Nat. Commun. 2020, 11, 6402. [Google Scholar] [CrossRef]
- Furuyama, W.; Shifflett, K.; Pinski, A.N.; Griffin, A.J.; Feldmann, F.; Okumura, A.; Gourdine, T.; Jankeel, A.; Lovaglio, J.; Hanley, P.W.; et al. Rapid protection from COVID-19 in nonhuman primates vaccinated intramuscularly but not intranasally with a single dose of a recombinant vaccine. bioRxiv Prepr. Serv. Biol. 2021. [Google Scholar] [CrossRef]
- Emanuel, J.; Callison, J.; Dowd, K.A.; Pierson, T.C.; Feldmann, H.; Marzi, A. A VSV-based Zika virus vaccine protects mice from lethal challenge. Sci. Rep. 2018, 8, 11043. [Google Scholar] [CrossRef]
- Li, A.; Yu, J.; Lu, M.; Ma, Y.; Attia, Z.; Shan, C.; Xue, M.; Liang, X.; Craig, K.; Makadiya, N.; et al. A Zika virus vaccine expressing premembrane-envelope-NS1 polyprotein. Nat. Commun. 2018, 9, 3067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.; Xue, M.; Attia, Z.; Yu, J.; Lu, M.; Shan, C.; Liang, X.; Gao, T.Z.; Shi, P.-Y.; Peeples, M.E.; et al. Vesicular Stomatitis Virus and DNA Vaccines Expressing Zika Virus Nonstructural Protein 1 Induce Substantial but Not Sterilizing Protection against Zika Virus Infection. J. Virol. 2020, 94, e00048-20. [Google Scholar] [CrossRef]
- Rodriguez, S.E.; Cross, R.W.; Fenton, K.A.; Bente, D.A.; Mire, C.E.; Geisbert, T.W. Vesicular Stomatitis Virus-Based Vaccine Protects Mice against Crimean-Congo Hemorrhagic Fever. Sci. Rep. 2019, 9, 7755. [Google Scholar] [CrossRef] [Green Version]
- Bishnoi, S.; Tiwari, R.; Gupta, S.; Byrareddy, S.N.; Nayak, D. Oncotargeting by Vesicular Stomatitis Virus (VSV): Advances in cancer therapy. Viruses 2018, 10, 90. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, G.; Cao, W.; Salawudeen, A.; Zhu, W.; Emeterio, K.; Safronetz, D.; Banadyga, L. Vesicular Stomatitis Virus: From Agricultural Pathogen to Vaccine Vector. Pathogens 2021, 10, 1092. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10091092
Liu G, Cao W, Salawudeen A, Zhu W, Emeterio K, Safronetz D, Banadyga L. Vesicular Stomatitis Virus: From Agricultural Pathogen to Vaccine Vector. Pathogens. 2021; 10(9):1092. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10091092
Chicago/Turabian StyleLiu, Guodong, Wenguang Cao, Abdjeleel Salawudeen, Wenjun Zhu, Karla Emeterio, David Safronetz, and Logan Banadyga. 2021. "Vesicular Stomatitis Virus: From Agricultural Pathogen to Vaccine Vector" Pathogens 10, no. 9: 1092. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10091092