
Comparative Genomics of Clinical and Environmental Isolates of Vibrio spp. of Colombia: Implications of Traits Associated with Virulence and Resistance

 , , and
, , and
Abstract
:1. Introduction
2. Results
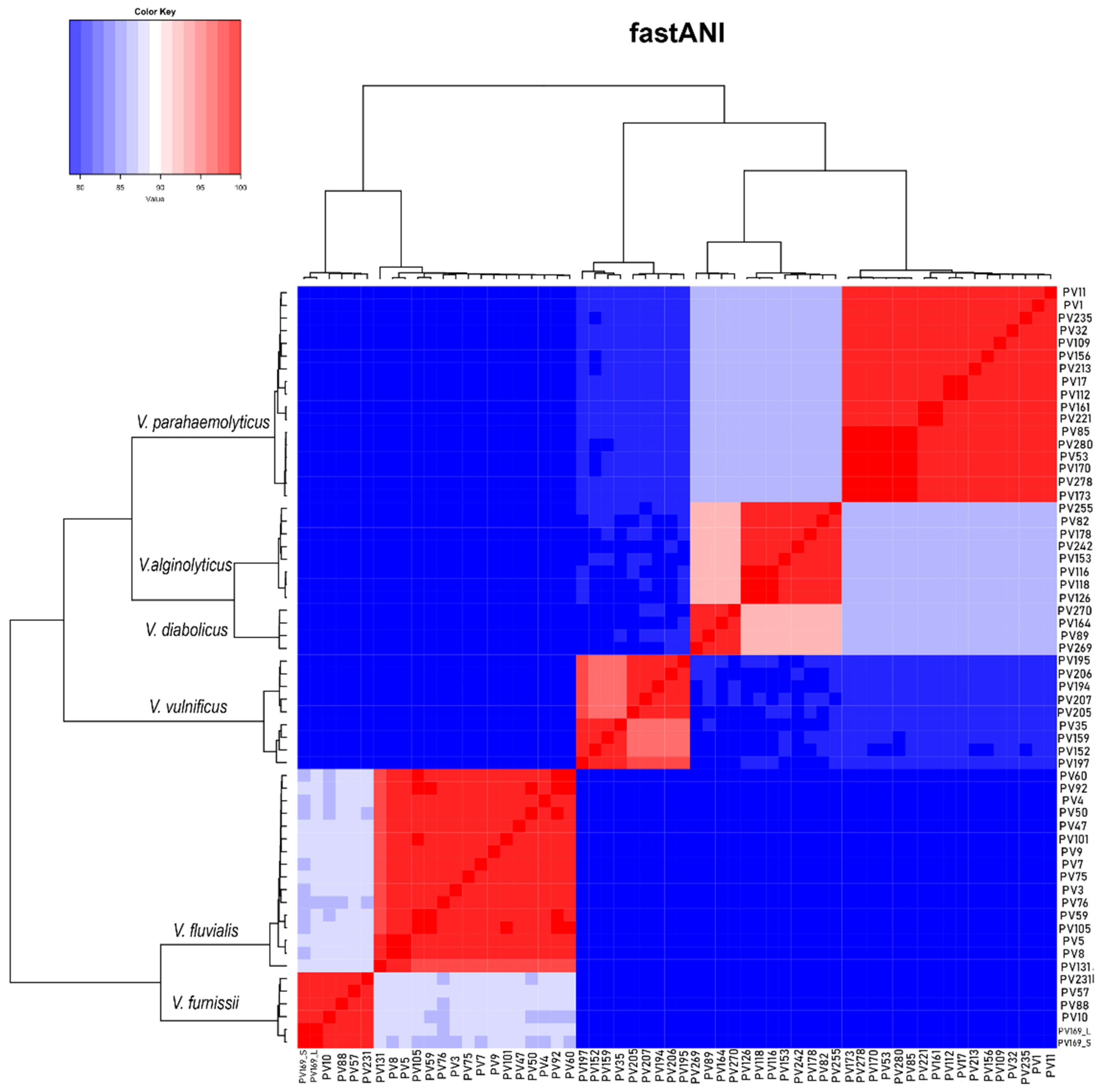
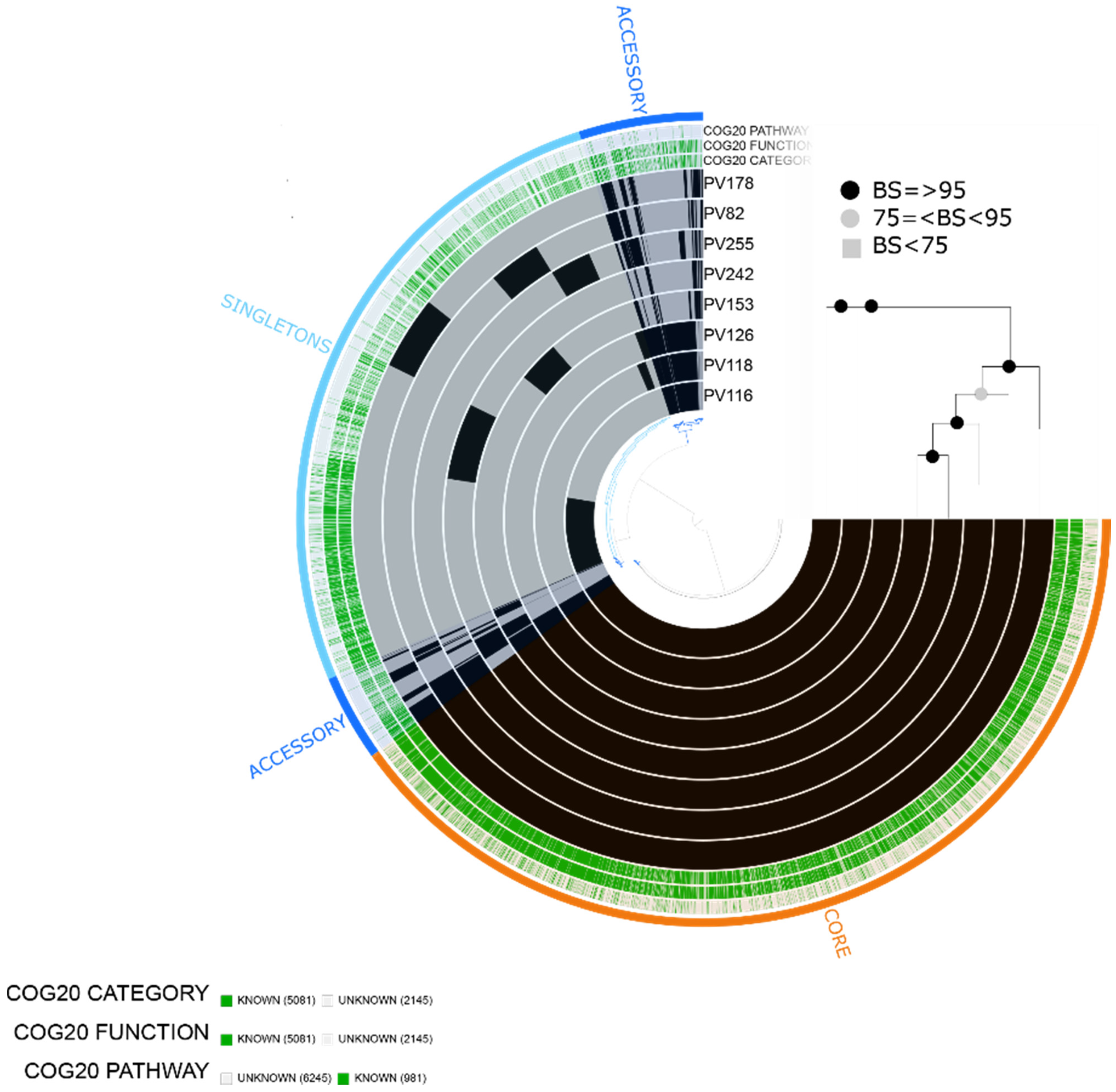
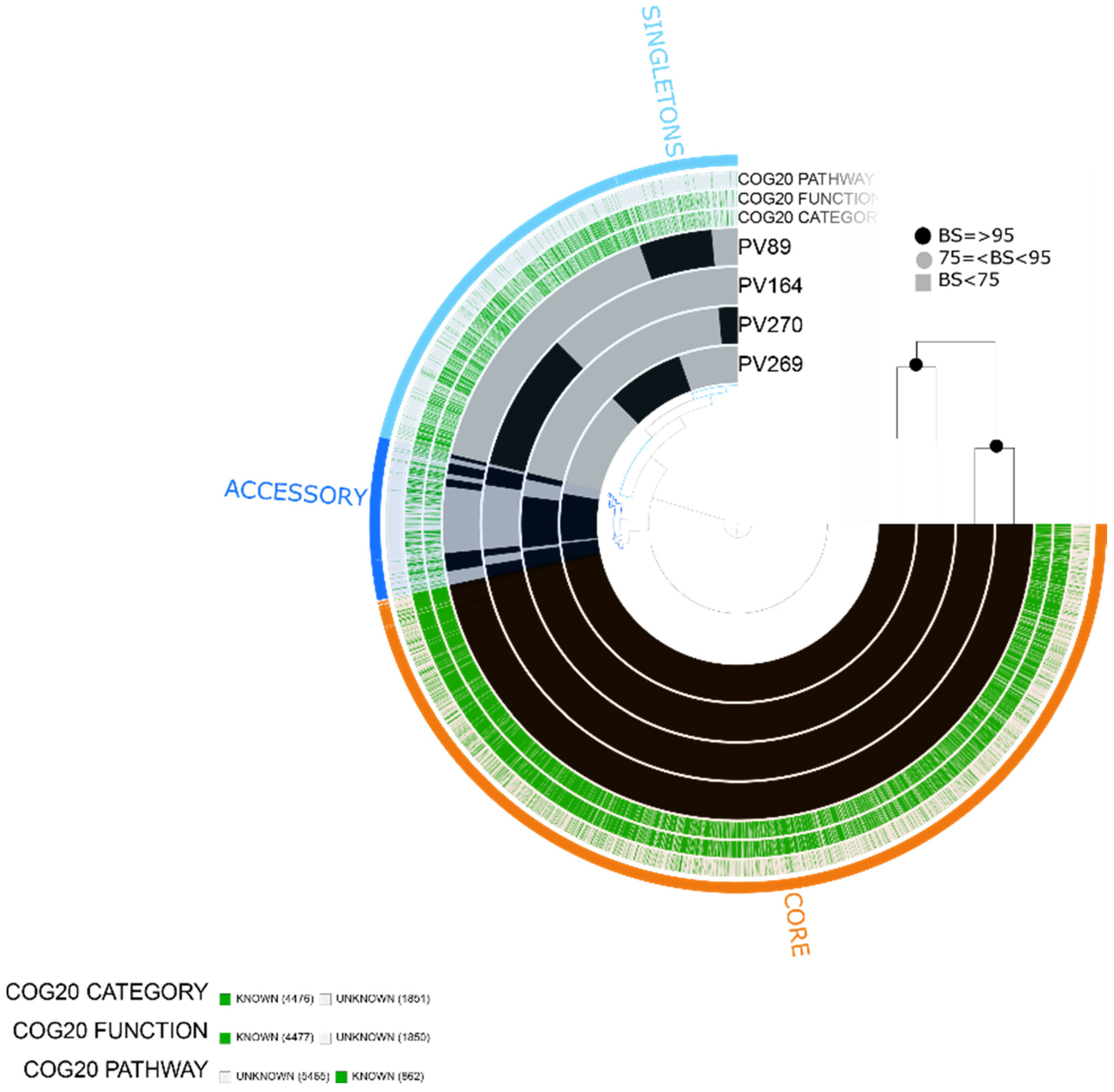
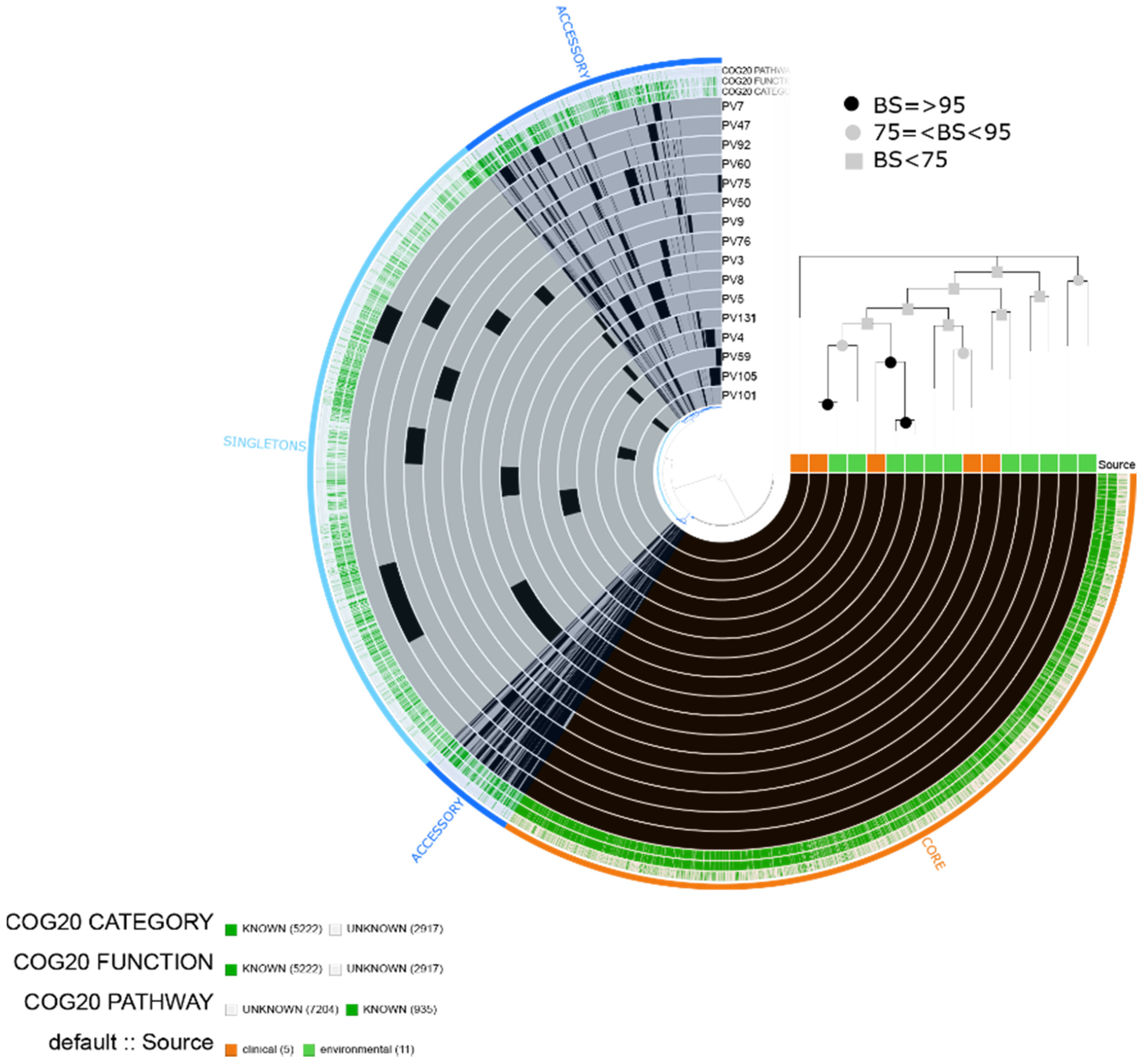
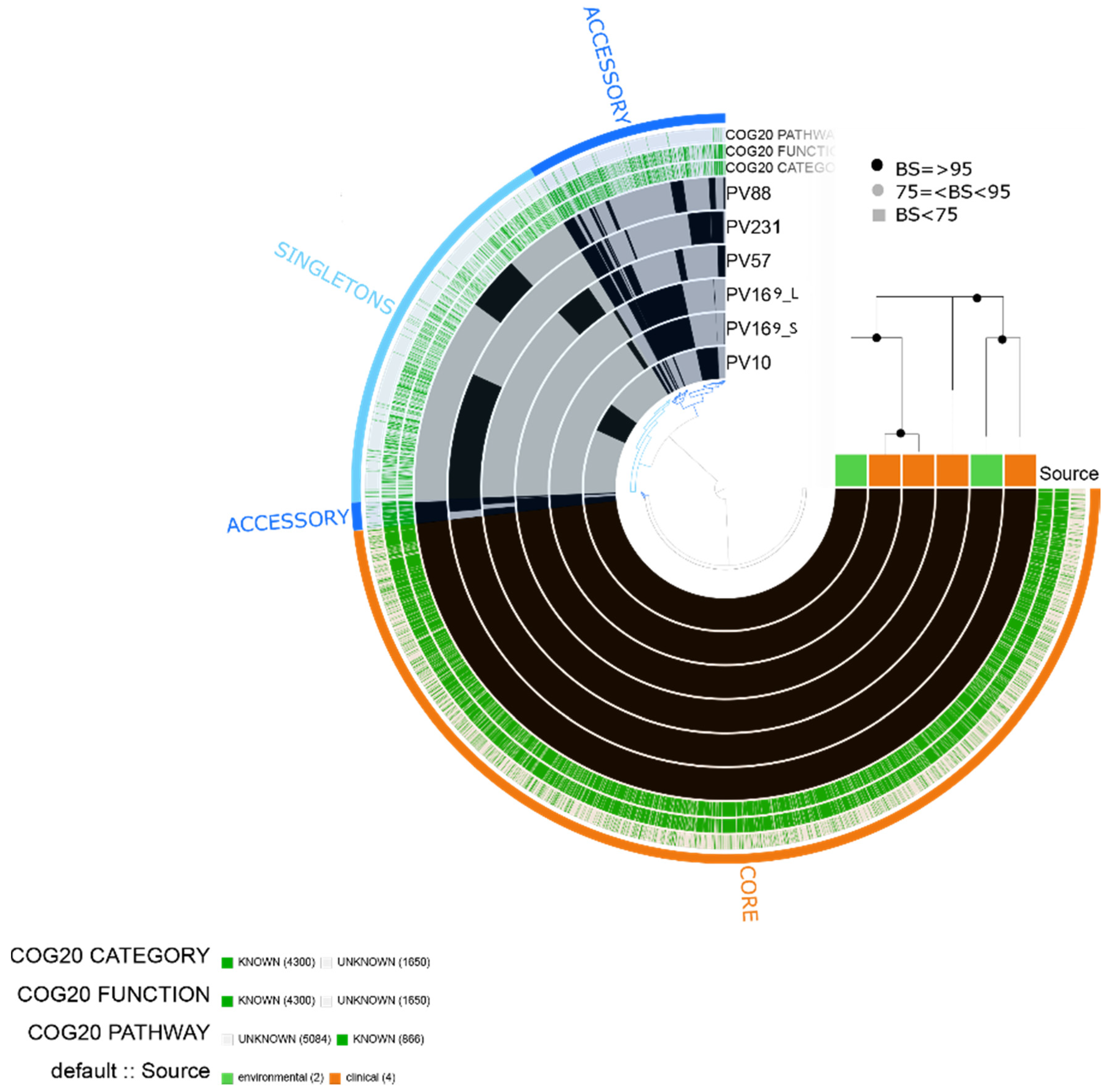
2.1. Characteristics of the Assembled Genomes of Vibrio spp.
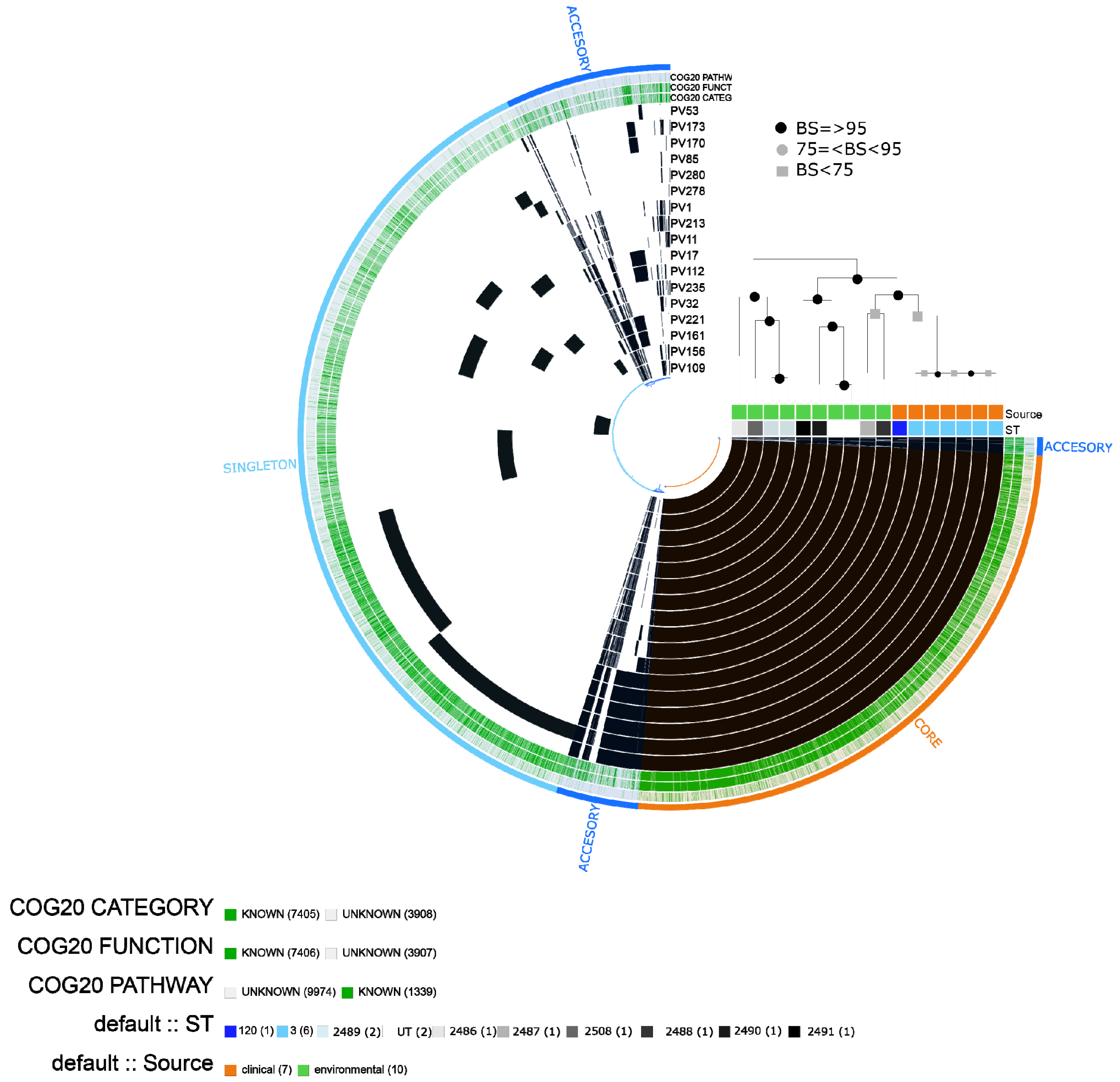
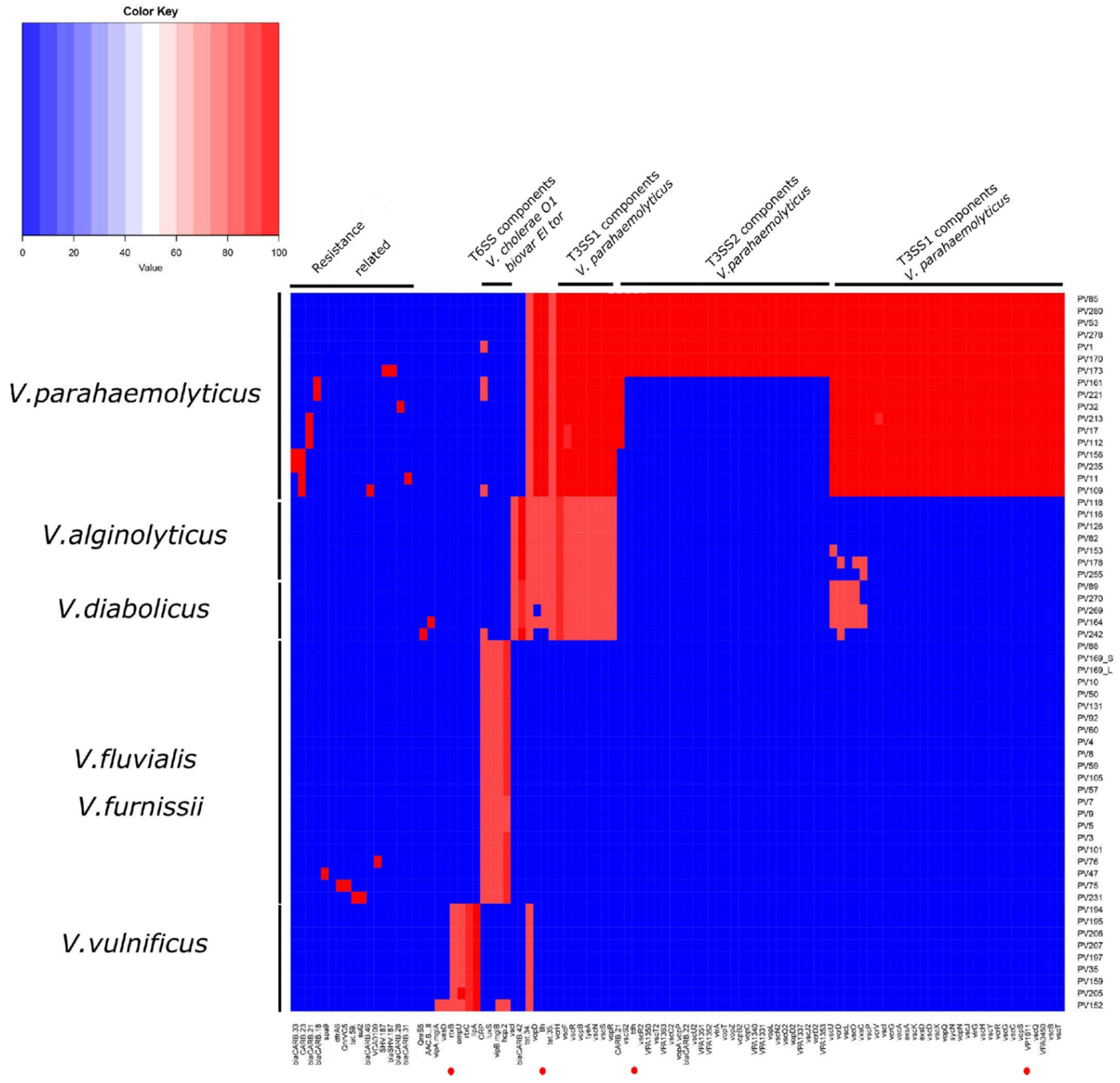
2.2. Vibrio parahaemolyticus
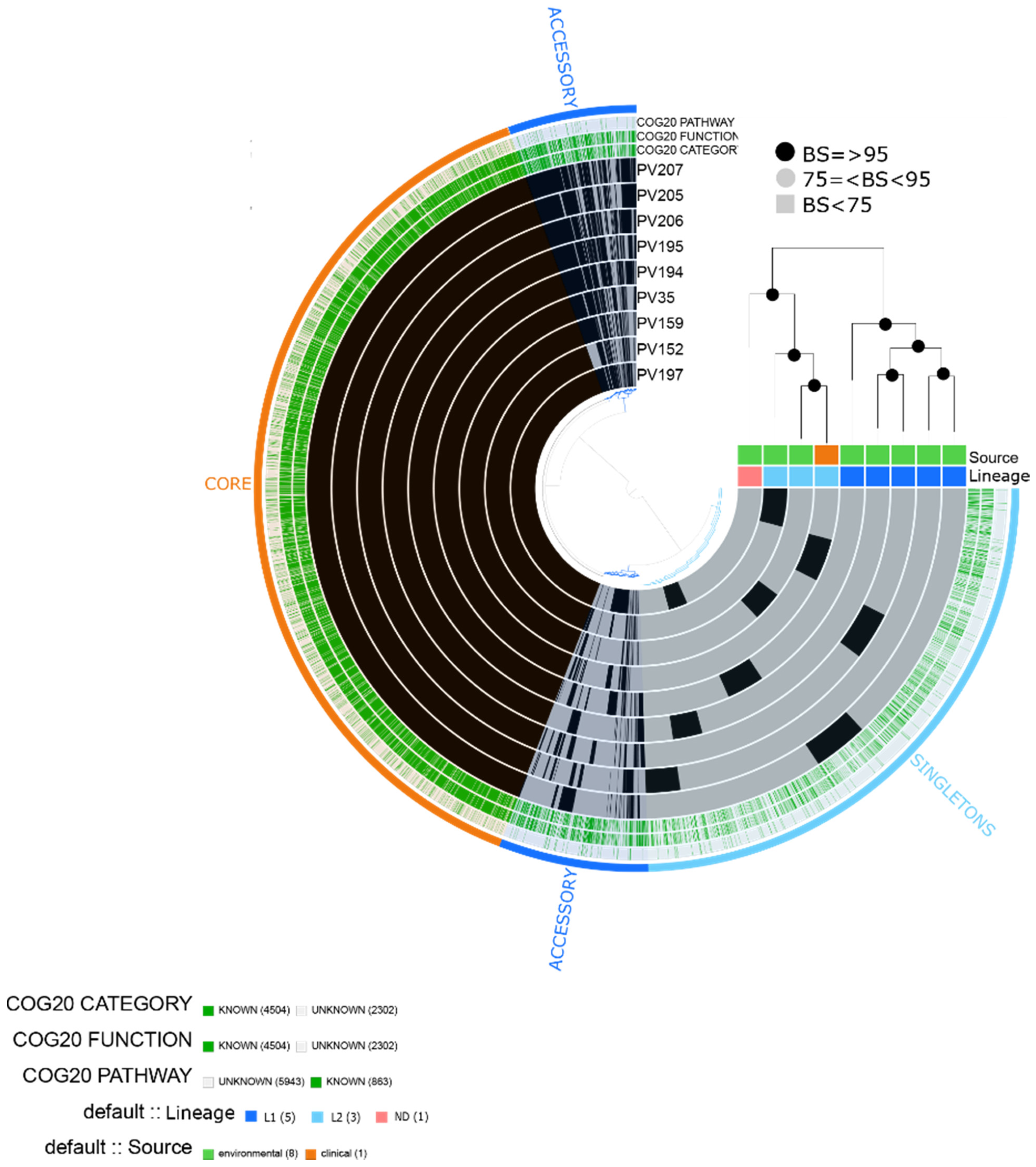
2.3. Vibrio vulnificus
2.4. Vibrio alginolyticus
2.5. Vibrio diabolicus
2.6. Vibrio fluvialis
2.7. Vibrio furnissii
3. Discussion
4. Materials and Methods
4.1. Study Area and Sampling
4.2. Whole Genome Sequencing, De Novo Assembly and Annotation
4.3. Characterization of the Most Important Species in Public Health
4.4. Pangenome and Phylogenomic Analyses
4.5. Identification of Virulence Factors and Antimicrobial Resistance Determinants
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baker-Austin, C.; Oliver, J.D.; Alam, M.; Ali, A.; Waldor, M.K.; Qadri, F.; Martinez-Urtaza, J. Vibrio spp. infections. Nat. Rev. Dis. Primers 2018, 4, 1–19. [Google Scholar] [CrossRef]
- OMS. Cólera. 2019. Available online: https://www.who.int/es/news-room/fact-sheets/detail/cholera (accessed on 25 January 2021).
- CDC. Vibrio Species Causing Vibriosis. 2020. Available online: https://www.cdc.gov/vibrio/index.html (accessed on 2 February 2021).
- Mok, J.S.; Ryu, A.; Kwon, J.Y.; Kim, B.; Park, K. Distribution of Vibrio species isolated from bivalves and bivalve culture environments along the Gyeongnam coast in Korea: Virulence and antimicrobial resistance of Vibrio parahaemolyticus isolates. Food Control 2019, 106, 106697. [Google Scholar] [CrossRef]
- Baker-Austin, C.; Trinanes, J.; Gonzalez-Escalona, N.; Martinez-Urtaza, J. Non-Cholera Vibrios: The Microbial Barometer of Climate Change. Trends Microbiol. 2017, 25, 76–84. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Flórez, C.E.; Cáceres-Manrique, F.D.M. Cólera, ¿se aproxima una nueva pandemia? Med. UIS 2014, 27, 67–83. Available online: http://www.scielo.org.co/scielo.php?script=sci_arttext&pid=S0121-03192014000200008 (accessed on 12 January 2021).
- Escobar, L.E.; Ryan, S.J.; Stewart-Ibarra, A.M.; Finkelstein, J.L.; King, C.A.; Qiao, H.; Polhemus, M.E. A global map of suitability for coastal Vibrio cholerae under current and future climate conditions. Acta Trop. 2015, 149, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Raszl, S.M.; Froelich, B.A.; Vieira, C.R.W.; Blackwood, A.D.; Noble, R.T. Vibrio parahaemolyticus and Vibrio vulnificus in South America: Water, seafood and human infections. J. Appl. Microbiol. 2016, 121, 1201–1222. [Google Scholar] [CrossRef]
- López, M. Acciones de Vigilancia Intensificada de Cólera Ante Posible Reintroducción en Los Componentes de Vigilancia Epidemiológica y Laboratorio, Colombia, 2011–2012. 2012. Available online: https://www.ins.gov.co/buscador-eventos/IQEN/IQEN%20vol%2018%202013%20num%2024.pdf (accessed on 11 November 2021).
- Instituto Nacional de Salud. Vigilancia Fenotípica y Genotípica de Vibrio Cholerae 2010–2013. 1–11. 2013. Available online: https://www.ins.gov.co/buscador/Informacin%20de%20laboratorio/Vigilancia%20C%C3%B3lera%20Colombia%202013.pdf (accessed on 10 October 2021).
- Mohamad, N.; Amal MN, A.; Saad, M.Z.; Yasin, I.S.M.; Zulkiply, N.A.; Mustafa, M.; Nasruddin, N.S. Virulence-associated genes and antibiotic resistance patterns of Vibrio spp. isolated from cultured marine fishes in Malaysia. BMC Vet. Res. 2019, 15, 1–13. [Google Scholar] [CrossRef]
- Bruto, M.; Labreuche, Y.; James, A.; Piel, D.; Chenivesse, S.; Petton, B.; Polz, M.F.; Le Roux, F. Ancestral gene acquisition as the key to virulence potential in environmental Vibrio populations. ISME J. 2018, 12, 2954–2966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, S.; Pipes, S.; Lovell, C.R. Occurrence and significance of pathogenicity and fitness islands in environmental vibrios. AMB Express 2018, 8, 177. [Google Scholar] [CrossRef]
- Nathamuni, S.; Jangam, A.K.; Katneni, V.K.; Selvaraj, A.; Krishnan, K.; Kumar, S.; Avunje, S.; Balasubramaniam, S.; Grover, M.; Alavandi, S.V.; et al. Insights on genomic diversity of Vibrio spp. through Pan-genome analysis. Ann. Microbiol. 2019, 69, 1547–1555. [Google Scholar] [CrossRef]
- Mok, J.S.; Ryu, A.; Kwon, J.Y.; Park, K.; Shim, K.B. Abundance, antimicrobial resistance, and virulence of pathogenic Vibrio strains from molluscan shellfish farms along the Korean coast. Mar. Pollut. Bull. 2019, 149, 110559. [Google Scholar] [CrossRef]
- Thompson, F.L.; Iida, T.; Swings, J. Biodiversity of Vibrios. J. Clin. Microbiol. 2004, 68, 403–431. [Google Scholar] [CrossRef] [Green Version]
- Castillo, D.; Kauffman, K.; Hussain, F.; Kalatzis, P.; Rørbo, N.; Polz, M.F.; Middelboe, M. Widespread distribution of prophage-encoded virulence factors in marine Vibrio communities. Sci. Rep. 2018, 8, 2–10. [Google Scholar] [CrossRef]
- Dobrindt, U.; Hochhut, B.; Hentschel, U.; Hacker, J. Genomic islands in pathogenic and environmental microorganisms. Nat. Rev. Microbiol. 2004, 2, 414–424. [Google Scholar] [CrossRef]
- Nishino, K.; Senda, Y.; Yamaguchi, A. CRP regulator modulates multidrug resistance of Escherichia coli by repressing the mdtEF multidrug efflux genes. J. Antibiot. 2008, 61, 120–127. [Google Scholar] [CrossRef] [Green Version]
- Roig, F.J.; González-Candelas, F.; Sanjuán, E.; Fouz, B.; Feil, E.J.; Llorens, C.; Baker-Austin, C.; Oliver, J.D.; Danin-Poleg, Y.; Gibas, C.J.; et al. Phylogeny of Vibrio vulnificus from the analysis of the core-genome: Implications for intra-species taxonomy. Front. Microbiol. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Thompson, C.C.; Vicente, A.C.P.; Souza, R.C.; Vasconcelos, A.T.R.; Vesth, T.; Alves, N.; Ussery, D.W.; Iida, T.; Thompson, F.L. Genomic taxonomy of vibrios. BMC Evol. Biol. 2009, 9, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavilan, R.G.; Zamudio, M.L.; Martinez-Urtaza, J. Molecular Epidemiology and Genetic Variation of Pathogenic Vibrio parahaemolyticus in Peru. PLoS Negl. Trop. Dis. 2013, 7, e2210. [Google Scholar] [CrossRef]
- Baker-Austin, C.; Jenkins, C.; Dadzie, J.; Mestanza, O.; Delgado, E.; Powell, A.; Bean, T.; Martinez-Urtaza, J. Genomic epidemiology of domestic and travel-associated Vibrio parahaemolyticus infections in the UK, 2008–2018. Food Control 2020, 115, 107244. [Google Scholar] [CrossRef]
- Gonzalez-Escalona, N.; Gavilan, R.G.; Toro, M.; Zamudio, M.L.; Martinez-Urtaza, J. Outbreak of Vibrio parahaemolyticus sequence type 120, Peru, 2009. Emerg. Infect. Dis. 2016, 22, 1235–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velazquez-Roman, J.; León-Sicairos, N.; Hernández-Díaz, L.D.J.; Canizalez-Roman, A. Pandemic Vibrio parahaemolyticus O3: K6 on the American continent. Front. Cell. Infect. Microbiol. 2014, 3, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Boyd, E.F.; Cohen, A.L.V.; Naughton, L.M.; Ussery, D.W.; Binnewies, T.T.; Stine, O.C.; Parent, M.A. Molecular analysis of the emergence of pandemic Vibrio parahaemolyticus. BMC Microbiol. 2008, 8, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.R.; Lee, S.E.; Kook, H.; Yeom, J.A.; Na, H.S.; Kim, S.Y.; Chung, S.S.; Choy, H.E.; Rhee, J.H. Vibrio vulnificus RTX toxin kills host cells only after contact of the bacteria with host cells. Cell. Microbiol. 2008, 10, 848–862. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.K.; Oliver, J.D. Vibrio vulnificus: Disease and pathogenesis. Infect. Immun. 2009, 77, 1723–1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Z.Y.; Hu, C.Q.; Chen, C.; Zhang, L.P.; Ren, C.H. Investigation of seven Vibrio virulence genes among Vibrio alginolyticus and Vibrio parahaemolyticus strains from the coastal mariculture systems in Guangdong, China. Lett. Appl. Microbiol. 2005, 41, 202–207. [Google Scholar] [CrossRef]
- Hernández-Robles, M.F.; Álvarez-Contreras, A.K.; Juárez-García, P.; Natividad-Bonifacio, I.; Curiel-Quesada, E.; Vázquez-Salinas, C.; Quiñones-Ramírez, E.I. Virulence factors and antimicrobial resistance in environmental strains of Vibrio alginolyticus. Int. Microbiol. 2016, 19, 191–198. [Google Scholar] [CrossRef]
- Osorio, C.R. T3SS effectors in Vibrios: Homology in sequence, diversity in biological functions? Virulence 2018, 9, 721–723. [Google Scholar] [CrossRef]
- Song, J.; Liu, X.; Wu, C.; Zhang, Y.; Fan, K.; Zhang, X.; Wei, Y. Isolation, identification and pathogenesis study of Vibrio diabolicus. Aquaculture 2021, 533, 736043. [Google Scholar] [CrossRef]
- Chibani, C.M.; Roth, O.; Liesegang, H.; Wendling, C.C. Genomic variation among closely related Vibrio alginolyticus strains is located on mobile genetic elements. BMC Genom. 2020, 21, 1–14. [Google Scholar] [CrossRef]
- Ramamurthy, T.; Chowdhury, G.; Pazhani, G.P.; Shinoda, S. Vibrio fluvialis: An emerging human pathogen. Front. Microbiol. 2014, 5, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Pan, J.; Gao, H.; Han, Y.; Zhang, A.; Huang, Y.; Liu, P.; Kan, B.; Liang, W. CqsA/LuxS-HapR Quorum sensing circuit modulates type VI secretion system VflT6SS2 in Vibrio fluvialis. Emerg. Microbes Infect. 2021, 10, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Li, X.; Xie, Y.; Bi, D.; Sun, J.; Li, J.; Tai, C.; Deng, Z.; Ou, H.Y. ICEberg 2.0: An updated database of bacterial integrative and conjugative elements. Nucleic Acids Res. 2019, 47, D660–D665. [Google Scholar] [CrossRef] [PubMed]
- Cattoir, V.; Poirel, L.; Mazel, D.; Soussy, C.J.; Nordmann, P. Vibrio splendidus as the source of plasmid-mediated QnrS-like quinolone resistance determinants. Antimicrob. Agents Chemother. 2007, 51, 2650–2651. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, A.; Morita, D.; Ghosh, A.; Chowdhury, G.; Mukhopadhyay, A.K.; Okamoto, K.; Ramamurthy, T. Altered Integrative and Conjugative Elements (ICEs) in Recent Vibrio cholerae O1 Isolated From Cholera Cases, Kolkata, India. Front. Microbiol. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Loo, K.Y.; Letchumanan, V.; Law, J.W.F.; Pusparajah, P.; Goh, B.H.; Ab Mutalib, N.S.; He, Y.W.; Lee, L.H. Incidence of antibiotic resistance in Vibrio spp. Rev. Aquac. 2020, 12, 2590–2608. [Google Scholar] [CrossRef]
- Paul, B.; Dixit, G.; Murali, T.S.; Satyamoorthy, K. Genome Based Taxonomic Classification. Genome 2019, 62, 1–17. [Google Scholar] [CrossRef]
- Lepuschitz, S.; Baron, S.; Larvor, E.; Granier, S.A.; Pretzer, C.; Mach, R.L.; Farnleitner, A.H.; Ruppitsch, W.; Pleininger, S.; Indra, A.; et al. Phenotypic and Genotypic Antimicrobial Resistance Traits of Vibrio cholerae Non-O1/Non-O139 Isolated From a Large Austrian Lake Frequently Associated With Cases of Human Infection. Front. Microbiol. 2019, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gennari, M.; Ghidini, V.; Caburlotto, G.; Lleo, M.M. Virulence genes and pathogenicity islands in environmental Vibrio strains nonpathogenic to humans. FEMS Microbiol. Ecol. 2012, 82, 563–573. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, L.P.; Martínez, M.; León, T.; Córdoba, T.; Díaz, P.; Calvo, M.; Montaño, A.; Escandón, P.; Narváez, S.; Vivas, J.; et al. Desarrollo e Implementación de una PCR Multiplex Para la Detección de Cuatro Especies de Vibrio spp. Biomédica 2019, 39. Available online: https://revistabiomedica.org/index.php/biomedica/issue/download/171/58 (accessed on 8 September 2021).
- Instituto Nacional de Salud. Manual de Procedimientos Para la Toma, Conservación y Envío de Muestras al Laboratorio Nacional de Referencia. Dirección Redes en Salud Pública. Available online: https://www.ins.gov.co/Direcciones/RedesSaludPublica/DocumentosdeInteresSRNL/Manual_toma_envio_muestras_INS-2019.pdf (accessed on 20 September 2021).
- Williams; Wilkins. Bergey’s Manual of Systematic Bacteriology. In The Proteobacteria, 2nd ed.; Garrity, G., Ed.; Springer: Berlin/Heidelberg, Germany, 1984; Volume 2. [Google Scholar]
- Instituto Nacional de Salud Dirección Redes en Salud Pública. Guía Para la Vigilancia por Laboratorio de Vibrio Cholerae (p. 17). Dirección Redes en Salud Pública; 2017. Available online: https://www.ins.gov.co/buscador-eventos/Informacin%20de%20laboratorio/Gu%C3%ADa%20para%20la%20vigilancia%20por%20laboratorio%20de%20Vibrio%20cholerae.pdf (accessed on 11 November 2021).
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef] [Green Version]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Seqtk. 2013. Available online: https://github.com/lh3/seqtk (accessed on 17 August 2021).
- Lischer, H.E.L.; Shimizu, K.K. Reference-guided de novo assembly approach improves genome reconstruction for related species. BMC Bioinform. 2017, 18, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. A J. Comput. Mol. Cell Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews; Simon. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2017. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 10 January 2021).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [Green Version]
- Pop, M.; Phillippy, A.; Delcher, A.L.; Salzberg, S.L. Comparative genome assembly. Brief. Bioinform. 2004, 5, 237–248. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Delcher, A.L.; Salzberg, S.L.; Phillippy, A.M. Using MUMmer to identify similar regions in large sequence sets. Curr. Protoc. Bioinform. 2003, 10.3.1–10.3.18. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [Green Version]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. Ggplot2. Wiley Interdiscip. Rev. Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- GitHub—Tseemann/Mlst: Scan Contig Files against PubMLST Typing Schemes. Available online: https://github.com/tseemann/mlst (accessed on 18 August 2021).
- Jolley, K.A.; Bray, J.E.; Maiden, M.C.J. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 2018, 3, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Treangen, T.J.; Ondov, B.D.; Koren, S.; Phillippy, A.M. The harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 2014, 15, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Release FigTree v1.4.4 · Rambaut/Figtree · GitHub. Available online: https://github.com/rambaut/figtree/releases/tag/v1.4.4 (accessed on 20 August 2021).
- Eren, A.M.; Esen, O.C.; Quince, C.; Vineis, J.H.; Morrison, H.G.; Sogin, M.L.; Delmont, T.O. Anvi’o: An advanced analysis and visualization platformfor’omics data. PeerJ 2015, 2015, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Shaiber, A.; Willis, A.D.; Delmont, T.O.; Roux, S.; Chen, L.X.; Schmid, A.C.; Yousef, M.; Watson, A.R.; Lolans, K.; Esen, Ö.C.; et al. Functional and genetic markers of niche partitioning among enigmatic members of the human oral microbiome. BioRxiv 2020, 1–35. [Google Scholar] [CrossRef] [PubMed]
- GitHub—Tseemann/Abricate: Mass Screening of Contigs for Antimicrobial and Virulence Genes. Available online: https://github.com/tseemann/abricate (accessed on 18 August 2021).
- Feldgarden, M.; Brover, V.; Haft, D.H.; Prasad, A.B.; Slotta, D.J.; Tolstoy, I.; Tyson, G.H.; Zhao, S.; Hsu, C.H.; McDermott, P.F.; et al. Validating the AMRFINder tool and resistance gene database by using antimicrobial resistance genotype-phenotype correlations in a collection of isolates. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [Green Version]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| COG20_Category | Enrichment_Score | Unadjusted_p_Value | Adjusted_q_Value | Associated_Isolate_Sources |
|---|---|---|---|---|
| Cell motility|Intracellular trafficking, secretion and vesicular transport | 17 | 3.74 × 10−5 | 3.03 × 10−3 | clinical |
| Cell cycle control, cell division, chromosome partitioning|Cell motility | 13.388 | 2.53 × 10−4 | 3.16 × 10−3 | environmental |
| Amino acid transport and metabolism|Nucleotide transport and metabolism | 13.246 | 2.73 × 10−4 | 3.16 × 10−3 | clinical |
| Cell cycle control, cell division, chromosome partitioning|Coenzyme transport and metabolism | 13.246 | 2.73 × 10−4 | 3.16 × 10−3 | clinical |
| Nucleotide transport and metabolism|Signal transduction mechanisms | 13.246 | 2.73 × 10−4 | 3.16 × 10−3 | clinical |
| Signal transduction mechanisms|Signal transduction mechanisms | 13.246 | 2.73 × 10−4 | 3.16 × 10−3 | clinical |
| Carbohydrate transport and metabolism|Transcription | 13.246 | 2.73 × 10−4 | 3.16 × 10−3 | environmental |
| Cell cycle control, cell division, chromosome partitioning | 10.578 | 1.14 × 10−3 | 1.03 × 10−2 | clinical |
| Signal transduction mechanisms|Intracellular trafficking, secretion and vesicular transport | 10.578 | 1.14 × 10−3 | 1.03 × 10−2 | clinical |
| Intracellular trafficking, secretion and vesicular transport|General function prediction only|Transcription | 9.745 | 1.80 × 10−3 | 1.03 × 10−2 | clinical |
| Nucleotide transport and metabolism | 9.745 | 1.80 × 10−3 | 1.03 × 10−2 | environmental |
| Species | Isolate | Type | GC (%) | Size (kbp) | Predicted Virulence Genes (Min e-Value 0.01) |
|---|---|---|---|---|---|
| V. parahaemolyticus | PV1 | Putative ICE | 44.45 | 195 | VP1611(MAM7), mshE, mshH, mshA, mshL, mshG, mshJ, tcpI |
| V. parahaemolyticus | PV173 | Putative IME | 39.38 | 82 | vcrD2, vscC2, vopL, vscN2, vopB2, vscU2, VPA1351, vopA/vopP, VPA1353, vopZ, vopD2, vopT, vscR2, vscT2, VPA1350, vscJ2, VPA1352, VPA1331, VPA1331, VPA1363, VPA1337, VPA1340, vscS2, vscQ2, acfD, tdh, flhA, yscV/lcrD, flhA, vcrD, yscN, mxiA, flhA |
| V. parahaemolyticus | PV278 | Putative IME | 40.6 | 35 | |
| V. fluvialis | PV3 | Putative ICE with T4SS | 52 | 166 | IlpA, motB, motA |
| V. fluvialis | PV4 | Putative IME | 46.86 | 74 | |
| V. fluvialis | PV9 | Putative IME | 50.94 | 40 | |
| V. fluvialis | PV50 | Putative ICE with T4SS | 48.48 | 233 | tcpI |
| V. fluvialis | PV50 | Putative IME | 45.8 | 77 | |
| V. fluvialis | PV59 | Putative ICE with T4SS | 47.8 | 215 | cqsA |
| V. fluvialis | PV59 | Putative ICE with T4SS | 48.85 | 231 | |
| V. fluvialis | PV60 | Putative ICE with T4SS | 48.4 | 296 | flaC,flaB,flgA,flgJ, flaD, flgT, flgP, flgI, flgL,flgC,flgO,flgF,flgB,cheR,cheV,flgK,flgD,flaA,flgH,flgG,tcpI,flgN |
| V. fluvialis | PV76 | Putative ICE with T4SS | 47.74 | 226 | cqsA |
| V. fluvialis | PV92 | Putative IME with T4SS | 47.99 | 183 | cqsA |
| V. fluvialis | PV92 | Putative IME | 50.62 | 48 | |
| V. fluvialis | PV105 | Putative ICE with T4SS | 49.15 | 165 | flgD |
| V. fluvialis | PV131 | Putative IME | 47.34 | 52 | |
| V. alginolyticus | PV126 | Putative IME | 41.8 | 75 | |
| V. alginolyticus | PV242 | Putative IME | 45.81 | 15 | |
| V. vulnificus | PV35 | Putative IME | 43.19 | 79 | |
| V. vulnificus | PV159 | Putative IME | 40.53 | 15 | |
| V. vulnificus | PV194 | Putative IME | 49.13 | 62 | |
| V. vulnificus | PV207 | Putative IME | 40.18 | 57 | |
| V. furnissii | PV10 | Putative ICE with T4SS | 53.14 | 173 | |
| V. furnissii | PV88 | Putative IME | 45.96 | 80 | |
| V. furnissii | PV169_L | Putative ICE with T4SS | 51.94 | 388 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Duque, A.; Gonzalez-Muñoz, A.; Arboleda-Valencia, J.; Vivas-Aguas, L.J.; Córdoba-Meza, T.; Rodriguez-Rey, G.T.; Díaz-Guevara, P.; Martinez-Urtaza, J.; Wiesner-Reyes, M. Comparative Genomics of Clinical and Environmental Isolates of Vibrio spp. of Colombia: Implications of Traits Associated with Virulence and Resistance. Pathogens 2021, 10, 1605. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10121605
Pérez-Duque A, Gonzalez-Muñoz A, Arboleda-Valencia J, Vivas-Aguas LJ, Córdoba-Meza T, Rodriguez-Rey GT, Díaz-Guevara P, Martinez-Urtaza J, Wiesner-Reyes M. Comparative Genomics of Clinical and Environmental Isolates of Vibrio spp. of Colombia: Implications of Traits Associated with Virulence and Resistance. Pathogens. 2021; 10(12):1605. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10121605
Chicago/Turabian StylePérez-Duque, Alejandra, Andrea Gonzalez-Muñoz, Jorge Arboleda-Valencia, Lizbeth Janet Vivas-Aguas, Tania Córdoba-Meza, Ghennie Tatiana Rodriguez-Rey, Paula Díaz-Guevara, Jaime Martinez-Urtaza, and Magdalena Wiesner-Reyes. 2021. "Comparative Genomics of Clinical and Environmental Isolates of Vibrio spp. of Colombia: Implications of Traits Associated with Virulence and Resistance" Pathogens 10, no. 12: 1605. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens10121605