
Subtyping Options for Microsporum canis Using Microsatellites and MLST: A Case Study from Southern Italy

 ,
,  , and
, and
Abstract
:1. Introduction
2. Results
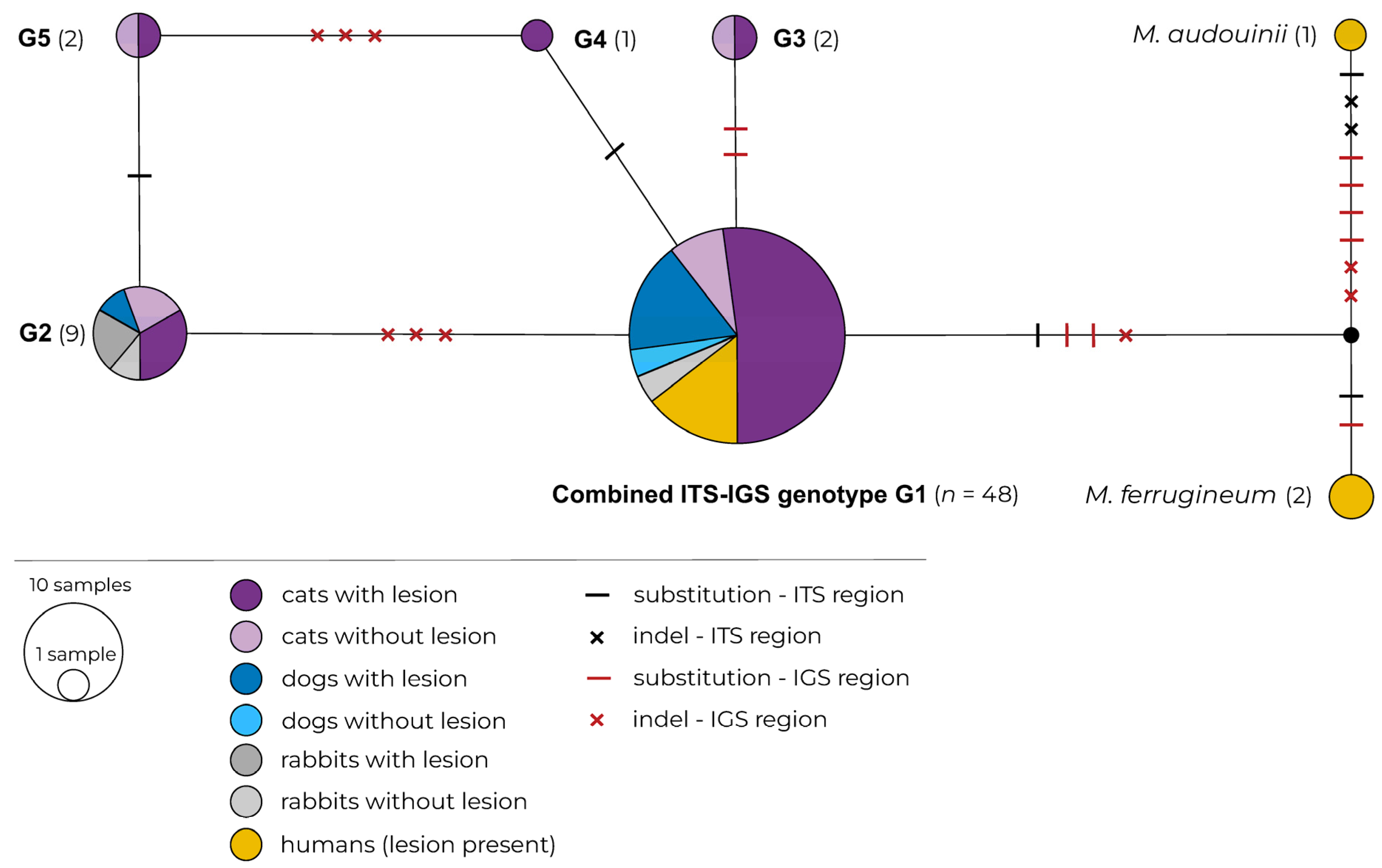
2.1. Multilocus Sequence Typing (MLST)
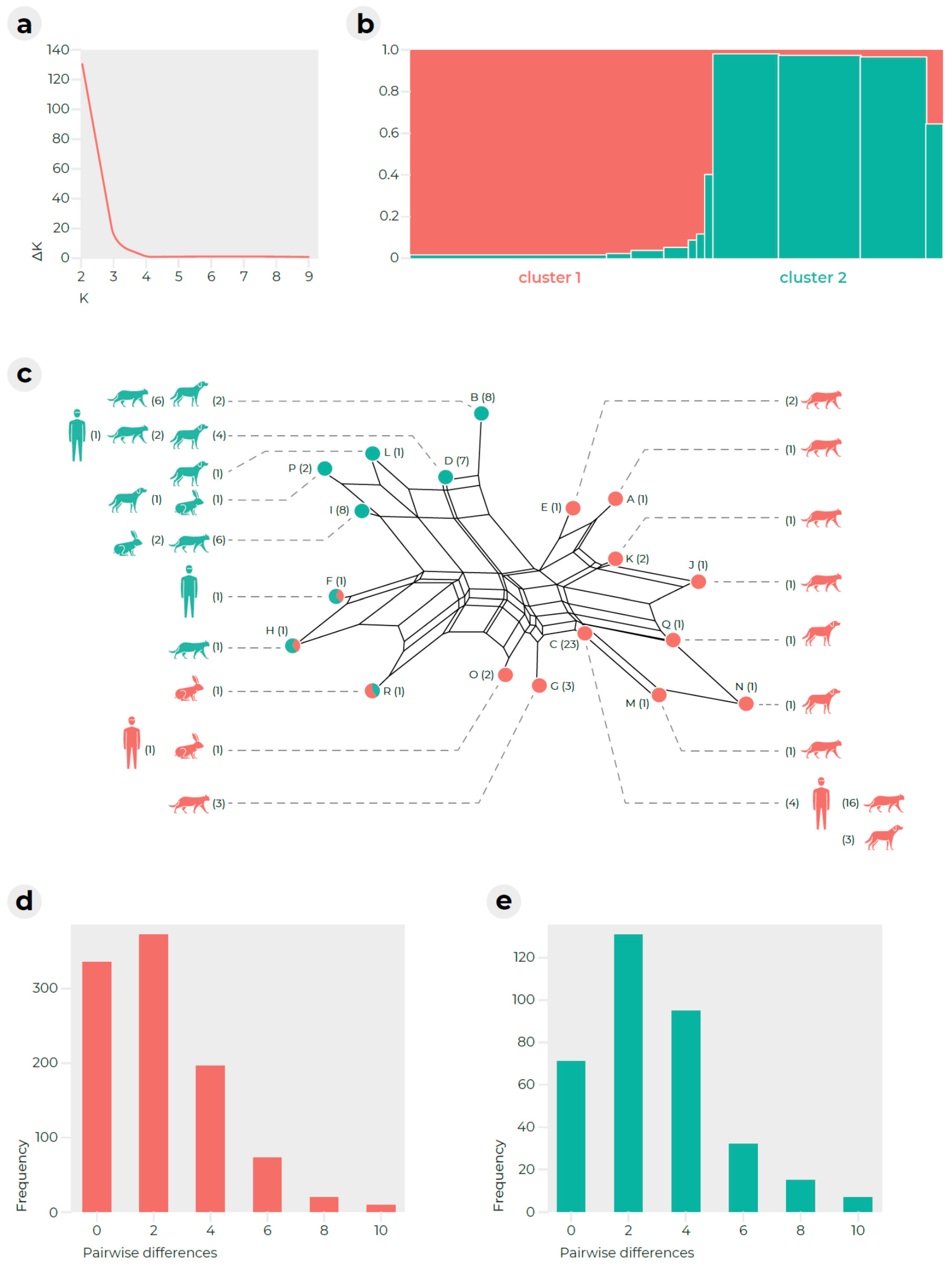
2.2. Multilocus Microsatellite Typing (MLMT)
3. Discussion
4. Materials and Methods
4.1. Source of Isolates
4.2. Multilocus Sequence Typing (MLST)
4.3. Development of Microsatellite Markers
4.4. Statistical Analysis of Microsatellite Data
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Uhrlaß, S.; Krüger, C.; Nenoff, P. Microsporum canis: Current data on the prevalence of the zoophilic dermatophyte in central Germany. Hautarzt 2015, 66, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Aneke, C.I.; Otranto, D.; Cafarchia, C. Therapy and antifungal susceptibility profile of Microsporum canis. J. Fungi 2018, 4, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, R.; De Hoog, G.S.; Presber, W.; Gräser, Y. Virulent genotype of Microsporum canis is responsible for the majority of human infections. J. Med. Microbiol. 2007, 56, 1377–1385. [Google Scholar] [CrossRef] [Green Version]
- Pasquetti, M.; Peano, A.; Soglia, D.; Min, A.R.M.; Pankewitz, F.; Ohst, T.; Gräser, Y. Development and validation of a microsatellite marker-based method for tracing infections by Microsporum canis. J. Dermatol. Sci. 2013, 70, 123–129. [Google Scholar] [CrossRef] [Green Version]
- Cafarchia, C.; Romito, D.; Capelli, G.; Guillot, J.; Otranto, D. Isolation of Microsporum canis from the hair coat of pet dogs and cats belonging to owners diagnosed with M. canis tinea corporis. Vet. Dermatol. 2006, 17, 327–333. [Google Scholar] [CrossRef]
- Gräser, Y.; Frohlich, J.; Presber, W.; de Hoog, G.S. Microsatellite markers reveal geographic population differentiation in Trichophyton rubrum. J. Med. Microbiol. 2007, 56, 1058–1065. [Google Scholar] [CrossRef]
- Abdel-Rahman, S.M. Strain differentiation of dermatophytes. Mycopathologia 2008, 166, 319–333. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, T.; Takeda, K.; Anzawa, K. Molecular markers useful for intraspecies subtyping and strain differentiation of dermatophytes. Mycopathologia 2017, 182, 57–65. [Google Scholar] [CrossRef]
- Taylor, J.W.; Hann-Soden, C.; Branco, S.; Sylvain, I.; Ellison, C.E. Clonal reproduction in fungi. Proc. Natl. Acad. Sci. USA 2015, 112, 8901–8908. [Google Scholar] [CrossRef] [Green Version]
- Dobrowolska, A.; Debska, J.; Kozlowska, M.; Staczek, P. Strains differentiation of Microsporum canis by RAPD analysis using (GACA)4 and (ACA)5 primers. Pol. J. Microbiol. 2011, 60, 145–148. [Google Scholar] [CrossRef]
- Čmoková, A.; Rezaei-Matehkolaei, A.; Kuklová, I.; Kolařík, M.; Shamsizadeh, F.; Ansari, S.; Gharaghani, M.; Miňovská, V.; Najafzadeh, M.J.; Nouripour-Sisakht, S. Discovery of new Trichophyton members, T. persicum and T. spiraliforme spp. nov., as a cause of highly inflammatory tinea cases in Iran and Czechia. Microbiol. Spectr. 2021, 2, e0028421. [Google Scholar] [CrossRef]
- Čmoková, A.; Kolařík, M.; Dobiáš, R.; Hoyer, L.L.; Janouškovcová, H.; Kano, R.; Kuklová, I.; Lysková, P.; Machová, L.; Maier, T.; et al. Resolving the taxonomy of emerging zoonotic pathogens in the Trichophyton benhamiae complex. Fungal Divers. 2020, 104, 333–387. [Google Scholar] [CrossRef]
- de Valk, H.A.; Meis, J.F.G.M.; Klaassen, C.H.W. Microsatellite based typing of Aspergillus fumigatus: Strengths, pitfalls and solutions. J. Microbiol. Methods 2007, 69, 268–272. [Google Scholar] [CrossRef] [PubMed]
- de Groot, T.; Puts, Y.; Berrio, I.; Chowdhary, A.; Meis, J.F. Development of Candida auris short tandem repeat typing and its application to a global collection of isolates. MBio 2020, 11, e02971-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Rezaei-Matehkolaei, A.; Makimura, K.; de Hoog, G.S.; Shidfar, M.R.; Satoh, K.; Najafzadeh, M.J.; Mirhendi, H. Multilocus differentiation of the related dermatophytes Microsporum canis, Microsporum ferrugineum and Microsporum audouinii. J. Med. Microbiol. 2012, 61, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Gräser, Y.; Scott, J.; Summerbell, R. The new species concept in dermatophytes—a polyphasic approach. Mycopathologia 2008, 166, 239–256. [Google Scholar] [CrossRef] [Green Version]
- White, T.C.; Oliver, B.G.; Gräser, Y.; Henn, M.R. Generating and testing molecular hypotheses in the dermatophytes. Eukaryot. Cell 2008, 7, 1238–1245. [Google Scholar] [CrossRef] [Green Version]
- da Costa, F.V.; Farias, M.R.; Bier, D.; de Andrade, C.P.; de Castro, L.A.; da Silva, S.C.; Ferreiro, L. Genetic variability in Microsporum canis isolated from cats, dogs and humans in Brazil. Mycoses 2013, 56, 582–588. [Google Scholar] [CrossRef]
- Watanabe, J.; Anzawa, K.; Mochizuki, T. Molecular Epidemiology of Japanese isolates of Microsporum canis based on multilocus microsatellite typing fragment analysis. Jpn. J. Infect. Dis. 2017, 70, 544–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubka, V.; Nováková, A.; Jurjević, Ž.; Sklenář, F.; Frisvad, J.C.; Houbraken, J.; Arendrup, M.C.; Jørgensen, K.M.; Siqueira, J.P.Z.; Gené, J.; et al. Polyphasic data support the splitting of Aspergillus candidus into two species; proposal of A. dobrogensis sp. nov. Int. J. Syst. Evol. Microbiol. 2018, 68, 995–1011. [Google Scholar] [CrossRef]
- Sklenář, F.; Jurjević, Ž.; Houbraken, J.; Kolařík, M.; Arendrup, M.C.; Jørgensen, K.M.; Siqueira, J.P.Z.; Gené, J.; Yaguchi, T.; Ezekiel, C.N.; et al. Re-examination of species limits in Aspergillus section Flavipedes using advanced species delimitation methods and proposal of four new species. Stud. Mycol. 2021, 99, 100120. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2017, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martins, W.S.; Lucas, D.C.S.; de Souza Neves, K.F.; Bertioli, D.J. WebSat-A web software for microsatellite marker development. Bioinformation 2009, 3, 282–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuelke, M. An economic method for the fuorescent labeling of PCR fragments. Nat. Biotechnol. 2000, 18, 233. [Google Scholar] [CrossRef]
- Alberto, F. MsatAllele_1.0: An R package to visualize the binning of microsatellite alleles. J. Hered. 2009, 100, 394–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huson, D.H. SplitsTree: Analyzing and visualizing evolutionary data. Bioinformatics 1998, 14, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrich, D. AFLPdat: A collection of R functions for convenient handling of AFLP data. Mol. Ecol. Notes 2006, 6, 603–604. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2016. [Google Scholar]
- Excofer, L.; Smouse, P.E.; Quattro, J.M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar] [CrossRef]
- Schneider, S.; Roessli, D.; Excofer, L. ARLEQUIN: A Software for Population Genetics Data Analysis; Version 2.000; University of Geneva: Geneva, Switzerland, 2000; Volume 2. [Google Scholar]
- Agapow, P.M.; Burt, A. Indices of multilocus linkage disequilibrium. Mol. Ecol. Notes 2001, 1, 101–102. [Google Scholar] [CrossRef]
- Burt, A.; Carter, D.A.; Koenig, G.L.; White, T.J.; Taylor, J.W. Molecular markers reveal cryptic sex in the human pathogen Coccidioides immitis. Proc. Natl. Acad. Sci. USA 1996, 93, 770–773. [Google Scholar] [CrossRef] [Green Version]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987. [Google Scholar]
- Kosman, E. Nei’s gene diversity and the index of average differences are identical measures of diversity within populations. Plant Pathol. 2003, 52, 533–535. [Google Scholar] [CrossRef]
- Schonswetter, P.; Tribsch, A. Vicariance and dispersal in the alpine perennial Bupleurum stellatum L. (Apiaceae). Taxon 2005, 54, 725–732. [Google Scholar] [CrossRef] [Green Version]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J.W.; Innis, M.A.; Gelfand, D.H.; Sninsky, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Academic Press Inc.: New York, NY, USA, 1990; pp. 315–322. [Google Scholar]
- Gardes, M.; Bruns, T.D. ITS primers with enhanced specificity for basidiomycetes-application to the identification of mycorrhizae and rusts. Mol. Ecol. 1993, 2, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Shukla, P.K. Use of PCR targeting of internal transcribed spacer regions and single-stranded conformation polymorphism analysis of sequence variation in different regions of rRNA genes in fungi for rapid diagnosis of mycotic keratitis. J. Clin. Microbiol. 2005, 43, 662–668. [Google Scholar] [CrossRef] [Green Version]
- Glass, N.L.; Donaldson, G.C. Development of primer sets designed for use with the PCR to amplify conserved genes from filamentous ascomycetes. Appl. Environ. Microbiol. 1995, 61, 1323–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirhendi, H.; Makimura, K.; de Hoog, G.S. Translation elongation factor 1-α gene as a potential taxonomic and identification marker in dermatophytes. Med. Mycol. 2015, 53, 215–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, S.W. Phylogenetic analysis of Aspergillus species using DNA sequences from four loci. Mycologia 2008, 100, 205–226. [Google Scholar] [CrossRef]
- Carbone, I.; Kohn, L.M. A method for designing primer sets for speciation studies in filamentous ascomycetes. Mycologia 1999, 91, 553–556. [Google Scholar] [CrossRef]
- Kawasaki, M.; Anzawa, K.; Ushigami, T.; Kawanishi, J.; Mochizuki, T. Multiple gene analyses are necessary to understand accurate phylogenetic relationships among Trichophyton species. Med. Mycol. J. 2011, 52, 245–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, I.; Crespo, A.; Divakar, P.K.; Fankhauser, J.D.; Herman-Sackett, E.; Kalb, K.; Nelsen, M.P.; Nelson, N.A.; Rivas-Plata, E.; Shimp, A.D.; et al. New primers for promising single-copy genes in fungal phylogenetics and systematics. Persoonia 2009, 23, 35–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Sample | Source | Lesion | Typing Using ITS and IGS Loci | Multilocus Microsatellite Typing | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ITS-GT | IGS-GT | MLST | TC10 | GT17C | AG12 | GT17B | CAT8 | GT14 | MLMT | Cluster | |||
| CD367 | dog | no | G1 | G1 | G1 | NA | NA | NA | NA | NA | NA | NA | NA |
| CD1131 | cat | yes | G1 | G1 | G1 | 109 | 370 | 374 | 108 | 396 | 102 | A | 1 |
| CD1133 | cat | yes | NA | NA | NA | 103 | 368 | 374 | 108 | 396 | 102 | C | 1 |
| CD1134 | cat | no | G1 | G3 | G3 | 103 | 368 | 374 | 108 | 396 | 102 | C | 1 |
| CD1149 | cat | yes | G1 | G1 | G1 | 103 | 368 | 374 | 108 | 396 | 102 | C | 1 |
| CD1150 | human | yes | G1 | G1 | G1 | 103 | 368 | 374 | 108 | 396 | 102 | C | 1 |
| CD1151 | cat | yes | G1 | G1 | G1 | 103 | 368 | 374 | 108 | 396 | 102 | C | 1 |
| CD1152 | human | yes | G1 | G1 | G1 | 103 | 368 | 374 | 108 | 396 | 102 | C | 1 |
| CD1171 | dog | yes | NA | NA | NA | 103 | 368 | 374 | 108 | 396 | 102 | C | 1 |
| CD1194 | cat | yes | G1 | G1 | G1 | 103 | 368 | 374 | 108 | 396 | 102 | C | 1 |
| CD1195 | cat | yes | G1 | G1 | G1 | 103 | 368 | 374 | 108 | 396 | 102 | C | 1 |
| CD1196 | human | yes | G1 | G1 | G1 | 103 | 368 | 374 | 108 | 396 | 102 | C | 1 |
| CD1211 | cat | yes | G1 | G1 | G1 | 103 | 368 | 374 | 108 | 396 | 102 | C | 1 |
| CD1233 | cat | yes | G1 | G1 | G1 | 103 | 368 | 374 | 108 | 396 | 102 | C | 1 |
| CD1595 | cat | no | G1 | G1 | G1 | 103 | 368 | 374 | 108 | 396 | 102 | C | 1 |
| CD1601 | cat | yes | G1 | G1 | G1 | 103 | 368 | 374 | 108 | 396 | 102 | C | 1 |
| CD1602 | cat | yes | G1 | G1 | G1 | 103 | 368 | 374 | 108 | 396 | 102 | C | 1 |
| CD368 | dog | yes | NA | NA | NA | 103 | 368 | 374 | 108 | 396 | 102 | C | 1 |
| CD382 | cat | yes | G1 | G1 | G1 | 103 | 368 | 374 | 108 | 396 | 102 | C | 1 |
| CD396 | human | yes | G1 | G1 | G1 | 103 | 368 | 374 | 108 | 396 | 102 | C | 1 |
| CD441 | dog | yes | G1 | G1 | G1 | 103 | 368 | 374 | 108 | 396 | 102 | C | 1 |
| CD975 | cat | yes | G1 | G1 | G1 | 103 | 368 | 374 | 108 | 396 | 102 | C | 1 |
| CD976 | cat | yes | G1 | G1 | G1 | 103 | 368 | 374 | 108 | 396 | 102 | C | 1 |
| CD979 | cat | yes | G1 | G1 | G1 | 103 | 368 | 374 | 108 | 396 | 102 | C | 1 |
| CD980 | cat | yes | G1 | G1 | G1 | 103 | 368 | 374 | 108 | 396 | 102 | C | 1 |
| CD1145 | cat | no | G1 | G1 | G1 | 109 | 368 | 374 | 108 | 396 | 102 | E | 1 |
| CD1191 | cat | yes | G1 | G1 | G1 | 103 | 368 | 374 | 108 | 394 | 102 | G | 1 |
| CD383 | cat | no | G1 | G1 | G1 | 103 | 368 | 374 | 108 | 394 | 102 | G | 1 |
| CD978 | cat | n0 | G1 | G1 | G1 | 103 | 368 | 374 | 108 | 394 | 102 | G | 1 |
| CD1235 | cat | yes | G1 | G1 | G1 | 103 | 370 | 374 | 108 | 396 | 104 | J | 1 |
| CD1242 | cat | yes | G1 | G1 | G1 | 103 | 370 | 374 | 108 | 396 | 102 | K | 1 |
| CD1289 | cat | yes | G2 | G1 | G4 | 103 | 370 | 374 | 108 | 396 | 102 | K | 1 |
| CD1565 | cat | yes | G1 | G1 | G1 | 103 | 368 | 368 | 108 | 396 | 102 | M | 1 |
| CD366 | dog | yes | NA | NA | NA | 103 | 368 | 368 | 108 | 396 | 104 | N | 1 |
| CD384 | human | yes | G1 | G1 | G1 | 103 | 368 | 374 | 106 | 396 | 102 | O | 1 |
| CD415 | rabbit | no | G1 | G1 | G1 | 103 | 368 | 374 | 106 | 396 | 102 | O | 1 |
| CD416 | dog | yes | G1 | G1 | G1 | 103 | 368 | 374 | 108 | 396 | 104 | Q | 1 |
| CD448 | rabbit | no | G1 | G1 | G1 | 107 | 368 | 374 | 106 | 394 | 102 | R | 1 |
| CD1132 | dog | yes | G1 | G1 | G1 | 105 | 368 | 374 | 110 | 396 | 104 | B | 2 |
| CD1135 | cat | yes | G1 | G1 | G1 | 105 | 368 | 374 | 110 | 396 | 104 | B | 2 |
| CD1146 | dog | yes | G1 | G1 | G1 | 105 | 368 | 374 | 110 | 396 | 104 | B | 2 |
| CD1148 | cat | yes | G1 | G1 | G1 | 105 | 368 | 374 | 110 | 396 | 104 | B | 2 |
| CD1320 | cat | yes | G1 | G1 | G1 | 105 | 368 | 374 | 110 | 396 | 104 | B | 2 |
| CD1598 | cat | yes | G1 | G3 | G3 | 105 | 368 | 374 | 110 | 396 | 104 | B | 2 |
| CD1600 | cat | yes | G1 | G1 | G1 | 105 | 368 | 374 | 110 | 396 | 104 | B | 2 |
| CD761 | cat | yes | G1 | G1 | G1 | 105 | 368 | 374 | 110 | 396 | 104 | B | 2 |
| CD1143 | human | yes | G1 | G1 | G1 | 105 | 368 | 374 | 110 | 396 | 102 | D | 2 |
| CD1153 | dog | yes | G1 | G1 | G1 | 105 | 368 | 374 | 110 | 396 | 102 | D | 2 |
| CD1229 | dog | yes | G1 | G1 | G1 | 105 | 368 | 374 | 110 | 396 | 102 | D | 2 |
| CD1230 | cat | yes | G1 | G1 | G1 | 105 | 368 | 374 | 110 | 396 | 102 | D | 2 |
| CD1231 | dog | no | G1 | G1 | G1 | 105 | 368 | 374 | 110 | 396 | 102 | D | 2 |
| CD1232 | cat | yes | G1 | G1 | G1 | 105 | 368 | 374 | 110 | 396 | 102 | D | 2 |
| CD1567 | dog | yes | G1 | G1 | G1 | 105 | 368 | 374 | 110 | 396 | 102 | D | 2 |
| CD1190 | human | yes | G1 | G1 | G1 | 107 | 370 | 374 | 112 | 396 | 102 | F | 2 |
| CD1192 | cat | no | G1 | G2 | G2 | 107 | 370 | 374 | 112 | 394 | 102 | H | 2 |
| CD1193 | cat | yes | G1 | G2 | G2 | 105 | 370 | 374 | 110 | 396 | 102 | I | 2 |
| CD1209 | cat | yes | G1 | G2 | G2 | 105 | 370 | 374 | 110 | 396 | 102 | I | 2 |
| CD1306 | cat | yes | G1 | G2 | G2 | 105 | 370 | 374 | 110 | 396 | 102 | I | 2 |
| CD1307 | cat | yes | G2 | G2 | G5 | 105 | 370 | 374 | 110 | 396 | 102 | I | 2 |
| CD1308 | cat | no | G2 | G2 | G5 | 105 | 370 | 374 | 110 | 396 | 102 | I | 2 |
| CD409 | rabbit | no | G1 | G2 | G2 | 105 | 370 | 374 | 110 | 396 | 102 | I | 2 |
| CD412 | rabbit | yes | G1 | G2 | G2 | 105 | 370 | 374 | 110 | 396 | 102 | I | 2 |
| CD760 | cat | no | G1 | G2 | G2 | 105 | 370 | 374 | 110 | 396 | 102 | I | 2 |
| CD1279 | dog | yes | G1 | G1 | G1 | 105 | 368 | 374 | 110 | 394 | 102 | L | 2 |
| CD387 | rabbit | yes | G1 | G2 | G2 | 105 | 370 | 374 | 110 | 394 | 102 | P | 2 |
| CD430 | dog | yes | G1 | G2 | G2 | 105 | 370 | 374 | 110 | 394 | 102 | P | 2 |
| Locus | Primer | Sequence (5′–3′) | 5′-Fluorescent Dye | Product Size (bp) | Reference |
|---|---|---|---|---|---|
| AG12 | forward | CCGAATCCCAAGAACAAGAAC | NED | 368–374 | this study |
| reverse | CATGACCTCCAAGACCATCAC | ||||
| TC10 | forward | TATACGATGTGTACGGCGAGAG | VIC | 103–109 | this study |
| reverse | GTTACAGAGGAACGAACAACCC | ||||
| CAT8 | forward | TTCAAGTCAAAGGAGAGCTGTG | PET | 394–396 | this study |
| reverse | TGCAGTGTATTTGGGTCAAGTC | ||||
| GT17B | foward | GAAGGAGGTATATATGGGTGTG | NED | 106–112 | [4] |
| reverse | GATAAGGTGTTTGGCACTGA | ||||
| GT17C | foward | AGGTGTTTGGCACTGAGC | VIC | 368–370 | [4] |
| reverse | CGAAGAGAAGGAGGTATATATGG | ||||
| GT14 | foward | GGTTTACACGCAGCATGA | PET | 102–104 | [4] |
| reverse | CGTGGCTGAAGAAGTCTACC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aneke, C.I.; Čmoková, A.; Hubka, V.; Rhimi, W.; Otranto, D.; Cafarchia, C. Subtyping Options for Microsporum canis Using Microsatellites and MLST: A Case Study from Southern Italy. Pathogens 2022, 11, 4. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11010004
Aneke CI, Čmoková A, Hubka V, Rhimi W, Otranto D, Cafarchia C. Subtyping Options for Microsporum canis Using Microsatellites and MLST: A Case Study from Southern Italy. Pathogens. 2022; 11(1):4. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11010004
Chicago/Turabian StyleAneke, Chioma Inyang, Adéla Čmoková, Vít Hubka, Wafa Rhimi, Domenico Otranto, and Claudia Cafarchia. 2022. "Subtyping Options for Microsporum canis Using Microsatellites and MLST: A Case Study from Southern Italy" Pathogens 11, no. 1: 4. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens11010004