
Role of TAM Receptors in Antimalarial Humoral Immune Response

1
Department of Veterinary Biochemistry, Kerala Veterinary and Animal Sciences University, Pookode 673576, Kerala, India
2
Center for Cancer Cell Biology, Immunology and Infection, Rosalind Franklin University of Medicine and Science, North Chicago, IL 60047, USA
3
Department of Microbiology and Immunology, Rosalind Franklin University of Medicine and Science, North Chicago, IL 60047, USA
*
Author to whom correspondence should be addressed.
Pathogens 2024, 13(4), 298; https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens13040298
Submission received: 15 March 2024
/
Revised: 29 March 2024
/
Accepted: 31 March 2024
/
Published: 2 April 2024
(This article belongs to the Special Issue Biology of Parasitism)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Immune response against malaria and the clearance of Plasmodium parasite relies on germinal-center-derived B cell responses that are temporally and histologically layered. Despite a well-orchestrated germinal center response, anti-Plasmodium immune response seldom offers sterilizing immunity. Recent studies report that certain pathophysiological features of malaria such as extensive hemolysis, hypoxia as well as the extrafollicular accumulation of short-lived plasmablasts may contribute to this suboptimal immune response. In this review, we summarize some of those studies and attempt to connect certain host intrinsic features in response to the malarial disease and the resultant gaps in the immune response.
1. Introduction
Malaria, transmitted by infected female Anopheles mosquitoes, is caused by an apicomplexan parasite, Plasmodium spp. The major symptoms of malaria range from fever, chills, to severe anemia, sometimes resulting in debilitating neurological symptoms to even death [1]. The disease is widespread in tropical and subtropical regions of the world, predominantly in Sub-Saharan Africa. Disease severity varies due to the differences in the geographical distribution of different strains of Plasmodium and the genetic peculiarities of the host population [2]. Despite decades of investigation, malaria remains a major global public health threat with an estimated 249 million cases and 608,000 related deaths in 2022 [3]. The burden of the disease is further exacerbated by Plasmodium-immune evasion strategies, continuing emergence of drug-resistant strains, and suboptimal humoral immune memory, resulting in repeat infections [4]. Persistent public health measures and interventions to eliminate malaria were showing promise (with incidence rates dropping nearly 18%) over the last decade; however, the COVID-19 pandemic redirected the focus of the scientific and medical community, reversing these gains at least partially.
A successful immune response should provide adequate protection from an ongoing infection and safeguard the host from repeat infections. Identifying molecules and mechanisms underlying the inefficient induction of humoral immune memory and investigating ways to rectify them using immunotherapeutic approaches is vital to reducing the malaria burden. As a part of the Malaria Vaccine Implementation Program, in October 2021, the World Health Organization recommended the roll out of the first ever malaria vaccine RTS, S/AS01, in 18 countries of the sub-Saharan region where malaria is endemic. To further complement this vaccination approach, a second malaria vaccine called R21/Matrix-M was added to the program in October 2023. It remains to be seen how these vaccines will be able to protect against repeat infections and whether the elusive sterile immunity against Plasmodium can be effectively generated.
Our major goal with this review is to bring to attention some cellular and molecular characteristics intrinsic to the pathophysiology of malaria that may be critical to the development of suboptimal effector and memory immune response following Plasmodium infection. We especially focus on the role of a membrane phospholipid, phosphatidylserine (PtS), that becomes exposed on the outer leaflet of the RBC membrane during malaria-induced hemolysis and how it may regulate anti-Plasmodium immune response.
2. Pathophysiology of Malaria
Plasmodium is one of the most complex eukaryotic pathogens completing its heteroxenous life cycle between a free-flying, blood-feeding dipteran vector and a vertebrate host [5]. Within the infected vector, the infectious forms of the parasite (sporozoites) reside in the salivary glands. During a blood meal, the motile sporozoites in the saliva gets injected into the skin of the primary host and rapidly migrate to the liver, infecting and multiplying in hepatocytes (Figure 1). This initiates the asymptomatic ‘liver stage’ of infection. Subsequently, invasive merozoites that are released from the hepatocytes proceed to infect red blood cells (RBCs), where they undergo repeated rounds of maturation, proliferation, egress, and invasion, initiating the ‘blood stage’ of the disease. This repeated cycle of the erythrocytic or blood stage of infection results in extensive hemolysis and anemia and is also the stage when most clinical symptoms of malaria are manifested.
Development of Plasmodium within the RBCs is marked by high demand for cholesterol and membrane phospholipids [6]. Parasites’ demand for membrane components leads to alterations in the RBC membrane, which is characterized by cholesterol depletion and modifications to membrane lipid composition [7,8]. The release of parasites and the parasite-induced membrane changes together contribute to the increased exposure of PtS on the outer membrane of the RBCs (Figure 1). In this review, we cite some of our ex vivo and in vivo studies using PtS blocking antibodies as well as bone marrow chimeras, showing that PtS exposed on the RBC membrane serves as an important immune modulator during Plasmodium infection [9]. We also discuss some of our published studies that show that detection and binding of PtS to certain receptors, predominantly the AXL receptor (detailed below) on B cells, may be critical to the proliferation and accumulation of a certain class of B cells called plasmablasts; these cells exercise an immunosuppressive role and may be key to the suboptimal immune response observed during malaria. Our future studies are aimed at elucidating the precise mechanism by which the pathophysiology of malarial disease regulates this novel pathway of plasmablast differentiation.
3. Anti-Malarial Immune Response
The immune response against malaria is complex and not yet completely understood. Among the different stages of infection, the liver stage is clinically silent compared to the blood stage of infection [10]. However, the liver stage of infection induces a strong innate immune response that is relevant for early control of parasite load and delaying progression to the blood stage. This response in the liver is dependent on interferon γ, type I interferon receptors, and natural killer T cells [11]. Unlike as in viral or bacterial infections, the type I interferon response elicited by malarial parasites has been shown to be independent of toll-like receptors or its downstream signaling, instead mediated by melanoma differentiation-associated gene 5 protein (MDA5) and mitochondrial antiviral signaling (MAVS) protein. The resulting interferon response was necessary for the recruitment of lymphoid cells to the liver [10,12]. Furthermore, studies also show that a fraction of the parasites injected into the skin following a mosquito bite are taken by CD11c+ dendritic cells in the draining lymph node, which eventually prime a potent CD8 T cell response against the sporozoite antigens [13]. These data show that although the liver stage shows minimal symptoms during malaria, it is far from immunologically inert.
The clinical signs observed during the blood stage of infection result from the release of inflammatory cytokines that follows a strong immune B-cell-centric adaptive immune response against Plasmodium [14]. Studies conducted in rodent malaria models, immuno-epidemiological studies, and even control human malaria infection studies have conclusively shown the importance of antibodies in protection from malaria [15,16,17]. Clearance of Plasmodium relies on spatially and temporally layered CD4 T-cell-dependent B cell activation events culminating in the production of parasite-specific antibodies [18,19,20]. As a blood-borne infection, immune activation takes place in and around distinct splenic regions called B cell follicles, which transform into more specialized structures called germinal centers (GC). The clearance of and protection from Plasmodium relies on GC-derived plasma cells (PC), which migrate to the bone marrow (BM) and secrete high-affinity parasite-specific antibodies, and memory B cells (MBCs) that remain in the spleen and circulation to later differentiate swiftly into PCs upon pathogen re-encounter [21]. However, several studies have shown that induction of MBCs following malaria is rather suboptimal and short-lived and even after being exposed to parasitic loads known to be sufficient to induce B cell responses [22,23]. Prior to the appearance of GC-derived PCs or MBCs, another class of B cells with a pronounced endoplasmic reticular (ER) network and Golgi complex accumulated outside the GCs [24] (Figure 2). These extrafollicular B cells called plasmablasts (PB) are short-lived and were believed to secrete low-affinity antibodies against Plasmodium, to essentially ‘buy the host time’ before the GC-derived high-affinity B cell response. Contrarily, our studies in rodent malaria models show that [24] these cells are in fact immunosuppressive and that their depletion enhanced humoral immune response, parasite control, and protection from reinfection (a sign of heightened MBC recall) [24]. Our studies also indicate that the immunosuppressive effect of PB is reversed by administering L-glutamine in drinking water to Plasmodium-infected mice. This was evidenced by enhanced parasite control, GC responses, and protection from lethal Plasmodium challenge along with heightened MBC-driven antibody responses in L-glutamine-administered mice [24]. Moreover, these effects of L-glutamine supplementation disappeared when PBs were depleted, showing that the immunosuppressive effects were indeed due to their (PB) functioning as a nutrient (L-glut) sink [24]. This paradigm shift on the role of early short-lived PB may have major implications, not only in malaria but also in other infections, such as COVID-19, Dengue, and Trypanosomiasis, in which accumulation of PB seems to not effectively control the pathogen or the disease [25,26,27,28]. In light of our published and unpublished observations, we discuss the putative role played by a family of receptor tyrosine kinases that comprises of Tyrosine protein kinase receptor 3(TYRO3), AXL and Membrane receptor tyrosine kinase (MERTK), together abbreviated as TAM (TYRO3, AXL, MERTK) in governing the outcome of GC responses during Plasmodium infection.
4. TAM Receptor Biology
The TAM receptor tyrosine kinase family plays an important role in the phagocytosis of apoptotic cells, innate immune response, autoimmunity, spermatogenesis, and a plethora of other biological functions [29,30,31]. Unlike many other receptors, TAM receptors utilize serum factors such as Protein-S (PROS1) and growth arrest-specific-6 (GAS-6), as bridging molecules to bind to their ligands [32,33] (Figure 3).
Protein-S (PROS1) and growth arrest-specific-6 (GAS-6) are vitamin K-dependent soluble proteins, sharing 43% of the amino acid identity among them. PROS1 was originally identified as a cofactor for Protein C, which negatively regulates blood coagulation by degrading clotting factors Va and VIIIa [34]. PROS1 is capable of activating TYRO3 and MERTK but not AXL [32,35,36,37,38,39]. GAS6, on the other hand, while capable of binding to all three receptors (TAM), binds to AXL with the highest affinity [40]. Among the multiple families of PtS-binding proteins, the most studied are those with a Gla (γ-carboxylated glutamic acid residues) domain [41,42,43]. Both PROS1 and GAS6 are Gla domain-containing proteins which are capable of binding to negatively charged phospholipid groups in a calcium- and vitamin K-dependent manner [44,45,46]. The ability of PROS1/GAS6 to activate TAM receptors depends on the carboxylation of the Gla domain and its binding to phosphatidylserine (PtS) [47]. Apart from the Gla domain, the SHBG domain (composed of two laminin G domains) and four EGF-related domains are also part of the receptor complex. The SHBG domain is positioned at the carboxy-terminal end, and the EGF-related domain is present in between the SHBG and Gla domains [30,32,48] (Figure 3).
Expression of TAM receptors vary based on the cell type, even within the same lineage. For example, bone marrow-derived dendritic cells show higher expression of AXL than MERTK, whereas the bone marrow-derived macrophage population shows the opposite trend [49]. Both cell populations show very low expression of TYRO3. The expression of AXL was controlled at the translational level, but MERTK was found to be regulated transcriptionally [49]. Intriguingly, AXL and GAS6 exhibit a peculiar relationship, where AXL specifically needs GAS6 for its activation, while GAS6 relies on AXL for its stable maintenance, showing a co-dependency between the receptor and its ligand [49].
All receptor tyrosine kinases except insulin receptors exist as monomers in the cell membrane [50]. Upon ligand binding, RTKs are dimerized and activated, although each receptor employs different mechanisms of receptor dimerization [51,52,53]. The extracellular amino-terminal regions comprising of an Ig-like domain of TAM proteins bind with the laminin G domain of SHBG in PROS1/GAS6, followed by tandem repeats of fibronectin-type repeats, a transmembrane domain, and an intracellular tyrosine kinase domain [54,55,56,57,58,59]. TAM ligand binding leads to receptor dimerization in two steps, with a high-affinity GAS6/TAM complex forming first and autophosphorylation of the intracellular kinase domain [55,60]. The TAM receptor, upon binding to their ligand, has been shown to both homo- or heterodimerize to initiate signaling [61,62]. AXL exerts its effects by signaling via PI3K, MAPKs, and EGFR/PKC/mTOR pathways [63,64], depending on the nature of dimerization. Heterodimerization is reported when one of the TAM receptors is overexpressed. When TYRO3 is overexpressed, AXL signaling shifts from its predominant PI3K pathway to the MAPK pathway [61], indicating that the dimerization pattern of TAM receptors affects the outcome of GAS6-mediated signaling.
Phosphatidylserine (PtS) is a phospholipid normally restricted to the inner and cytoplasm-facing leaflets of the plasma membrane [65]. The caspase-mediated cleavage of flippases, a P4-type ATPase that restricts PtS to the inner leaflet, and the activation of a Ca2+-dependent enzyme scramblase lead to redistribution of PtS to the outer leaflet. The presence of PtS on the outer leaflet of the plasma membrane acts as an ‘eat me’ signal for the phagocytic immune cells [41,66,67,68]. Phagocytosis of the apoptotic cells that expose PtS on their outer membrane is carried out by MERTK-mediated signaling, which is elicited by either GAS6 or PROS1. Inflammatory signals promote PtS–GAS6-mediated phosphorylation of AXL and enhancement of AXL-mediated phagocytosis. This demonstrates that AXL and MERTK participate in phagocytosis under varying conditions, as they respond to different stimuli by interacting with different ligands [49]. This suggests that pathogens utilize the AXL–GAS6 pathway to gain entry into cells and/or even hide from the host immune response preventing their clearance [24,69,70,71,72,73,74,75].
5. TAM Receptors and Infections
5.1. AXL and Viral Infections
Several studies focusing on viral infections have documented that the immunosuppressive role of AXL occurs mainly through inhibition of the interferon-stimulating genes (ISG) [31,71,76,77]. Many enveloped viruses utilize PtS on their envelope to exercise apoptotic mimicry (‘eat me signal’) to attach to the host GAS6 molecule and thus AXL to elicit a signaling cascade facilitating AXL-mediated cell entry and inhibition of ISGs. This interference in the natural anti-viral response disrupts the innate immune response, facilitating the establishment of infection [78]. This hijacking of the GAS6–AXL signaling axis is reported in Ebola virus [79], Zika virus [71], Respiratory Syncytial Virus (RSV) [80], SARS-CoV-2 [74], and Parvovirus [75].
Region-specific deletion/inhibition studies in AXL revealed that the intracellular kinase domain is essential for the internalization of the virus [69,81]. While the presence or absence of TAM receptors (especially AXL) did not seem to affect Zika virus cell entry [70], AXL seems to attenuate the Zika virus-induced activation of type I interferon genes, thereby promoting virus infection [71]. This was confirmed by gene knock-out studies involving AXL and IFNαR [72]. In contrast, experiments in human glioblastoma cell lines using CRISPR-Cas-mediated knockdown of AXL showed its requirement for Zika virus cell entry and infection [73]. Thus, the role of AXL receptor appears to be different depending on the cell types they are expressed on.
Recent reports also suggest that AXL may act as a receptor to facilitate SARS-CoV-2 virus entry into pulmonary and bronchial epithelial cells. Interestingly, SARS-CoV-2 binding to AXL is independent of GAS6 or the virus envelope-associated PtS [74]. Human parvovirus B19 also shows similarities in AXL utilization, where AXL acts as a co-receptor for infection in human erythroid progenitor cells [75].
AXL appears to also have an indirect effect during RSV infection. RSV infection drives increased production of GAS6, leading to heightened AXL stimulation. This, in turn, impairs antibacterial immunity by attenuating caspase 1 activation and IL-18 release, rendering patients vulnerable to secondary pneumococcal infections. Elevated GAS6 levels also cause polarization of M0 to M2 instead to an M1 phenotype, further attenuating the antibacterial activity [82]. On the other hand, the absence of AXL promotes IL-1α from pyroptotic macrophages, compromising the integrity of the blood–brain barrier and permitting the neural invasion of Japanese encephalitis virus (JEV) [83]. Contrary to studies showing an inhibitory effect of AXL on ISGs, some studies have shown that AXL enhances interferon-stimulated genes, allowing for increased vulnerability to JEV infection of neurons [84], further reiterating the cell-specific role of AXL in regulating the immune response.
These contrasting findings in various infectious disease settings not only highlight the diverse roles played by AXL in modulating immune response and disease outcomes but also emphasize the importance of further investigating this seemingly ubiquitous PtS–AXL–GAS6 signaling axis.
5.2. AXL and Malaria
The role of AXL in malaria is poorly understood and investigated. Plasmodium parasites are not known to utilize TAM receptors to gain entry into the host cells like viruses do (discussed in Section 5.1). The only report connecting AXL and malaria stems from our studies showing that AXL ligation and signaling may be key to the accumulation of immunosuppressive pre-GC plasmablasts [9].
As mentioned earlier (Section 3), plasmablasts induced by Plasmodium infection express high levels of amino acid transporter molecules and act as a glutamine sink [24]. Our studies also revealed that plasmablast accumulation may require polyclonal activation signals, but it is largely independent of antigen-specific mechanisms and may still rely on certain pathophysiological features related to hemolysis. In fact, hemolysis-mediated exposure of PtS on RBCs during malaria has been previously reported [85]. Furthermore, the observation that Plasmodium-induced plasmablasts preferentially secrete anti-PtS antibodies led us to speculate that PtS may be involved in the differentiation of these cells. Importantly, our RNA sequencing studies revealed that resting and activated B cells that appear before the accumulation of plasmablasts expressed higher levels of many PtS receptors including AXL. Further studies using ex vivo stimulation approaches and in vivo blocking studies revealed that by binding to PtS, AXL may play a pivotal role in the expansion of polyclonal plasmablast [9] during Plasmodium infection. The direct cell-to-cell contacts required between PtS-exposed RBCs and AXL-expressing B cells for driving plasmablasts differentiation would also mean that plasmablasts may be derived from B cells located in the marginal zone rather than from the splenic white pulp due to their close proximity to one another. After all, marginal zone B cells localized in the red pulp have been shown to interact with RBCs and are considered important for antibody responses against blood-borne pathogens [86]. It is also important to note that, while B cell activation, differentiation, and proliferation preferentially happen in the white pulp, foci of antibody-secreting B cells have been observed in the red pulp [87,88,89]. Whether these B cells are indeed short-lived plasmablasts or are germinal-center-derived needs to be validated.
Splenic red-pulp macrophages that have been shown to play a major role in RBC clearance may also act as a bridge between PtS-exposed RBCs and B cells that are destined to become plasmablasts. Expression of TAM receptors, especially AXL, has been documented in macrophages [33], although there is no evidence to suggest that this may regulate anti-Plasmodium humoral immune response. However, the gross disruption of splenic architecture as well as the formation of the blood–spleen barrier [89] early during Plasmodium infections likely prevents the interaction of red-pulp macrophages and RBCs. These notions regarding the role of marginal zone B cells as the precursors of polyclonal immunosuppressive plasmablasts or lack of role played by red-pulp macrophages in this phenomenon need to be further validated.
Nevertheless, most of our data indicate that accumulation of plasmablasts that are detrimental to an upcoming or ongoing germinal center response are unique to malaria and, to some extent, Trypanosoma infection [9]. However, what pathophysiological factors specific to malaria contribute to this phenomenon is still not clear. Hemolysis and systemic hypoxia develop as early as four days post infection with Plasmodium [90,91]; higher levels of HIF1α were also noticed in placental malaria cases [92]. Targeted metabolomics data of Plasmodium-infected mouse serum showed elevated orotate and lactate levels (unpublished data), a characteristic feature of systemic hypoxia [93]. Resting and activated B cells in the spleen of the Plasmodium-infected mice also showed a more hypoxic signature in comparison with plasmablasts, indicating that proliferation of plasmablasts is favored by a cellular hypoxic environment. Studies have also independently shown that alleviation of the hypoxic status increases the probability of survival in a mouse cerebral malaria model [94,95]. Understanding how systemic hypoxia promotes the progression of malaria is crucial in designing strategies to improve anti-malarial immunity.
There have been several attempts to establish a molecular link between AXL and hypoxia-inducible factors. Studies have shown that hypoxia and hypoxia-inducible factor (HIF)1 can stabilize AXL expression and signaling [96,97] as well as support plasma cell differentiation [98,99]. HIF1 and HIF2 are also known to directly bind to the hypoxia response elements in the AXL promoter region and enhance its expression [97]. Furthermore, exogenous supplementation of GAS6 was shown to downregulate AXL protein expression without affecting the transcription of Axl mRNA in in vitro tumor environment [96]. This indicates a regulatory role of GAS6 over AXL at the translational level, although reports supporting this claim are few and far between. There are also reports that AXL increases HIF levels during hypoxia [100]. To further add complexity to the already sophisticated signaling axis, overexpression of AXL has been shown to increase translation of HIF1α without altering the mRNA levels, and this effect of AXL was dependent on PI3K/Akt/p70S6K signaling cascade [101]. Thus, there are different levels of regulation of the AXL–GAS6–PtS signaling axis, and hypoxia appears to have a significant yet underappreciated role in his phenomenon. Given that malarial infection is characterized by (Figure 4) (i) the appearance of immunosuppressive plasmablasts, (ii) hemolytic events, (iii) systemic hypoxia, and (iv) PtS exposure, more mechanistic studies aimed at investigating these pathophysiological features may reveal critical new information to devise immunomodulatory therapies to enhance anti-malarial immunity.
6. Summary
Studies show that gradual acquisition of protective or clinical immunity is evident following multiple malarial episodes; however, sterilizing immunity is rarely achieved. This makes the older population in malaria endemic regions relatively asymptomatic during the disease, but still acting as critical reservoirs of the pathogen, facilitating inadvertent transmission. Among the many factors that may result in the suppression of an optimal anti-malarial immune response (so that sterilizing immunity is achieved), we identified that the pre-GC extrafollicular plasmablast may also contribute to this phenomenon. This population of B cells that appear early during Plasmodium infection is short-lived, yet metabolically hyperactive and seem to deprive the GC B cells from key nutrients, especially L-glutamine. Our recent studies also show that a key member of the TAM receptor tyrosine kinases, AXL, plays a significant role in the differentiation of these immunosuppressive cells by recognizing and binding to PtS exposed on the surface of RBCs. Hemolytic anemia and the ensuing hypoxia during malaria may likely stabilize AXL, thereby facilitating the cascade of events leading to the differentiation of plasmablasts. While the role of AXL in tumor progression, phagocytosis, cell proliferation, immune cell interactions, and autoimmunity is well studied, its role in B-cell-mediated immune response is not yet fully understood. Moreover, given that all three TAM receptors bind to similar ligand(s), it is possible that TYRO3 and MERTK may also have synergistic or redundant roles in modulating anti-malarial immune responses. Further mechanistic insight can be achieved by investigating their temporal and cell-specific role during infection. Such studies will not only aid in the development of specific inhibitors of these receptors (some are already in clinical trials) but will also help in exploring the possibility of using them in combinatorial therapeutics alongside anti-malarial ones.
Author Contributions
Writing—original draft preparation, L.J., writing—review and editing, L.J. and R.V. All authors have read and agreed to the published version of the manuscript.
Funding
The research was funded by RFUMS startup funds to R.V. and an RFUMS-DePaul pilot grant.
Acknowledgments
The authors thank Fabio Re for the critical feedback. The figures in the manuscript were prepared using BioRender.com.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Milner, D.A., Jr. Malaria Pathogenesis. Cold Spring Harb. Perspect. Med. 2018, 8, a025569. [Google Scholar] [CrossRef] [PubMed]
- Ashley, E.A.; Pyae Phyo, A.; Woodrow, C.J. Malaria. Lancet 2018, 391, 1608–1621. [Google Scholar] [CrossRef] [PubMed]
- WHO. World Malaria Report 2023; World Health Organization: Geneva, Switzerland, 2023. [Google Scholar]
- Tran, T.M.; Li, S.; Doumbo, S.; Doumtabe, D.; Huang, C.Y.; Dia, S.; Bathily, A.; Sangala, J.; Kone, Y.; Traore, A.; et al. An intensive longitudinal cohort study of Malian children and adults reveals no evidence of acquired immunity to Plasmodium falciparum infection. Clin. Infect. Dis. 2013, 57, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Perkins, S.L. Malaria’s Many Mates: Past, Present, and Future of the Systematics of the Order Haemosporida. J. Parasitol. 2014, 100, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, L.L.; Howard, R.J.; Aikawa, M.; Taraschi, T.F. Modification of host cell membrane lipid composition by the intra-erythrocytic human malaria parasite Plasmodium falciparum. Biochem. J. 1991, 274 Pt 1, 121–132. [Google Scholar] [CrossRef]
- Tokumasu, F.; Crivat, G.; Ackerman, H.; Hwang, J.; Wellems, T.E. Inward cholesterol gradient of the membrane system in P. falciparum-infected erythrocytes involves a dilution effect from parasite-produced lipids. Biol. Open 2014, 3, 529–541. [Google Scholar] [CrossRef]
- Hernandez-Castaneda, M.A.; Lavergne, M.; Casanova, P.; Nydegger, B.; Merten, C.; Subramanian, B.Y.; Matthey, P.; Lannes, N.; Mantel, P.Y.; Walch, M. A Profound Membrane Reorganization Defines Susceptibility of Plasmodium falciparum Infected Red Blood Cells to Lysis by Granulysin and Perforin. Front. Immunol. 2021, 12, 643746. [Google Scholar] [CrossRef]
- Vijay, R.; Guthmiller, J.J.; Sturtz, A.J.; Crooks, S.; Johnson, J.T.; Li, L.; Lan, L.Y.; Pope, R.L.; Chen, Y.; Rogers, K.J.; et al. Hemolysis-associated phosphatidylserine exposure promotes polyclonal plasmablast differentiation. J. Exp. Med. 2021, 218, e20202359. [Google Scholar] [CrossRef]
- Holz, L.E.; Fernandez-Ruiz, D.; Heath, W.R. Protective immunity to liver-stage malaria. Clin. Transl. Immunol. 2016, 5, e105. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.L.; Sack, B.K.; Baldwin, M.; Vaughan, A.M.; Kappe, S.H.I. Interferon-mediated innate immune responses against malaria parasite liver stages. Cell Rep. 2014, 7, 436–447. [Google Scholar] [CrossRef]
- Liehl, P.; Zuzarte-Luis, V.; Chan, J.; Zillinger, T.; Baptista, F.; Carapau, D.; Konert, M.; Hanson, K.K.; Carret, C.; Lassnig, C.; et al. Host-cell sensors for Plasmodium activate innate immunity against liver-stage infection. Nat. Med. 2014, 20, 47–53. [Google Scholar] [CrossRef]
- Kurup, S.P.; Anthony, S.M.; Hancox, L.S.; Vijay, R.; Pewe, L.L.; Moioffer, S.J.; Sompallae, R.; Janse, C.J.; Khan, S.M.; Harty, J.T. Monocyte-Derived CD11c(+) Cells Acquire Plasmodium from Hepatocytes to Prime CD8 T Cell Immunity to Liver-Stage Malaria. Cell Host Microbe 2019, 25, 565–577 e566. [Google Scholar] [CrossRef] [PubMed]
- Angulo, I.; Fresno, M. Cytokines in the pathogenesis of and protection against malaria. Clin. Vaccine Immunol. 2002, 9, 1145–1152. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Butcher, G.A.; Crandall, R.B. Action of malarial antibody in vitro. Nature 1969, 223, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.Y.; Hodder, A.N.; Lin, C.S.; Hill, D.L.; Li Wai Suen, C.S.; Schofield, L.; Siba, P.M.; Mueller, I.; Cowman, A.F.; Hansen, D.S. Antibodies to the Plasmodium falciparum Proteins MSPDBL1 and MSPDBL2 Opsonize Merozoites, Inhibit Parasite Growth, and Predict Protection From Clinical Malaria. J. Infect. Dis. 2015, 212, 406–415. [Google Scholar] [CrossRef]
- Richards, J.S.; Stanisic, D.I.; Fowkes, F.J.; Tavul, L.; Dabod, E.; Thompson, J.K.; Kumar, S.; Chitnis, C.E.; Narum, D.L.; Michon, P.; et al. Association between naturally acquired antibodies to erythrocyte-binding antigens of Plasmodium falciparum and protection from malaria and high-density parasitemia. Clin. Infect. Dis. 2010, 51, e50–e60. [Google Scholar] [CrossRef]
- Cohen, S.; Mc, G.I.; Carrington, S. Gamma-globulin and acquired immunity to human malaria. Nature 1961, 192, 733–737. [Google Scholar] [CrossRef]
- Akter, J.; Khoury, D.S.; Aogo, R.; Lansink, L.I.M.; SheelaNair, A.; Thomas, B.S.; Laohamonthonkul, P.; Pernold, C.P.S.; Dixon, M.W.A.; Soon, M.S.F.; et al. Plasmodium-specific antibodies block in vivo parasite growth without clearing infected red blood cells. PLoS Pathog. 2019, 15, e1007599. [Google Scholar] [CrossRef]
- Kana, I.H.; Singh, S.K.; Garcia-Senosiain, A.; Dodoo, D.; Singh, S.; Adu, B.; Theisen, M. Breadth of Functional Antibodies Is Associated With Plasmodium falciparum Merozoite Phagocytosis and Protection Against Febrile Malaria. J. Infect. Dis. 2019, 220, 275–284. [Google Scholar] [CrossRef]
- Stephens, R.; Ndungu, F.M.; Langhorne, J. Germinal centre and marginal zone B cells expand quickly in a second Plasmodium chabaudi malaria infection producing mature plasma cells. Parasite Immunol. 2009, 31, 20–31. [Google Scholar] [CrossRef]
- Weiss, G.E.; Traore, B.; Kayentao, K.; Ongoiba, A.; Doumbo, S.; Doumtabe, D.; Kone, Y.; Dia, S.; Guindo, A.; Traore, A.; et al. The Plasmodium falciparum-specific human memory B cell compartment expands gradually with repeated malaria infections. PLoS Pathog. 2010, 6, e1000912. [Google Scholar] [CrossRef] [PubMed]
- Wendel, B.S.; He, C.; Qu, M.; Wu, D.; Hernandez, S.M.; Ma, K.Y.; Liu, E.W.; Xiao, J.; Crompton, P.D.; Pierce, S.K.; et al. Accurate immune repertoire sequencing reveals malaria infection driven antibody lineage diversification in young children. Nat. Commun. 2017, 8, 531. [Google Scholar] [CrossRef] [PubMed]
- Vijay, R.; Guthmiller, J.J.; Sturtz, A.J.; Surette, F.A.; Rogers, K.J.; Sompallae, R.R.; Li, F.; Pope, R.L.; Chan, J.A.; de Labastida Rivera, F.; et al. Infection-induced plasmablasts are a nutrient sink that impairs humoral immunity to malaria. Nat. Immunol. 2020, 21, 790–801. [Google Scholar] [CrossRef] [PubMed]
- Bockstal, V.; Guirnalda, P.; Caljon, G.; Goenka, R.; Telfer, J.C.; Frenkel, D.; Radwanska, M.; Magez, S.; Black, S.J. T. brucei infection reduces B lymphopoiesis in bone marrow and truncates compensatory splenic lymphopoiesis through transitional B-cell apoptosis. PLoS Pathog. 2011, 7, e1002089. [Google Scholar] [CrossRef] [PubMed]
- Obishakin, E.; de Trez, C.; Magez, S. Chronic Trypanosoma congolense infections in mice cause a sustained disruption of the B-cell homeostasis in the bone marrow and spleen. Parasite Immunol. 2014, 36, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Radwanska, M.; Guirnalda, P.; De Trez, C.; Ryffel, B.; Black, S.; Magez, S. Trypanosomiasis-induced B cell apoptosis results in loss of protective anti-parasite antibody responses and abolishment of vaccine-induced memory responses. PLoS Pathog. 2008, 4, e1000078. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, M.C.; Ramonell, R.P.; Haddad, N.S.; Anam, F.A.; Rudolph, M.E.; Walker, T.A.; Truong, A.D.; Dixit, A.N.; Han, J.E.; Cabrera-Mora, M.; et al. Dysregulated naive B cells and de novo autoreactivity in severe COVID-19. Nature 2022, 611, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Gore, M.; Zhang, Q.; Camenisch, T.; Boast, S.; Casagranda, F.; Lai, C.; Skinner, M.K.; Klein, R.; Matsushima, G.K.; et al. Tyro-3 family receptors are essential regulators of mammalian spermatogenesis. Nature 1999, 398, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G. Biology of the TAM receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a009076. [Google Scholar] [CrossRef] [PubMed]
- Rothlin, C.V.; Ghosh, S.; Zuniga, E.I.; Oldstone, M.B.; Lemke, G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 2007, 131, 1124–1136. [Google Scholar] [CrossRef]
- Stitt, T.N.; Conn, G.; Gore, M.; Lai, C.; Bruno, J.; Radziejewski, C.; Mattsson, K.; Fisher, J.; Gies, D.R.; Jones, P.F.; et al. The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro 3/Axl family of receptor tyrosine kinases. Cell 1995, 80, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Rothlin, C.V.; Carrera-Silva, E.A.; Bosurgi, L.; Ghosh, S. TAM receptor signaling in immune homeostasis. Annu. Rev. Immunol. 2015, 33, 355–391. [Google Scholar] [CrossRef] [PubMed]
- Dahlback, B.; Villoutreix, B.O. Regulation of blood coagulation by the protein C anticoagulant pathway: Novel insights into structure-function relationships and molecular recognition. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Manfioletti, G.; Brancolini, C.; Avanzi, G.; Schneider, C. The protein encoded by a growth arrest-specific gene (gas6) is a new member of the vitamin K-dependent proteins related to protein S, a negative coregulator in the blood coagulation cascade. Mol. Cell. Biol. 1993, 13, 4976–4985. [Google Scholar] [CrossRef] [PubMed]
- Morizono, K.; Xie, Y.; Olafsen, T.; Lee, B.; Dasgupta, A.; Wu, A.M.; Chen, I.S. The soluble serum protein Gas6 bridges virion envelope phosphatidylserine to the TAM receptor tyrosine kinase Axl to mediate viral entry. Cell Host Microbe 2011, 9, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Nagata, K.; Ohashi, K.; Nakano, T.; Arita, H.; Zong, C.; Hanafusa, H.; Mizuno, K. Identification of the product of growth arrest-specific gene 6 as a common ligand for Axl, Sky, and Mer receptor tyrosine kinases. J. Biol. Chem. 1996, 271, 30022–30027. [Google Scholar] [CrossRef]
- Lew, E.D.; Oh, J.; Burrola, P.G.; Lax, I.; Zagorska, A.; Traves, P.G.; Schlessinger, J.; Lemke, G. Differential TAM receptor-ligand-phospholipid interactions delimit differential TAM bioactivities. eLife 2014, 3, e03385. [Google Scholar] [CrossRef] [PubMed]
- Tsou, W.I.; Nguyen, K.Q.; Calarese, D.A.; Garforth, S.J.; Antes, A.L.; Smirnov, S.V.; Almo, S.C.; Birge, R.B.; Kotenko, S.V. Receptor tyrosine kinases, TYRO3, AXL, and MER, demonstrate distinct patterns and complex regulation of ligand-induced activation. J. Biol. Chem. 2014, 289, 25750–25763. [Google Scholar] [CrossRef]
- Hafizi, S.; Dahlback, B. Gas6 and protein S. Vitamin K-dependent ligands for the Axl receptor tyrosine kinase subfamily. FEBS J. 2006, 273, 5231–5244. [Google Scholar] [CrossRef]
- Penberthy, K.K.; Ravichandran, K.S. Apoptotic cell recognition receptors and scavenger receptors. Immunol. Rev. 2016, 269, 44–59. [Google Scholar] [CrossRef]
- Bandyopadhyay, P.K. Vitamin K-dependent gamma-glutamylcarboxylation: An ancient posttranslational modification. Vitam. Horm. 2008, 78, 157–184. [Google Scholar] [CrossRef] [PubMed]
- Berkner, K.L. Vitamin K-dependent carboxylation. Vitam. Horm. 2008, 78, 131–156. [Google Scholar] [CrossRef] [PubMed]
- Perera, L.; Li, L.; Darden, T.; Monroe, D.M.; Pedersen, L.G. Prediction of solution structures of the Ca2+-bound gamma-carboxyglutamic acid domains of protein S and homolog growth arrest specific protein 6: Use of the particle mesh Ewald method. Biophys. J. 1997, 73, 1847–1856. [Google Scholar] [CrossRef] [PubMed]
- Stafford, D.W. The vitamin K cycle. J. Thromb. Haemost. 2005, 3, 1873–1878. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Rigby, A.C.; Morelli, X.; Grant, M.A.; Huang, G.; Furie, B.; Seaton, B.; Furie, B.C. Structural basis of membrane binding by Gla domains of vitamin K-dependent proteins. Nat. Struct. Biol. 2003, 10, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, K.; Nagata, K.; Ohashi, K.; Nakano, T.; Arita, H.; Mizuno, K. Roles of gamma-carboxylation and a sex hormone-binding globulin-like domain in receptor-binding and in biological activities of Gas6. FEBS Lett. 1997, 408, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Nyberg, P.; He, X.; Hardig, Y.; Dahlback, B.; Garcia de Frutos, P. Stimulation of Sky tyrosine phosphorylation by bovine protein S--domains involved in the receptor-ligand interaction. Eur. J. Biochem. 1997, 246, 147–154. [Google Scholar] [CrossRef]
- Zagorska, A.; Traves, P.G.; Lew, E.D.; Dransfield, I.; Lemke, G. Diversification of TAM receptor tyrosine kinase function. Nat. Immunol. 2014, 15, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Hunter, T. Receptor signaling: When dimerization is not enough. Curr. Biol. 1999, 9, R568–R571. [Google Scholar] [CrossRef]
- Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef]
- Wiesmann, C.; Muller, Y.A.; de Vos, A.M. Ligand-binding sites in Ig-like domains of receptor tyrosine kinases. J. Mol. Med. 2000, 78, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Himanen, J.P.; Rajashankar, K.R.; Lackmann, M.; Cowan, C.A.; Henkemeyer, M.; Nikolov, D.B. Crystal structure of an Eph receptor-ephrin complex. Nature 2001, 414, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Heiring, C.; Dahlback, B.; Muller, Y.A. Ligand recognition and homophilic interactions in Tyro3: Structural insights into the Axl/Tyro3 receptor tyrosine kinase family. J. Biol. Chem. 2004, 279, 6952–6958. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Knyazev, P.G.; Clout, N.J.; Cheburkin, Y.; Gohring, W.; Ullrich, A.; Timpl, R.; Hohenester, E. Structural basis for Gas6-Axl signalling. EMBO J. 2006, 25, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Lemke, G.; Rothlin, C.V. Immunobiology of the TAM receptors. Nat. Rev. Immunol. 2008, 8, 327–336. [Google Scholar] [CrossRef] [PubMed]
- O’Bryan, J.P.; Frye, R.A.; Cogswell, P.C.; Neubauer, A.; Kitch, B.; Prokop, C.; Espinosa, R., 3rd; Le Beau, M.M.; Earp, H.S.; Liu, E.T. axl, a transforming gene isolated from primary human myeloid leukemia cells, encodes a novel receptor tyrosine kinase. Mol. Cell. Biol. 1991, 11, 5016–5031. [Google Scholar] [CrossRef] [PubMed]
- Powell, N.A.; Kohrt, J.T.; Filipski, K.J.; Kaufman, M.; Sheehan, D.; Edmunds, J.E.; Delaney, A.; Wang, Y.; Bourbonais, F.; Lee, D.Y.; et al. Novel and selective spiroindoline-based inhibitors of Sky kinase. Bioorg. Med. Chem. Lett. 2012, 22, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, C.; Simpson, C.; Deryckere, D.; Van Deusen, A.; Miley, M.J.; Kireev, D.; Norris-Drouin, J.; Sather, S.; Hunter, D.; et al. Discovery of Novel Small Molecule Mer Kinase Inhibitors for the Treatment of Pediatric Acute Lymphoblastic Leukemia. ACS Med. Chem. Lett. 2012, 3, 129–134. [Google Scholar] [CrossRef]
- Ling, L.; Templeton, D.; Kung, H.J. Identification of the major autophosphorylation sites of Nyk/Mer, an NCAM-related receptor tyrosine kinase. J. Biol. Chem. 1996, 271, 18355–18362. [Google Scholar] [CrossRef]
- Brown, J.E.; Krodel, M.; Pazos, M.; Lai, C.; Prieto, A.L. Cross-phosphorylation, signaling and proliferative functions of the Tyro3 and Axl receptors in Rat2 cells. PLoS ONE 2012, 7, e36800. [Google Scholar] [CrossRef]
- Adam-Artigues, A.; Arenas, E.J.; Arribas, J.; Prat, A.; Cejalvo, J.M. AXL—A new player in resistance to HER2 blockade. Cancer Treat. Rev. 2023, 121, 102639. [Google Scholar] [CrossRef] [PubMed]
- Elkabets, M.; Pazarentzos, E.; Juric, D.; Sheng, Q.; Pelossof, R.A.; Brook, S.; Benzaken, A.O.; Rodon, J.; Morse, N.; Yan, J.J.; et al. AXL mediates resistance to PI3Kalpha inhibition by activating the EGFR/PKC/mTOR axis in head and neck and esophageal squamous cell carcinomas. Cancer Cell 2015, 27, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Zhang, Z.; Miao, L.; Yang, Z.; Yang, J.; Wang, Y.; Qian, D.; Cai, H.; Wang, Y. Anexelekto (AXL) Increases Resistance to EGFR-TKI and Activation of AKT and ERK1/2 in Non-Small Cell Lung Cancer Cells. Oncol. Res. 2016, 24, 295–303. [Google Scholar] [CrossRef]
- Leventis, P.A.; Grinstein, S. The distribution and function of phosphatidylserine in cellular membranes. Annu. Rev. Biophys. 2010, 39, 407–427. [Google Scholar] [CrossRef] [PubMed]
- Krahling, S.; Callahan, M.K.; Williamson, P.; Schlegel, R.A. Exposure of phosphatidylserine is a general feature in the phagocytosis of apoptotic lymphocytes by macrophages. Cell Death Differ. 1999, 6, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Shiratsuchi, A.; Osada, S.; Kanazawa, S.; Nakanishi, Y. Essential role of phosphatidylserine externalization in apoptosing cell phagocytosis by macrophages. Biochem. Biophys. Res. Commun. 1998, 246, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Birge, R.B.; Boeltz, S.; Kumar, S.; Carlson, J.; Wanderley, J.; Calianese, D.; Barcinski, M.; Brekken, R.A.; Huang, X.; Hutchins, J.T.; et al. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. 2016, 23, 962–978. [Google Scholar] [CrossRef]
- Chen, X.; Zheng, J.; Li, T.; Liu, C.; Bao, M.; Wang, X.; Li, X.; Li, J.; Huang, L.; Zhang, Z.; et al. Coreceptor AXL Facilitates African Swine Fever Virus Entry via Apoptotic Mimicry. J. Virol. 2023, 97, e0061623. [Google Scholar] [CrossRef]
- Hastings, A.K.; Yockey, L.J.; Jagger, B.W.; Hwang, J.; Uraki, R.; Gaitsch, H.F.; Parnell, L.A.; Cao, B.; Mysorekar, I.U.; Rothlin, C.V.; et al. TAM Receptors Are Not Required for Zika Virus Infection in Mice. Cell Rep. 2017, 19, 558–568. [Google Scholar] [CrossRef]
- Chen, J.; Yang, Y.F.; Yang, Y.; Zou, P.; Chen, J.; He, Y.; Shui, S.L.; Cui, Y.R.; Bai, R.; Liang, Y.J.; et al. AXL promotes Zika virus infection in astrocytes by antagonizing type I interferon signalling. Nat. Microbiol. 2018, 3, 302–309. [Google Scholar] [CrossRef]
- Hastings, A.K.; Hastings, K.; Uraki, R.; Hwang, J.; Gaitsch, H.; Dhaliwal, K.; Williamson, E.; Fikrig, E. Loss of the TAM Receptor Axl Ameliorates Severe Zika Virus Pathogenesis and Reduces Apoptosis in Microglia. iScience 2019, 13, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Zwernik, S.D.; Adams, B.H.; Raymond, D.A.; Warner, C.M.; Kassam, A.B.; Rovin, R.A.; Akhtar, P. AXL receptor is required for Zika virus strain MR-766 infection in human glioblastoma cell lines. Mol. Ther. Oncolytics 2021, 23, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Qiu, Z.; Hou, Y.; Deng, X.; Xu, W.; Zheng, T.; Wu, P.; Xie, S.; Bian, W.; Zhang, C.; et al. AXL is a candidate receptor for SARS-CoV-2 that promotes infection of pulmonary and bronchial epithelial cells. Cell Res. 2021, 31, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Ning, K.; Zou, W.; Xu, P.; Cheng, F.; Zhang, E.Y.; Zhang-Chen, A.; Kleiboeker, S.; Qiu, J. Identification of AXL as a co-receptor for human parvovirus B19 infection of human erythroid progenitors. Sci. Adv. 2023, 9, eade0869. [Google Scholar] [CrossRef] [PubMed]
- Read, S.A.; Tay, E.S.; Shahidi, M.; O’Connor, K.S.; Booth, D.R.; George, J.; Douglas, M.W. Hepatitis C Virus Driven AXL Expression Suppresses the Hepatic Type I Interferon Response. PLoS ONE 2015, 10, e0136227. [Google Scholar] [CrossRef] [PubMed]
- Strange, D.P.; Jiyarom, B.; Pourhabibi Zarandi, N.; Xie, X.; Baker, C.; Sadri-Ardekani, H.; Shi, P.Y.; Verma, S. Axl Promotes Zika Virus Entry and Modulates the Antiviral State of Human Sertoli Cells. mBio 2019, 10, 10-1128. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Zagorska, A.; Lew, E.D.; Shrestha, B.; Rothlin, C.V.; Naughton, J.; Diamond, M.S.; Lemke, G.; Young, J.A. Enveloped viruses disable innate immune responses in dendritic cells by direct activation of TAM receptors. Cell Host Microbe 2013, 14, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Shimojima, M.; Ikeda, Y.; Kawaoka, Y. The mechanism of Axl-mediated Ebola virus infection. J. Infect. Dis. 2007, 196 (Suppl. S2), S259–S263. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhao, Y.; Wang, L.; You, X.; Li, J.; Zhang, G.; Hou, Y.; Wang, H.; He, S.; Li, E. Axl Mediates Resistance to Respiratory Syncytial Virus Infection Independent of Cell Attachment. Am. J. Respir. Cell Mol. Biol. 2022, 67, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Militerno, G.; Gugenheim, J.; Cuomo, O.; Hofman, P.; Mouiel, J.; Tovey, M. Synergistic interaction between anti-IFN alpha/beta antibody and low doses of cyclosporine therapy prolongs heart transplants in rats. Transplant. Proc. 1994, 26, 3050–3051. [Google Scholar]
- Shibata, T.; Makino, A.; Ogata, R.; Nakamura, S.; Ito, T.; Nagata, K.; Terauchi, Y.; Oishi, T.; Fujieda, M.; Takahashi, Y.; et al. Respiratory syncytial virus infection exacerbates pneumococcal pneumonia via Gas6/Axl-mediated macrophage polarization. J. Clin. Investig. 2020, 130, 3021–3037. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Zhen, Z.D.; Fan, D.Y.; Qin, C.F.; Han, D.S.; Zhou, H.N.; Wang, P.G.; An, J. Axl Deficiency Promotes the Neuroinvasion of Japanese Encephalitis Virus by Enhancing IL-1alpha Production from Pyroptotic Macrophages. J. Virol. 2020, 94, 10-1128. [Google Scholar] [CrossRef]
- Yang, J.; Li, M.; Yuan, M.; Bian, P.; Dong, Y.; Zhang, H.; Luo, C.; Xue, Z.; Wang, Y.; Zhang, F.; et al. Axl(−/−) neurons promote JEV infection by dampening the innate immunity. Virus Res. 2022, 307, 198605. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Tian, L.; Yu, X.; Pattaradilokrat, S.; Li, J.; Wang, M.; Yu, W.; Qi, Y.; Zeituni, A.E.; Nair, S.C.; et al. Strain-specific innate immune signaling pathways determine malaria parasitemia dynamics and host mortality. Proc. Natl. Acad. Sci. USA 2014, 111, E511–E520. [Google Scholar] [CrossRef]
- Liu, D.; Winer, B.Y.; Chou, M.Y.; Tam, H.; Xu, Y.; An, J.; Gardner, J.M.; Cyster, J.G. Dynamic encounters with red blood cells trigger splenic marginal zone B cell retention and function. Nat. Immunol. 2024, 25, 142–154. [Google Scholar] [CrossRef]
- Urban, B.C.; Hien, T.T.; Day, N.P.; Phu, N.H.; Roberts, R.; Pongponratn, E.; Jones, M.; Mai, N.T.; Bethell, D.; Turner, G.D.; et al. Fatal Plasmodium falciparum malaria causes specific patterns of splenic architectural disorganization. Infect. Immun. 2005, 73, 1986–1994. [Google Scholar] [CrossRef] [PubMed]
- Achtman, A.H.; Khan, M.; MacLennan, I.C.; Langhorne, J. Plasmodium chabaudi chabaudi infection in mice induces strong B cell responses and striking but temporary changes in splenic cell distribution. J. Immunol. 2003, 171, 317–324. [Google Scholar] [CrossRef]
- Weiss, L.; Geduldig, U.; Weidanz, W. Mechanisms of splenic control of murine malaria: Reticular cell activation and the development of a blood-spleen barrier. Am. J. Anat. 1986, 176, 251–285. [Google Scholar] [CrossRef]
- Hioki, A.; Yoshino, M.; Kano, S.; Ohtomo, H. Pathophysiology of hypoxia in mice infected with Plasmodium berghei. Parasitol. Res. 1987, 73, 298–302. [Google Scholar] [CrossRef]
- Park, M.K.; Ko, E.J.; Jeon, K.Y.; Kim, H.; Jo, J.O.; Baek, K.W.; Kang, Y.J.; Choi, Y.H.; Hong, Y.; Ock, M.S.; et al. Induction of Angiogenesis by Malarial Infection through Hypoxia Dependent Manner. Korean J. Parasitol. 2019, 57, 117–125. [Google Scholar] [CrossRef]
- Boeuf, P.; Tan, A.; Romagosa, C.; Radford, J.; Mwapasa, V.; Molyneux, M.E.; Meshnick, S.R.; Hunt, N.H.; Rogerson, S.J. Placental hypoxia during placental malaria. J. Infect. Dis. 2008, 197, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Kucharzewska, P.; Christianson, H.C.; Belting, M. Global profiling of metabolic adaptation to hypoxic stress in human glioblastoma cells. PLoS ONE 2015, 10, e0116740. [Google Scholar] [CrossRef] [PubMed]
- Blanco, Y.C.; Farias, A.S.; Goelnitz, U.; Lopes, S.C.; Arrais-Silva, W.W.; Carvalho, B.O.; Amino, R.; Wunderlich, G.; Santos, L.M.; Giorgio, S.; et al. Hyperbaric oxygen prevents early death caused by experimental cerebral malaria. PLoS ONE 2008, 3, e3126. [Google Scholar] [CrossRef] [PubMed]
- Hempel, C.; Combes, V.; Hunt, N.H.; Kurtzhals, J.A.; Grau, G.E. CNS hypoxia is more pronounced in murine cerebral than noncerebral malaria and is reversed by erythropoietin. Am. J. Pathol. 2011, 179, 1939–1950. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Wang, J.; Shiozawa, Y.; McGee, S.; Kim, J.; Jung, Y.; Joseph, J.; Berry, J.E.; Havens, A.; Pienta, K.J.; et al. Hypoxia stabilizes GAS6/Axl signaling in metastatic prostate cancer. Mol. Cancer Res. 2012, 10, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Fuh, K.C.; Castellini, L.; Viswanathan, K.; Finger, E.C.; Diep, A.N.; LaGory, E.L.; Kariolis, M.S.; Chan, A.; Lindgren, D.; et al. Direct regulation of GAS6/AXL signaling by HIF promotes renal metastasis through SRC and MET. Proc. Natl. Acad. Sci. USA 2014, 111, 13373–13378. [Google Scholar] [CrossRef] [PubMed]
- Vandyke, K.; Zeissig, M.N.; Hewett, D.R.; Martin, S.K.; Mrozik, K.M.; Cheong, C.M.; Diamond, P.; To, L.B.; Gronthos, S.; Peet, D.J.; et al. HIF-2alpha Promotes Dissemination of Plasma Cells in Multiple Myeloma by Regulating CXCL12/CXCR4 and CCR1. Cancer Res. 2017, 77, 5452–5463. [Google Scholar] [CrossRef] [PubMed]
- Schoenhals, M.; Jourdan, M.; Bruyer, A.; Kassambara, A.; Klein, B.; Moreaux, J. Hypoxia favors the generation of human plasma cells. Cell Cycle 2017, 16, 1104–1117. [Google Scholar] [CrossRef] [PubMed]
- Nalwoga, H.; Ahmed, L.; Arnes, J.B.; Wabinga, H.; Akslen, L.A. Strong Expression of Hypoxia-Inducible Factor-1alpha (HIF-1alpha) Is Associated with Axl Expression and Features of Aggressive Tumors in African Breast Cancer. PLoS ONE 2016, 11, e0146823. [Google Scholar] [CrossRef] [PubMed]
- Zou, P.-Y.; Liu, Y.-W.; Zha, X.-N.; Tong, S.; Zhang, R.; He, X.-X.; Shan, S.-S.; Wang, K.; Liu, C.-Y. Overexpression of Axl reverses endothelial cells dysfunction in high glucose and hypoxia. J. Cell. Biochem. 2019, 120, 11831–11841. [Google Scholar] [CrossRef]
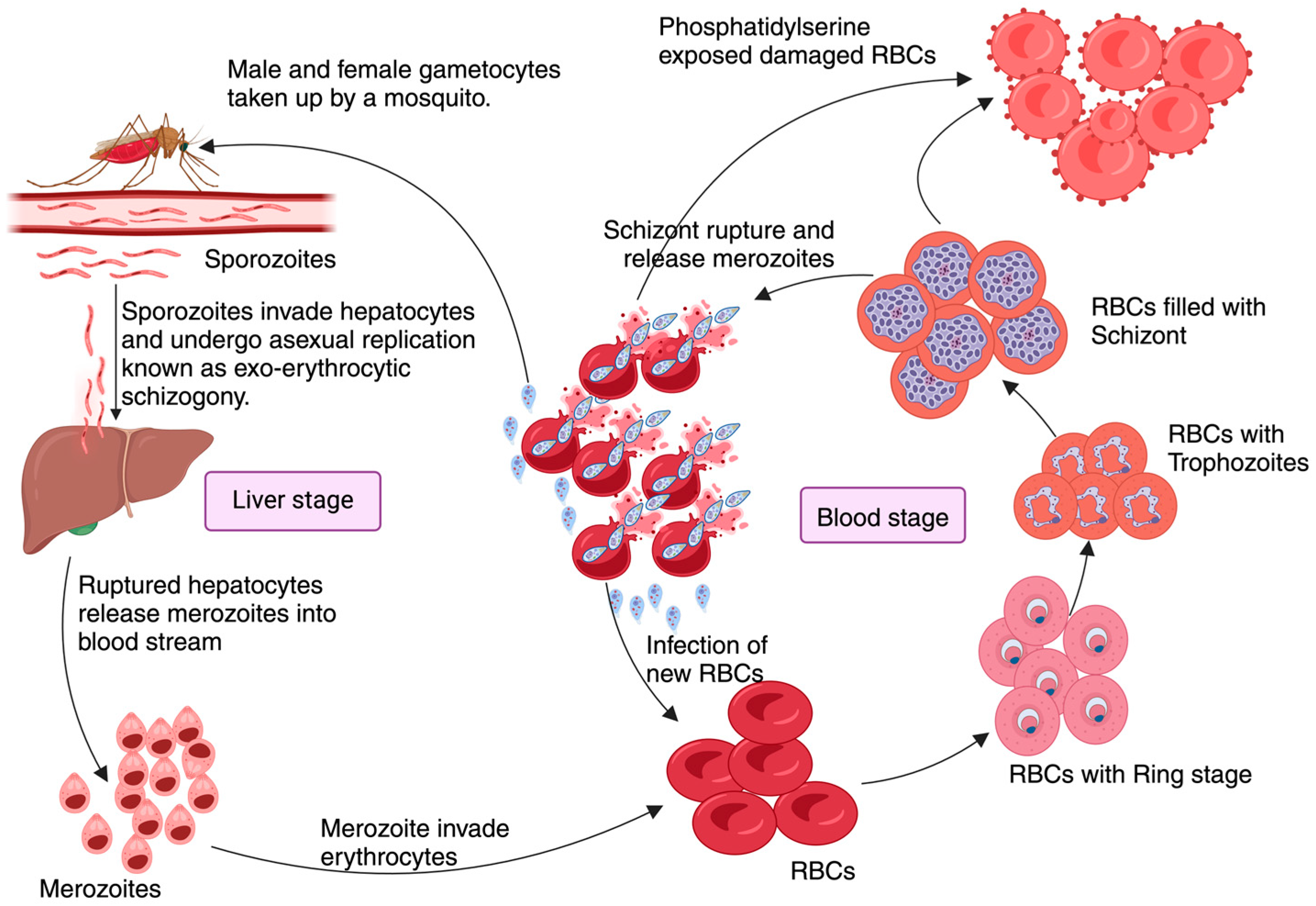
Figure 1.
Pathophysiology of Malaria: Plasmodium pathogenesis begins when an infected female Anopheles mosquito transfers motile sporozoites during a blood meal. These sporozoites invade hepatocytes after a brief resting period at the injection site initiating the ‘Liver stage’ of infection. Here, they undergo several cycles of asexual reproduction known as exo-erythrocytic schizogony, which usually takes approximately 9–16 days, depending on the Plasmodium species. Infected hepatocytes release merozoites into the blood which will invade the RBCs, initiating the ‘Blood stage’ of infection. Here, merozoites transform into various intra-erythrocytic forms such as the ring stage, trophozoite, and schizonts. The rupture of RBCs loaded with schizonts releases cellular content, parasites, and metabolic products into circulation, triggering immune reactions. The merozoites that are then released from the schizonts re-infect RBCs, perpetuating the blood stage of infection. Meanwhile, male and female gametocytes, that will be taken by another Anopheles mosquito seeking a blood meal, will fuse to form zygotes in the gut of the mosquito, to later develop into ocysts, completing the cycle when ingested by a female Anopheles mosquito during another blood meal.
Figure 1.
Pathophysiology of Malaria: Plasmodium pathogenesis begins when an infected female Anopheles mosquito transfers motile sporozoites during a blood meal. These sporozoites invade hepatocytes after a brief resting period at the injection site initiating the ‘Liver stage’ of infection. Here, they undergo several cycles of asexual reproduction known as exo-erythrocytic schizogony, which usually takes approximately 9–16 days, depending on the Plasmodium species. Infected hepatocytes release merozoites into the blood which will invade the RBCs, initiating the ‘Blood stage’ of infection. Here, merozoites transform into various intra-erythrocytic forms such as the ring stage, trophozoite, and schizonts. The rupture of RBCs loaded with schizonts releases cellular content, parasites, and metabolic products into circulation, triggering immune reactions. The merozoites that are then released from the schizonts re-infect RBCs, perpetuating the blood stage of infection. Meanwhile, male and female gametocytes, that will be taken by another Anopheles mosquito seeking a blood meal, will fuse to form zygotes in the gut of the mosquito, to later develop into ocysts, completing the cycle when ingested by a female Anopheles mosquito during another blood meal.

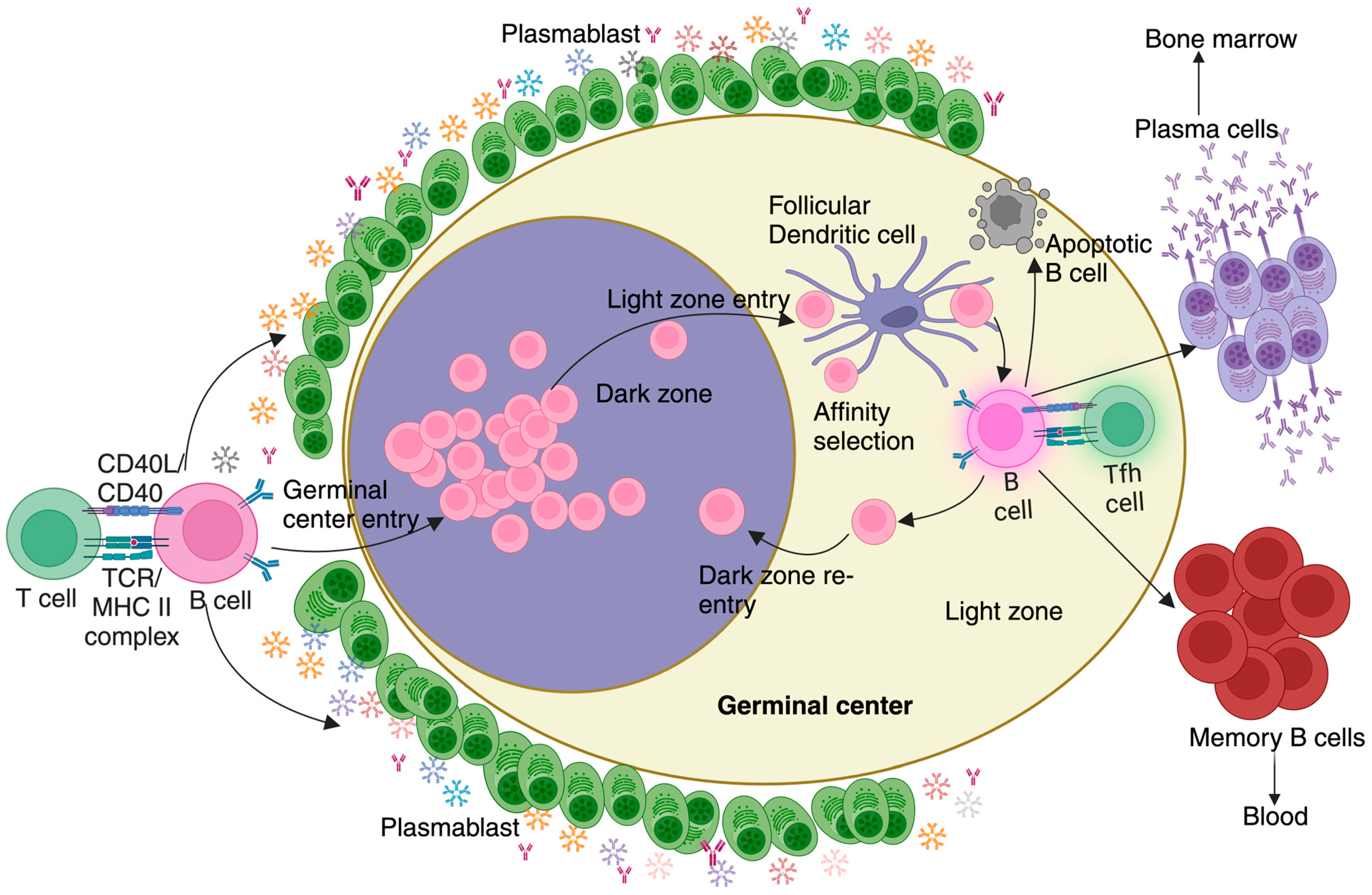
Figure 2.
Germinal center reaction during Malaria: An effective T-dependent B cell response involves the antigen-exposed activated B cells undergoing a series of maturation steps processes known as the germinal center reaction. This reaction occurs within specialized areas of B cell follicles known as germinal centers, which consist of distinct dark and light zones. Following entry into the germinal center, B cells progress through several stages, including somatic hypermutation, affinity selection, and maturation. B cells that successfully navigate these selection processes differentiate into plasma cells, responsible for producing pathogen-specific, high-affinity antibodies and residing primarily in the bone marrow, and memory B cells, which rapidly differentiate into plasma cells upon re-exposure to the same antigen and are released into and retained in circulation. Before the initiation of the germinal center reaction, a small population of rapidly proliferating B cells secreting predominantly IgM, known as plasmablasts, appear outside the B cell follicle. In malaria, these hypermetabolic plasmablasts are disproportionately numerous. The high metabolic demands of plasmablasts successfully compete for key nutrients, effectively acting as a nutrient sink to deprive GC B cells from sourcing them. This metabolic constraint ultimately contributes to a suboptimal immune response against malaria.
Figure 2.
Germinal center reaction during Malaria: An effective T-dependent B cell response involves the antigen-exposed activated B cells undergoing a series of maturation steps processes known as the germinal center reaction. This reaction occurs within specialized areas of B cell follicles known as germinal centers, which consist of distinct dark and light zones. Following entry into the germinal center, B cells progress through several stages, including somatic hypermutation, affinity selection, and maturation. B cells that successfully navigate these selection processes differentiate into plasma cells, responsible for producing pathogen-specific, high-affinity antibodies and residing primarily in the bone marrow, and memory B cells, which rapidly differentiate into plasma cells upon re-exposure to the same antigen and are released into and retained in circulation. Before the initiation of the germinal center reaction, a small population of rapidly proliferating B cells secreting predominantly IgM, known as plasmablasts, appear outside the B cell follicle. In malaria, these hypermetabolic plasmablasts are disproportionately numerous. The high metabolic demands of plasmablasts successfully compete for key nutrients, effectively acting as a nutrient sink to deprive GC B cells from sourcing them. This metabolic constraint ultimately contributes to a suboptimal immune response against malaria.

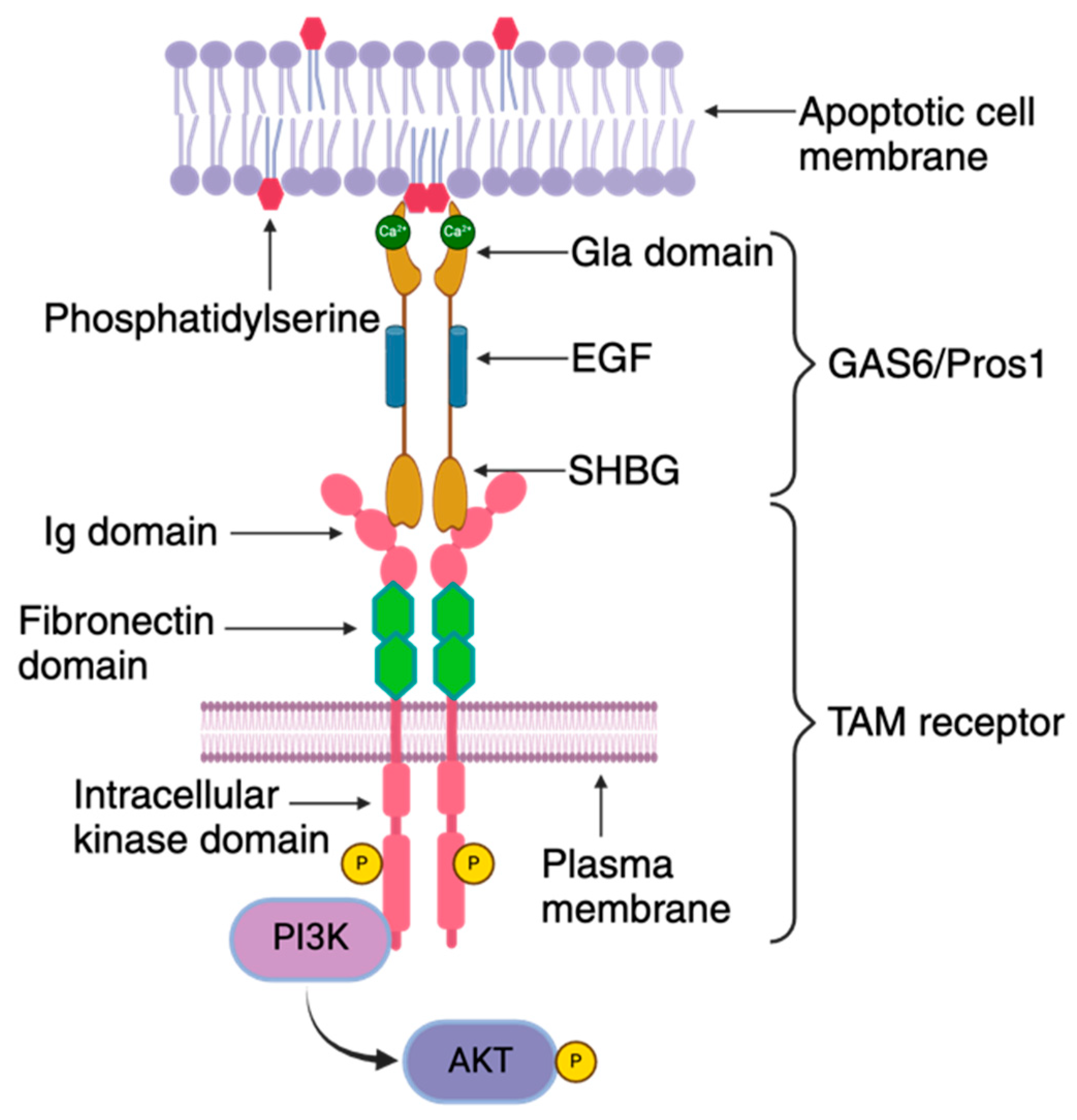
Figure 3.
TAM receptor organization: The TAM receptor is comprised of distinct structural elements, including an extracellular domain, a transmembrane segment, and an intracellular kinase domain. The extracellular region features an immunoglobulin domain responsible for binding to its ligands GAS6/PROS1, followed by tandem fibronectin type III domains. This is followed by a single-pass transmembrane domain and an intracellular protein tyrosine kinase domain. The TAM ligands, GAS6/PROS1, consist of a Gla domain, an EGF domain, and a sex hormone-binding globulin (SHBG) domain. The Gla domain is notable for its γ-carboxylated glutamic acid residues, which enables binding to exposed phosphatidylserine on the outer leaflet of the RBC membrane in a Ca2+- and vitamin K-dependent manner. The SHBG domain contains two laminin G domains that bind to the immunoglobulin domain of the TAM receptor. These domains are separated by four epidermal growth factor (EGF)-related domains. Interaction between GAS6/PROS1 and the TAM receptor occurs in a 1:1 stoichiometry, leading to receptor dimerization and subsequent activation of the intracellular protein tyrosine kinase. This activation is coupled to the activation of the phosphoinositide 3 kinase (PI3K)/AKT pathway.
Figure 3.
TAM receptor organization: The TAM receptor is comprised of distinct structural elements, including an extracellular domain, a transmembrane segment, and an intracellular kinase domain. The extracellular region features an immunoglobulin domain responsible for binding to its ligands GAS6/PROS1, followed by tandem fibronectin type III domains. This is followed by a single-pass transmembrane domain and an intracellular protein tyrosine kinase domain. The TAM ligands, GAS6/PROS1, consist of a Gla domain, an EGF domain, and a sex hormone-binding globulin (SHBG) domain. The Gla domain is notable for its γ-carboxylated glutamic acid residues, which enables binding to exposed phosphatidylserine on the outer leaflet of the RBC membrane in a Ca2+- and vitamin K-dependent manner. The SHBG domain contains two laminin G domains that bind to the immunoglobulin domain of the TAM receptor. These domains are separated by four epidermal growth factor (EGF)-related domains. Interaction between GAS6/PROS1 and the TAM receptor occurs in a 1:1 stoichiometry, leading to receptor dimerization and subsequent activation of the intracellular protein tyrosine kinase. This activation is coupled to the activation of the phosphoinositide 3 kinase (PI3K)/AKT pathway.

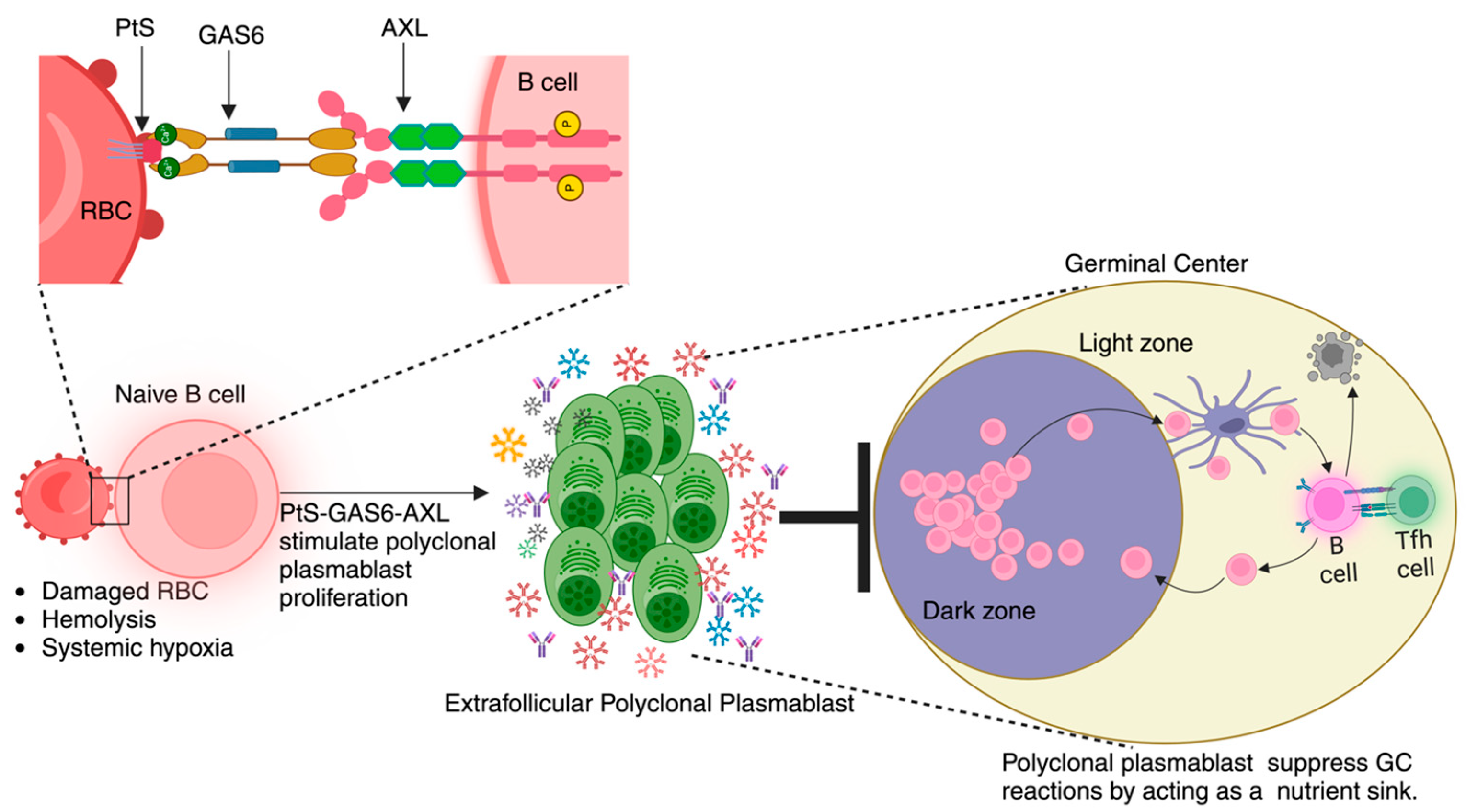
Figure 4.
Working model: We present a theoretical framework outlining the processes that occur during the development of malaria. The model centers around the interactions between Plasmodium-triggered surface exposure of phosphatidylserine (PtS) on red blood cells (RBCs), hemolysis-driven hypoxic state, and AXL receptor expression on B cells. Upon binding, this PtS–GAS6–AXL signaling cascade promotes the proliferation of polyclonal plasmablast population. The heightened metabolic activity of plasmablasts induces a state of nutrient deprivation and creates a competitive environment for key nutrients, which, in turn, limits the availability of resources for germinal center cells. This nutrient deprivation compromises the efficiency of germinal center reactions, ultimately leading to a dampened immune response against the pathogen.
Figure 4.
Working model: We present a theoretical framework outlining the processes that occur during the development of malaria. The model centers around the interactions between Plasmodium-triggered surface exposure of phosphatidylserine (PtS) on red blood cells (RBCs), hemolysis-driven hypoxic state, and AXL receptor expression on B cells. Upon binding, this PtS–GAS6–AXL signaling cascade promotes the proliferation of polyclonal plasmablast population. The heightened metabolic activity of plasmablasts induces a state of nutrient deprivation and creates a competitive environment for key nutrients, which, in turn, limits the availability of resources for germinal center cells. This nutrient deprivation compromises the efficiency of germinal center reactions, ultimately leading to a dampened immune response against the pathogen.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
John, L.; Vijay, R. Role of TAM Receptors in Antimalarial Humoral Immune Response. Pathogens 2024, 13, 298. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens13040298
AMA Style
John L, Vijay R. Role of TAM Receptors in Antimalarial Humoral Immune Response. Pathogens. 2024; 13(4):298. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens13040298
Chicago/Turabian StyleJohn, Lijo, and Rahul Vijay. 2024. "Role of TAM Receptors in Antimalarial Humoral Immune Response" Pathogens 13, no. 4: 298. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens13040298
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.