Role of Epstein-Barr Virus and Human Papillomavirus Coinfection in Cervical Cancer: Epidemiology, Mechanisms and Perspectives

Abstract
:1. Introduction
2. EBV Replication and Role in Cancer
3. HPV in Cervical Cancer
4. Frequency of HPV and EBV Coinfection in Uterine Cervix
5. EBV Infection in Tumor-Infiltrating Lymphocytes from Cervical Carcinomas
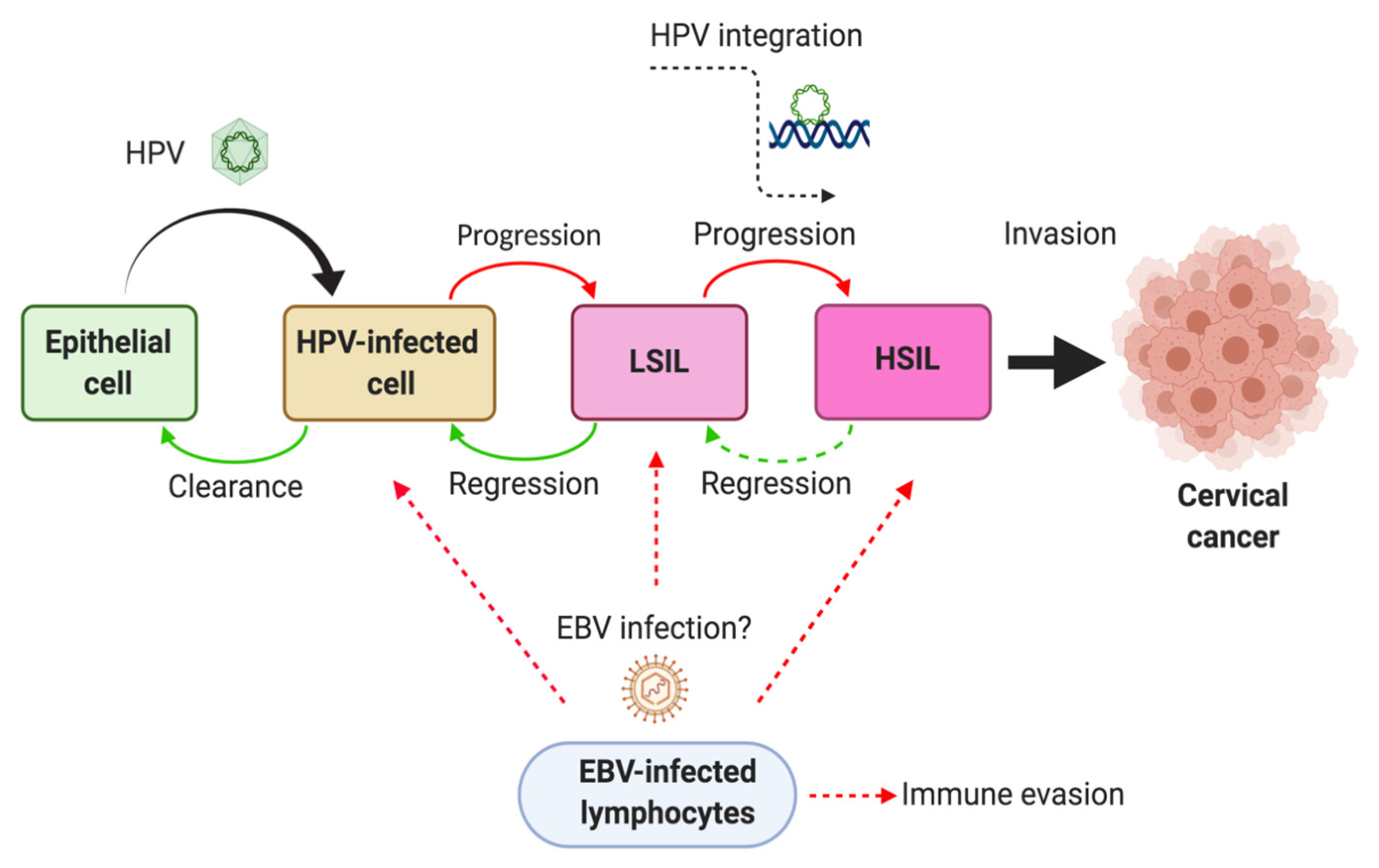
6. Mechanisms of HPV/EBV-Mediated Cervical Carcinogenesis
7. Conclusions and Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Arbyn, M.; Weiderpass, E.; Bruni, L.; de Sanjose, S.; Saraiya, M.; Ferlay, J.; Bray, F. Estimates of incidence and mortality of cervical cancer in 2018: A worldwide analysis. Lancet Glob. Health 2020, 8, e191–e203. [Google Scholar] [CrossRef] [Green Version]
- Bray, F.; Ferlay, J.; Serjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walboomers, J.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.; Peto, J.; Meijer, C.J.; Munoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 1999, 189, 12–19. [Google Scholar] [CrossRef]
- Ciapponi, A.; Bardach, A.; Glujovsky, D.; Gibbons, L.; Piconni, M.A. Type-specific HPV prevalence in cervical cancer and high-grade lesions in Latin America and the Caribbean: Systematic review and meta-analysis. PLoS ONE 2011, 6, e25493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard, E.; Pons-Salort, M.; Favre, M.; Heard, I.; Delarocque-Astagneau, E.; Guillemot, D.; Thiebaut, A.C.M. Comparing human papillomavirus prevalences in women with normal cytology or invasive cervical cancer to rank genotypes according to their oncogenic potential: A meta-analysis of observational studies. BMC Infect. Dis. 2013, 13, 373. [Google Scholar] [CrossRef] [PubMed]
- Howard, N.; Gallagher, K.E.; Mounier-Jack, S.; Burchett, H.E.D.; Kabakama, S.; Lamontagne, D.S.; Watson-Jones, D. What works for human papillomavirus vaccine introduction in low and middle-income countries? Papillomavirus Res. (Amst. Neth.) 2017, 4, 22–25. [Google Scholar] [CrossRef]
- Murillo, R.; Reyes, C.O. Human papillomavirus (HPV) vaccination: From clinical studies to immunization programs. Int. J. Gynecol. Cancer 2019, 29, 1317–1326. [Google Scholar] [CrossRef]
- Schlecht, N.F.; Platt, R.W.; Duarte-Franco, E.; Costa, M.C.; Sobrinho, J.P.; Prado, J.C.M.; Ferenczy, A.; Rohan, T.E.; Villa, L.L.; Franco, E. Human papillomavirus infection and time to progression and regression of cervical intraepithelial neoplasia. J. Natl. Cancer Inst. 2003, 95, 1336–1343. [Google Scholar] [CrossRef] [Green Version]
- Silveira, F.; Almeida, G.; Furtado, Y.; Cavalcanti, S.M.B.; Silva, K.; Maldonado, P.; Carvalho, M. The association of HPV genotype with the regression, persistence or progression of low-grade squamous intraepithelial lesions. Exp. Mol. Pathol. 2015, 99, 702–706. [Google Scholar] [CrossRef]
- Prayitno, A. Cervical cancer with Human Papilloma Virus and Epstein-Barr Virus positive. J. Carcinog. 2006, 5, 13. [Google Scholar] [CrossRef]
- Szostek, S.; Zawilinska, B.; Kopec, J.; Kosz-Vnenchak, M. Herpesviruses as possible cofactors in HPV-16-related oncogenesis. Acta Biochim. Pol. 2009, 56, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahla, S.; Oueslati, S.; Achour, M.; Kochbati, L.; Chanoufi, M.B.; Maalej, M.; Oueslati, R. Correlation between ebv co-infection and HPV16 genome integrity in Tunisian cervical cancer patients. Braz. J. Microbiol. 2012, 43, 744–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khenchouche, A.; Sadouki, N.; Boudriche, A.; Houali, K.; Graba, A.; Ooka, T.; Bouguermouh, A. Human Papillomavirus and Epstein-Barr virus co-infection in Cervical Carcinoma in Algerian women. Virol. J. 2013, 10, 340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aromseree, S.; Pientong, C.; Swangphon, P.; Chaiwongkot, A.; Patarapadungkit, N.; Kleebkaow, P.; Tangsiriwatthana, T.; Kongyingyoes, B.; Vendrig, T.; Middeldorp, J.M.; et al. Possible contributing role of Epstein-Barr virus (EBV) as a cofactor in human papillomavirus (HPV)-associated cervical carcinogenesis. J. Clin. Virol. 2015, 73, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Farrell, P.J. Epstein-Barr virus and cancer. Annu. Rev. Pathol. 2019, 14, 29–53. [Google Scholar] [CrossRef]
- Grünewald, K.; Desai, P.; Winkler, D.C.; Heymann, J.B.; Belnap, D.M.; Baumeister, W.; Steven, A.C. Three-dimensional structure of herpes simplex virus from cryo-electron tomography. Science 2003, 302, 1396–1398. [Google Scholar] [CrossRef] [Green Version]
- Murata, T.; Tsurumi, T. Switching of EBV cycles between latent and lytic states. Rev. Med Virol. 2013, 24, 142–153. [Google Scholar] [CrossRef]
- Barth, S.; Meister, G.; Grässer, F.A. EBV-encoded miRNAs. Biochim. Biophys. Acta 2011, 1809, 631–640. [Google Scholar] [CrossRef]
- Kang, M.S.; Kieff, E. Epstein-Barr virus latent genes. Exp. Mol. Med. 2015, 47, e131. [Google Scholar] [CrossRef] [Green Version]
- Babcock, G.J.; Hochberg, D.; Thorley-Lawson, D.A. The expression pattern of epstein-barr virus latent genes in vivo is dependent upon the differentiation stage of the infected b cell. Immunity 2000, 13, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Klein, E.; Nagy, N.; Rasul, A.E. EBV genome carrying B lymphocytes that express the nuclear protein EBNA-2 but not LMP-1. OncoImmunology 2013, 2, e23035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, A.M.; Luftig, M.A. To Be or Not IIb: A Multi-Step Process for Epstein-Barr Virus Latency Establishment and Consequences for B Cell Tumorigenesis. PLoS Pathog. 2015, 11, e1004656. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.; Koizumi, S.; Sugiura, M.; Tokunaga, M.; Uemura, Y.; Yamamoto, N.; Tanaka, S.; Sato, E.; Osato, T. Gastric carcinoma: Monoclonal epithelial malignant cells expressing Epstein-Barr virus latent infection protein. Proc. Natl. Acad. Sci. USA 1994, 91, 9131–9135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neparidze, N.; Lacy, J. Malignancies associated with -epstein-barr virus: Pathobiology, clinical features, and evolving treatments. Clin. Adv. Hematol. Oncol. 2014, 12, 358–371. [Google Scholar] [PubMed]
- Dugan, J.P.; Coleman, B.P.; Haverkos, B. Opportunities to target the life cycle of epstein-barr virus (EBV) in EBV-associated lymphoproliferative disorders. Front. Oncol. 2019, 9, 127. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Longnecker, R. Epithelial cell infection by Epstein-Barr virus. FEMS Microbiol. Rev. 2019, 43, 674–683. [Google Scholar] [CrossRef]
- Grywalska, E.; Rolinski, J. Epstein-Barr virus-associated lymphomas. Semin. Oncol. 2015, 42, 291–303. [Google Scholar] [CrossRef] [Green Version]
- Sivachandran, N.; Wang, X.; Frappier, L. Functions of the Epstein-Barr virus EBNA1 protein in viral reactivation and lytic infection. J. Virol. 2012, 86, 6146–6158. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Tian, W.-D.; Xu, X.; Nie, B.; Lu, J.; Liu, X.; Zhang, B.; Dong, Q.; Sunwoo, J.B.; Li, G.; et al. Epstein-Barr virus nuclear antigen 1 (EBNA1) protein induction of epithelial-mesenchymal transition in nasopharyngeal carcinoma cells. Cancer 2013, 120, 363–372. [Google Scholar] [CrossRef]
- O’Neil, J.D.; Owen, T.J.; Wood, V.H.J.; Date, K.L.; Valentine, R.; Chukwuma, M.B.; Arrand, J.R.; Dawson, C.W.; Young, L.S. Epstein-Barr virus-encoded EBNA1 modulates the AP-1 transcription factor pathway in nasopharyngeal carcinoma cells and enhances angiogenesis in vitro. J. Gen. Virol. 2008, 89, 2833–2842. [Google Scholar] [CrossRef]
- Yin, L.; Liao, W.; Deng, X.; Tang, M.; Gu, H.; Li, X.; Yi, W.; Cao, Y. LMP1 activates NF-kappa B via degradation of I kappa B alpha in nasopharyngeal carcinoma cells. Chin. Med. J. 2001, 114, 718–722. [Google Scholar] [PubMed]
- Yang, Y.; Shi, Y.; Yuan, Q.; Liu, X.; Yan, B.; Chen, L.; Tao, Y.; Cao, Y.; Song, X. Heterodimer formation between c-Jun and Jun B proteins mediated by Epstein-Barr virus encoded latent membrane protein 1. Cell Sign. 2004, 16, 1153–1162. [Google Scholar]
- Chen, H.; Lee, J.; Zong, Y.; Borowitz, M.; Ng, M.H.; Ambinder, R.F.; Hayward, S.D. Linkage between STAT regulation and Epstein-Barr virus gene expression in tumors. J. Virol. 2001, 75, 2929–2937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seto, E.; Yang, L.; Middeldorp, J.M.; Sheen, T.-S.; Chen, J.-Y.; Fukayama, M.; Eizuru, Y.; Ooka, T.; Takada, K. Epstein-Barr virus (EBV)-encodedBARF1 gene is expressed in nasopharyngeal carcinoma and EBV-associated gastric carcinoma tissues in the absence of lytic gene expression. J. Med. Virol. 2005, 76, 82–88. [Google Scholar] [CrossRef]
- Hoebe, E.K.; Le Large, T.Y.S.; Greijer, A.E.; Middeldorp, J.M. BamHI-A rightward frame 1, an Epstein-Barr virus-encoded oncogene and immune modulator. Rev. Med. Virol. 2013, 23, 367–383. [Google Scholar] [CrossRef] [Green Version]
- Elegheert, J.; Bracke, N.; Pouliot, P.; Gutsche, I.; Shkumatov, A.V.; Tarbouriech, N.; Verstraete, K.; Bekaert, A.; Burmeister, W.P.; Svergun, D.I.; et al. Allosteric competitive inactivation of hematopoietic CSF-1 signaling by the viral decoy receptor BARF1. Nat. Struct. Mol. Boil. 2012, 19, 938–947. [Google Scholar] [CrossRef] [Green Version]
- Wiech, T.; Nikolopoulos, E.; Lassman, S.; Heidt, T.; Schöpflin, A.; Sarbia, M.; Werner, M.; Shimizu, Y.; Sakka, E.; Ooka, T.; et al. Cyclin D1 expression is induced by viral BARF1 and is overexpressed in EBV-associated gastric cancer. Virchows Arch. 2008, 452, 621–627. [Google Scholar] [CrossRef]
- Chang, M.S.; Kim, D.H.; Roh, J.K.; Middeldorp, J.M.; Kim, Y.S.; Kim, S.; Han, S.; Kim, C.W.; Lee, B.L.; Kim, W.H.; et al. Epstein-Barr Virus-Encoded BARF1 Promotes Proliferation of Gastric Carcinoma Cells through Regulation of NF-B. J. Virol. 2013, 87, 10515–10523. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xu, M.; Zhang, X.; Chu, F.; Zhou, T. MAPK/c-Jun signaling pathway contributes to the upregulation of the anti-apoptotic proteins Bcl-2 and Bcl-xL induced by Epstein-Barr virus-encoded BARF1 in gastric carcinoma cells. Oncol. Lett. 2018, 15, 7537–7544. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ohyashiki, J.H.; Takaku, T.; Shimizu, N.; Ohyashiki, K. Transcriptional profiling of Epstein-Barr virus (EBV) genes and host cellular genes in nasal NK/T-cell lymphoma and chronic active EBV infection. Br. J. Cancer 2006, 94, 599–608. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.I.; Lekstrom, K. Epstein-Barr virus BARF1 protein is dispensable for B-cell transformation and inhibits alpha interferon secretion from mononuclear cells. J. Virol. 1999, 73, 7627–7632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, R.; Cabras, G.; Sheng, W.; Zeng, Y.; Ooka, T. Synergism of BARF1 with ras induces malignant transformation in primary primate epithelial cells and human nasopharyngeal epithelial cells. Neoplasia 2009, 11, 964–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, D.P.; Brink, A.A.; Vervoort, M.B.; Middeldorp, J.M.; Meijer, C.J.; van den Brule, A.J. Expression of Epstein-Barr virus (EBV) transcripts encoding homologues to important human proteins in diverse EBV associated diseases. Mol. Pathol. 1999, 52, 97–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinidis, P.; Tsikouras, P.; Iatrakis, G.; Zervoudis, S.; Koukouli, Z.; Bothou, A.; Galazios, G.; Vladareanu, S. Human papilloma virus’ life cycle and carcinogenesis. Maedica (Buchar.) 2016, 11, 48–54. [Google Scholar] [PubMed]
- Muñoz, N.; Bosch, F.X.; de Sanjosé, S.; Herrero, R.; Castellsagué, X.; Shah, K.V.; Snijders, P.J.; Meijer, C.J.; Group, I.A.f.R.o.C.M.C.C.S. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 2003, 348, 518–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Villiers, E.M.; Fauquet, C.; Broker, T.R.; Bernard, H.U.; zur Hausen, H. Classification of papillomaviruses. Virology 2004, 324, 17–27. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S.; Hwang, E.S.; Park, S.N.; Ahn, H.K.; Um, S.J.; Kim, C.J.; Kim, S.J.; Namkoong, S.E. Physical status and expression of hpv genes in cervical cancers. Gynecol. Oncol. 1997, 65, 121–129. [Google Scholar] [CrossRef]
- Li, K.; Jin, X.; Fang, Y.; Wang, C.; Gong, M.; Chen, P.; Liu, J.; Deng, D.; Ai, J. Correlation between physical status of human papilloma virus and cervical carcinogenesis. J. Huazhong Univ. Sci. Technol. Med. Sci. 2012, 32, 97–102. [Google Scholar] [CrossRef]
- Klaes, R.; Woerner, S.M.; Ridder, R.; Wentzensen, N.; Duerst, M.; Schneider, A.; Lotz, B.; Melsheimer, P.; von Knebel Doeberitz, M. Detection of high-risk cervical intraepithelial neoplasia and cervical cancer by amplification of transcripts derived from integrated papillomavirus oncogenes. Cancer Res. 1999, 59, 6132–6136. [Google Scholar]
- Hopman, A.H.; Smedts, F.; Dignef, W.; Ummelen, M.; Sonke, G.; Mravunac, M.; Vooijs, G.P.; Speel, E.J.; Ramaekers, F.C. Transition of high-grade cervical intraepithelial neoplasia to micro-invasive carcinoma is characterized by integration of HPV 16/18 and numerical chromosome abnormalities. J. Pathol. 2004, 202, 23–33. [Google Scholar] [CrossRef]
- Münger, K.; Baldwin, A.; Edwards, K.M.; Hayakawa, H.; Nguyen, C.L.; Owens, M.; Grace, M.; Huh, K. Mechanisms of human papillomavirus-induced oncogenesis. J. Virol. 2004, 78, 11451–11460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.; Laura, R.; Hepner, K.; Guccione, E.; Sawyers, C.; Lasky, L.; Banks, L. Oncogenic human papillomavirus E6 proteins target the MAGI-2 and MAGI-3 proteins for degradation. Oncogene 2002, 21, 5088–5096. [Google Scholar] [CrossRef] [PubMed]
- Pflaum, J.; Schlosser, S.; Müller, M. p53 family and cellular stress responses in cancer. Front. Oncol. 2014, 4, 285. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, O.; Veeraraghavalu, K.; Tergaonkar, V.; Liu, Y.; Androphy, E.J.; Stanley, M.A.; Krishna, S. Human papillomavirus type 16 E6 amino acid 83 variants enhance E6-mediated MAPK signaling and differentially regulate tumorigenesis by notch signaling and oncogenic Ras. J. Virol. 2004, 78, 5934–5945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spangle, J.M.; Münger, K. The human papillomavirus type 16 E6 oncoprotein activates mTORC1 signaling and increases protein synthesis. J. Virol. 2010, 84, 9398–9407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garnett, T.O.; Duerksen-Hughes, P.J. Modulation of apoptosis by human papillomavirus (HPV) oncoproteins. Arch Virol. 2006, 151, 2321–2335. [Google Scholar] [CrossRef] [Green Version]
- Yuan, C.H.; Filippova, M.; Tungteakkhun, S.S.; Duerksen-Hughes, P.J.; Krstenansky, J.L. Small molecule inhibitors of the HPV16-E6 interaction with caspase 8. Bioorg. Med. Chem. Lett. 2012, 22, 2125–2129. [Google Scholar] [CrossRef] [Green Version]
- Ohlenschläger, O.; Seiboth, T.; Zengerling, H.; Briese, L.; Marchanka, A.; Ramachandran, R.; Baum, M.; Korbas, M.; Meyer-Klaucke, W.; Dürst, M.; et al. Solution structure of the partially folded high-risk human papilloma virus 45 oncoprotein E7. Oncogene 2006, 25, 5953–5959. [Google Scholar] [CrossRef] [Green Version]
- Boyer, S.N.; Wazer, D.E.; Band, V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res 1996, 56, 4620–4624. [Google Scholar]
- Veldman, T.; Liu, X.; Yuan, H.; Schlegel, R. Human papillomavirus E6 and Myc proteins associate in vivo and bind to and cooperatively activate the telomerase reverse transcriptase promoter. Proc. Natl. Acad. Sci. USA 2003, 100, 8211–8216. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.W.; Chang, H.S.; Lin, C.H.; Yu, W.C. HPV-18 E7 conjugates to c-Myc and mediates its transcriptional activity. Int. J. Biochem. Cell Boil. 2007, 39, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Hellner, K.; Mar, J.; Fang, F.; Quackenbush, J.; Münger, K. HPV16 E7 oncogene expression in normal human epithelial cells causes molecular changes indicative of an epithelial to mesenchymal transition. Virology 2009, 391, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, E.A.; Kramer, R.E.; Tan, M.J.; Hayes, S.D.; Harper, J.W.; Howley, P.M. Comprehensive analysis of host cellular interactions with human papillomavirus E6 proteins identifies new E6 binding partners and reflects viral diversity. J. Virol. 2012, 86, 13174–13186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oyervides-Muñoz, M.A.; Pérez-Maya, A.A.; Rodríguez-Gutiérrez, H.F.; Gómez-Macias, G.S.; Fajardo-Ramírez, O.R.; Treviño, V.; Barrera-Saldaña, H.A.; Garza-Rodríguez, M.L. Understanding the HPV integration and its progression to cervical cancer. Infect. Genet. Evol. 2018, 61, 134–144. [Google Scholar] [CrossRef]
- Williams, V.M.; Filippova, M.; Soto, U.; Duerksen-Hughes, P.J. HPV-DNA integration and carcinogenesis: Putative roles for inflammation and oxidative stress. Future Virol. 2011, 6, 45–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deacon, J.M.; Evans, C.D.; Yule, R.; Desai, M.; Binns, W.; Taylor, C.; Peto, J. Sexual behaviour and smoking as determinants of cervical HPV infection and of CIN3 among those infected: A case–control study nested within the Manchester cohort. Br. J. Cancer 2000, 83, 1565–1572. [Google Scholar] [CrossRef] [Green Version]
- Peña, N.; Carrillo, D.; Muñoz, J.P.; Chnaiderman, J.; Urzúa, U.; León, O.; Tornesello, M.L.; Corvalán, A.H.; Soto-Rifo, R.; Aguayo, F. Tobacco smoke activates human papillomavirus 16 p97 promoter and cooperates with high-risk E6/E7 for oxidative DNA damage in lung cells. PLoS ONE 2015, 10, e0123029. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, J.P.; González, C.; Parra, B.; Corvalán, A.H.; Tornesello, M.L.; Eizuru, Y.; Aguayo, F. Functional interaction between human papillomavirus type 16 E6 and E7 oncoproteins and cigarette smoke components in lung epithelial cells. PLoS ONE 2012, 7, e38178. [Google Scholar] [CrossRef]
- Muñoz, J.P.; Carrillo-Beltrán, D.; Aedo-Aguilera, V.; Calaf, G.M.; León, O.; Maldonado, E.; Tapia, J.C.; Boccardo, E.; Ozbun, M.A.; Aguayo, F. Tobacco exposure enhances human papillomavirus 16 oncogene expression via EGFR/PI3K/Akt/c-Jun signaling pathway in cervical cancer cells. Front. Microbiol. 2018, 9, 3022. [Google Scholar] [CrossRef]
- Se Thoe, S.Y.; Wong, K.K.; Pathmanathan, R.; Sam, C.K.; Cheng, H.M.; Prasad, U. Elevated secretory IgA antibodies to Epstein-Barr virus (EBV) and presence of EBV DNA and EBV receptors in patients with cervical carcinoma. Gynecol. Oncol. 1993, 50, 168–172. [Google Scholar] [CrossRef]
- Landers, R.J.; O’Leary, J.J.; Crowley, M.; Healy, I.; Annis, P.; Burke, L.; O’Brien, D.; Hogan, J.; Kealy, W.F.; Lewis, F.A. Epstein-Barr virus in normal, pre-malignant, and malignant lesions of the uterine cervix. J. Clin. Pathol. 1993, 46, 931–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasagawa, T.; Shimakage, M.; Nakamura, M.; Sakaike, J.; Ishikawa, H.; Inoue, M. Epstein-Barr virus (EBV) genes expression in cervical intraepithelial neoplasia and invasive cervical cancer: A comparative study with human papillomavirus (HPV) infection. Hum. Pathol. 2000, 31, 318–326. [Google Scholar] [CrossRef]
- Shimakage, M.; Sasagawa, T. Detection of Epstein-Barr virus-determined nuclear antigen-2 mRNA by in situ hybridization. J. Virol. Methods 2001, 93, 23–32. [Google Scholar] [CrossRef]
- Szkaradkiewicz, A.; Wal, M.; Kuch, A.; Pieta, P. Human papillomavirus (HPV) and Epstein-Barr virus (EBV) cervical infections in women with normal and abnormal cytology. Pol. J. Microbiol. 2004, 53, 95–99. [Google Scholar] [PubMed]
- Hilton, D.A.; Brown, L.J.; Pringle, J.H.; Nandha, H. Absence of Epstein-Barr virus in carcinoma of the cervix. Cancer 1993, 72, 1946–1948. [Google Scholar] [CrossRef]
- Shoji, Y.; Saegusa, M.; Takano, Y.; Hashimura, M.; Okayasu, I. Detection of the Epstein-Barr virus genome in cervical neoplasia is closely related to the degree of infiltrating lymphoid cells: A polymerase chain reaction and in situ hybridization approach. Pathol. Int. 1997, 47, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Elgui de Oliveira, D.; Furtado Monteiro, T.A.; Alencar de Melo, W.; Amaral Rebouças Moreira, M.; Alvarenga, M.; Bacchi, C.E. Lack of Epstein-Barr virus infection in cervical carcinomas. Arch. Pathol. Lab. Med. 1999, 123, 1098–1100. [Google Scholar]
- Yang, Y.Y.; Koh, L.W.; Tsai, J.H.; Tsai, C.H.; Wong, E.F.; Lin, S.J.; Yang, C.C. Correlation of viral factors with cervical cancer in Taiwan. J. Microbiol. Immunol. Infect. 2004, 37, 282–287. [Google Scholar]
- Seo, S.S.; Kim, W.H.; Song, Y.S.; Kim, S.H.; Kim, J.W.; Park, N.H.; Kang, S.B.; Lee, H.P. Epstein-Barr virus plays little role in cervical carcinogenesis in Korean women. Int. J. Gynecol. Cancer 2005, 15, 312–318. [Google Scholar]
- Wei, Z.; Shunqian, J.; Junyao, L.; Xiao, L.; Lihua, M.; Xiaohong, W.; Ming, S.; Airu, W.; Jianheng, S.; Xixia, W.; et al. The infection of EBV for cervical epithelium, a new causative agent in the development of cervical carcinomas. Chin. J. Cancer Res. 1992, 4, 23–29. [Google Scholar]
- McCormick, T.M.; Canedo, N.H.; Furtado, Y.L.; Silveira, F.A.; de Lima, R.J.; Rosman, A.D.; Almeida Filho, G.L.; da C Carvalho, M.D.G. Association between human papillomavirus and Epstein-Barr virus DNA and gene promoter methylation of RB1 and CDH1 in the cervical lesions: A transversal study. Diagn. Pathol. 2015, 10, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lattario, F.; Furtado, Y.L.; Fonseca, R.; Silveira, F.A.; do Val, I.C.; Almeida, G.; Carvalho, M.G. Analysis of human papillomavirus and Epstein-Barr virus infection and aberrant death-associated protein kinase methylation in high-grade squamous intraepithelial lesions. Int. J. Gynecol. Cancer 2008, 18, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Cameron, J.E.; Rositch, A.F.; Vielot, N.A.; Mugo, N.R.; Kwatampora, J.K.L.; Waweru, W.; Gilliland, A.E.; Hagensee, M.E.; Smith, J.S. Epstein-Barr virus, high-risk human papillomavirus and abnormal cervical cytology in a prospective cohort of african female sex workers. Sex. Transm. Dis. 2018, 45, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Cameron, J.E.; Dennis, D.C.; Herrel, N.R.; Chapple, A.G.; Hagensee, M.E. Risk of abnormal cervical cytology in HIV-infected women testing positive for both human papillomavirus and Epstein-Barr virus in genital tract specimens. Cancer Causes Control. 2020, 31, 365–375. [Google Scholar] [CrossRef]
- Ammatuna, P.; Giovannelli, L.; Giambelluca, D.; Mancuso, S.; Rubino, E.; Colletti, P.; Mazzola, G.; Belfiore, P.; Lima, R. Presence of human papillomavirus and Epstein-Barr virus in the cervix of women infected with the human immunodeficiency virus. J. Med. Virol. 2000, 62, 410–415. [Google Scholar] [CrossRef]
- Smith, J.R.; Kitchen, V.S.; Botcherby, M.; Hepburn, M.; Wells, C.; Gor, D.; Forster, S.M.; Harris, J.R.; Steer, P.; Mason, P. Is HIV infection associated with an increase in the prevalence of cervical neoplasia? Br. J. Obstet. Gynaecol. 1993, 100, 149–153. [Google Scholar] [CrossRef]
- Abudoukadeer, A.; Niyazi, M.; Aikula, A.; Kamilijian, M.; Sulaiman, X.; Mutailipu, A.; Abudula, A. Association of EBV and HPV co-infection with the development of cervical cancer in ethnic Uyghur women. Eur. J. Gynaecol. Oncol. 2015, 36, 546–550. [Google Scholar]
- Al-Thawadi, H.; Ghabreau, L.; Aboulkassim, T.; Yasmeen, A.; Vranic, S.; Batist, G.; Al Moustafa, A.E. Co-Incidence of Epstein-Barr Virus and High-Risk Human Papillomaviruses in Cervical Cancer of Syrian W omen. Front. Oncol. 2018, 8, 250. [Google Scholar] [CrossRef] [Green Version]
- Schindl, M.; Oberhuber, G.; Obermair, A.; Schoppmann, S.F.; Karner, B.; Birner, P. Overexpression Overexpression of Id-1 protein is a marker for unfavorable prognosis in early-stage cervical cancer. Cancer Res. 2001, 61, 5703–5706. [Google Scholar]
- Darnel, A.D.; Wang, D.; Ghabreau, L.; Yasmeen, A.; Sami, S.; Akil, N.; Al Moustafa, A.E. Correlation between the presence of high-risk human papillomaviruses and Id gene expression in Syrian women with cervical cancer. Clin. Microbiol. Infect. 2010, 16, 262–266. [Google Scholar] [CrossRef] [Green Version]
- Resende Rodrigues, F.; Lourenco MIranda, N.; Carvalho da Fonseca, E.; Rodrigues Cordovil Pires, A.; Pedra Dias, E. Investigação da LMP1 do EBV e a coinfeçcão do HPV em lesões genitais de pacientes infectados ou não pelo HIV. Bras. Patol. Med. Lab. 2010, 46, 6. [Google Scholar] [CrossRef] [Green Version]
- Albanese, M.; Tagawa, T.; Bouvet, M.; Maliqi, L.; Lutter, D.; Hoser, J.; Hastreiter, M.; Hayes, M.; Sugden, B.; Martin, L.; et al. Epstein-Barr virus microRNAs reduce immune surveillance by virus-specific CD8+ T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E6467–E6475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jochum, S.; Moosmann, A.; Lang, S.; Hammerschmidt, W.; Zeidler, R. The EBV Immunoevasins vIL-10 and BNLF2a Protect Newly Infected B Cells from Immune Recognition and Elimination. PLoS Pathog. 2012, 8, e1002704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitano, Y.; Fujisaki, S.; Nakamura, N.; Miyazaki, K.; Katsuki, T.; Okamura, H. Immunological Disorder against the Epstein-Barr Virus Infection and Prognosis in Patients with Cervical Carcinoma. Gynecol. Oncol. 1995, 57, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Shimabuku, T.; Tamanaha, A.; Kitamura, B.; Tanabe, Y.; Tawata, N.; Ikehara, F.; Arakaki, K.; Kinjo, T. Dual expression of Epstein-Barr virus, latent membrane protein-1 and human papillomavirus-16 E6 transform primary mouse embryonic fibroblasts through NF-κB signaling. Int. J. Clin. Exp. Pathol. 2014, 7, 1920–1934. [Google Scholar] [PubMed]
- Wang, L.W.; Jiang, S.; Gewurz, B.E. Epstein-Barr Virus LMP1-Mediated Oncogenicity. J. Virol. 2017, 91, 21. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.L.; Li, H.P.; Lu, Y.J.; Hsueh, C.; Liang, Y.; Chen, C.L.; Tsao, S.W.; Tse, K.P.; Yu, J.S.; Chang, Y.S. Activation of DNA methyltransferase 1 by EBV LMP1 involves c-Jun NH2-terminal kinase signaling. Cancer Res. 2006, 66, 11668–11676. [Google Scholar] [CrossRef] [Green Version]
- Horikawa, T.; Yang, J.; Kondo, S.; Yoshizaki, T.; Joab, I.; Furukawa, M.; Pagano, J.S. Twist and epithelial-mesenchymal transition are induced by the EBV oncoprotein latent membrane protein 1 and are Associated with metastatic nasopharyngeal carcinoma. Cancer Res. 2007, 67, 1970–1978. [Google Scholar] [CrossRef] [Green Version]
- Horikawa, T.; Yoshizaki, T.; Kondo, S.; Furukawa, M.; Kaizaki, Y.; Pagano, J.S. Epstein-Barr Virus latent membrane protein 1 induces Snail and epithelial–mesenchymal transition in metastatic nasopharyngeal carcinoma. Br. J. Cancer 2011, 104, 1160–1167. [Google Scholar] [CrossRef]
- Al Moustafa, A.E.; Al-Antary, N.; Aboulkassim, T.; Akil, N.; Batist, G.; Yasmeen, A. Co-prevalence of Epstein-Barr virus and high-risk human papillomaviruses in Syrian women with breast cancer. Hum. Vaccines Immunother. 2016, 12, 1936–1939. [Google Scholar] [CrossRef] [Green Version]
- Moon, J.W.; Kong, S.K.; Kim, B.S.; Kim, H.J.; Lim, H.; Noh, K.; Kim, Y.; Choi, J.W.; Lee, J.H.; Kim, Y.S. IFNγ induces PD-L1 overexpression by JAK2/STAT1/IRF-1 signaling in EBV-positive gastric carcinoma. Sci. Rep. 2017, 7, 17810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Dakic, A.; Chen, R.; Dai, Y.; Schlegel, R.; Liu, X. Direct HPV E6/Myc interactions induce histone modifications, Pol II phosphorylation, and hTERT promoter activation. Oncotarget 2017, 8, 96323–96339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurray, H.R.; McCance, D.J. Human papillomavirus type 16 E6 activates TERT gene transcription through induction of c-Myc and release of USF-mediated repression. J. Virol. 2003, 77, 9852–9861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, A.; Månér, S.; Betz, R.; Angström, T.; Stendahl, U.; Bergman, F.; Zetterberg, A.; Wallin, K.L. Genetic alterations in cervical carcinomas: Frequent low-level amplifications of oncogenes are associated with human papillomavirus infection. Int. J. Cancer 2002, 101, 427–433. [Google Scholar] [CrossRef]
- Makielski, K.R.; Lee, D.; Lorenz, L.D.; Nawandar, D.M.; Chiu, Y.F.; Kenney, S.C.; Lambert, P.F. Human papillomavirus promotes Epstein-Barr virus maintenance and lytic reactivation in immortalized oral keratinocytes. Virology 2016, 495, 52–62. [Google Scholar] [CrossRef]
- Guidry, J.T.; Myers, J.E.; Bienkowska-Haba, M.; Songock, W.K.; Ma, X.; Shi, M.; Nathan, C.O.; Bodily, J.M.; Sapp, M.J.; Scott, R.S. Inhibition of Epstein-Barr Virus Replication in Human Papillomavirus-Immortalized Keratinocytes. J. Virol. 2018, 93, 2. [Google Scholar] [CrossRef] [Green Version]
- Aromseree, S.; Middeldorp, J.M.; Pientong, C.; van Eijndhoven, M.; Ramayanti, O.; Lougheed, S.M.; Pegtel, D.M.; Steenbergen, R.D.; Ekalaksananan, T. High Levels of EBV-Encoded RNA 1 (EBER1) Trigger Interferon and Inflammation-Related Genes in Keratinocytes Expressing HPV16 E6/E7. PLoS ONE 2017, 12, e0169290. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Zhang, W.; Jin, M.; Zhang, J.; Li, S.; Tong, F.; Zhou, Y. Differential expression of EBV proteins LMP1 and BHFR1 in EBV-associated gastric and nasopharyngeal cancer tissues. Mol. Med. Rep. 2016, 13, 4151–4158. [Google Scholar] [CrossRef] [Green Version]
- Jiang, R.; Ekshyyan, O.; Moore-Medlin, T.; Rong, X.; Nathan, S.; Gu, X.; Abreo, F.; Rosenthal, E.L.; Shi, M.; Guidry, J.T.; et al. Association between human papilloma virus/Epstein-Barr virus coinfection and oral carcinogenesis. J. Oral Pathol. Med. 2014, 44, 28–36. [Google Scholar] [CrossRef]
- Hoebe, E.; Wille, C.; Hagemeier, S.; Kenney, S.; Greijer, A.; Middeldorp, J. Epstein-Barr Virus Gene BARF1 expression is regulated by the epithelial differentiation factor ΔNp63α in undifferentiated nasopharyngeal carcinoma. Cancers (Basel) 2018, 10, 76. [Google Scholar] [CrossRef] [Green Version]
- Temple, R.M.; Meyers, C.; Sample, C.E. Generation and infection of organotypic cultures with Epstein-Barr virus. Methods Mol. Biol. 2017, 1532, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Temple, R.M.; Zhu, J.; Budgeon, L.; Christensen, N.D.; Meyers, C.; Sample, C.E. Efficient replication of Epstein-Barr virus in stratified epithelium in vitro. Proc. Natl. Acad. Sci. USA 2014, 111, 16544–16549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, C.M.; Deng, W.; Yip, Y.L.; Zeng, M.S.; Lo, K.W.; Tsao, S.W. Epstein-Barr virus infection and persistence in nasopharyngeal epithelial cells. Chin. J. Cancer 2014, 33, 549–555. [Google Scholar] [PubMed]
- De Lima, M.A.P.; Neto, P.J.N.; Lima, L.P.M.; Gonçalves Júnior, J.; Teixeira Junior, A.G.; Teodoro, I.P.P.; Facundo, H.T.; da Silva, C.G.L.; Lima, M.V.A. Association between Epstein-Barr virus (EBV) and cervical carcinoma: A meta-analysis. Gynecol. Oncol. 2018, 148, 317–328. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Ref. | EBV | HPV | EBV/HPV Coinfection | ||
|---|---|---|---|---|---|
| Methods | Results | Methods | Results | ||
| [70] | ISH of BamHI O/K | Normal cervix = 0/15 (0%) | - | - | - |
| CIN I = 1/1 (100%) | |||||
| Cervical cancer = 5/8 (62.5%) | |||||
| [71] | ISH of BamHI W | Normal cervix = 0/25 (0%) | - | - | - |
| CIN I = 0/25 (0%) | |||||
| CIN II = 2/25 (8.0%) | |||||
| CIN III = 2/25 (8.0%) | |||||
| SCC = 5/18 (27.8%) | |||||
| [72] | ISH of EBER1 | Normal cervix = 0/5 (0%) | PCR for E6/E7 | Normal cervix = 0/5 (0%) | Normal cervix = 0/5 (0%) |
| CIN II = 1/5 (20.0%) | CIN II = 1/3 (33.3%) | CIN II = 0/3 (0%) | |||
| CIN III = 5/12 (41.7%) | CIN III = 6/8 (75.0%) | CIN III = 2/8 (25.0%) | |||
| Cervical cancer = 7/14 (50.0%) | Cervical cancer = 10/13 (76.9%) | Cervical cancer = 4/13 (30.8%) | |||
| ISH of BamHI W | Normal cervix = 0/5 (%) | Normal cervix = 0/5 (0%) | |||
| CIN II = 4/5 (80.0%) | CIN II = 0/3 (0%) | ||||
| CIN III = 8/12 (66.7%) | CIN III = 3/8 (37.5%) | ||||
| Cervical cancer = 12/14 (85.7%) | Cervical cancer = 9/13 (69.2%) | ||||
| IFI for EBNA2 | Normal cervix = 0/3 (0%) | Normal cervix = 0/3 (0%) | |||
| CIN III = 6/8 (75.0%) | CIN III = 3/6 (50.0%) | ||||
| Cervical cancer = 8/9 (88.9%) | Cervical cancer = 5/8 (62.5%) | ||||
| IFI for LMP1 | Normal cervix = 0/3 (0%) | Normal cervix = 0/3 (0%) | |||
| CIN III = 4/8 (50.0%) | CIN III = 2/6 (33.3%) | ||||
| Cervical cancer = 6/9 (66.7%) | Cervical cancer = 4/8 (50.0%) | ||||
| [73] | ISH of BamHI W | Normal cervix = 0/2 (0%) | - | - | - |
| CIN I = 2/2 (100%) | |||||
| CIN II-III = 2/2 (100%) | |||||
| Cervical cancer = 10/10 (100%) | |||||
| ISH of EBNA2 | Normal cervix = 0/3 (0%) | ||||
| CIN I = 2/2 (100%) | |||||
| CIN II-III = 2/3 (66.7%) | |||||
| Cervical cancer = 14/16 (87.5%) | |||||
| IFI for EBNA2 | Normal cervix = 0/3 (0%) | ||||
| CIN I = 0/2 (0%) | |||||
| CIN II-III = 1/3 (33.3%) | |||||
| Cervical cancer = 11/16 (68.7%) | |||||
| [74] | ISH of EBERs | CIN I-II = 4/12 (33.3%) | PCR-ELISA for MY09/MY11 | CIN-negative = 2/26 (7.7%) | CIN I-II = 3/12 (25.0%) |
| CIN III = 7/10 (70.0%) | CIN I-II = 5/12 (41.7%) | CIN III = 4/10 (40.0%) | |||
| CIN III = 7/10 (70.0%) | |||||
| [91] | IHC for LMP1 | CIN I = 1/10 (10.0%) | IHC for HPV | CIN I = 3/10 (30.0%) | CIN I = 3/10 (30.0%) |
| CIN III = 3/3 (100%) | CIN II = 2/6 (33.3%) | CIN II = 4/6 (66.7%) | |||
| [13] | IHC for EBNA1 | SCC = 8/23 (34.8%) | PCR/Hybrid Capture 2 (HC2) | Normal cervix = 2/14 (14.3%) | - |
| IHC for LMP1 | SCC = 6/23 (26.1%) | CIN I = 12/16 (75.0%) | |||
| CIN II-III = 20/21 (95.2%) | |||||
| SCC = 51/58 (87.9%) | |||||
| [88] | IHC for LMP1 | SCC = 15/44 (34.1%) | PCR for E6/E7 | SCC = 42/44 (95.5%) | SCC = 15/44 (34.1%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blanco, R.; Carrillo-Beltrán, D.; Osorio, J.C.; Calaf, G.M.; Aguayo, F. Role of Epstein-Barr Virus and Human Papillomavirus Coinfection in Cervical Cancer: Epidemiology, Mechanisms and Perspectives. Pathogens 2020, 9, 685. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens9090685
Blanco R, Carrillo-Beltrán D, Osorio JC, Calaf GM, Aguayo F. Role of Epstein-Barr Virus and Human Papillomavirus Coinfection in Cervical Cancer: Epidemiology, Mechanisms and Perspectives. Pathogens. 2020; 9(9):685. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens9090685
Chicago/Turabian StyleBlanco, Rancés, Diego Carrillo-Beltrán, Julio C. Osorio, Gloria M Calaf, and Francisco Aguayo. 2020. "Role of Epstein-Barr Virus and Human Papillomavirus Coinfection in Cervical Cancer: Epidemiology, Mechanisms and Perspectives" Pathogens 9, no. 9: 685. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens9090685