Orthohantaviruses, Emerging Zoonotic Pathogens


 , , , , and
, , , , and
Abstract
:1. Introduction
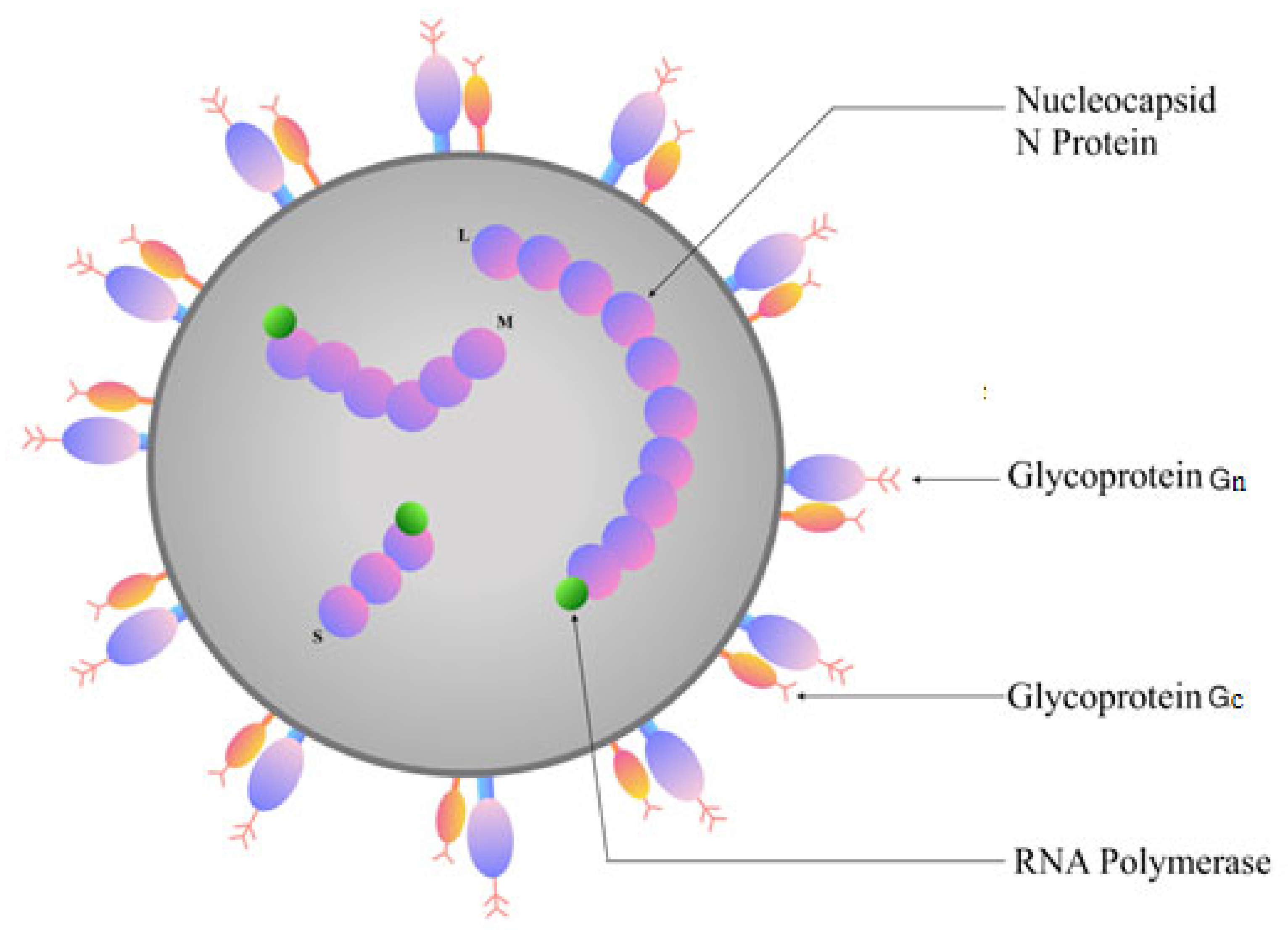
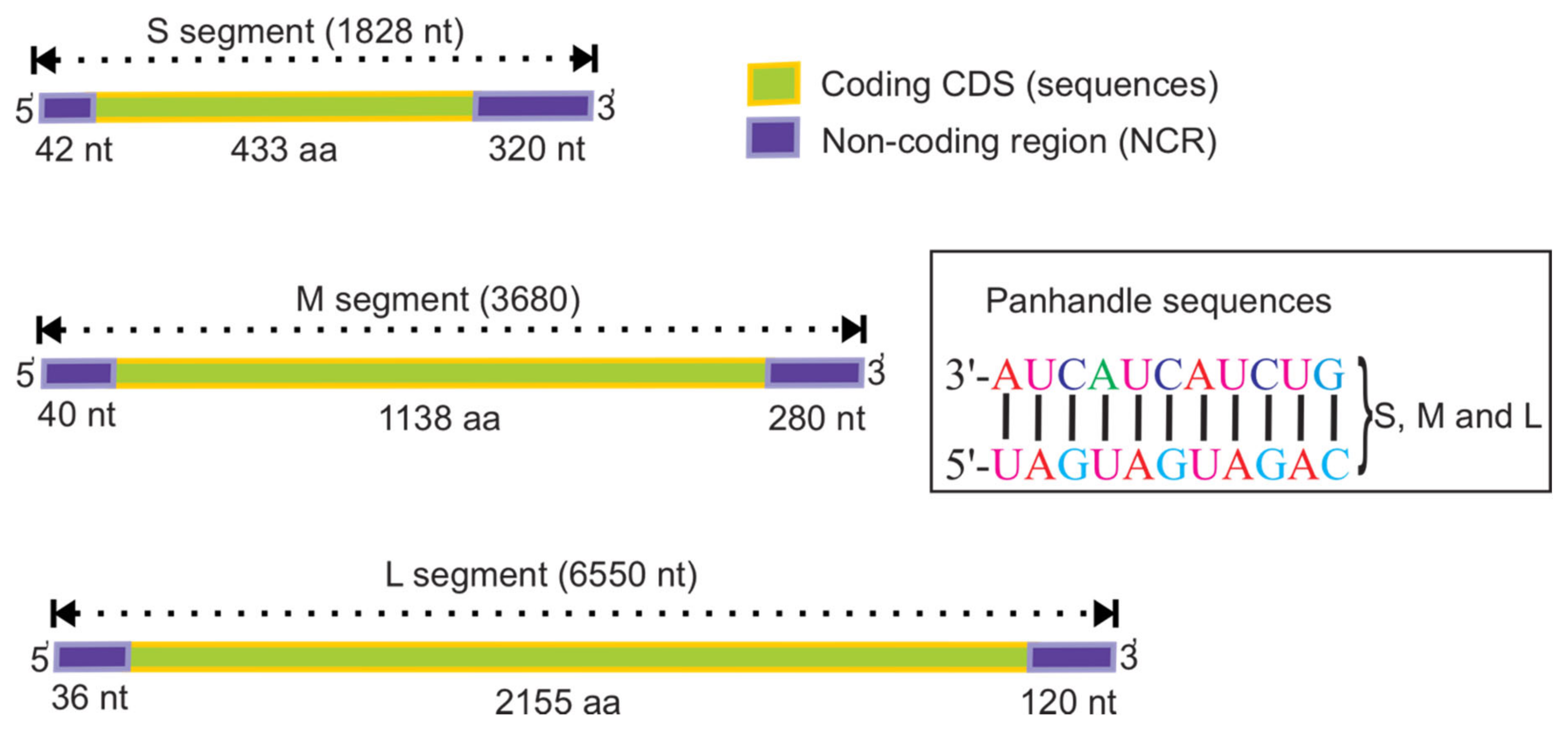
2. Genome Structure of Orthohantaviruses
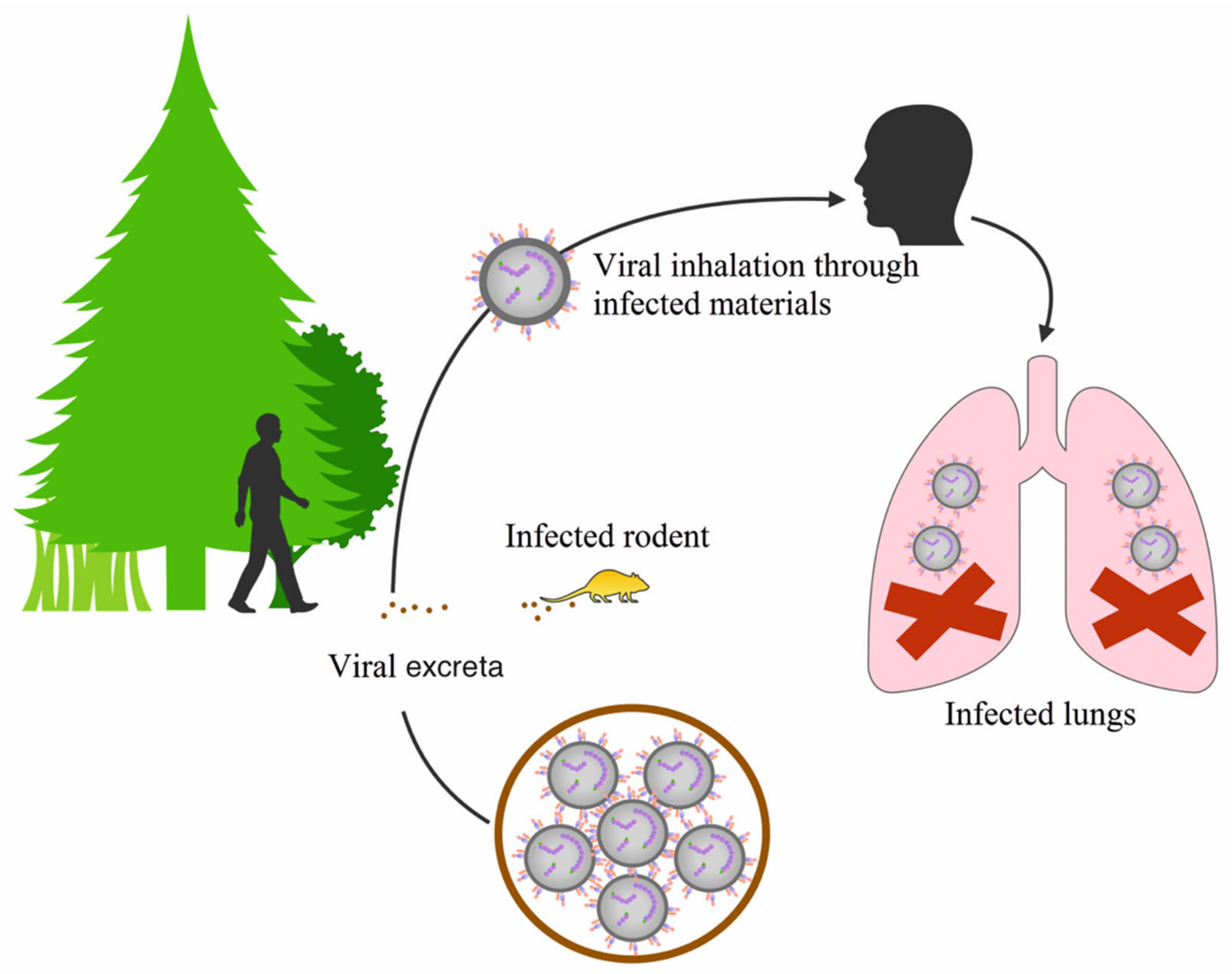
3. Distribution of Orthohantaviruses and the Diseases They Cause
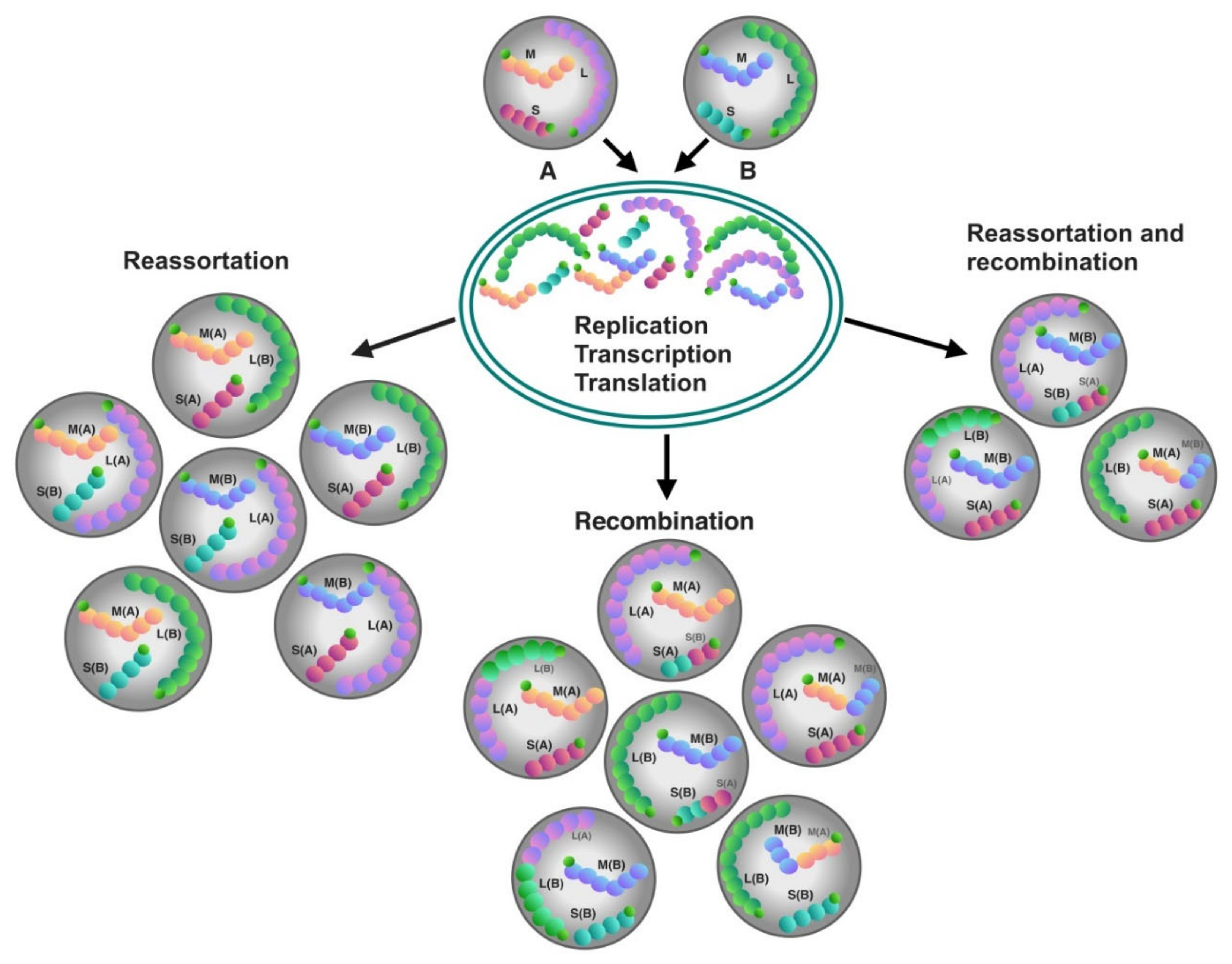
4. Gene Exchange between Orthohantaviruses
4.1. Recombination
4.2. Reassortment
5. Orthohantavirus Evolution
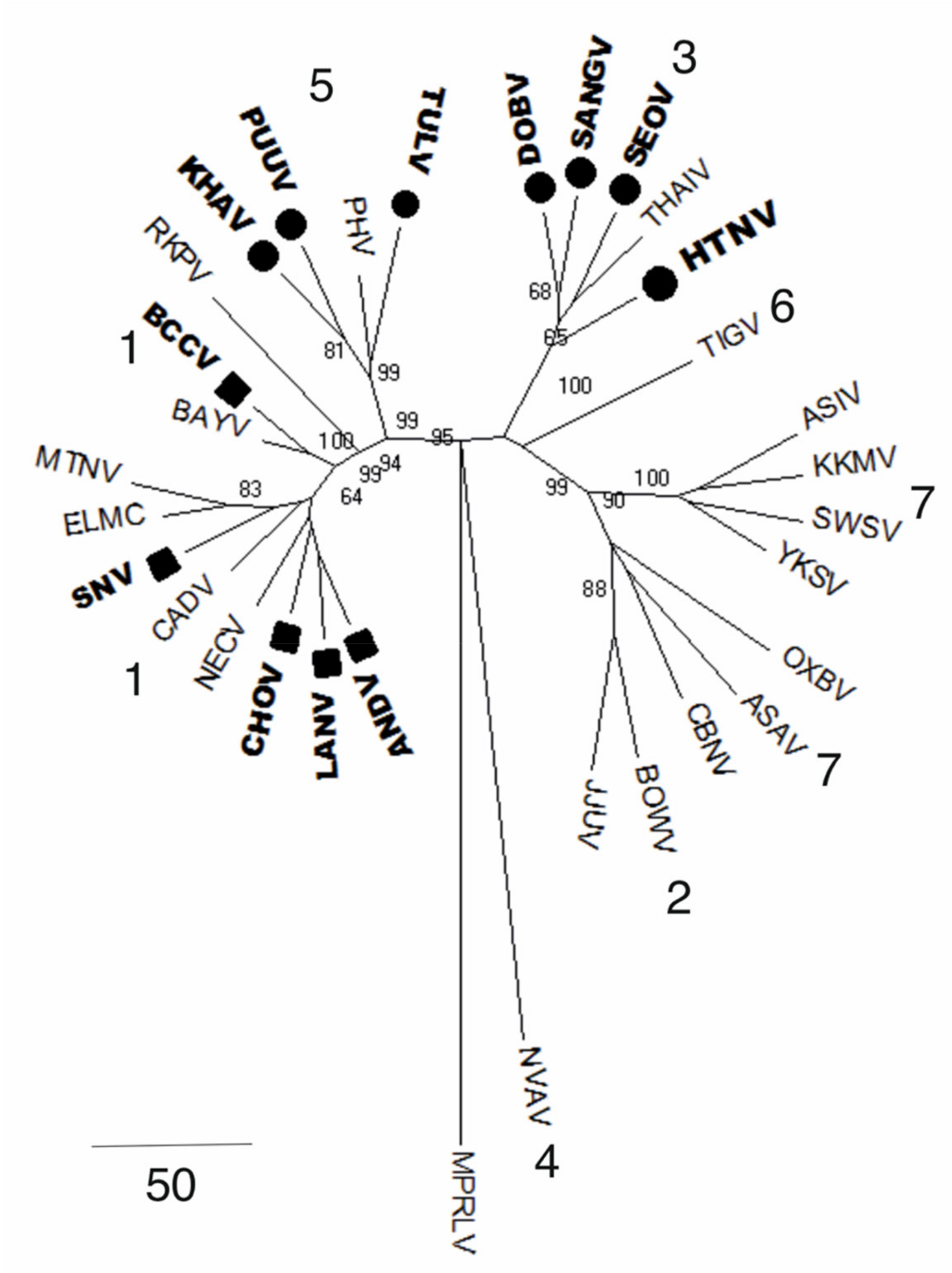
6. Genetic Diversity of Orthohantaviruses
Genetic Diversity of Puumala Orthohantaviruses
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Reynes, J.M.; Carli, D.; Boukezia, N.; Debruyne, M.; Herti, S. Tula hantavirus infection in a hospitalised patient, France, June 2015. Eurosurveillance 2015, 20, 30095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelená, H.; Mrázek, J.; Kuhn, T. Tula hantavirus infection in immunocompromised host, Czech Republic. Emerg. Infect. Dis. 2013, 19, 1873. [Google Scholar] [CrossRef] [PubMed]
- Khaiboullina, S.F.; Morzunov, S.; St Jeor, S.C. Hantaviruses: Molecular biology, evolution and pathogenesis. Curr. Mol. Med. 2005, 5, 773–790. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Valdebenito, C.; Calvo, M.; Vial, C.; Mansilla, R.; Marco, C.; Palma, R.E.; Vial, P.A.; Valdivieso, F.; Mertz, G.; Ferrés, M. Person-to-person household and nosocomial transmission of Andes hantavirus, Southern Chile, 2011. Emerg. Infect. Dis. 2014, 20, 1629. [Google Scholar] [CrossRef] [PubMed]
- Bi, Z.; Formenty, P.B.; Roth, C.E. Hantavirus infection: A review and global update. J. Infect. Dev. Ctries. 2008, 2, 003–023. [Google Scholar] [CrossRef]
- Morozov, V.G.; Ishmukhametov, A.A.; Dzagurova, T.K.; Tkachenko, E.A. Clinical manifestations of hemorrhagic fever with renal syndrome in Russia. Meditsinskiy Sov. Med. Counc. 2017, 5, 156–161. (In Russian) [Google Scholar] [CrossRef]
- Tkachenko, E.; Morozov, V.; Dzagurova, T.; Yunicheva, Y.V.; Pilikova, O.; Zavora, D.; Ishmukhametov, A.; Gorodin, V.; Bakhtina, V.; Zagidullin, I. Etiologic and clinical epidemiological features of hemorrhagic fever with renal syndrome (HFRS) in the Krasnodar Krai. Epidemiol. Infect. Dis. 2016, 21, 22–30. [Google Scholar]
- International Committee on Taxonomy of Viruses (ICTV). Available online: https://talk.ictvonline.org/ (accessed on 20 January 2014).
- Chandy, S.; Mathai, D. Globally emerging hantaviruses: An overview. Indian J. Med Microbiol. 2017, 35, 165. [Google Scholar]
- Klempa, B.; Tkachenko, E.A.; Dzagurova, T.K.; Yunicheva, Y.V.; Morozov, V.G.; Okulova, N.M.; Slyusareva, G.P.; Smirnov, A.; Kruger, D.H. Hemorrhagic fever with renal syndrome caused by 2 lineages of Dobrava hantavirus, Russia. Emerg. Infect. Dis. 2008, 14, 617–625. [Google Scholar] [CrossRef]
- Klempa, B. Reassortment events in the evolution of hantaviruses. Virus Genes 2018, 54, 638–646. [Google Scholar] [CrossRef] [Green Version]
- Kolodziej, M.; Melgies, A.; Joniec-Wiechetek, J.; Michalski, A.; Nowakowska, A.; Pitucha, G.; Niemcewicz, M. First molecular characterization of dobrava-belgrade virus found in apodemus flavicollis in poland. Ann. Agric. Environ. Med. 2018, 25, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Korva, M.; Saksida, A.; Kejzar, N.; Schmaljohn, C.; Avŝiĉ-Zupanc, T. Viral load and immune response dynamics in patients with haemorrhagic fever with renal syndrome. Clin. Microbiol. Infect. 2013, 19, e358–e366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papa, A.; Vaheri, A.; LeDuc, J.W.; Krüger, D.H.; Avšič-Županc, T.; Arikawa, J.; Song, J.-W.; Markotić, A.; Clement, J.; Liang, M. Meeting report: Tenth international conference on hantaviruses. Antivir. Res. 2016, 133, 234–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clement, J.; Maes, P.; Van Ypersele de Strihou, C.; Van Der Groen, G.; Barrios, J.M.; Verstraeten, W.W.; Van Ranst, M. Beechnuts and outbreaks of nephropathia epidemica (NE): Of mast, mice and men. Nephrol. Dial. Transplant. 2010, 25, 1740–1746. [Google Scholar] [CrossRef] [Green Version]
- Voutilainen, L.; Kallio, E.R.; Niemimaa, J.; Vapalahti, O.; Henttonen, H. Temporal dynamics of Puumala hantavirus infection in cyclic populations of bank voles. Sci. Rep. 2016, 6, 1–15. [Google Scholar]
- Tersago, K.; Verhagen, R.; Vapalahti, O.; Heyman, P.; Ducoffre, G.; Leirs, H. Hantavirus outbreak in Western Europe: Reservoir host infection dynamics related to human disease patterns. Epidemiol. Infect. 2011, 139, 381–390. [Google Scholar] [CrossRef] [Green Version]
- Razzauti, M.; Plyusnina, A.; Niemimaa, J.; Henttonen, H.; Plyusnin, A. Co-circulation of two Puumala hantavirus lineages in Latvia: A Russian lineage described previously and a novel Latvian lineage. J. Med. Virol. 2012, 84, 314–318. [Google Scholar] [CrossRef]
- Khan, A.S.; Khabbaz, R.F.; Armstrong, L.R.; Holman, R.C.; Bauer, S.P.; Graber, J.; Strine, T.; Miller, G.; Reef, S.; Tappero, J. Hantavirus pulmonary syndrome: The first 100 US cases. J. Infect. Dis. 1996, 173, 1297–1303. [Google Scholar] [CrossRef]
- Sironen, T.; Vaheri, A.; Plyusnin, A. Molecular evolution of Puumala hantavirus. J. Virol. 2001, 75, 11803–11810. [Google Scholar] [CrossRef] [Green Version]
- Plyusnin, A.; Vapalahti, O.; Lehväslaiho, H.; Apekina, N.; Mikhailova, T.; Gavrilovskaya, I.; Laakkonen, J.; Niemimaa, J.; Henttonen, H.; Brummer-Korvenkontio, M. Genetic variation of wild Puumala viruses within the serotype, local rodent populations and individual animal. Virus Res. 1995, 38, 25–41. [Google Scholar] [CrossRef]
- De Vries, A.; Vennema, H.; Bekker, D.; Maas, M.; Adema, J.; Opsteegh, M.; van der Giessen, J.; Reusken, C. Characterization of Puumala hantavirus in bank voles from two regions in the Netherlands where human cases occurred. J. Gen. Virol. 2016, 97, 1500–1510. [Google Scholar] [CrossRef] [PubMed]
- Razzauti, M.; Plyusnina, A.; Henttonen, H.; Plyusnin, A. Accumulation of point mutations and reassortment of genomic RNA segments are involved in the microevolution of Puumala hantavirus in a bank vole (Myodes glareolus) population. J. Gen. Virol. 2008, 89, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Castel, G.; Couteaudier, M.; Sauvage, F.; Pons, J.-B.; Murri, S.; Plyusnina, A.; Pontier, D.; Cosson, J.-F.; Plyusnin, A.; Marianneau, P. Complete genome and phylogeny of Puumala hantavirus isolates circulating in France. Viruses 2015, 7, 5476–5488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yashina, L.N.; Abramov, S.A.; Dupal, T.A.; Danchinova, G.A.; Malyshev, B.S.; Hay, J.; Gu, S.H.; Yanagihara, R. Hokkaido genotype of Puumala virus in the grey red-backed vole (Myodes rufocanus) and northern red-backed vole (Myodes rutilus) in Siberia. Infect. Genet. Evol. 2015, 33, 304–313. [Google Scholar] [CrossRef] [Green Version]
- Razzauti, M.; Plyusnina, A.; Henttonen, H.; Plyusnin, A. Microevolution of Puumala hantavirus during a complete population cycle of its host, the bank vole (Myodes glareolus). PLoS ONE 2013, 8, e64447. [Google Scholar] [CrossRef] [Green Version]
- Hörling, J.; Chizhikov, V.; Lundkvist, Å.; Jonsson, M.; Ivanov, L.; Dekonenko, A.; Niklasson, B.; Dzagurova, T.; Peters, C.J.; Tkachenko, E. Khabarovsk virus: A phylogenetically and serologically distinct hantavirus isolated from Microtus fortis trapped in far-east Russia. J. Gen. Virol. 1996, 77, 687–694. [Google Scholar] [CrossRef]
- Asikainen, K.; Hänninen, T.; Henttonen, H.; Niemimaa, J.; Laakkonen, J.; Andersen, H.K.; Bille, N.; Leirs, H.; Vaheri, A.; Plyusnin, A. Molecular evolution of Puumala hantavirus in Fennoscandia: Phylogenetic analysis of strains from two recolonization routes, Karelia and Denmark. J. Gen. Virol. 2000, 81, 2833–2841. [Google Scholar] [CrossRef]
- Johansson, P.; Olsson, G.E.; Low, H.-T.; Bucht, G.; Ahlm, C.; Juto, P.; Elgh, F. Puumala hantavirus genetic variability in an endemic region (Northern Sweden). Infect. Genet. Evol. 2008, 8, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Monchatre-Leroy, E.; Murri, S.; Castel, G.; Calavas, D.; Boue, F.; Henaux, V.; Marianneau, P. First insights into Puumala orthohantavirus circulation in a rodent population in Alsace, France. Zoonoses Public Health 2018, 65, 540–551. [Google Scholar] [CrossRef]
- Evander, M.; Eriksson, I.; Pettersson, L.; Juto, P.; Ahlm, C.; Olsson, G.E.; Bucht, G.; Allard, A. Puumala hantavirus viremia diagnosed by real-time reverse transcriptase PCR using samples from patients with hemorrhagic fever and renal syndrome. J. Clin. Microbiol. 2007, 45, 2491–2497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynes, J.-M.; Carli, D.; Thomas, D.; Castel, G. Puumala hantavirus genotypes in humans, France, 2012–2016. Emerg. Infect. Dis. 2019, 25, 140. [Google Scholar] [CrossRef]
- Kramski, M.; Meisel, H.; Klempa, B.; Kruger, D.H.; Pauli, G.; Nitsche, A. Detection and typing of human pathogenic hantaviruses by real-time reverse transcription-PCR and pyrosequencing. Clin. Chem. 2007, 53, 1899–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razzauti, M.; Plyusnina, A.; Sironen, T.; Henttonen, H.; Plyusnin, A. Analysis of Puumala hantavirus in a bank vole population in northern Finland: Evidence for co-circulation of two genetic lineages and frequent reassortment between strains. J. Gen. Virol. 2009, 90, 1923–1931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekonenko, A.; Yakimenko, V.; Ivanov, A.; Morozov, V.; Nikitin, P.; Khasanova, S.; Dzagurova, T.; Tkachenko, E.; Schmaljohn, C. Genetic similarity of Puumala viruses found in Finland and western Siberia and of the mitochondrial DNA of their rodent hosts suggests a common evolutionary origin. Infect. Genet. Evol. 2003, 3, 245–257. [Google Scholar] [CrossRef]
- Lee, S.-H.; Kim, W.-K.; No, J.S.; Kim, J.-A.; Kim, J.I.; Gu, S.H.; Kim, H.-C.; Klein, T.A.; Park, M.-S.; Song, J.-W. Dynamic circulation and genetic exchange of a shrew-borne hantavirus, Imjin virus, in the Republic of Korea. Sci. Rep. 2017, 7, 44369. [Google Scholar] [CrossRef] [Green Version]
- Mir, M.A. Hantaviruses. Clin. Lab. Med. 2010, 30, 67–91. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, C.B.; Figueiredo, L.T.M.; Vapalahti, O. A global perspective on hantavirus ecology, epidemiology, and disease. Clin. Microbiol. Rev. 2010, 23, 412–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmaljohn, C.S.; Hasty, S.E.; Harrison, S.A.; Dalrymple, J.M. Characterization of Hantaan virions, the prototype virus of hemorrhagic fever with renal syndrome. J. Infect. Dis. 1983, 148, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Vaheri, A.; Henttonen, H.; Voutilainen, L.; Mustonen, J.; Sironen, T.; Vapalahti, O. Hantavirus infections in Europe and their impact on public health. Rev. Med. Virol. 2013, 23, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Davidyuk, Y.; Shamsutdinov, A.; Kabwe, E.; Ismagilova, R.; Martynova, E.; Belyaev, A.; Shuralev, E.; Trifonov, V.; Savitskaya, T.; Isaeva, G. Prevalence of the Puumala orthohantavirus Strains in the Pre-Kama Area of the Republic of Tatarstan, Russia. Pathogens 2020, 9, 540. [Google Scholar] [CrossRef]
- Flick, K.; Hooper, J.W.; Schmaljohn, C.S.; Pettersson, R.F.; Feldmann, H.; Flick, R. Rescue of Hantaan virus minigenomes. Virology 2003, 306, 219–224. [Google Scholar] [CrossRef] [Green Version]
- Jääskeläinen, K.M.; Kaukinen, P.; Minskaya, E.S.; Plyusnina, A.; Vapalahti, O.; Elliott, R.M.; Weber, F.; Vaheri, A.; Plyusnin, A. Tula and Puumala hantavirus NSs ORFs are functional and the products inhibit activation of the interferon-beta promoter. J. Med. Virol. 2007, 79, 1527–1536. [Google Scholar] [CrossRef]
- Okuno, Y.; Yamanishi, K.; Takahashi, Y.; Tanishita, O.; Nagai, T.; Dantas, J.R., Jr.; Okamoto, Y.; Tadano, M.; Takahashi, M. Haemagglutination-inhibition test for haemorrhagic fever with renal syndrome using virus antigen prepared from infected tissue culture fluid. J. Gen. Virol. 1986, 67, 149–156. [Google Scholar] [CrossRef]
- Kolakofsky, D.; Hacker, D. Bunyavirus RNA synthesis: Genome transcription and replication. In Bunyaviridae; Springer: Berlin/Heidelberg, Germany, 1991; pp. 143–159. [Google Scholar]
- Kukkonen, S.K.; Vaheri, A.; Plyusnin, A. L protein, the RNA-dependent RNA polymerase of hantaviruses. Arch. Virol. 2005, 150, 533–556. [Google Scholar] [CrossRef]
- Pljusnin, A.; Elliott, R.M. Bunyaviridae: Molecular and Cellular Biology; Caister Academic Press: Norfolk, UK, 2011. [Google Scholar]
- Binder, F.; Reiche, S.; Roman-Sosa, G.; Saathoff, M.; Ryll, R.; Trimpert, J.; Kunec, D.; Höper, D.; Ulrich, R.G. Isolation and characterization of new Puumala orthohantavirus strains from Germany. Virus Genes 2020, 56, 448–460. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Wang, Y.; Mu, D.; Xu, Z.; Qian, Q.; Chen, G.; Wen, L.; Yin, W.; Li, S.; Zhang, W. The Impacts of Climatic Factors and Vegetation on Hemorrhagic Fever with Renal Syndrome Transmission in China: A Study of 109 Counties. Int. J. Environ. Res. Public Health 2019, 16, 3434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, H.; Tian, H.-Y.; Cazelles, B.; Li, X.-J.; Tong, S.-L.; Gao, L.-D.; Qin, J.-X.; Lin, X.-L.; Liu, H.-N.; Zhang, X.-X. Atmospheric moisture variability and transmission of hemorrhagic fever with renal syndrome in Changsha City, Mainland China, 1991–2010. PLoS Negl. Trop. Dis. 2013, 7, e2260. [Google Scholar] [CrossRef] [PubMed]
- Nafeev, A.A.e.; Eremeeva, N.; Nafeyev, A.; Eremeyeva, N. Epidemic manifestations of hemorrhagic fever with renal syndrome in an active natural focus. Epidemiol. Infect. Dis. 2011, 16, 40–42. [Google Scholar]
- Fang, L.; Yan, L.; Liang, S.; de Vlas, S.J.; Feng, D.; Han, X.; Zhao, W.; Xu, B.; Bian, L.; Yang, H. Spatial analysis of hemorrhagic fever with renal syndrome in China. BMC Infect. Dis. 2006, 6, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, X.; Huang, C. Study farther on hemorrhagic fever with renal syndrome. Chin. J. Infect. Dis. 2002, 20, 197–198. [Google Scholar]
- Jameson, L.; Logue, C.; Atkinson, B.; Baker, N.; Galbraith, S.; Carroll, M.; Brooks, T.; Hewson, R. The continued emergence of hantaviruses: Isolation of a Seoul virus implicated in human disease, United Kingdom, October 2012. Eurosurveillance 2013, 18, 20344. [Google Scholar] [PubMed]
- Marcotic, A. Clinic and laboratory findings of HFRS patients in South-East Europe. In Proceedings of the IX International Conference of HFRS, HPS and Hantaviruses, Beijing, China, 7 June 2013; p. 13. [Google Scholar]
- Sanada, T.; Ozaki, Y.; Seto, T. Isolation and characterization of Hokkaido virus, genus Hantavirus. In Proceedings of the IX International Conference of HFRS, HPS and Hantaviruses, Beijing, China, 7 June 2013; p. 20. [Google Scholar]
- Charbonnel, N.; Sironen, T.; Henttonen, H.; Vapalahti, O.; Mustonen, J.; Vaheri, A. Immunogenetic factors affecting susceptibility of humans and rodents to hantaviruses and the clinical course of hantaviral disease in humans. Viruses 2014, 6, 2214–2241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brummer-Korvenkontio, M.; Vaheri, A.; Hovi, T.; Von Bonsdorff, C.-H.; Vuorimies, J.; Manni, T.; Penttinen, K.; Oker-Blom, N.; Lähdevirta, J. Nephropathia epidemica: Detection of antigen in bank voles and serologic diagnosis of human infection. J. Infect. Dis. 1980, 141, 131–134. [Google Scholar] [CrossRef]
- Sibold, C.; Meisel, H.; Krüger, D.H.; Labuda, M.; Lysy, J.; Kozuch, O.; Pejcoch, M.; Vaheri, A.; Plyusnin, A. Recombination in Tula hantavirus evolution: Analysis of genetic lineages from Slovakia. J. Virol. 1999, 73, 667–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomason, A.G.; Begon, M.; Bradley, J.E.; Paterson, S.; Jackson, J.A. Endemic hantavirus in field voles, northern England. Emerg. Infect. Dis. 2017, 23, 1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chappell, J.G.; Tsoleridis, T.; Onianwa, O.; Drake, G.; Ashpole, I.; Dobbs, P.; Edema, W.; Kumi-Ansah, F.; Bennett, M.; Tarlinton, R.E. Retrieval of the Complete Coding Sequence of the UK-Endemic Tatenale Orthohantavirus Reveals Extensive Strain Variation and Supports Its Classification as a Novel Species. Viruses 2020, 12, 454. [Google Scholar] [CrossRef] [Green Version]
- Zdolnik, T.D.; Baranova, N.Y.; Kostyrko, V.I.; Kharlamov, V.V. On the circulation of HFRS pathogens in the territory of the Ryazan Region. Vopr Virusol 2012, 7, 276–279. [Google Scholar]
- Kiryakov, V.Y.; Reshetnyak, E.A. Some features of the course of the epidemic process of hemorrhagic fever with renal syndrome in the Primorsky Territory. Health Med. Ecol. Sci. 2015, 62, 118–121. [Google Scholar]
- Dzagurova, T.K.; Klempa, B.; Tkachenko, E.A.; Slyusareva, G.P.; Morozov, V.G.; Auste, B.; Kruger, D.H. Molecular diagnostics of hemorrhagic fever with renal syndrome during a Dobrava virus infection outbreak in the European part of Russia. J. Clin. Microbiol. 2009, 47, 4029–4036. [Google Scholar] [CrossRef] [Green Version]
- Plyusnin, A.; Sironen, T. Evolution of hantaviruses: Co-speciation with reservoir hosts for more than 100 MYR. Virus Res. 2014, 187, 22–26. [Google Scholar] [CrossRef]
- Bennett, S.N.; Gu, S.H.; Kang, H.J.; Arai, S.; Yanagihara, R. Reconstructing the evolutionary origins and phylogeography of hantaviruses. Trends Microbiol. 2014, 22, 473–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, Y.K.; Milligan, B.; Owen, R.D.; Goodin, D.G.; Jonsson, C.B. Phylogenetic and geographical relationships of hantavirus strains in eastern and western Paraguay. Am. J. Trop. Med. Hyg. 2006, 75, 1127–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klempa, B.; Witkowski, P.T.; Popugaeva, E.; Auste, B.; Koivogui, L.; Fichet-Calvet, E.; Strecker, T.; ter Meulen, J.; Krüger, D.H. Sangassou virus, the first hantavirus isolate from Africa, displays genetic and functional properties distinct from those of other murinae-associated hantaviruses. J. Virol. 2012, 86, 3819–3827. [Google Scholar] [CrossRef] [Green Version]
- Klempa, B.; Fichet-Calvet, E.; Lecompte, E.; Auste, B.; Aniskin, V.; Meisel, H.; Denys, C.; Koivogui, L.; ter Meulen, J.; Krüger, D.H. Hantavirus in African wood mouse, Guinea. Emerg. Infect. Dis. 2006, 12, 838. [Google Scholar] [CrossRef] [PubMed]
- Maxema, I.G.; Kompanets, G.G.; Iunikhina, O.V.; Kushnareva, T.V.; Slonova, R.A. Characteristics of the incidence of hemorrhagic fever with renal syndrome in Primorsky Krai in 1999–2008. Pac. Med. J. 2010, 3, 41. [Google Scholar]
- Khaiboullina, S.F. Molecular and Cellular Mechanisms of Pathogenesis of Hantavirus Infections. Ph.D.Thesis, Kayan State Medical University, Kazan, Russia, 2015. [Google Scholar]
- Ravkov, E.V.; Rollin, P.E.; Ksiazek, T.G.; Peters, C.J.; Nichol, S.T. Genetic and serologic analysis of Black Creek Canal virus and its association with human disease and Sigmodon hispidus infection. Virology 1995, 210, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M.J.; Quiroz, E.; Gracia, F.; Sanchez, A.J.; Ksiazek, T.G.; Kitsutani, P.T.; Ruedas, L.A.; Tinnin, D.S.; Caceres, L.; Garcia, A. Hantavirus pulmonary syndrome in Panama: Identification of novel hantaviruses and their likely reservoirs. Virology 2000, 277, 14–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salazar-Bravo, J.; Armién, B.; Suzán, G.; Armién, A.; Ruedas, L.A.; Avila, M.; Zaldívar, Y.; Pascale, J.M.; Gracia, F.; Yates, T.L. Serosurvey of wild rodents for hantaviruses in Panama, 2000–2002. J. Wildl. Dis. 2004, 40, 103–109. [Google Scholar] [CrossRef] [Green Version]
- da Rosa, E.S.T.; Medeiros, D.B.; Nunes, M.R.; Simith, D.B.; Pereira, A.d.S.; Elkhoury, M.R.; Santos, E.D.; Lavocat, M.; Marques, A.A.; Via, A.V. Molecular epidemiology of Laguna Negra virus, Mato Grosso State, Brazil. Emerg. Infect. Dis. 2012, 18, 982. [Google Scholar] [CrossRef]
- Arai, S.; Gu, S.H.; Baek, L.J.; Tabara, K.; Bennett, S.N.; Oh, H.-S.; Takada, N.; Kang, H.J.; Tanaka-Taya, K.; Morikawa, S. Divergent ancestral lineages of newfound hantaviruses harbored by phylogenetically related crocidurine shrew species in Korea. Virology 2012, 424, 99–105. [Google Scholar] [CrossRef] [Green Version]
- Heyman, P.; Ceianu, C.; Christova, I.; Tordo, N.; Beersma, M.; Alves, M.J.; Lundkvist, Å.; Hukic, M.; Papa, A.; Tenorio, A. A five-year perspective on the situation of haemorrhagic fever with renal syndrome and status of the hantavirus reservoirs in Europe, 2005–2010. Eurosurveillance 2011, 16, 19961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, S.H.; Markowski, J.; Kang, H.J.; Hejduk, J.; Sikorska, B.; Liberski, P.P.; Yanagihara, R. Boginia virus, a newfound hantavirus harbored by the Eurasian water shrew (Neomys fodiens) in Poland. Virol. J. 2013, 10, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulhorst, C.F.; Monroe, M.C.; Salas, R.A.; Duno, G.; Utrera, A.; Ksiazek, T.G.; Nichol, S.T.; De Manzione, N.M.; Tovar, D.; Tesh, R.B. Isolation, characterization and geographic distribution of Cano Delgadito virus, a newly discovered South American hantavirus (family Bunyaviridae). Virus Res. 1997, 51, 159–171. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.-P.; Lin, X.-D.; Wang, W.; Tian, J.-H.; Cong, M.-L.; Zhang, H.-L.; Wang, M.-R.; Zhou, R.-H.; Wang, J.-B.; Li, M.-H. Phylogeny and origins of hantaviruses harbored by bats, insectivores, and rodents. PLoS Pathog. 2013, 9, e1003159. [Google Scholar] [CrossRef] [Green Version]
- Song, J.-W.; Kang, H.J.; Song, K.-J.; Truong, T.T.; Bennett, S.N.; Arai, S.; Truong, N.U.; Yanagihara, R. Newfound hantavirus in Chinese mole shrew, Vietnam. Emerg. Infect. Dis. 2007, 13, 1784. [Google Scholar] [CrossRef] [PubMed]
- Hjelle, B.; Jenison, S.; Torrez-Martinez, N.; Yamada, T.; Nolte, K.; Zumwalt, R.; MacInnes, K.; Myers, G. A novel hantavirus associated with an outbreak of fatal respiratory disease in the southwestern United States: Evolutionary relationships to known hantaviruses. J. Virol. 1994, 68, 592–596. [Google Scholar] [CrossRef] [Green Version]
- Kariwa, H.; Tkachenko, E.A.; Morozov, V.G.; Seto, T.; Tanikawa, Y.; Kolominov, S.I.; Belov, S.N.; Nakamura, I.; Hashimoto, N.; Balakiev, A.E. Epidemiological study of hantavirus infection in the Samara Region of European Russia. J. Vet. Med Sci. 2009, 71, 1569–1578. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.J.; Arai, S.; Hope, A.G.; Cook, J.A.; Yanagihara, R. Novel hantavirus in the flat-skulled shrew (Sorex roboratus). Vector Borne Zoonotic Dis. 2010, 10, 593–597. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yuan, J.; Yang, X.; Zhou, J.; Yang, W.; Peng, C.; Zhang, H.-L.; Shi, Z. A novel hantavirus detected in Yunnan red-backed vole (Eothenomys miletus) in China. J. Gen. Virol. 2011, 92, 1454–1457. [Google Scholar] [CrossRef]
- Saasa, N.; Sánchez-Hernández, C.; de Lourdes Romero-Almaraz, M.; Guerrero-Ibarra, E.; Almazán-Catalán, A.; Yoshida, H.; Miyashita, D.; Ishizuka, M.; Sanada, T.; Seto, T. Ecology of hantaviruses in Mexico: Genetic identification of rodent host species and spillover infection. Virus Res. 2012, 168, 88–96. [Google Scholar] [CrossRef]
- Kang, H.J.; Bennett, S.N.; Dizney, L.; Sumibcay, L.; Arai, S.; Ruedas, L.A.; Song, J.-W.; Yanagihara, R. Host switch during evolution of a genetically distinct hantavirus in the American shrew mole (Neurotrichus gibbsii). Virology 2009, 388, 8–14. [Google Scholar] [CrossRef] [Green Version]
- Arthur, R.R.; Lofts, R.S.; Gomez, J.; Glass, G.E.; Leduc, J.W.; Childs, J.E. Grouping of hantaviruses by small (S) genome segment polymerase chain reaction and amplification of viral RNA from wild-caught rats. Am. J. Trop. Med. Hyg. 1992, 47, 210–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.J.; Bennett, S.N.; Hope, A.G.; Cook, J.A.; Yanagihara, R. Shared ancestry between a newfound mole-borne hantavirus and hantaviruses harbored by cricetid rodents. J. Virol. 2011, 85, 7496–7503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yashina, L.N.; Abramov, S.A.; Gutorov, V.V.; Dupal, T.A.; Krivopalov, A.V.; Panov, V.V.; Danchinova, G.A.; Vinogradov, V.V.; Luchnikova, E.M.; Hay, J. Seewis virus: Phylogeography of a shrew-borne hantavirus in Siberia, Russia. Vector Borne Zoonotic Dis. 2010, 10, 585–591. [Google Scholar] [CrossRef] [Green Version]
- Meheretu, Y.; Čížková, D.; Těšíková, J.; Welegerima, K.; Tomas, Z.; Kidane, D.; Girmay, K.; Schmidt-Chanasit, J.; Bryja, J.; Günther, S. High diversity of RNA viruses in rodents, Ethiopia. Emerg. Infect. Dis. 2012, 18, 2047. [Google Scholar] [CrossRef] [PubMed]
- Elwell, M.R.; Ward, G.S.; Tingpalapong, M.; LeDuc, J. Serologic evidence of Hantaan-like virus in rodents and man in Thailand. Southeast Asian J. Trop. Med. Public Health 1985, 16, 349–354. [Google Scholar]
- Xiao, S.-Y.; Leduc, J.W.; Chu, Y.K.; Schmaljohn, C.S. Phylogenetic analyses of virus isolates in the genus Hantavirus, family Bunyaviridae. Virology 1994, 198, 205–217. [Google Scholar] [CrossRef]
- Yashina, L.N. Hantavirus and its natural carriers in Siberia. Bull. East Sib. Sci. Cent. Sib. Branch Russ. Acad. Med Sci. 2012, 5, 351–355. [Google Scholar]
- Froissart, R.; Roze, D.; Uzest, M.; Galibert, L.; Blanc, S.; Michalakis, Y. Recombination every day: Abundant recombination in a virus during a single multi-cellular host infection. PLoS Biol. 2005, 3, e89. [Google Scholar] [CrossRef] [Green Version]
- Fleischmann, W.R., Jr. Viral genetics. In Medical Microbiology, 4th ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996. [Google Scholar]
- Plyusnin, A. Genetics of hantaviruses: Implications to taxonomy. Arch. Virol. 2002, 147, 665–682. [Google Scholar] [CrossRef]
- Kim, J.-A.; Kim, W.-k.; No, J.S.; Lee, S.-H.; Lee, S.-Y.; Kim, J.H.; Kho, J.H.; Lee, D.; Song, D.H.; Gu, S.H. Genetic diversity and reassortment of Hantaan virus tripartite RNA genomes in nature, the Republic of Korea. PLoS Negl. Trop. Dis. 2016, 10, e0004650. [Google Scholar] [CrossRef] [PubMed]
- Kirsanovs, S.; Klempa, B.; Franke, R.; Lee, M.-H.; Schönrich, G.; Rang, A.; Kruger, D.H. Genetic reassortment between high-virulent and low-virulent Dobrava-Belgrade virus strains. Virus Genes 2010, 41, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Schmaljohn, A.L.; Anderson, K.; Schmaljohn, C.S. Complete nucleotide sequences of the M and S segments of two hantavirus isolates from California: Evidence for reassortment in nature among viruses related to hantavirus pulmonary syndrome. Virology 1995, 206, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Chandler, L.J.; Hogge, G.; Endres, M.; Jacoby, D.R.; Nathanson, N.; Beaty, B.J. Reassortment of La Crosse and Tahyna bunyaviruses in Aedes triseriatus mosquitoes. Virus Res. 1991, 20, 181–191. [Google Scholar] [CrossRef]
- Henderson, W.W.; Monroe, M.C.; JEOR, S.C.S.; Thayer, W.P.; Rowe, J.E.; Peters, C.; Nichol, S.T. Naturally occurring Sin Nombre virus genetic reassortants. Virology 1995, 214, 602–610. [Google Scholar] [CrossRef] [Green Version]
- Handke, W.; Oelschlegel, R.; Franke, R.; Wiedemann, L.; Krüger, D.H.; Rang, A. Generation and characterization of genetic reassortants between Puumala and Prospect Hill hantavirus in vitro. J. Gen. Virol. 2010, 91, 2351–2359. [Google Scholar] [CrossRef]
- Beaty, B.J.; Sundin, D.R.; Chandler, L.J.; Bishop, D. Evolution of bunyaviruses by genome reassortment in dually infected mosquitoes (Aedes triseriatus). Science 1985, 230, 548–550. [Google Scholar] [CrossRef]
- Rizvanov, A.A.; Khaiboullina, S.F.; Jeor, S.S. Development of reassortant viruses between pathogenic hantavirus strains. Virology 2004, 327, 225–232. [Google Scholar] [CrossRef] [Green Version]
- Yanagihara, R.; Gu, S.H.; Arai, S.; Kang, H.J.; Song, J.-W. Hantaviruses: Rediscovery and new beginnings. Virus Res. 2014, 187, 6–14. [Google Scholar] [CrossRef] [Green Version]
- Schlegel, M.; Radosa, L.; Rosenfeld, U.M.; Schmidt, S.; Triebenbacher, C.; Löhr, P.-W.; Fuchs, D.; Heroldová, M.; Jánová, E.; Stanko, M. Broad geographical distribution and high genetic diversity of shrew-borne Seewis hantavirus in Central Europe. Virus Genes 2012, 45, 48–55. [Google Scholar] [CrossRef]
- Hughes, A.L.; Friedman, R. Evolutionary diversification of protein-coding genes of hantaviruses. Mol. Biol. Evol. 2000, 17, 1558–1568. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, C.; Holmes, E.C.; Charleston, M.A. Hantavirus evolution in relation to its rodent and insectivore hosts: No evidence for codivergence. Mol. Biol. Evol. 2009, 26, 143–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, W.C.; Doty, J.B.; Hughes, M.T.; Beaty, B.J.; Calisher, C.H. Temporal and geographic evidence for evolution of Sin Nombre virus using molecular analyses of viral RNA from Colorado, New Mexico and Montana. Virol. J. 2009, 6, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adkins, R.M.; Gelke, E.L.; Rowe, D.; Honeycutt, R.L. Molecular phylogeny and divergence time estimates for major rodent groups: Evidence from multiple genes. Mol. Biol. Evol. 2001, 18, 777–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souza, W.; Bello, G.; Amarilla, A.; Alfonso, H.; Aquino, V.; Figueiredo, L. Phylogeography and evolutionary history of rodent-borne hantaviruses. Infect. Genet. Evol. 2014, 21, 198–204. [Google Scholar] [CrossRef]
- Cao, S.; Ma, J.; Cheng, C.; Ju, W.; Wang, Y. Genetic characterization of hantaviruses isolated from rodents in the port cities of Heilongjiang, China, in 2014. BMC Vet. Res. 2016, 12, 69. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yoshimatsu, K.; Ebihara, H.; Ogino, M.; Araki, K.; Kariwa, H.; Wang, Z.; Luo, Z.; Li, D.; Hang, C. Genetic diversity of hantaviruses isolated in China and characterization of novel hantaviruses isolated from Niviventer confucianus and Rattus rattus. Virology 2000, 278, 332–345. [Google Scholar] [CrossRef] [Green Version]
- Avsic-Zupanc, T.; Nemirov, K.; Petrovec, M.; Trilar, T.; Poljak, M.; Vaheri, A.; Plyusnin, A. Genetic analysis of wild-type Dobrava hantavirus in Slovenia: Co-existence of two distinct genetic lineages within the same natural focus. J. Gen. Virol. 2000, 81, 1747–1755. [Google Scholar] [CrossRef]
- Wójcik, J.; Kawałko, A.; Marková, S.; Searle, J.; Kotlík, P. Phylogeographic signatures of northward post-glacial colonization from high-latitude refugia: A case study of bank voles using museum specimens. J. Zool. 2010, 281, 249–262. [Google Scholar] [CrossRef]
- Castel, G.; Tordo, N.; Plyusnin, A. Estimation of main diversification time-points of hantaviruses using phylogenetic analyses of complete genomes. Virus Res. 2017, 233, 60–69. [Google Scholar] [CrossRef]
- Ali, H.S.; Drewes, S.; de Melo, V.W.; Schlegel, M.; Freise, J.; Groschup, M.H.; Heckel, G.; Ulrich, R.G. Complete genome of a Puumala virus strain from Central Europe. Virus Genes 2015, 50, 292–298. [Google Scholar] [CrossRef]
- Han, G.-Z.; Worobey, M. Homologous recombination in negative sense RNA viruses. Viruses 2011, 3, 1358–1373. [Google Scholar] [CrossRef] [PubMed]
- Garanina, S.; Platonov, A.; Zhuravlev, V.; Murashkina, A.; Yakimenko, V.; Korneev, A.; Shipulin, G. Genetic diversity and geographic distribution of hantaviruses in Russia. Zoonoses Public Health 2009, 56, 297–309. [Google Scholar] [CrossRef]
- Löber, C.; Anheier, B.; Lindow, S.; Klenk, H.-D.; Feldmann, H. The Hantaan virus glycoprotein precursor is cleaved at the conserved pentapeptide WAASA. Virology 2001, 289, 224–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Oliveira, R.C.; Guterres, A.; Fernandes, J.; D’Andrea, P.S.; Bonvicino, C.R.; De Lemos, E.R.S. Hantavirus reservoirs: Current status with an emphasis on data from Brazil. Viruses 2014, 6, 1929–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Li, A.; Liu, Y.; Wu, W.; Li, C.; Yu, D.; Zhu, Y.; Li, J.; Li, D.; Wang, S. Genetic diversity and evolution of Hantaan virus in China and its neighbors. bioRxiv 2020. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genetic Lineage (Designation) | Geographical Location | Reference | |
|---|---|---|---|
| 1 | Central European (CE) | France, Belgium, Germany, Slovakia, Netherland | [19,20,21,23,27,28,29] |
| 2 | Alpe-Adrian (ALAD) | Austria, Slovenia, Croatia, Hungary | |
| 3 | Danish (DAN) | Denmark | |
| 4 | South Scandinavian (S-SCAN) | Norway, Southern Sweden | |
| 5 | North Scandinavian (N-SCAN) | Northern Sweden | |
| 6 | Finnish (FIN) | Finland, Russian Karelia and Western Siberia | |
| 7 | Russian (RUS) | Central Russia, Estonia, Latvia | |
| 8 | Latvian (LAT) | Latvia, North-East Poland | [18,22] |
| Orthohantavirus (Diseases) | Rodents, Natural Reservoir | Area | Reference |
|---|---|---|---|
| Dobrava-Belgrade (HFRS) | Apodemus flavicollis | Europe (Balkan), European part of Russia | [7] |
| Hantaan (HFRS) | Apodemus agrarius | Eastern Asia | |
| Khabarovsk (HFRS) | Microtus fortis | Asia (Siberia, Far East of Russia) | |
| Puumala (HFRS) | Myodes glareolus | Western, Central and Northern Europe, European part of Russia | [58] |
| Sangassou (HFRS) | Hylomyscus alleni | Africa (Guinea) | [68,69] |
| Seoul (HFRS) | Rattus norvegicus | Worldwide | [70] |
| Tula (HFRS) | Microtus arvalis | Europe | [62] |
| Andes (HPS) | Oligoryzomys longicaudatus | South America (Argentina, Chile) | [71] |
| Black Creek Canal (HPS) | Sigmodon hispidus | USA | [72] |
| Choclo (HPS) | Zygodontomys brevicauda | South America (Colombia, French Guiana and Panama) | [73,74] |
| Laguna Negra (HPS) | Calomys callidus, Akodon simulator, Calomys laucha | South America (Bolivia, Argentina and Paraguay; parts of Brazil) | [75] |
| Sin Nombre (HPS) | Peromyscus maniculatus | Northern America | [71] |
| Asama (unknown) | Urotrichus talpoides | Asia (Japan) | [76] |
| Asikkala (unknown) | Sorex minutus | Europe (Czech Republic, Finland, Germany and Slovakia) | [77] |
| Bowe (unknown) | Crocidura douceti | Africa (Guinea) | [78] |
| Cano Delgadito (unknown) | Sigmodon alstoni | South America (Venezuela) | [79] |
| Cao Bang (unknown) | Anourosorex squamipes | Asia (China and Vietnam) | [80,81] |
| El Moro Canyon (unknown) | Reithrodontomys megalotis | USA and Mexico | [82,83] |
| Jeju (unknown) | Crocidura shantungensis | Asia (Korea) | [76] |
| Kenkeme (unknown) | Sorex roboratus | Russia (Siberia) | [84] |
| Luxi (unknown) | Eothenomys miletus | Asia (China) | [85] |
| Maporal (unknown) | Sigmodon alstoni | South America (Venezuela) | [79] |
| Montano (unknown) | Peromyscus aztecus | Northern America (Mexico) | [86] |
| Oxbow (unknown) | Neurotrichus gibbsii | USA | [87] |
| Prospect Hill virus (unknown) | Microtus pennsylvanicus | USA (Maryland) | [88] |
| Rockport (unknown) | Scalopus aquaticus | USA | [89] |
| Seewis (unknown) | Sorex daphaenodon | Russia (Siberia) | [90] |
| Tatenale (unknown) | Microtus agrestis | United Kingdom (England). | [60,61] |
| Tigray (unknown) | Stenocephalemys albipes | Africa (Ethiopia) | [91] |
| Thailand (unknown) | Bandicota indica | Asia (Thailand) | [92,93] |
| Yakeshi (unknown) | Sorex isodon | Asia (China) | [80] |
| Necocli, Fusong, Fugong, Dabieshan, Bruges | unknown | unknown | [8] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kabwe, E.; Davidyuk, Y.; Shamsutdinov, A.; Garanina, E.; Martynova, E.; Kitaeva, K.; Malisheni, M.; Isaeva, G.; Savitskaya, T.; Urbanowicz, R.A.; et al. Orthohantaviruses, Emerging Zoonotic Pathogens. Pathogens 2020, 9, 775. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens9090775
Kabwe E, Davidyuk Y, Shamsutdinov A, Garanina E, Martynova E, Kitaeva K, Malisheni M, Isaeva G, Savitskaya T, Urbanowicz RA, et al. Orthohantaviruses, Emerging Zoonotic Pathogens. Pathogens. 2020; 9(9):775. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens9090775
Chicago/Turabian StyleKabwe, Emmanuel, Yuriy Davidyuk, Anton Shamsutdinov, Ekaterina Garanina, Ekaterina Martynova, Kristina Kitaeva, Moffat Malisheni, Guzel Isaeva, Tatiana Savitskaya, Richard A. Urbanowicz, and et al. 2020. "Orthohantaviruses, Emerging Zoonotic Pathogens" Pathogens 9, no. 9: 775. https://0-doi-org.brum.beds.ac.uk/10.3390/pathogens9090775