
Current and Future Therapeutic Strategies for Limb Girdle Muscular Dystrophy Type R1: Clinical and Experimental Approaches



Abstract
:1. Introduction
2. Current Clinical and Experimental Studies
3. Future Therapy Strategies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AAV | Adeno-associated virus |
| AAVrh74.tMCK.hCAPN3 | AAV rhesus 74 truncated muscle creatine kinase human CAPN3 |
| AMBMP | 2-amino-4-(3,4-(methylenedioxy)benzylamino)-6-(3-methoxyphenyl) pyrimidine |
| ATF6 | Activating transcription factor 6 |
| C3KO | Calpain 3 knock out |
| CaMKII | Ca2+/calmodulin-dependent protein kinase II |
| CaMKII | Ca2+/calmodulin (CaM)-dependent protein kinase II |
| CAPN3 | Calpain 3 |
| Cas9 | Native Cas9 nuclease |
| CHOP | C/-EBP homologous protein |
| CRISPR-Cas9 | Clustered regularly interspaced short palindromic repeats CRISPR-associated proteins 9 |
| CSQ | Calsequestrin |
| DMD | Duchenne muscular dystrophy |
| elF2α | Eukaryotic initiation factor 2α |
| ER | Endoplasmic reticulum |
| ERAD | ER-associated protein degradation |
| FRZB | Frizzled-related protein |
| GRP78 | Glucose-regulated protein |
| IPSC | Induced pluripotent stem cell |
| IRE1α | Inositol-requiring enzyme 1α |
| LGMDR1 | Limb girdle muscular dystrophy R1 |
| LIM | Lin-11 Isl-1 Mec-3 |
| Mss51 | Mitochondrial translational activator |
| MuRF1 | Muscle RING-finger protein-1 |
| MYO-029 | Stamulumab |
| MyoD | Myogenic differentiation antigen |
| NCX3 | Na+-Ca2+ exchanger 3 |
| pAAV-CMV-mSeAPpropmyoD76A | Plasmid AAV-cytomegalovirus- murine-secreted alkaline phosphatase myogenic differentiation antigen murine-secreted alkaline phosphatase |
| Pax3/Pax7 | Paired box gene 3/Paired box gene 7 |
| PERK | PKR-like ER kinase |
| RyR1 | Ryanodine receptor 1 |
| SERCA | Sarcoplasmic/endoplasmic reticulum Ca2+ ATPase |
| TCF/LEF | T-cell factor/lymphoid enhancer factor |
| TRIM32 | Tripartite motif-containing protein 32 |
| TUDCA | Tauroursodeoxycholic acid |
| UPR | Unfolded protein response |
| WNT | Wingless-related integration site |
| XBP1 | X-box binding protein 1 |
References
- Richard, I.; Hogrel, J.Y.; Stockholm, D.; Payan, C.A.M.; Fougerousse, F.; Eymard, B.; Mignard, C.; Lopez de Munain, A.; Fardeau, M.; Urtizberea, J.A. Natural history of LGMD2A for delineating outcome measures in clinical trials. Ann. Clin. Transl. Neurol. 2016, 3, 248–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- OMIM—Online Mendelian Inheritance in Man. Available online: https://www.omim.org/ (accessed on 20 October 2020).
- Richard, I.; Broux, O.; Allamand, V.; Fougerousse, F.; Chiannilkulchai, N.; Bourg, N.; Brenguier, L.; Devaud, C.; Pasturaud, P.; Roudaut, C.; et al. Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell 1995, 81, 27–40. [Google Scholar] [CrossRef] [Green Version]
- Vissing, J.; Barresi, R.; Witting, N.; Van Ghelue, M.; Gammelgaard, L.; Bindoff, L.A.; Straub, V.; Lochmüller, H.; Hudson, J.; Wahl, C.M.; et al. A heterozygous 21-bp deletion in CAPN3 causes dominantly inherited limb girdle muscular dystrophy. Brain 2016, 139, 2154–2163. [Google Scholar] [CrossRef] [Green Version]
- Lasa-Elgarresta, J.; Mosqueira-Martín, L.; Naldaiz-Gastesi, N.; Sáenz, A.; de Munain, A.L.; Vallejo-Illarramendi, A. Calcium Mechanisms in Limb-Girdle Muscular Dystrophy with CAPN3 Mutations. Int. J. Mol. Sci. 2019, 20, 4548. [Google Scholar] [CrossRef] [Green Version]
- Calpainopathy-GeneReviews®-NCBI Bookshelf. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK1313/#lgmd2a.Molecular_Genetics (accessed on 4 May 2021).
- The CAPN3 gene homepage—Global Variome shared LOVD. Available online: https://databases.lovd.nl/shared/genes/CAPN3 (accessed on 10 October 2020).
- Sorimachi, H.; Imajoh-Ohmi, S.; Emori, Y.; Kawasaki, H.; Ohno, S.; Minami, Y.; Suzuki, K. Molecular cloning of a novel mammalian calcium-dependent protease distinct from both m- and μ-types. Specific expression of the mRNA in skeletal muscle. J. Biol. Chem. 1989, 264, 20106–20111. [Google Scholar] [CrossRef]
- Ojima, K.; Ono, Y.; Ottenheijm, C.; Hata, S.; Suzuki, H.; Granzier, H.; Sorimachi, H. Non-proteolytic functions of calpain-3 in sarcoplasmic reticulum in skeletal muscles. J. Mol. Biol. 2011, 407, 439–449. [Google Scholar] [CrossRef] [Green Version]
- Toral-Ojeda, I.; Aldanondo, G.; Lasa-Elgarresta, J.; Lasa-Fernandez, H.; Vesga-Castro, C.; Mouly, V.; de Munain, A.L.; Vallejo-Illarramendi, A. A Novel Functional In Vitro Model that Recapitulates Human Muscle Disorders. In Muscle Cell and Tissue-Current Status of Research Field; InTech: London, UK, 2018. [Google Scholar]
- Kramerova, I.; Kudryashova, E.; Ermolova, N.; Saenz, A.; Jaka, O.; López de munain, A.; Spencer, M.J. Impaired calcium calmodulin kinase signaling and muscle adaptation response in the absence of calpain 3. Hum. Mol. Genet. 2012, 21, 3193–3204. [Google Scholar] [CrossRef] [Green Version]
- Michel, L.Y.M.; Hoenderop, J.G.J.; Bindels, R.J.M. Calpain-3-mediated regulation of the Na+-Ca2+ exchanger isoform 3. Pflugers Arch. Eur. J. Physiol. 2016, 468, 243–255. [Google Scholar] [CrossRef] [Green Version]
- Ono, Y.; Ojima, K.; Torii, F.; Takaya, E.; Doi, N.; Nakagawa, K.; Hata, S.; Abe, K.; Sorimachi, H. Skeletal muscle-specific calpain is an intracellular Na+- dependent protease. J. Biol. Chem. 2010, 285, 22986–22998. [Google Scholar] [CrossRef] [Green Version]
- Ono, Y.; Ojima, K.; Shinkai-Ouchi, F.; Hata, S.; Sorimachi, H. An eccentric calpain, CAPN3/p94/calpain-3. Biochimie 2016, 122, 169–187. [Google Scholar] [CrossRef] [Green Version]
- Beckmann, J.S.; Spencer, M. Calpain 3, the “gatekeeper” of proper sarcomere assembly, turnover and maintenance. Neuromuscul. Disord. 2008, 18, 913–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorimachi, H.; Toyama-Sorimachi, N.; Saido, T.C.; Kawasaki, H.; Sugita, H.; Miyasaka, M.; Arahata, K.I.; Ishiura, S.; Suzuki, K. Muscle-specific calpain, p94, is degraded by autolysis immediately after translation, resulting in disappearance from muscle. J. Biol. Chem. 1993, 268, 10593–10605. [Google Scholar] [CrossRef]
- de Paula, F.; Vainzof, M.; Passos-Bueno, M.R.; Pavanello, R.d.C.; Matioli, S.R.; Anderson, L.V.B.; Nigro, V.; Zatz, M. Clinical variability in calpainopathy: What makes the difference? Eur. J. Hum. Genet. 2002, 10, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Schessl, J.; Walter, M.C.; Schreiber, G.; Schara, U.; Müller, C.R.; Lochmüller, H.; Bönnemann, C.G.; Korinthenberg, R.; Kirschner, J. Phenotypic variability in siblings with calpainopathy (LGMD2A). Acta Myol. 2008, 27, 54–58. [Google Scholar] [PubMed]
- de Albuquerque, M.A.V.; Abath Neto, O.; da Silva, F.M.A.; Zanoteli, E.; Reed, U.C. Limb-girdle muscular dystrophy type 2A in Brazilian children. Arq. Neuropsiquiatr. 2015, 73, 993–997. [Google Scholar] [CrossRef] [Green Version]
- Weekly Steroids in Muscular Dystrophy-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04054375 (accessed on 12 October 2020).
- Merlini, L.; Cicognani, A.; Malaspina, E.; Gennari, M.; Gnudi, S.; Talim, B.; Franzoni, E. Early prednisone treatment in Duchenne muscular dystrophy. Muscle Nerve 2003, 27, 222–227. [Google Scholar] [CrossRef]
- Mesa, L.E.; Dubrovsky, A.L.; Corderi, J.; Marco, P.; Flores, D. Steroids in duchenne muscular dystrophy—Deflazacort trial. Neuromuscul. Disord. 1991, 1, 261–266. [Google Scholar] [CrossRef]
- Hussein, M.R.; Hamed, S.A.; Mostafa, M.G.; Abu-Dief, E.E.; Kamel, N.F.; Kandil, M.R. The effects of glucocorticoid therapy on the inflammatory and Dendritic cells in muscular dystrophies. Int. J. Exp. Pathol. 2006, 87, 451–461. [Google Scholar] [CrossRef]
- Spencer, M.J.; Tidball, J.G. Do immune cells promote the pathology of dystrophin-deficient myopathies? Neuromuscul. Disord. 2001, 11, 556–564. [Google Scholar] [CrossRef]
- Gaud, A.; Simon, J.M.; Witzel, T.; Carre-Pierrat, M.; Wermuth, C.G.; Ségalat, L. Prednisone reduces muscle degeneration in dystrophin-deficient Caenorhabditis elegans. Neuromuscul. Disord. 2004, 14, 365–370. [Google Scholar] [CrossRef]
- Wagner, K.R.; Fleckenstein, J.L.; Amato, A.A.; Barohn, R.J.; Bushby, K.; Escolar, D.M.; Flanigan, K.M.; Pestronk, A.; Tawil, R.; Wolfe, G.I.; et al. A phase I/II trial of MYO-029 in adult subjects with muscular dystrophy. Ann. Neurol. 2008, 63, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, M.; Poupiot, J.; Vulin, A.; Fougerousse, F.; Arandel, L.; Daniele, N.; Roudaut, C.; Noulet, F.; Garcia, L.; Danos, O.; et al. AAV-mediated delivery of a mutated myostatin propeptide ameliorates calpain 3 but not α-sarcoglycan deficiency. Gene Ther. 2007, 14, 733–740. [Google Scholar] [CrossRef] [Green Version]
- Kramerova, I.; Marinov, M.; Owens, J.; Lee, S.J.; Becerra, D.; Spencer, M.J. Myostatin inhibition promotes fast fibre hypertrophy but causes loss of AMP-activated protein kinase signalling and poor exercise tolerance in a model of limb-girdle muscular dystrophy R1/2A. J. Physiol. 2020, 598, 3927–3939. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, M.; Ishida, J.; Ebner, N.; Anker, S.D.; Von Haehling, S. Myostatin inhibitors as pharmacological treatment for muscle wasting and muscular dystrophy. JCSM Clin. Rep. 2017, 2, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Mariot, V.; Joubert, R.; Hourdé, C.; Féasson, L.; Hanna, M.; Muntoni, F.; Maisonobe, T.; Servais, L.; Bogni, C.; Le Panse, R.; et al. Downregulation of myostatin pathway in neuromuscular diseases may explain challenges of anti-myostatin therapeutic approaches. Nat. Commun. 2017, 8, 1–8. [Google Scholar] [CrossRef]
- Roudaut, C.; Le Roy, F.; Suel, L.; Poupiot, J.; Charton, K.; Bartoli, M.; Richard, I. Restriction of calpain3 expression to the skeletal muscle prevents cardiac toxicity and corrects pathology in a murine model of limb-girdle muscular dystrophy. Circulation 2013, 128, 1094–1104. [Google Scholar] [CrossRef] [Green Version]
- Yalvac, M.E.; Amornvit, J.; Braganza, C.; Chen, L.; Hussain, S.R.A.; Shontz, K.M.; Montgomery, C.L.; Flanigan, K.M.; Lewis, S.; Sahenk, Z. Impaired regeneration in calpain-3 null muscle is associated with perturbations in mTORC1 signaling and defective mitochondrial biogenesis. Skelet. Muscle 2017, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, M.; Roudaut, C.; Martin, S.; Fougerousse, F.; Suel, L.; Poupiot, J.; Gicquel, E.; Noulet, F.; Danos, O.; Richard, I. Safety and efficacy of AAV-mediated calpain 3 gene transfer in a mouse model of limb-girdle muscular dystrophy Type 2A. Mol. Ther. 2006, 13, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Lostal, W.; Roudaut, C.; Faivre, M.; Charton, K.; Suel, L.; Bourg, N.; Best, H.; Smith, J.E.; Gohlke, J.; Corre, G.; et al. Titin splicing regulates cardiotoxicity associated with calpain 3 gene therapy for limb-girdle muscular dystrophy type 2A. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Chicoine, L.G.; Al-Zaidy, S.A.; Sahenk, Z.; Lehman, K.; Lowes, L.; Miller, N.; Alfano, L.; Galliers, B.; Lewis, S.; et al. Gene Delivery for Limb-Girdle Muscular Dystrophy Type 2D by Isolated Limb Infusion. Hum. Gene Ther. 2019, 30, 794–801. [Google Scholar] [CrossRef]
- Chicoine, L.G.; Rodino-Klapac, L.R.; Shao, G.; Xu, R.; Bremer, W.G.; Camboni, M.; Golden, B.; Montgomery, C.L.; Shontz, K.; Heller, K.N.; et al. Vascular delivery of rAAVrh74.MCK.GALGT2 to the gastrocnemius muscle of the rhesus macaque stimulates the expression of dystrophin and laminin α2 surrogates. Mol. Ther. 2014, 22, 713–724. [Google Scholar] [CrossRef] [Green Version]
- 1135—Biopotency and Biodistribution/ Toxicology Studies Following Systemic Gene Therapy with AAVrh74.tMCK.hCAPN3 in the Mouse Model for LGMD2A-ASGCT 23rd Annual Meeting. Available online: https://cslide-us.ctimeetingtech.com/asgct23/attendee/eposter/poster/466 (accessed on 16 October 2020).
- Li, C.; Samulski, R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020, 21, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Jaka, O.; Casas-Fraile, L.; Azpitarte, M.; Aiastui, A.; López de Munain, A.; Sáenz, A. FRZB and melusin, overexpressed in LGMD2A, regulate integrin β1D isoform replacement altering myoblast fusion and the integrin-signalling pathway. Expert Rev. Mol. Med. 2017, 19. [Google Scholar] [CrossRef] [Green Version]
- Kramerova, I.; Kudryashova, E.; Wu, B.; Spencer, M.J. Regulation of the M-Cadherin-β-Catenin Complex by Calpain 3 during Terminal Stages of Myogenic Differentiation. Mol. Cell. Biol. 2006, 26, 8437–8447. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wu, X.; Mitchell, B.; Kintner, C.; Ding, S.; Schultz, P.G. A small-molecule agonist of the Wnt signaling pathway. Angew. Chem. Int. Ed. 2005, 44, 1987–1990. [Google Scholar] [CrossRef]
- Lim, J.C.; Kania, K.D.; Wijesuriya, H.; Chawla, S.; Sethi, J.K.; Pulaski, L.; Romero, I.A.; Couraud, P.O.; Weksler, B.B.; Hladky, S.B.; et al. Activation of β-catenin signalling by GSK-3 inhibition increases p-glycoprotein expression in brain endothelial cells. J. Neurochem. 2008, 106, 1855–1865. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Campagna, J.; John, V.; Damoiseaux, R.; Mokhonova, E.; Becerra, D.; Meng, H.; McNally, E.M.; Pyle, A.D.; Kramerova, I.; et al. A Small-Molecule Approach to Restore a Slow-Oxidative Phenotype and Defective CaMKIIβ Signaling in Limb Girdle Muscular Dystrophy. Cell Rep. Med. 2020, 1. [Google Scholar] [CrossRef]
- Alenzi, F.Q.; Lotfy, M.; Tamimi, W.G.; Wyse, R.K.H. Review: Stem cells and gene therapy. Lab. Hematol. 2010, 16, 53–73. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, S.; Dhoke, N.R.; Kiley, J.; Mateos-Aierdi, A.J.; Tungtur, S.; Mondragon-Gonzalez, R.; Killeen, G.; Oliveira, V.K.P.; López de Munain, A.; Perlingeiro, R.C.R. Gene Correction of LGMD2A Patient-Specific iPSCs for the Development of Targeted Autologous Cell Therapy. Mol. Ther. 2019, 27, 2147–2157. [Google Scholar] [CrossRef]
- Peng, G.-Y.; Lin, Y.; Li, J.-J.; Wang, Y.; Huang, H.-Y.; Shen, Z.-Y. The Application of Induced Pluripotent Stem Cells in Pathogenesis Study and Gene Therapy for Vascular Disorders: Current Progress and Future Challenges. Stem Cells Int. 2019, 2019, 9613258. [Google Scholar] [CrossRef]
- Nilsson, M.I.; Macneil, L.G.; Kitaoka, Y.; Alqarni, F.; Suri, R.; Akhtar, M.; Haikalis, M.E.; Dhaliwal, P.; Saeed, M.; Tarnopolsky, M.A. Redox state and mitochondrial respiratory chain function in skeletal muscle of LGMD2A patients. PLoS ONE 2014, 9, e102549. [Google Scholar] [CrossRef] [PubMed]
- Kramerova, I.; Kudryashova, E.; Wu, B.; Germain, S.; Vandenborne, K.; Romain, N.; Haller, R.G.; Verity, M.A.; Spencer, M.J. Mitochondrial abnormalities, energy deficit and oxidative stress are features of calpain 3 deficiency in skeletal muscle. Hum. Mol. Genet. 2009, 18, 3194–3205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moyer, A.L.; Wagner, K.R. Mammalian Mss51 is a Skeletal Muscle-Specific Gene Modulating Cellular Metabolism. J. Neuromuscul. Dis. 2015, 2, 371–385. [Google Scholar] [CrossRef] [Green Version]
- Research–Coalition to Cure Calpain 3. Available online: https://www.curecalpain3.org/research/ (accessed on 16 October 2020).
- Fanin, M.; Nascimbeni, A.C.; Angelini, C. Muscle atrophy in Limb Girdle Muscular Dystrophy 2A: A morphometric and molecular study. Neuropathol. Appl. Neurobiol. 2013, 39, 762–771. [Google Scholar] [CrossRef]
- CAPN3 Protein (Human)-STRING İnteraction Network. Available online: https://string-db.org/network/9606.ENSP00000380349 (accessed on 6 February 2021).
- Kaneko, M.; Imaißumi, K.; Saito, A.; Kanemoto, S.; Asada, R.; Matsuhisa, K.; Ohtake, Y. ER stress and disease: Toward prevention and treatment. Biol. Pharm. Bull. 2017, 40, 1337–1343. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Kaufman, R.J. Signaling the unfolded protein response from the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 25935–25938. [Google Scholar] [CrossRef] [Green Version]
- Kincaid, M.M.; Cooper, A.A. ERADicate ER stress or die trying. Antioxid. Redox Signal. 2007, 9, 2373–2387. [Google Scholar] [CrossRef]
- Toral-Ojeda, I.; Aldanondo, G.; Lasa-Elgarresta, J.; Lasa-Fernández, H.; Fernández-Torrón, R.; De Munain, A.L.; Vallejo-Illarramendi, A. Calpain 3 deficiency affects SERCA expression and function in the skeletal muscle. Expert Rev. Mol. Med. 2016, 18. [Google Scholar] [CrossRef] [Green Version]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. The endoplasmic reticulum and the unfolded protein response. Semin. Cell Dev. Biol. 2007, 18, 716–731. [Google Scholar] [CrossRef] [Green Version]
- Nakka, V.P.; Prakash-babu, P.; Vemuganti, R. Crosstalk Between Endoplasmic Reticulum Stress, Oxidative Stress, and Autophagy: Potential Therapeutic Targets for Acute CNS Injuries. Mol. Neurobiol. 2016, 53, 532–544. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, S.J.; Ron, D. Endoplasmic reticulum stress signaling in disease. Physiol. Rev. 2006, 86, 1133–1149. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, N. ER and aging-Protein folding and the ER stress response. Ageing Res. Rev. 2009, 8, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Sarvani, C.; Sireesh, D.; Ramkumar, K.M. Unraveling the role of ER stress inhibitors in the context of metabolic diseases. Pharmacol. Res. 2017, 119, 412–421. [Google Scholar] [CrossRef]
- Liu, M.Q.; Chen, Z.; Chen, L.X. Endoplasmic reticulum stress: A novel mechanism and therapeutic target for cardiovascular diseases. Acta Pharmacol. Sin. 2016, 37, 425–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, H.; Zhou, C.; Chi, J.; Pan, S.; Lin, H.; Gao, F.; Ni, T.; Meng, L.; Zhang, J.; Jiang, C.; et al. The Role of Tauroursodeoxycholic Acid on Dedifferentiation of Vascular Smooth Muscle Cells by Modulation of Endoplasmic Reticulum Stress and as an Oral Drug Inhibiting In-Stent Restenosis. Cardiovasc. Drugs Ther. 2019, 33, 25–33. [Google Scholar] [CrossRef]
- Mesbah Moosavi, Z.S.; Hood, D.A. The unfolded protein response in relation to mitochondrial biogenesis in skeletal muscle cells. Am. J. Physiol. Cell Physiol. 2017, 312, C583–C594. [Google Scholar] [CrossRef]
- Romero-Ramírez, L.; Nieto-Sampedro, M.; Barreda-Manso, M.A. All roads go to salubrinal: Endoplasmic reticulum stress, neuroprotection and glial scar formation. Neural Regen. Res. 2015, 10, 1926–1927. [Google Scholar] [CrossRef]
- Foltz, S.J.; Luan, J.; Call, J.A.; Patel, A.; Peissig, K.B.; Fortunato, M.J.; Beedle, A.M. Four-week rapamycin treatment improves muscular dystrophy in a fukutin-deficient mouse model of dystroglycanopathy. Skelet. Muscle 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Bibee, K.P.; Cheng, Y.J.; Ching, J.K.; Marsh, J.N.; Li, A.J.; Keeling, R.M.; Connolly, A.M.; Golumbek, P.T.; Myerson, J.W.; Hu, G.; et al. Rapamycin nanoparticles target defective autophagy in muscular dystrophy to enhance both strength and cardiac function. FASEB J. 2014, 28, 2047–2061. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, Y.; Hambright, W.S.; Takayama, K.; Mu, X.; Lu, A.; Cummins, J.H.; Matsumoto, T.; Yurube, T.; Kuroda, R.; Kurosaka, M.; et al. Rapamycin Rescues Age-Related Changes in Muscle-Derived Stem/Progenitor Cells from Progeroid Mice. Mol. Ther. Methods Clin. Dev. 2019, 14, 64–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurevich, E.V.; Gurevich, V.V. Therapeutic potential of small molecules and engineered proteins. Handb. Exp. Pharmacol. 2014, 219, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.; Dahl, R.; Hsieh, W.; Shin, A.; Zsebo, K.M.; Buettner, C.; Hajjar, R.J.; Lebeche, D. Small molecular allosteric activator of the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) attenuates diabetes and metabolic disorders. J. Biol. Chem. 2016, 291, 5185–5198. [Google Scholar] [CrossRef] [Green Version]
- Dahl, R. A new target for Parkinson’s disease: Small molecule SERCA activator CDN1163 ameliorates dyskinesia in 6-OHDA-lesioned rats. Bioorg. Med. Chem. 2017, 25, 53–57. [Google Scholar] [CrossRef]
- Nelson, B.R.; Makarewich, C.A.; Anderson, D.M.; Winders, B.R.; Troupes, C.D.; Wu, F.; Reese, A.L.; McAnally, J.R.; Chen, X.; Kavalali, E.T.; et al. Muscle physiology: A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science 2016, 351, 271–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]




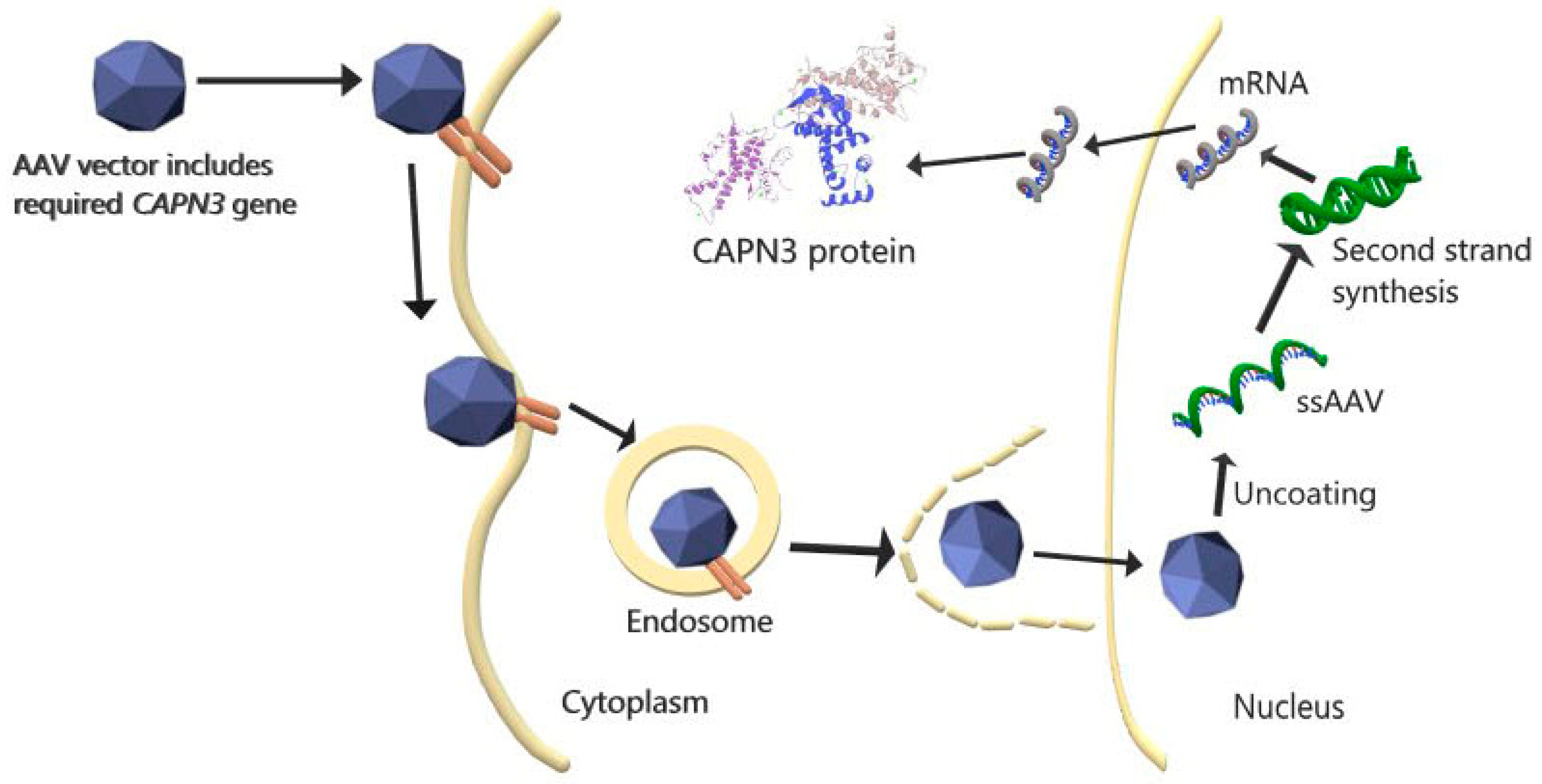{kind=link}
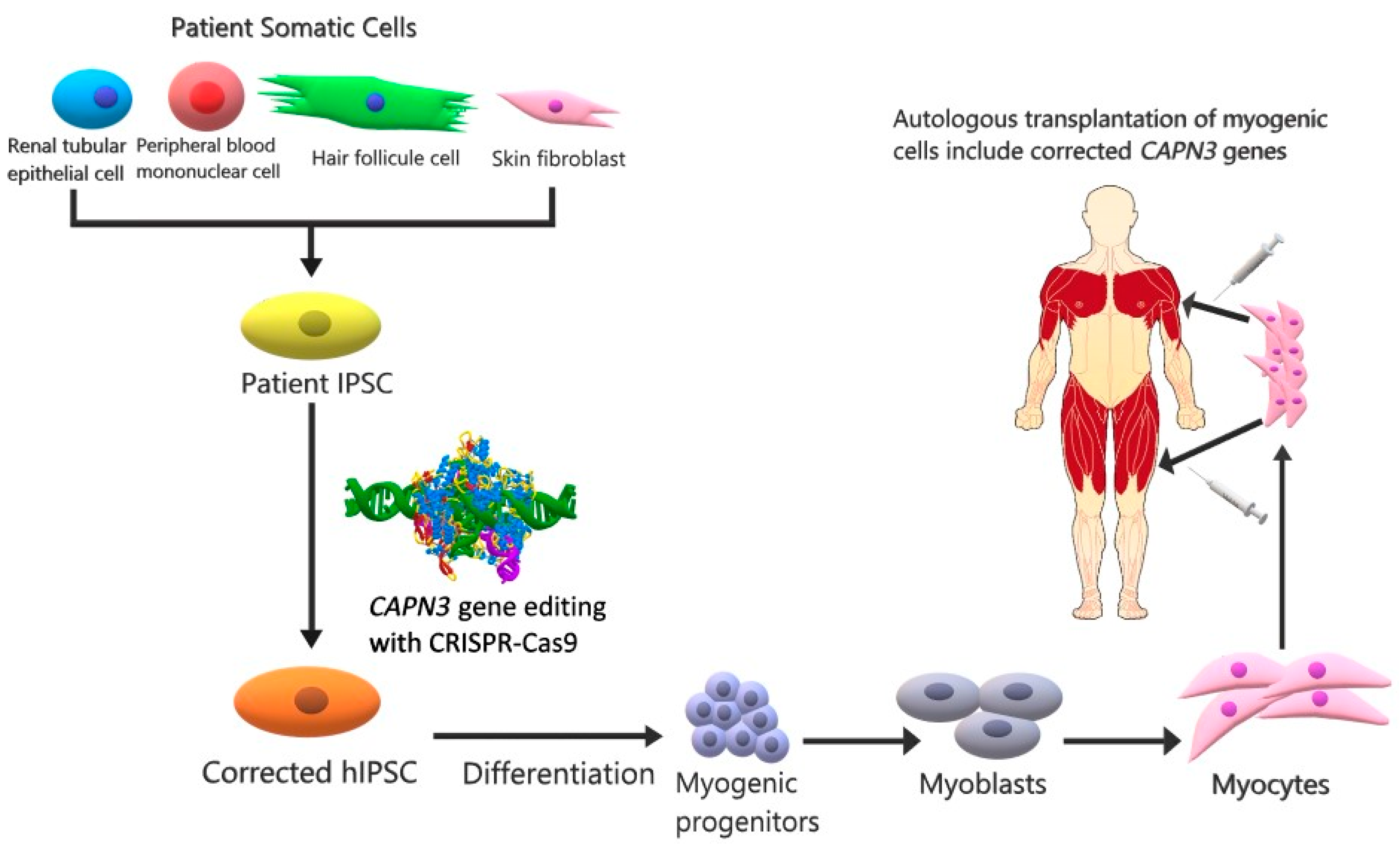{kind=link}
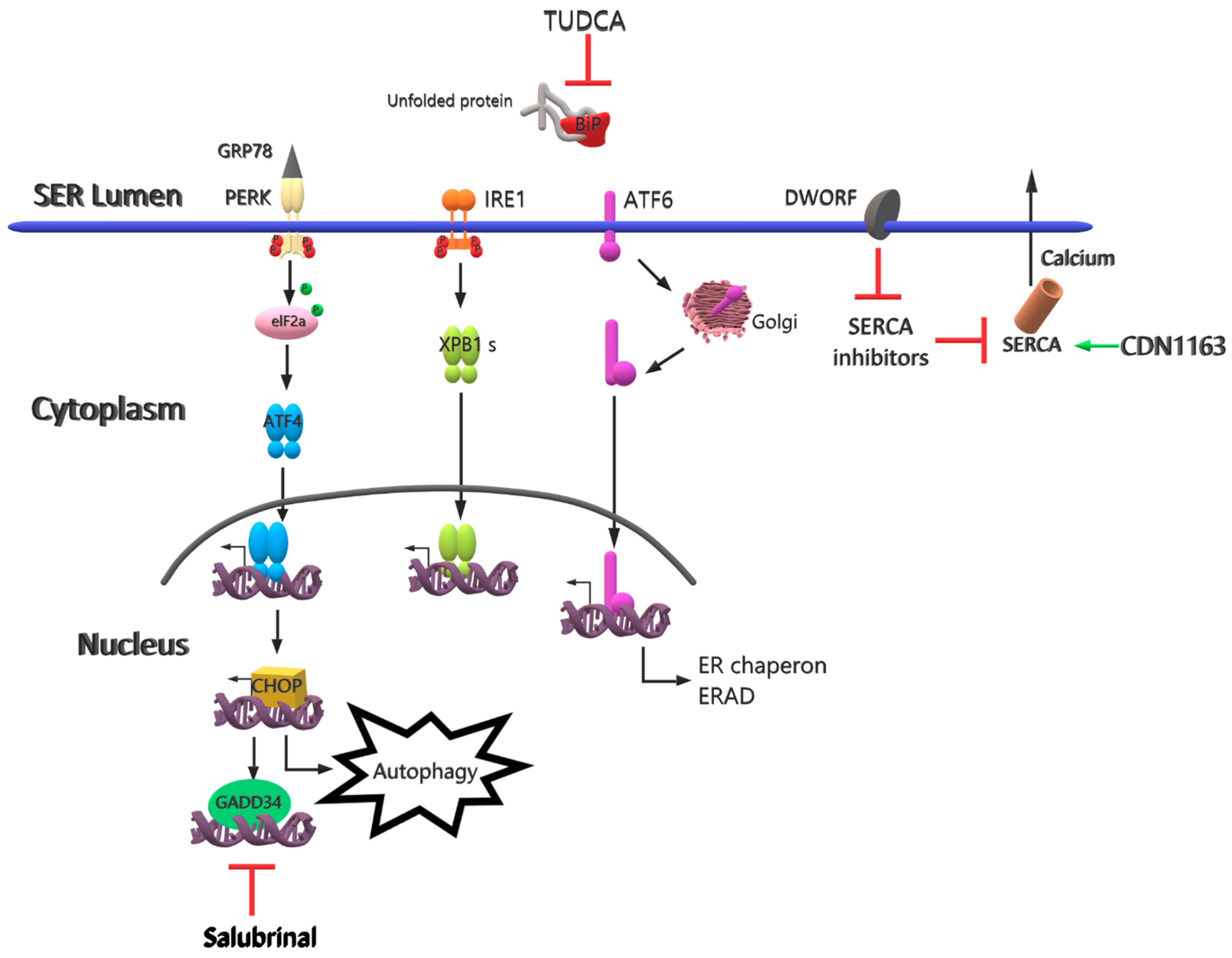{kind=link}
| Type | Administration | Expectation | Stage | Comment | Ref. | |
|---|---|---|---|---|---|---|
| Drug Therapy | ||||||
| Prednisone | Glucocorticoid steroid | Taking orally | Reduce inflammatory response | Phase I/II study | Undesirable situations may occur due to suppressing the immune system. | [20] |
| MYO-029 | Antibody | Injected intravenously | Neutralize myostatin protein | Phase I/II study | Myostatin inhibition resulted in a minor improvement in muscle. | [26] |
| Anti-myostatin antibody | Antibody | Injected intraperitoneally | Inhibition of follistatin, which is an endogenous inhibitor of myostatin | Experimental study on a murine model | Increase in muscle mass but not in functional muscle. | [28] |
| AMBMP | Small molecule | Injected intraperitoneally | As a Wnt agonist activates CaMKII | Experimental study on a murine model | Induction of slow oxidative genes. | [43] |
| Gene Therapy | ||||||
| pAAV-CMV-mSeAPpropmyoD76A vector | Plasmid DNA | Injected intramuscularly | Inhibition of myostatin | Experimental study on a murine model | Increase in muscle mass and absolute power | [27] |
| CAPN3 gene transfer via AAV vector, | Plasmid DNA | Systemic injection | Replacement of functional CAPN3 gene | Experimental study on a murine model | CAPN3 overexpression caused cardiac toxicity. | [31] |
| CAPN3 gene, and cardiac-specific microRNA-208a transfer via AAV | Plasmid DNA | Systemic injection | Replacement of functional CAPN3 gene and overcoming cardiac toxicity | Experimental study on a murine model | CAPN3 expression and no cardiac toxicity were achieved. | [31] |
| AAVrh74.tMCK.hCAPN3 vector | Plasmid DNA | Injected intravenously | Replacement of functional CAPN3 gene, overcoming off-target and toxic effects | Experimental study on a primate model | CAPN3 expression, no toxicity, and skeletal-muscle-specific vector were achieved. | [37] |
| rAAV-C3+miRT and rAAV-C3 | Plasmid DNA | Injected intravascularly and intramuscularly | Replacement of functional CAPN3 gene and overcoming cardiac toxicity | Experimental study on a primate model | In murine models, overexpression of CAPN3 is more prone to cardiac toxicity than in primates, due to physiological differences. CAPN3 expression increased in both applications and no cardiac toxicity was observed. | [34] |
| Combined Therapy (Cell- and Gene-Based) | ||||||
| IPSCs | CRISPR-Cas9 and stem cell | Injected intramuscularly | Replacement of functional CAPN3 in myogenic progenitor and mature muscle cells expressing CAPN3 | Experimental study on a murine model | CAPN3 mRNA levels were increased. | [44] |
| Type | Application | Expectation | Ref. | |
|---|---|---|---|---|
| Mss51 | Muscle-specific protein | Inhibition of Mss51 gene | Energy production increases and mitochondrial activity improves | [50] |
| TUDCA | The chemical chaperone mimetic drug | Different applications of TUDCA | Reduces effects on ER stress-related molecules | [65] |
| Salubrinal | A small molecule for selective inhibition of eIF2α | Different applications of salubrinal | Induces degradation of non-translated ER-targeted protein mRNAs | [66] |
| Rapamycin | Drug | Oral gavage | Provides inhibition of mTORC1, decrease in ER stress and inflammation, Improves muscle strength | [67] |
| CDN1163 | A small molecule as a SERCA2 activator | Injected intraperitoneally | Reduces ER stress and maintains Ca+2 homeostasis | [71] |
| DWORF | Muscle-specific long non-coding RNA | Upregulate of DWORF gene | Inhibits SERCA inhibitors and increases SERCA activity | [73] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Şahin, İ.O.; Özkul, Y.; Dündar, M. Current and Future Therapeutic Strategies for Limb Girdle Muscular Dystrophy Type R1: Clinical and Experimental Approaches. Pathophysiology 2021, 28, 238-249. https://0-doi-org.brum.beds.ac.uk/10.3390/pathophysiology28020016
Şahin İO, Özkul Y, Dündar M. Current and Future Therapeutic Strategies for Limb Girdle Muscular Dystrophy Type R1: Clinical and Experimental Approaches. Pathophysiology. 2021; 28(2):238-249. https://0-doi-org.brum.beds.ac.uk/10.3390/pathophysiology28020016
Chicago/Turabian StyleŞahin, İzem Olcay, Yusuf Özkul, and Munis Dündar. 2021. "Current and Future Therapeutic Strategies for Limb Girdle Muscular Dystrophy Type R1: Clinical and Experimental Approaches" Pathophysiology 28, no. 2: 238-249. https://0-doi-org.brum.beds.ac.uk/10.3390/pathophysiology28020016