Discovering Genotype Variants in an Infant with VACTERL through Clinical Exome Sequencing: A Support for Personalized Risk Assessment and Disease Prevention

 , , ,
, , ,  ,
,
Abstract
:1. Introduction
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Solomon, B.D. The etiology of VACTERL association: Current knowledge and hypotheses. Am. J. Med. Genet. C Semin. Med. Genet. 2018, 178, 440–446. [Google Scholar] [CrossRef]
- Solomon, B.D. VACTERL/VATER Association. Orphanet. J. Rare Dis. 2011, 16, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldacci, S.; Gorini, F.; Santoro, M.; Pierini, A.; Minichilli, F.; Bianchi, F. Environmental and individual exposure and the risk of congenital anomalies: Areview of recent epidemiological evidence. Epidemiol. Prev. 2018, 42, 1–34. [Google Scholar] [PubMed]
- Norwood, M.S.; Lupo, P.J.; Chow, E.J.; Scheurer, M.E.; Plon, S.E.; Danysh, H.E.; Spector, L.G.; Carozza, S.E.; Doody, D.R.; Mueller, B.A. Childhood cancer risk in those with chromosomal and non-chromosomal congenital anomalies in Washington State: 1984–2013. PLoS ONE 2017, 12, e0179006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charles, M.A.; Delpierre, C.; Bréant, B. Developmental origin of health and adult diseases (DOHaD): Evolution of a concept over three decades. Med. Sci. 2016, 32, 15–20. [Google Scholar]
- Kim, D.; Kobayashi, T.; Voisin, B.; Jo, J.H.; Sakamoto, K.; Jin, S.P.; Kelly, M.; Pasieka, H.B.; Naff, J.L.; Meyerle, J.H.; et al. Targeted therapy guided by single-cell transcriptomic analysis in drug-induced hypersensitivity syndrome: A case report. Nat. Med. 2002, 26, 236–243. [Google Scholar] [CrossRef]
- Sandhu, C.; Qureshi, A.; Emili, A. Panomics for Precision Medicine. Trends Mol. Med. 2018, 24, 85–101. [Google Scholar] [CrossRef]
- Manzoni, C.; Kia, D.A.; Vandrovcova, J.; Hardy, J.; Wood, N.W.; Lewis, P.A.; Ferrari, R. Genome, transcriptome and proteome: The rise of omics data and their integration in biomedical sciences. Brief. Bioinform. 2018, 19, 286–302. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Hobbs, C.A.; Chowdhury, S.; Cleves, M.A.; Erickson, S.; MacLeod, S.L.; Shaw, G.M.; Shete, S.; Witte, J.S.; Tycko, B. Genetic epidemiology and nonsyndromic structural birth defects: From candidate genes to epigenetics. JAMA Pediatr. 2014, 168, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Tatsuguchi, M.; Furutani, M.; Hinagata, J.; Tanaka, T.; Furutani, Y.; Imamura, S.; Kawana, M.; Masaki, H.; Kasanuki, H.; Sawamura, T.; et al. Oxidized LDL receptor gene (OLR1) is associated with the risk of myocardial infarction. Biochem. Biophys. Res. Commun. 2003, 303, 247–250. [Google Scholar] [CrossRef]
- Ozaki, K.; Sato, H.; Iida, A.; Mizuno, H.; Nakamura, T.; Miyamoto, Y.; Takahashi, A.; Tsunoda, T.; Ikegawa, S.; Kamatani, N.; et al. A functional SNP in PSMA6 confers risk of myocardial infarction in the Japanese population. Nat. Genet. 2006, 38, 921–925. [Google Scholar] [CrossRef] [PubMed]
- Hinohara, K.; Nakajima, T.; Sasaoka, T.; Sawabe, M.; Lee, B.S.; Ban, J.; Park, J.E.; Izumi, T.; Kimura, A. Replication studies for the association of PSMA6 polymorphism with coronary artery disease in East Asian populations. J. Hum. Genet. 2009, 54, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Kammerer, S.; Burns-Hamuro, L.L.; Ma, Y.; Hamon, S.C.; Canaves, J.M.; Shi, M.M.; Nelson, M.R.; Sing, C.F.; Cantor, C.R.; Taylor, S.S.; et al. Amino acid variant in the kinase binding domain of dual-specific A kinase-anchoring protein 2: A disease susceptibility polymorphism. Proc. Natl. Acad. Sci. USA 2003, 100, 4066–4071. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Yasue, H.; Yoshimura, M.; Nakamura, S.; Nakayama, M.; Shimasaki, Y.; Harada, E.; Mizuno, Y.; Kawano, H.; Ogawa, H. Paraoxonase gene Gln192Arg (Q192R) polymorphism is associated with coronary artery spasm. Hum. Genet. 2002, 110, 89–94. [Google Scholar] [CrossRef]
- Fornage, M.; Papanicolaou, G.; Lewis, C.E.; Boerwinkle, E.; Siscovick, D.S. Common INSIG2 polymorphisms are associated with age-related changes in body size and high-density lipoprotein cholesterol from young adulthood to middle age. Metabolism 2010, 59, 1084–1091. [Google Scholar] [CrossRef] [Green Version]
- Ohtoshi, K.; Kaneto, H.; Node, K.; Nakamura, Y.; Shiraiwa, T.; Matsuhisa, M.; Yamasaki, Y. Association of soluble epoxide hydrolase gene polymorphism with insulin resistance in type 2 diabetic patients. Biochem. Biophys. Res. Commun. 2005, 331, 347–350. [Google Scholar] [CrossRef]
- Müller, T.D.; Nogueiras, R.; Andermann, M.L.; Andrews, Z.B.; Anker, S.D.; Argente, J.; Batterham, R.L.; Benoit, S.C.; Bowers, C.Y.; Broglio, F.; et al. Ghrelin. Mol. Metab. 2015, 4, 437–460. [Google Scholar] [CrossRef]
- Imaizumi, T.; Ando, M.; Nakatochi, M.; Yasuda, Y.; Honda, H.; Kuwatsuka, Y.; Kato, S.; Kondo, T.; Iwata, M.; Nakashima, T.; et al. Effect of dietary energy and polymorphisms in BRAP and GHRL on obesity and metabolic traits. Obes. Res. Clin. Pract. 2018, 12, 39–48. [Google Scholar] [CrossRef]
- Carozza, S.E.; Langlois, P.H.; Miller, E.A.; Canfield, M. Are children with birth defects at higher risk of child- hood cancers? Am. J. Epidemiol. 2012, 175, 1217–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasle, H.; Clemmensen, I.H.; Mikkelsen, M. Risks of leukaemia and solid tumours in individuals with Down’s syndrome. Lancet 2000, 355, 165–169. [Google Scholar] [CrossRef]
- Motegi, T.; Kaga, M.; Yanagawa, Y.; Kadowaki, H.; Watanabe, K.; Inoue, A.; Komatsu, M.; Minoda, K. A recognizable pattern of the midface of retinoblastoma patients with interstitial deletion of 13q. Hum. Genet. 1983, 64, 160–162. [Google Scholar] [CrossRef] [PubMed]
- Mita, Y.; Yasuda, Y.; Sakai, A.; Yamamoto, H.; Toyooka, S.; Gunduz, M.; Tanabe, S.; Naomoto, Y.; Ouchida, M.; Shimizu, K. Missense polymorphisms of PTPRJ and PTPN13 genes affect susceptibility to a variety of human cancers. J. Cancer Res. Clin. Oncol. 2010, 136, 249–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hryhorowicz, S.; Ziemnicka, K.; Kaczmarek-Ryś, M.; Hoppe-Gołębiewska, J.; Pławski, A.; Skrzypczak-Zielińska, M.; Szkudlarek, M.; Gołąb, M.; Budny, B.; Ruchała, M.; et al. CCND1 gene polymorphic variants in patients with differentiated thyroid carcinoma. Oncol. Lett. 2015, 9, 442–448. [Google Scholar] [CrossRef] [Green Version]
- Weinhold, N.; Johnson, D.C.; Chubb, D.; Chen, B.; Försti, A.; Hosking, F.J.; Broderick, P.; Ma, Y.P.; Dobbins, S.E.; Hose, D.; et al. The CCND1 c.870G>A polymorphism is a risk factor for t(11;14)(q13;q32) multiple myeloma. Nat. Genet. 2013, 45, 522–525. [Google Scholar] [CrossRef] [Green Version]
- Absenger, G.; Benhaim, L.; Szkandera, J.; Zhang, W.; Yang, D.; Labonte, M.J.; Pichler, M.; Stotz, M.; Samonigg, H.; Renner, W.; et al. The cyclin D1 (CCND1) rs9344 G>A polymorphism predicts clinical outcome in colon cancer patients treated with adjuvant 5-FU-based chemotherapy. Pharmacogenom. J. 2014, 14, 130–134. [Google Scholar] [CrossRef]
- Ewart-Toland, A.; Briassouli, P.; de Koning, J.P.; Mao, J.H.; Yuan, J.; Chan, F.; MacCharthy-Morrogh, L.; Ponder, B.A.J.; Nagase, H.; Burn, J.; et al. Identification of Stk6/STK15 as a candidate low- penetrance tumor-susceptibility gene in mouse and human. Nat. Genet. 2003, 34, 403–412. [Google Scholar] [CrossRef]
- Dubash, A.D.; Koetsier, J.L.; Amargo, E.V.; Najor, N.A.; Harmon, R.M.; Green, K.J. The GEF Bcr activates RhoA/MAL signaling to promote keratinocyte differentiation via desmoglein-1. J Cell Biol. 2013, 202, 653–666. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Toruner, G.A.; Tang, G.; Cameron, Y.C.; Wang, W.; Hu, S.; Thakral, B.; Wang, S.A.; Miranda, R.N.; Khoury, J.D.; et al. Chronic myeloid leukemia with insertion- derived BCR-ABL1 fusion: Redefining complex chromosomal abnormalities by correlation of FISH and karyotype predicts prognosis. Mod. Pathol. [CrossRef]
- Huang, D.; Nagata, Y.; Grossmann, V.; Radivoyevitch, T.; Okuno, Y.; Nagae, G.; Hosono, N.; Schnittger, S.; Sanada, M.; Przychodzen, B.; et al. BRCC3 mutations in myeloid neoplasms. Haematologica 2015, 100, 1051–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, T.; Jahn, N.; Lindner, S.; Röhner, L.; Dolnik, A.; Weber, D.; Scheffold, A.; Köpff, S.; Paschka, P.; Gaidzik, V.I.; et al. Functional characterization of BRCC3 mutations in acute myeloid leukemia with t(8;21)(q22;q22.1). Leukemia 2020, 34, 404–415. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Nandi, S.; Chitu, V.; Yeung, Y.G.; Yu, W.; Huang, M.; Williams, L.T.; Lin, H.; Stanley, E.R. Functional overlap but differential expression of CSF-1 and IL-34 in their CSF-1 receptor-mediated regulation of myeloid cells. J. Leukoc. Biol. 2010, 88, 495–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamimi, R.M.; Brugge, J.S.; Freedman, M.L.; Miron, A.; Iglehart, J.D.; Colditz, G.A.; Hankinson, S.E. Circulating colony stimulating factor-1 and breast cancer risk. Cancer Res. 2008, 68, 18–21. [Google Scholar] [CrossRef] [Green Version]
- Oosterhof, N.; Chang, I.J.; Karimiani, E.G.; Kuil, L.E.; Jensen, D.M.; Daza, R.; Young, E.; Astle, L.; van der Linde, H.C.; Shivaram, G.M.; et al. Homozygous Mutations in CSF1R Cause a Pediatric-Onset Leukoencephalopathy and Can Result in Congenital Absence of Microglia. Am. J. Hum. Genet. 2019, 104, 936–947. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Bertola, D.R.; Takanohashi, A.; Saito, A.; Segawa, Y.; Yokota, T.; Ishibashi, S.; Nishida, Y.; Yamamoto, G.L.; Franco, J.F.D.S.; et al. Bi-allelic CSF1R Mutations Cause Skeletal Dysplasia of Dysosteosclerosis-Pyle Disease Spectrum and Degenerative Encephalopathy with Brain Malformation. Am. J. Hum. Genet. 2019, 104, 925–935. [Google Scholar] [CrossRef] [Green Version]
- Mollenhauer, J.; Wiemann, S.; Scheurlen, W.; Korn, B.; Hayashi, Y.; Wilgenbus, K.K.; von Deimling, A.; Poustka, A. DMBT1, a new member of the SRCR superfamily, on chromosome 10q25.3-26.1 is deleted in malignant brain tumours. Nat. Genet. 1997, 17, 32–39. [Google Scholar] [CrossRef]
- Holmskov, U.; Mollenhauer, J.; Madsen, J.; Vitved, L.; Gronlund, J.; Tornoe, I.; Kliem, A.; Reid, K.B.M.; Poutska, A.; Skjødt, K. Cloning of gp-340, a putative opsonin receptor for lung surfactant protein D. Proc. Natl. Acad. Sci. USA 1999, 96, 10794–10799. [Google Scholar] [CrossRef] [Green Version]
- Mollenhauer, J.; Herbertz, S.; Helmke, B.; Kollender, G.; Krebs, I.; Madsen, J.; Holmskov, U.; Sorger, K.; Schmitt, L.; Wiemann, S.; et al. Deleted in Malignant Brain Tumors 1 is a versatile mucin-like molecule likely to play a differential role in digestive tract cancer. Cancer Res. 2001, 61, 8880–8886. [Google Scholar]
- Mollenhauer, J.; Herbertz, S.; Holmskov, U.; Tolnay, M.; Krebs, I.; Merlo, A.; Schrøder, H.D.; Maier, D.; Breitling, F.; Wiemann, S.; et al. DMBT1 encodes a protein involved in the immune defense and in epithelial differentiation and is highly unstable in cancer. Cancer Res. 2000, 60, 1704–1710. [Google Scholar]
- End, C.; Bikker, F.; Renner, M.; Bergmann, G.; Lyer, S.; Blaich, S.; Hudler, M.; Helmke, B.; Gassler, N.; Autschbach, F.; et al. DMBT1 functions as pattern-recognition molecule for poly-sulfated and poly-phosphorylated ligands. Eur. J. Immunol. 2009, 39, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Pan, Z.Z.; Devaux, Y.; Ray, P. p21-activated protein kinase 4 (PAK4) interacts with the keratinocyte growth factor receptor and participates in keratinocyte growth factor-mediated inhibition of oxidant-induced cell death. J. Biol. Chem. 2003, 278, 10374–10380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaabeche, K.; Lemonnier, J.; Le Mée, S.; Caverzasio, J.; Marie, P.J. Cbl-mediated degradation of Lyn and Fyn induced by constitutive fibroblast growth factor receptor-2 activation supports osteoblast differentiation. J. Biol. Chem. 2004, 279, 36259–36267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceridono, M.; Belleudi, F.; Ceccarelli, S.; Torrisi, M.R. Tyrosine 769 of the keratinocyte growth factor receptor is required for receptor signaling but not endocytosis. Biochem. Biophys. Res. Commun. 2005, 327, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Kurosu, H.; Choi, M.; Ogawa, Y.; Dickson, A.S.; Goetz, R.; Eliseenkova, A.V.; Mohammadi, M.; Rosenblatt, K.P.; Kliewer, S.A.; Kuro-o, M. Tissue-specific expression of betaKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. J. Biol. Chem. 2007, 282, 26687–26695. [Google Scholar] [CrossRef] [Green Version]
- Linger, R.M.; Keating, A.K.; Earp, H.S.; Graham, D.K. TAM receptor tyrosine kinases: Biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv. Cancer Res. 2008, 100, 35–83. [Google Scholar]
- Yang, R.M.; Nanayakkara, D.; Kalimutho, M.; Mitra, P.; Khanna, K.K.; Dray, E.; Gonda, T.J. MYB regulates the DNA damage response and components of the homology-directed repair pathway in human estrogen receptor-positive breast cancer cells. Oncogene 2019, 38, 5239–5249. [Google Scholar] [CrossRef]
- Xu, L.H.; Zhao, F.; Yang, W.W.; Chen, C.W.; Du, Z.H.; Fu, M.; Ge, X.Y.; Li, S.L. MYB promotes the growth and metastasis of salivary adenoid cystic carcinoma. Int. J. Oncol. 2019, 54, 1579–1590. [Google Scholar] [CrossRef] [Green Version]
- Alhopuro, P.; Phichith, D.; Tuupanen, S.; Sammalkorpi, H.; Nybondas, M.; Saharinen, J.; Robinson, J.P.; Yang, Z.; Chen, L.Q.; Orntoft, T.; et al. Unregulated smooth-muscle myosin in human intestinal neoplasia. Proc. Natl. Acad. Sci. USA 2008, 105, 5513–5518. [Google Scholar] [CrossRef] [Green Version]
- Jo, Y.S.; Kim, M.S.; Yoo, N.J.; Lee, S.H. Somatic Mutations and Intratumoral Heterogeneity of MYH11 Gene in Gastric and Colorectal Cancers. Appl. Immunohistochem. Mol. Morphol. 2018, 26, 562–566. [Google Scholar] [CrossRef]
- Shoumariyeh, K.; Hussung, S.; Niemöller, C.; Bleul, S.; Veratti, P.; Follo, M.; Riba, J.; Philipp, U.; Palmer, J.M.; Pfeifer, D.; et al. Blastic transformation of BCR-ABL1 positive chronic myeloid leukaemia through acquisition of CBFB-MYH11 and mutant KIT. Br. J. Haematol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Dammermann, A.; Merdes, A. Assembly of centrosomal proteins and microtubule organization depends on PCM-1. J. Cell Biol. 2002, 159, 255–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baer, C.; Muehlbacher, V.; Kern, W.; Haferlach, C.; Haferlach, T. Molecular genetic characterization of myeloid/lymphoid neoplasms associated with eosinophilia and rearrangement of PDGFRA, PDGFRB, FGFR1 or PCM1-JAK2. Haematologica 2018, 103, e348–e350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.M.; Lee, J.; Han, E.; Kim, M.; Kim, Y.; Han, K.; Kim, H.J. PCM1-JAK2 Fusion in a Patient With Acute Myeloid Leukemia. Ann. Lab. Med. 2018, 38, 492–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torroja, C.; Gorfinkiel, N.; Guerrero, I. Patched controls the Hedgehog gradient by endocytosis in a dynamin-dependent manner, but this internalization does not play a major role in signal transduction. Development 2004, 131, 2395–2408. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, L.A.; Quiezi, R.G.; Nascimento, A.; Bertolacini, C.P.; Richieri-Costa, A. Holoprosencephaly and holoprosencephaly-like phenotype and GAS1 DNA sequence changes: Report of four Brazilian patients. Am. J. Med. Genet. A 2010, 152A, 1688–1694. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, G.; Shi, M.; Liu, Z.; Xiao, M.; Fu, S.; Gong, X.; Shi, X. A novel splicing mutation of PTCH1 in a Chinese family with nevoid basal cell carcinoma syndrome. Med. Mol. Morphol. 2019, 52, 235–237. [Google Scholar] [CrossRef]
- Jones, D.T.; Jäger, N.; Kool, M.; Zichner, T.; Hutter, B.; Sultan, M.; Cho, Y.J.; Pugh, T.J.; Hovestadt, V.; Stütz, A.M.; et al. Dissecting the genomic complexity underlying medulloblastoma. Nature 2012, 488, 100–105. [Google Scholar] [CrossRef]
- Morselli, E.; Shen, S.; Ruckenstuhl, C.; Bauer, M.A.; Mariño, G.; Galluzzi, L.; Criollo, A.; Michaud, M.; Maiuri, M.C.; Chano, T. p53 inhibits autophagy by interacting with the human ortholog of yeast Atg17, RB1CC1/FIP200. Cell Cycle 2011, 10, 2763–2769. [Google Scholar] [CrossRef] [Green Version]
- Bae, H.; Guan, J.L. Suppression of autophagy by FIP200 deletion impairs DNA damage repair and increases cell death upon treatments with anticancer agents. Mol. Cancer Res. 2011, 9, 1232–1241. [Google Scholar] [CrossRef] [Green Version]
- Kontani, K.; Chano, T.; Ozaki, Y.; Tezuka, N.; Sawai, S.; Fujino, S.; Saeki, Y.; Okabe, H. RB1CC1 suppresses cell cycle progression through RB1 expression in human neoplastic cells. Int. J. Mol. Med. 2003, 12, 767–769. [Google Scholar] [CrossRef] [PubMed]
- Charest, A.; Wilker, E.W.; McLaughlin, M.E.; Lane, K.; Gowda, R.; Coven, S.; McMahon, K.; Kovach, S.; Feng, Y.; Yaffe, M.B.; et al. ROS fusion tyrosine kinase activates a SH2 domain-containing phosphatase-2/phosphatidylinositol 3-kinase/mammalian target of rapamycin signaling axis to form glioblastoma in mice. Cancer Res. 2006, 66, 7473–74781. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.; Sachdev, P.; Yan, L.; Chan, J.L.; Trenkle, T.; McClelland, M.; Welsh, J.; Wang, L.H. Vav3 mediates receptor protein tyrosine kinase signaling, regulates GTPase activity, modulates cell morphology, and induces cell transformation. Mol. Cell. Biol. 2000, 20, 9212–9224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berge, E.M.; Doebele, R.C. Targeted therapies in non-small cell lung cancer: Emerging oncogene targets following the success of epidermal growth factor receptor. Semin. Oncol. 2014, 41, 110–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birchmeier, C.; Sharma, S.; Wigler, M. Expression and rearrangement of the ROS1 gene in human glioblastoma cells. Proc. Natl. Acad. Sci. USA 1987, 84, 9270–9274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, K.D.; Doebele, R.C. Molecular pathways: ROS1 fusion proteins in cancer. Clin. Cancer Res. 2013, 19, 4040–4045. [Google Scholar] [CrossRef] [Green Version]



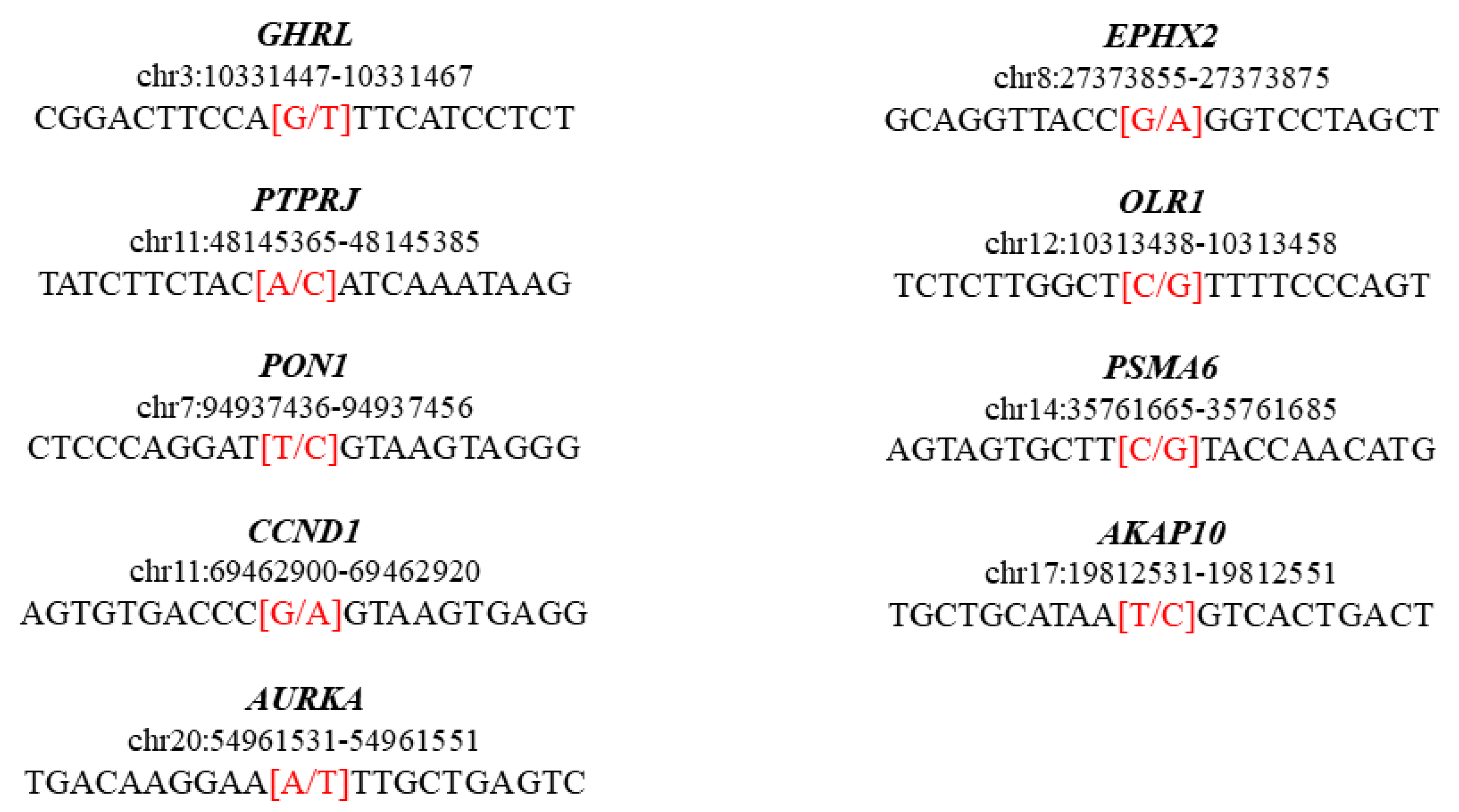{kind=link}
{kind=link}
| Chromosome | Gene | Position | Rs ID | Reference | Alternative | Variant Type | Consequence | HGVS.c | HGVS.p | Condition | Clinical Significance |
|---|---|---|---|---|---|---|---|---|---|---|---|
| chr3 | GHRL | 10331457 | rs696217 | G | T | SNP HET | Missense variant | c.214C>A | p.Leu72Met | Metabolic syndrome, susceptibility to obesity, age at onset of | Pathogenic, risk factor |
| chr11 | PTPRJ | 48145375 | rs1566734 | A | C | SNP HET | Missense variant | c.827A>C | p.Gln276Pro | Carcinoma of colon | Pathogenic |
| chr7 | PON1 | 94937446 | rs662 | T | C | SNP HOM | Missense variant | c.575A>G | p.Gln192Arg | Enzyme activity finding coronary artery disease, susceptibility to|Coronary artery spasm 2, susceptibility to | Risk factor |
| chr11 | CCND1 | 69462910 | rs9344 | G | A | SNP HOM | Splice region variant and synonymous variant | c.723G>A | p.Pro241Pro | Von Hippel–Lindau syndrome, modifier of colorectal cancer, susceptibility to multiple myeloma, translocation 11,14 type | Risk factor |
| chr20 | AURKA | 54961541 | rs2273535 | A | T | SNP HOM | Missense variant | c.91T>A | p.Phe31Ile | Colon cancer, susceptibility to | Risk factor |
| chr8 | EPHX2 | 27373865 | rs751141 | G | A | SNP HET | Missense variant | c.860G>A | p.Arg287Gln | Familial hypercholesterolemia 1 | Risk factor |
| chr12 | OLR1 | 10313448 | rs11053646 | C | G | SNP HET | Missense variant | c.501G>C | p.Lys167Asn | Myocardial infarction | Risk factor |
| chr14 | PSMA6 | 35761675 | rs1048990 | C | G | SNP HET | 5 prime UTR premature start codon gain variant | c.-8C>G | . | Myocardial infarction | Risk factor |
| chr17 | AKAP10 | 19812541 | rs203462 | T | C | SNP HET | Missense variant | c.1936A>G | p.Ile646Val | Cardiac conduction defect, susceptibility to | Risk factor |
| Chromosome | Gene | Position | Rs ID | Reference | Alternative | Variant Type | Consequence | HGVS.c | HGVS.p |
|---|---|---|---|---|---|---|---|---|---|
| chr2 | MERTK | 112766060 | rs112541306 | C | T | SNP HET | Splice region variant and intron variant | c.1960+8C>T | . |
| chr5 | CSF1R | 149456893 | rs3829986 | C | T | SNP HET | Missense variant | c.835G>A | p.Val279Met |
| chr5 | SDHA | 256483 | rs112307877 | CTT | C | DEL HET | Frameshift variant | c.1945 | 1946delTT |
| chr6 | MYB | 135518255 | rs182817536 | C | T | SNP HET | Missense variant | c.1360C>T | p.Arg454Cys |
| chr6 | ROS1 | 117717348 | rs79119625 | T | C | SNP HET | Splice region variant and intron variant | c.856+3A>G | . |
| chr8 | PCM1 | 17796382 | rs754721723 | AC | GT | MNP HET | Missense variant | c.476_477delACinsGT | p.Asn159Ser |
| chr8 | RB1CC1 | 53543090 | rs770160515 | TA | T | DEL HET | Splice region variant and intron variant | c.4441-3delT | . |
| chr8 | RB1CC1 | 53586746 | rs77653001 | C | T | SNP HET | Missense variant | c.661G>A | p.Asp221Asn |
| chr9 | PTCH1 | 98270531 | rs143494325 | C | A | SNP HET | Missense variant | c.113G>T | p.Gly38Val |
| chr10 | DMBT1 | 124329672 | rs34118835 | TC | CT | MNP HOM | Splice region variant and intron variant | c.92-6_92-5delTCinsCT | . |
| chr10 | FGFR2 | 123274705 | rs772986332 | T | C | SNP HET | Missense variant | c.1216A>G | p.Lys406Glu |
| chr16 | MYH11 | 15808856 | rs79129097 | T | C | SNP HET | Missense variant | c.5717A>G | p.Asn1906Ser |
| chr16 | MYH11 | 15831299 | rs201955317 | C | T | SNP HET | Splice region variant and intron variant | c.3314+7G>A | . |
| chr22 | BCR | 23654017 | rs879255379 | G | A | SNP HET | Missense variant | c.3316G>A | p.Asp1106Asn |
| chrX | BRCC3 | 154348267 | rs2290069 | A | G | SNP HET | Splice region variant and intron variant | c.800-7A>G | . |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pelizzo, G.; Chiricosta, L.; Mazzon, E.; Zuccotti, G.V.; Avanzini, M.A.; Croce, S.; Lima, M.; Bramanti, P.; Calcaterra, V. Discovering Genotype Variants in an Infant with VACTERL through Clinical Exome Sequencing: A Support for Personalized Risk Assessment and Disease Prevention. Pediatr. Rep. 2021, 13, 45-56. https://0-doi-org.brum.beds.ac.uk/10.3390/pediatric13010006
Pelizzo G, Chiricosta L, Mazzon E, Zuccotti GV, Avanzini MA, Croce S, Lima M, Bramanti P, Calcaterra V. Discovering Genotype Variants in an Infant with VACTERL through Clinical Exome Sequencing: A Support for Personalized Risk Assessment and Disease Prevention. Pediatric Reports. 2021; 13(1):45-56. https://0-doi-org.brum.beds.ac.uk/10.3390/pediatric13010006
Chicago/Turabian StylePelizzo, Gloria, Luigi Chiricosta, Emanuela Mazzon, Gian Vincenzo Zuccotti, Maria Antonietta Avanzini, Stefania Croce, Mario Lima, Placido Bramanti, and Valeria Calcaterra. 2021. "Discovering Genotype Variants in an Infant with VACTERL through Clinical Exome Sequencing: A Support for Personalized Risk Assessment and Disease Prevention" Pediatric Reports 13, no. 1: 45-56. https://0-doi-org.brum.beds.ac.uk/10.3390/pediatric13010006