Pediatric Autoimmune Hepatitis


1
Division of Gastroenterology, Hepatology & Nutrition, Boston Children’s Hospital and Harvard Medical School, Boston, MA 02115, USA
2
Pediatric Department and Transplantation, ISMETT, 90133 Palermo, Italy
3
Division of Pathology, Bambino Gesù Children’s Hospital, IRCCS, 00165 Rome, Italy
4
Division of Hepatogastroenterology, and Liver Transplant, ERN RARE LIVER, Bambino Gesù Children’s Hospital, IRCCS, 00165 Rome, Italy
*
Author to whom correspondence should be addressed.
Pediatr. Rep. 2024, 16(1), 110-113; https://0-doi-org.brum.beds.ac.uk/10.3390/pediatric16010011
Submission received: 23 October 2023
/
Accepted: 11 January 2024
/
Published: 1 February 2024
Pediatric autoimmune hepatitis (PAIH) is a rare necro-inflammatory disease of the liver of unknown etiology thought to derive from the dysregulation of the immune response upon exposure to environmental triggers in genetically predisposed individuals. PAIH is severe and progressive and, if untreated, leads to cirrhosis and liver failure [1,2,3,4]. Two types of PAIH have been characterized based on the presence of specific circulating antibodies: PAIH type 1, in which smooth muscle antibodies (SMAs) and/or antinuclear antibodies (ANAs) are present, and PAIH type 2, which is identified using liver kidney microsomal type 1 antibodies (anti-LKM-1s) and/or anti-liver cytosol type 1 antibodies (anti-LC1s) [1]. PAIH can present with a variety of clinical scenarios, including acute hepatitis and liver failure, but it can also present with non-specific symptoms such as fatigue and abdominal pain or with an incidental finding of elevated liver enzymes or advanced liver disease (Table 1).
Once other causes of liver disease have been excluded (e.g., viral hepatitis, drug-induced liver injury, hereditary, metabolic), PAIH should be suspected in the presence of three main laboratory and histologic features: (1) hypergammaglobulinemia, (2) specific circulating autoantibodies, as described above, and (3) interface hepatitis with dense portal lymphoplasmacytic infiltrate on liver biopsy (Figure 1).
A simplified diagnostic scoring system for AIH was developed in 2008 on the basis of the scoring system created by the International Autoimmune Hepatitis Group (IAIHG) in 1993 [5,6]. The simplified criteria show high specificity (95%) but only moderate sensitivity (77%) for the diagnosis of autoimmune hepatitis in children, making the scoring system a helpful tool although not sufficient to rule out PAIH [7]. Due to the severe and progressive nature of PAIH, immunosuppressive treatment should be started promptly at diagnosis with the goal to halt inflammation and ultimately prevent the development of cirrhosis and end-stage liver disease. First-line treatment consists of a combination of prednisolone or prednisone (1–2 mg/kg daily) in association with azathioprine (an initial dose of 1 mg/kg/day which can be further increased up to 2.5 mg/kg/day). Calcineurin inhibitors (cyclosporine and tacrolimus) and mycophenolate are the most well-known alternative therapies used in difficult-to-treat cases, such as in those with failure to induce complete remission with first-line treatment and in those with multiple relapses during tapering or discontinuation attempts [1,2,3,4]. Overall, none of these treatments can restore immune tolerance and side effects are not negligeable; however, at least multiple studies have shown high rates of remission with first-line therapy of up to 80–90%. After starting immunosuppressive treatment, patients are followed with serial blood draws which include monitoring transaminases, gamma glutamyl transferase, bilirubin, albumin, prothrombin ratio/INR, and the immunoglobulin G level, as well as the specific circulating antibodies. The aim of the treatment is to achieve complete biochemical remission, which is defined as a strict normalization of serum activities of ALT/AST and immunoglobulin G values and negative or very low-titer autoantibodies in addition to the normalization of hepatic function [1,2]. Despite the high first remission rate mentioned above, PAIH is burdened by an equally high risk of relapse reported to occur in 60–80% of children after treatment withdrawal [8,9]. Additionally, up to 8–16% of pediatric patients with PAIH have been reported to require a liver transplant, with the main indications being acute liver failure unresponsive to salvage therapy, acute chronic liver disease, complications of end-stage liver disease, and hepatocellular carcinoma [10,11].
We recently published a long-term study on a large group of 117 children with type 1 or type 2 PAIH with a 20-year median follow-up in survivors, which provided additional insight in the progression of this disease [12]. The patients in our study were mainly treated with a combination of prednisone and azathioprine, and treatment withdrawal was attempted when transaminases had been persistently normal. In a subset of patients managed before 1981, withdrawal was performed after a liver biopsy had been carried out; however, once it was noted that histologic remission did not reliably predict the risk of relapse, a biopsy was no longer considered necessary for treatment withdrawal. We found that overall, the normalization of aminotransferase was observed in more than 90% of children but particular attention needs to be paid to the prothrombin ratio as the lack of its normalization or its decline after initial normalization was found to be independently associated with a lower probability of overall and native liver survivals. The probabilities of overall and native liver survival at 30 years were 81% and 61%, respectively. It is well known that AIH is a rapidly progressive disease, and it was not surprising to see that the presence of cirrhosis, the lack of normalization of the prothrombin ratio while on treatment, gastroesophageal varices, and gastrointestinal bleeding from varices are related to a longer interval between first symptoms and diagnosis. This was especially significant since cirrhosis was present in close to half of the children seen within 3 months of the first sign of liver disease. Relapse-free treatment withdrawal ≥4 years was seen in 19% of the overall population (including patients who self-discontinued treatment) and in 42% of patients in whom withdrawal was undertaken under medical supervision. In the majority of patients, the criteria for withdrawal included persistently normal alanine aminotransferase activity without an assessment of liver histology. This finding aligns with the recent AASLD guidelines, which state that liver tissue examination is not mandatory prior to treatment withdrawal in adults and suggest that similar criteria could be applied to the pediatric population [3]. Additionally, our data did not show any difference in the outcome of treatment withdrawal under medical supervision in patients at the time of puberty compared to at other ages. This result is particularly interesting given the recommendation to avoid withdrawal attempts at the time of puberty based on the notion that hormonal changes may increase the risk of immune perturbations. Moreover, although PAIH type 2 has long been thought to be more severe than type 1, our results did not show any differences between the two types of PAIH in regard to sustained alanine aminotransferase normalization on azathioprine monotherapy, successful treatment withdrawal, and overall long-term native liver survival [12,13].
In conclusion, PAIH is a severe and rapidly progressive liver disease for which providers need to maintain a high index of clinical suspicion, expedite work when suspected, and promptly initiate treatment upon diagnosis to halt disease progression. Unfortunately, current treatments do not restore immune tolerance, but recent results offer hope of a 20% possibility of relatively long-term treatment-free survival throughout childhood and early adulthood with present-day treatments.
Author Contributions
Conceptualization, S.N. and G.M.; validation, S.N., G.M. and M.S.; writing—original draft preparation, S.N.; writing—review and editing, M.S., P.F.; supervision, G.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Maggiore, G.; Nastasio, S.; Sciveres, M. Juvenile autoimmune hepatitis: Spectrum of the disease. World J. Hepatol. 2014, 6, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Mieli-Vergani, G.; Vergani, D.; Baumann, U.; Czubkowski, P.; Debray, D.; Dezsofi, A.; Fischler, B.; Gupte, G.; Hierro, L.; Indolfi, G.; et al. Diagnosis and Management of Pediatric Autoimmune Liver Disease: ESPGHAN Hepatology Committee Position Statement. J. Pediatr. Gastroenterol. Nutr. 2018, 66, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Mack, C.L.; Adams, D.; Assis, D.N.; Kerkar, N.; Manns, M.P.; Mayo, M.J.; Vierling, J.M.; Alsawas, M.; Murad, M.H.; Czaja, A.J. Diagnosis and Management of Autoimmune Hepatitis in Adults and Children: 2019 Practice Guidance and Guidelines from the American Association for the Study of Liver Diseases. Hepatology 2020, 72, 671–722. [Google Scholar] [CrossRef] [PubMed]
- Sciveres, M.; Nastasio, S.; Maggiore, G. Novel Diagnostic and Therapeutic Strategies in Juvenile Autoimmune Hepatitis. Front. Pediatr. 2019, 7, 382. [Google Scholar] [CrossRef] [PubMed]
- Hennes, E.M.; Zeniya, M.; Czaja, A.J.; Parés, A.; Dalekos, G.N.; Krawitt, E.L.; Bittencourt, P.L.; Porta, G.; Boberg, K.M.; Hofer, H.; et al. Simplified criteria for the diagnosis of autoimmune hepatitis. Hepatology 2008, 48, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, F.; Berg, P.A.; Bianchi, F.B.; Bianchi, L.; Burroughs, A.K.; Cancado, E.L.; Chapman, R.W.; Cooksley, W.G.E.; Czaja, A.J.; Desmet, V.J.; et al. International Autoimmune Hepatitis Group Report: Review of criteria for diagnosis of autoimmune hepatitis. J. Hepatol. 1999, 31, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Arcos-Machancoses, J.V.; Busoms, C.M.; Tatis, E.J.; Bovo, M.V.; de Carpi, J.M. Accuracy of the Simplified Criteria for Autoimmune Hepatitis in Children: Systematic Review and Decision Analysis. J. Clin. Exp. Hepatol. 2019, 9, 147–155. [Google Scholar] [CrossRef] [PubMed]
- van Gerven, N.M.; Verwer, B.J.; Witte, B.I.; van Hoek, B.; Coenraad, M.J.; van Erpecum, K.J.; Beuers, U.; van Buuren, H.R.; de Man, R.A.; Drenth, J.P.; et al. Relapse is almost universal after withdrawal of immunosuppressive medication in patients with autoimmune hepatitis in remission. J. Hepatol. 2013, 58, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Deneau, M.; Book, L.S.; Guthery, S.L.; Jensen, M.K. Outcome after Discontinuation of Immunosuppression in Children with Autoimmune Hepatitis: A Population-Based Study. J. Pediatr. 2014, 164, 714–719.e2. [Google Scholar] [CrossRef] [PubMed]
- Saadah, O.I.; Smith, A.L.; Hardikar, W. Long-term outcome of autoimmune hepatitis in children. J. Gastroenterol. Hepatol. 2001, 16, 1297–1302. [Google Scholar] [CrossRef] [PubMed]
- Gregorio, G.V.; Portmann, B.; Reid, F.; Donaldson, P.T.; Doherty, D.G.; McCartney, M.; Mowat, A.P.; Vergani, D.; Mieli-Vergani, G. Autoimmune hepatitis in childhood: A 20-year experience. Hepatology 1997, 25, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Maggiore, G.; Bernard, O.; Mosca, A.; Ballot, E.; Johanet, C.; Jacquemin, E. Long-term outcomes of patients with type 1 or 2 autoimmune hepatitis presenting in childhood. J. Hepatol. 2023, 78, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Lohse, A.W.; Hørby Jørgensen, M.J. Paediatric autoimmune hepatitis: Time to change the textbooks? J. Hepatol. 2023, 78, 893–895. [Google Scholar] [CrossRef] [PubMed]
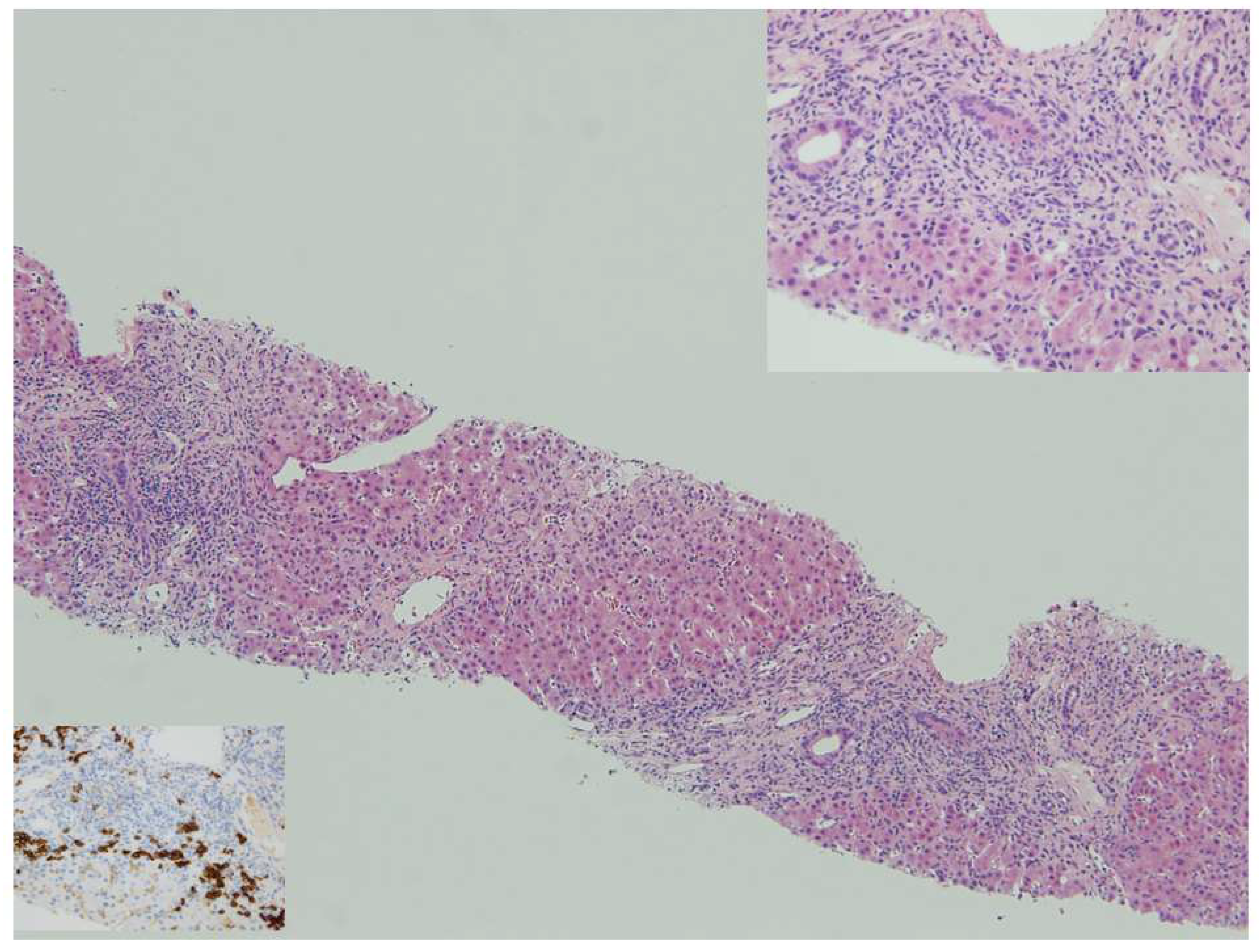
Figure 1.
Autoimmune hepatitis: panacinar hepatitis with bridging necrosis. Lymphoplasmacytic portal and periportal infiltrates with active interface hepatitis, HE 10x. Top insert: Plasma cells in clusters are typically abundant at the interface, HE 40x. Bottom insert: anti-CD38 shows numerous plasma cells at the progression front of the inflammatory process, anti-CD38 40x.
Figure 1.
Autoimmune hepatitis: panacinar hepatitis with bridging necrosis. Lymphoplasmacytic portal and periportal infiltrates with active interface hepatitis, HE 10x. Top insert: Plasma cells in clusters are typically abundant at the interface, HE 40x. Bottom insert: anti-CD38 shows numerous plasma cells at the progression front of the inflammatory process, anti-CD38 40x.


{kind=link}
Table 1.
Most common presentations of JAIH.
| Type of Presentation | Clinical Characteristics | Frequency | Additional Notes |
|---|---|---|---|
| Acute onset | Anorexia, nausea, vomiting, and abdominal pain followed by jaundice, similar to acute viral hepatitis. | Most common presentation | Some patients go on to develop acute liver failure with encephalopathy |
| Insidious onset | Progressive fatigue, anorexia, and intermittent jaundice lasting for several months/years before diagnosis | ~30% of patients | All patients have clinical evidence of chronic liver disease and/or cirrhosis at diagnosis |
| Asymptomatic | No symptoms | ~10–15% of patients | Incidental finding of clinical signs of chronic liver disease or elevated transaminases |
| Symptomatic portal hypertension | Variceal bleed; ascites | Rare | |
| JIAH presenting with symptoms related to an extrahepatic autoimmune disease | Symptoms related to autoimmune thrombocytopenia, autoimmune hemolytic anemia, type 1 diabetes, autoimmune thyroiditis, vitiligo, cutaneous vasculitis, uveitis, glomerulonephritis, juvenile chronic arthritis, systemic lupus erythematosus, Sjögren’s syndrome, celiac disease, and inflammatory bowel disease | Rare |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Nastasio, S.; Sciveres, M.; Francalanci, P.; Maggiore, G. Pediatric Autoimmune Hepatitis. Pediatr. Rep. 2024, 16, 110-113. https://0-doi-org.brum.beds.ac.uk/10.3390/pediatric16010011
AMA Style
Nastasio S, Sciveres M, Francalanci P, Maggiore G. Pediatric Autoimmune Hepatitis. Pediatric Reports. 2024; 16(1):110-113. https://0-doi-org.brum.beds.ac.uk/10.3390/pediatric16010011
Chicago/Turabian StyleNastasio, Silvia, Marco Sciveres, Paola Francalanci, and Giuseppe Maggiore. 2024. "Pediatric Autoimmune Hepatitis" Pediatric Reports 16, no. 1: 110-113. https://0-doi-org.brum.beds.ac.uk/10.3390/pediatric16010011