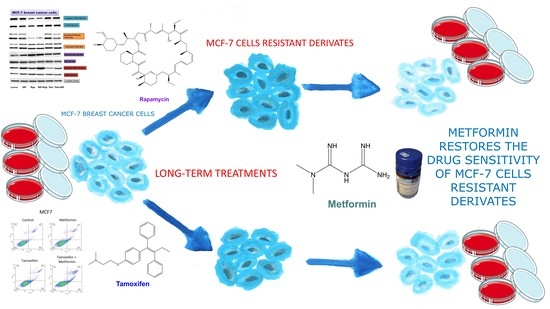
Metformin Restores the Drug Sensitivity of MCF-7 Cells Resistant Derivates via the Cooperative Modulation of Growth and Apoptotic-Related Pathways

 ,
,  ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
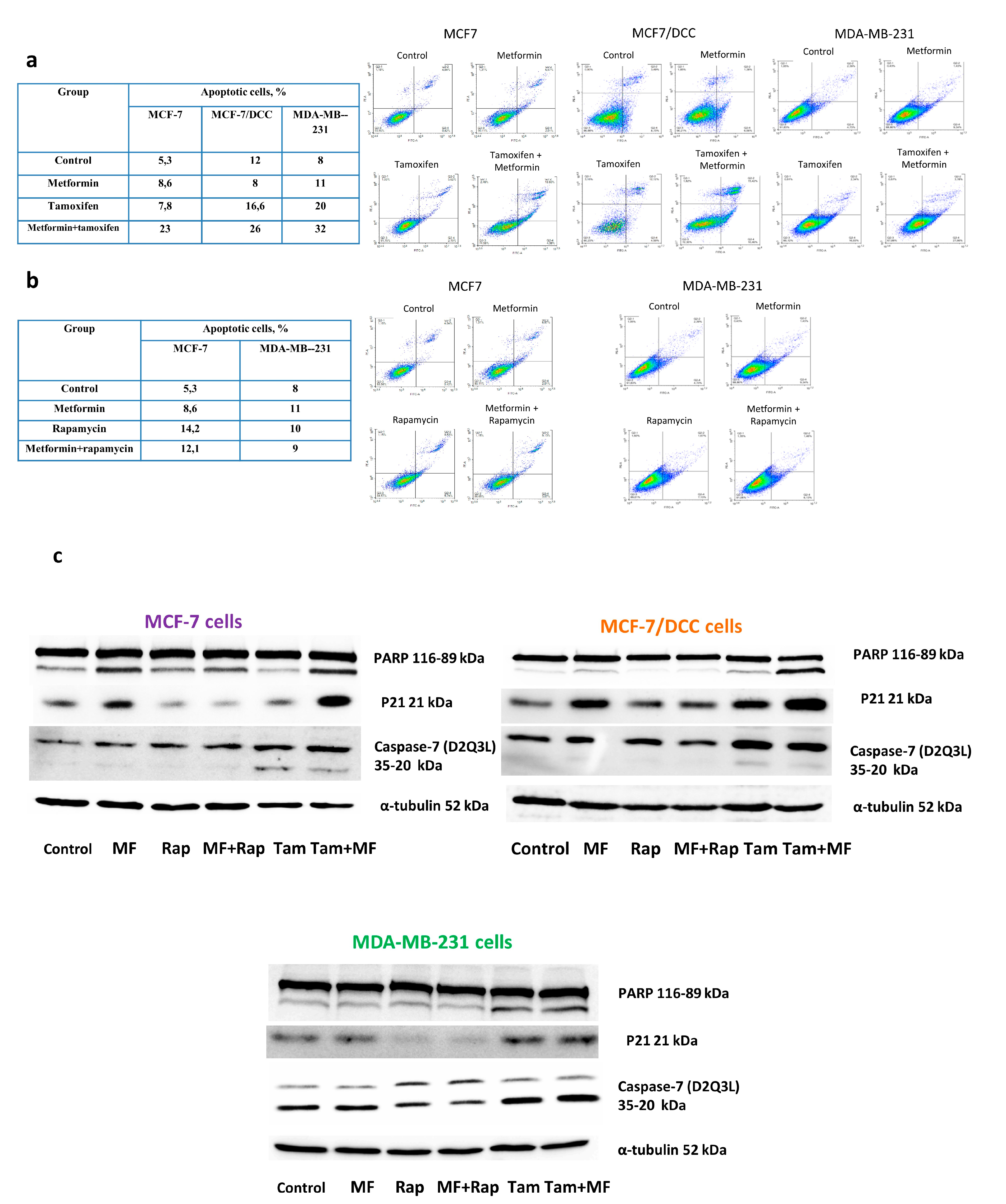
2. Results
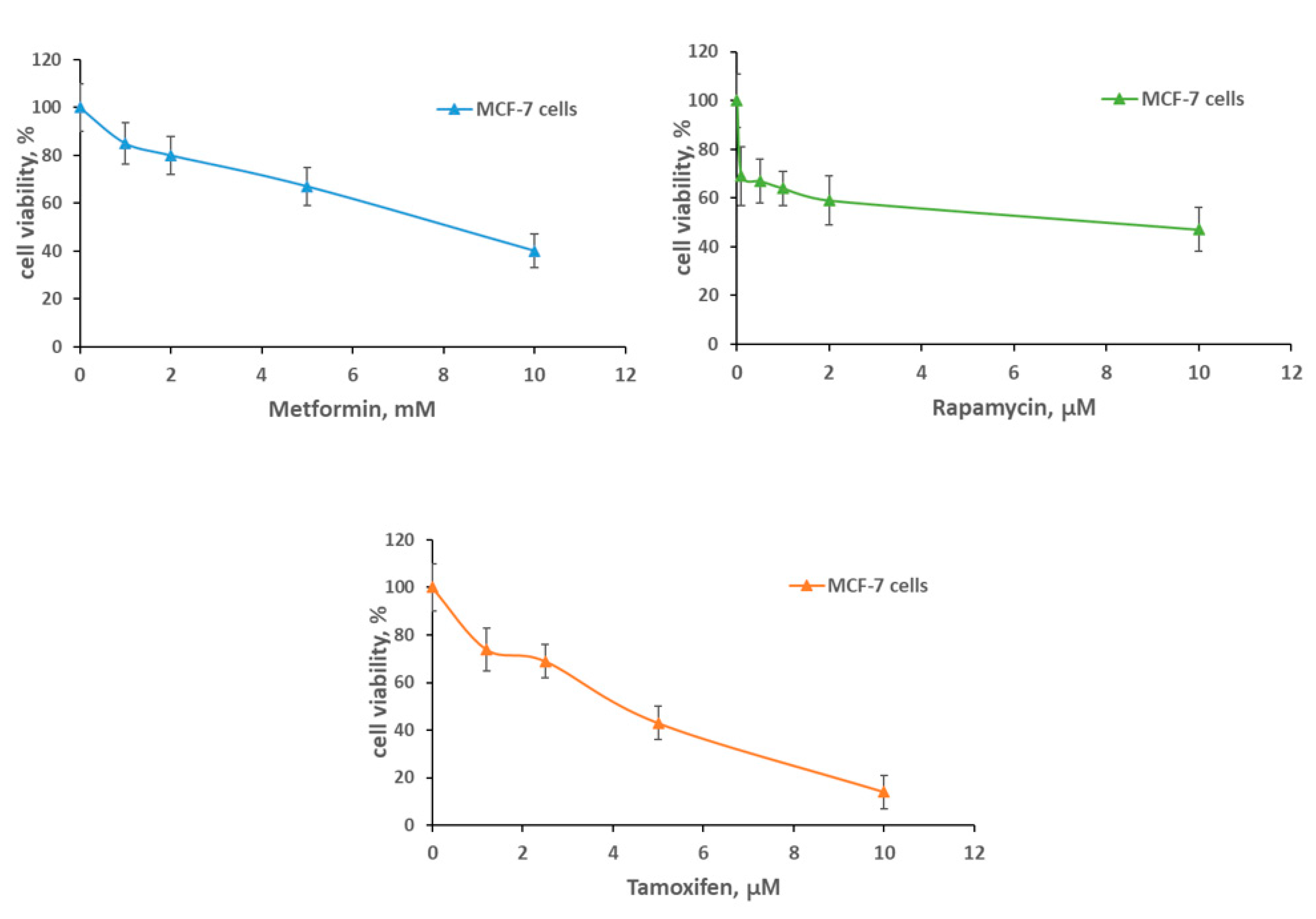
2.1. Metformin and MCF-7 Cells Response to Cytostatic Drugs
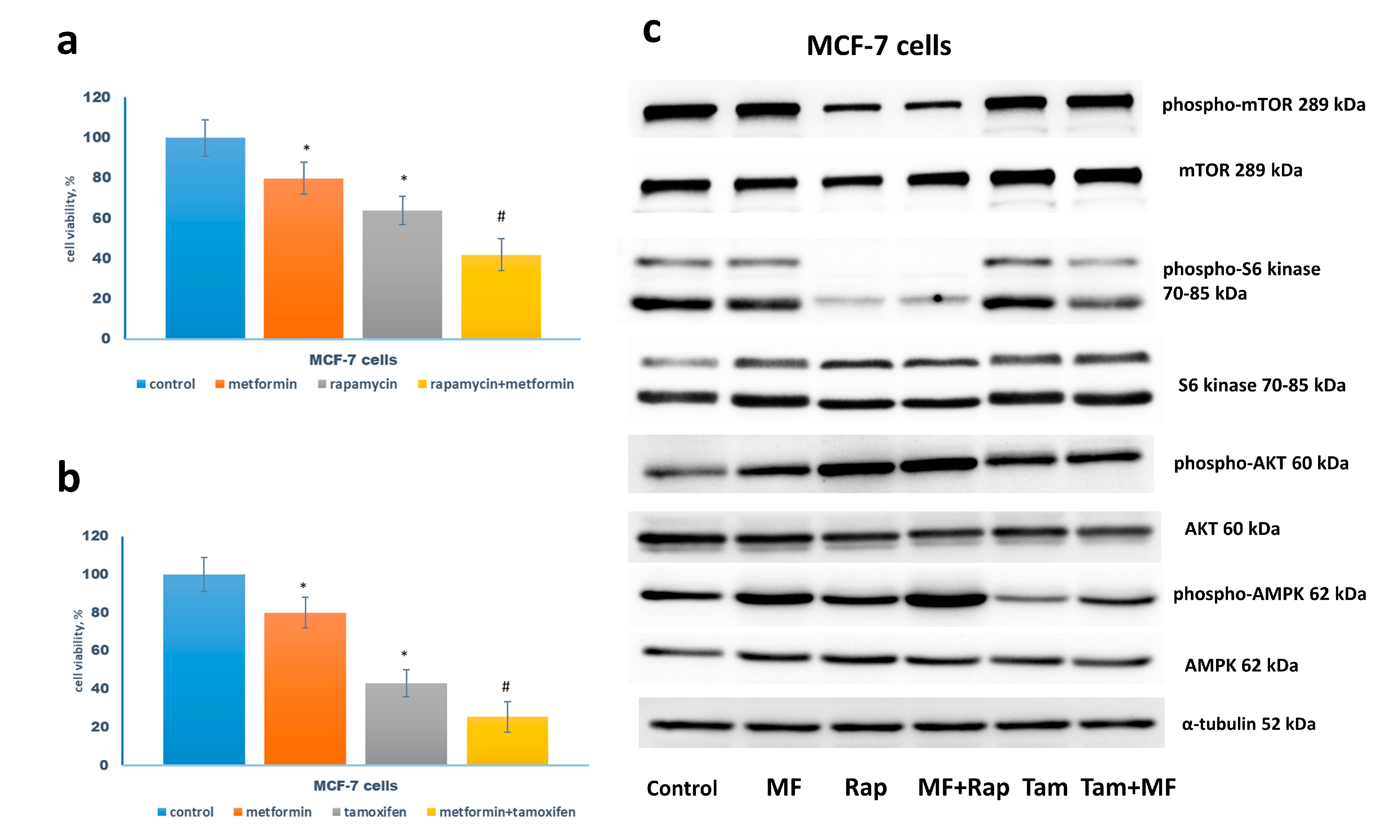
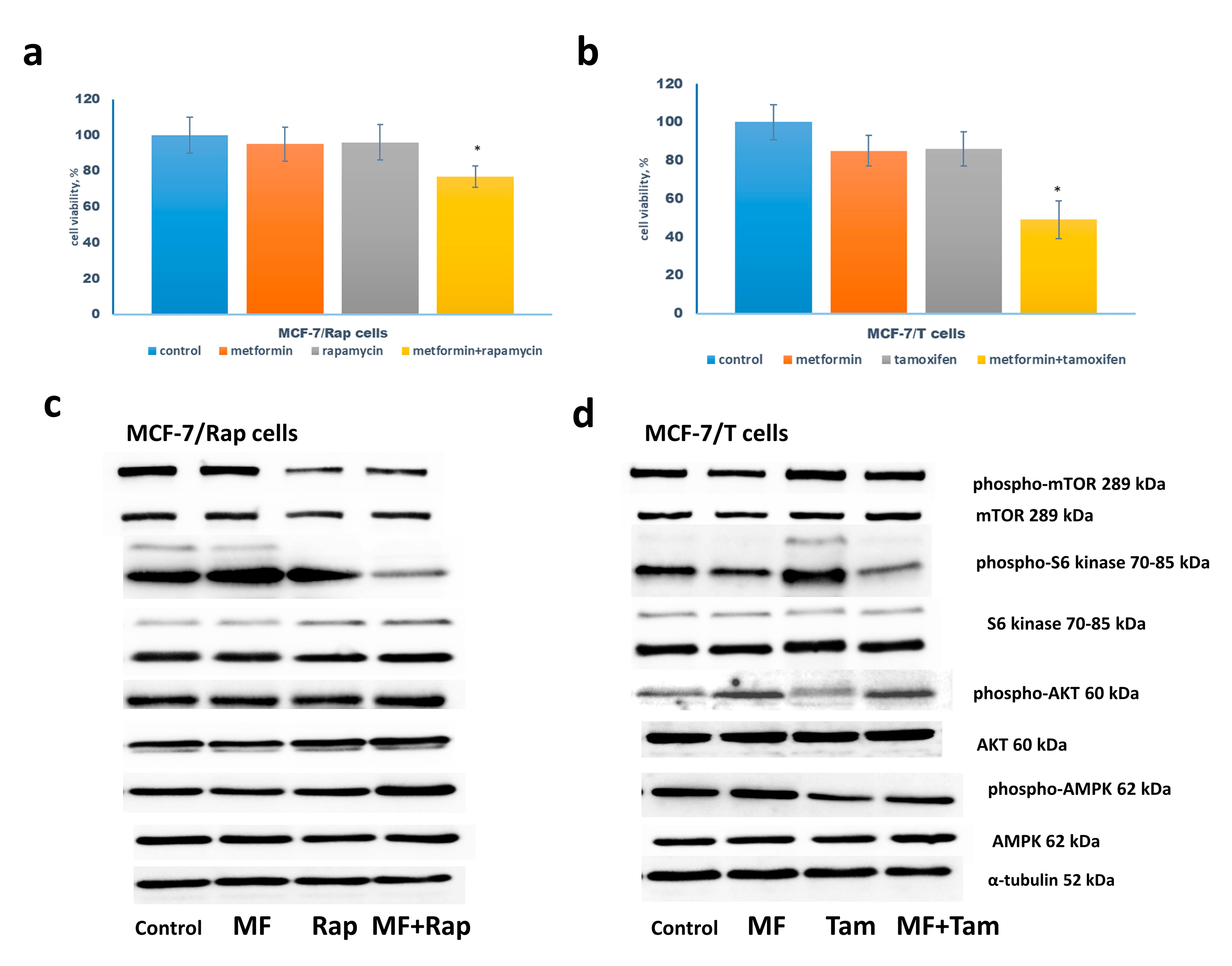
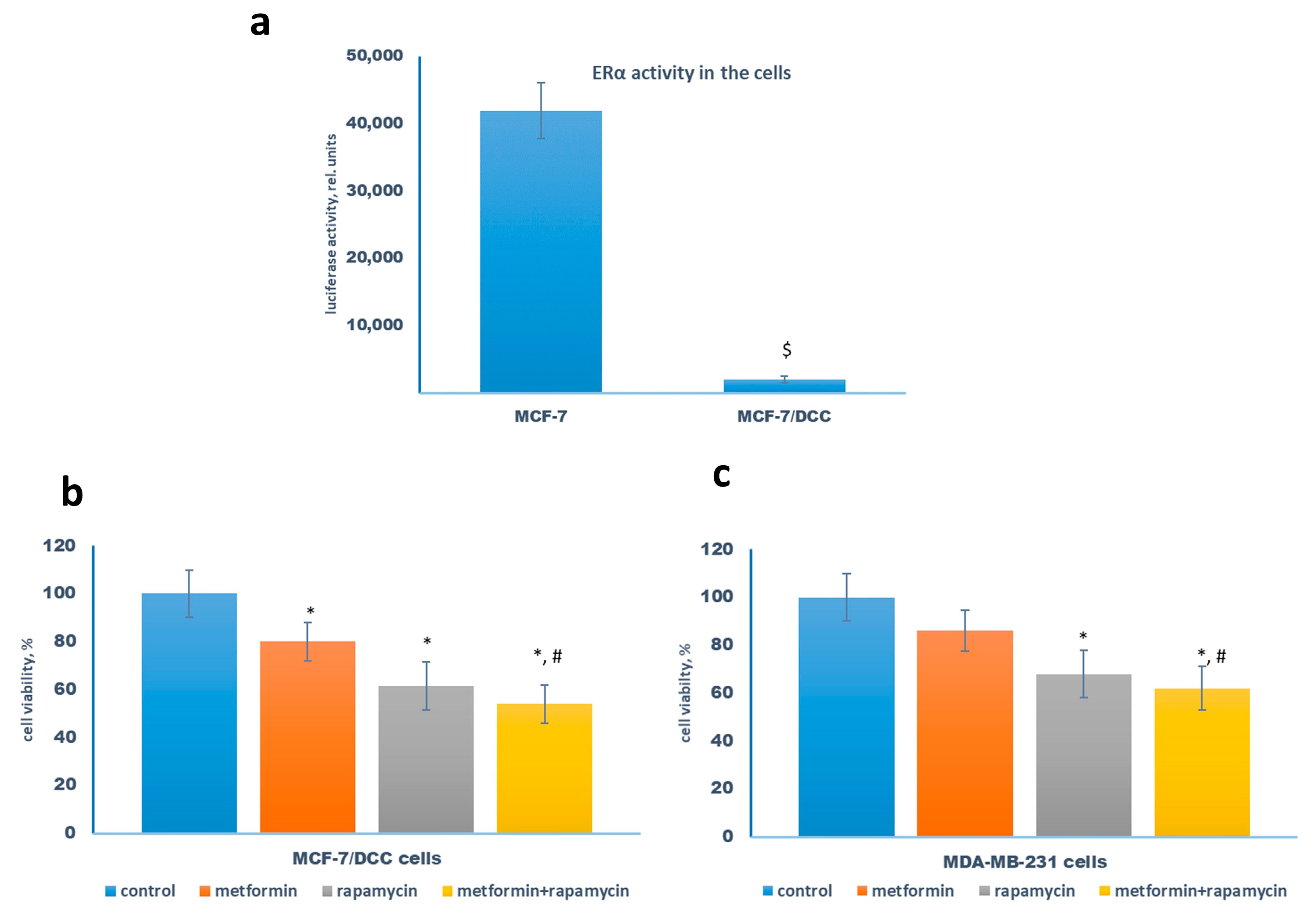
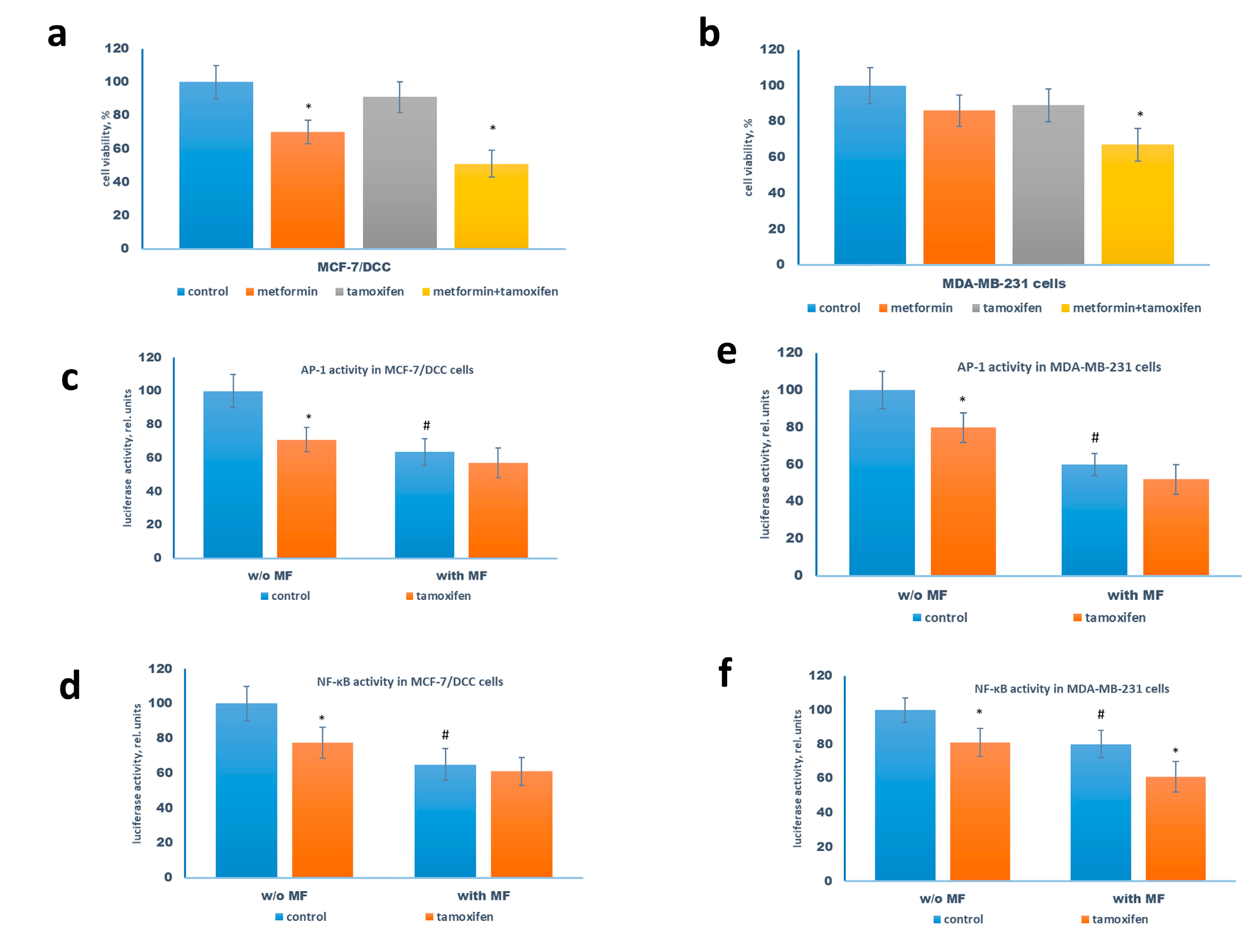
2.2. Metformin Increases the Sensitivity to Treatment of MCF-7 Cells Resistant Derivates
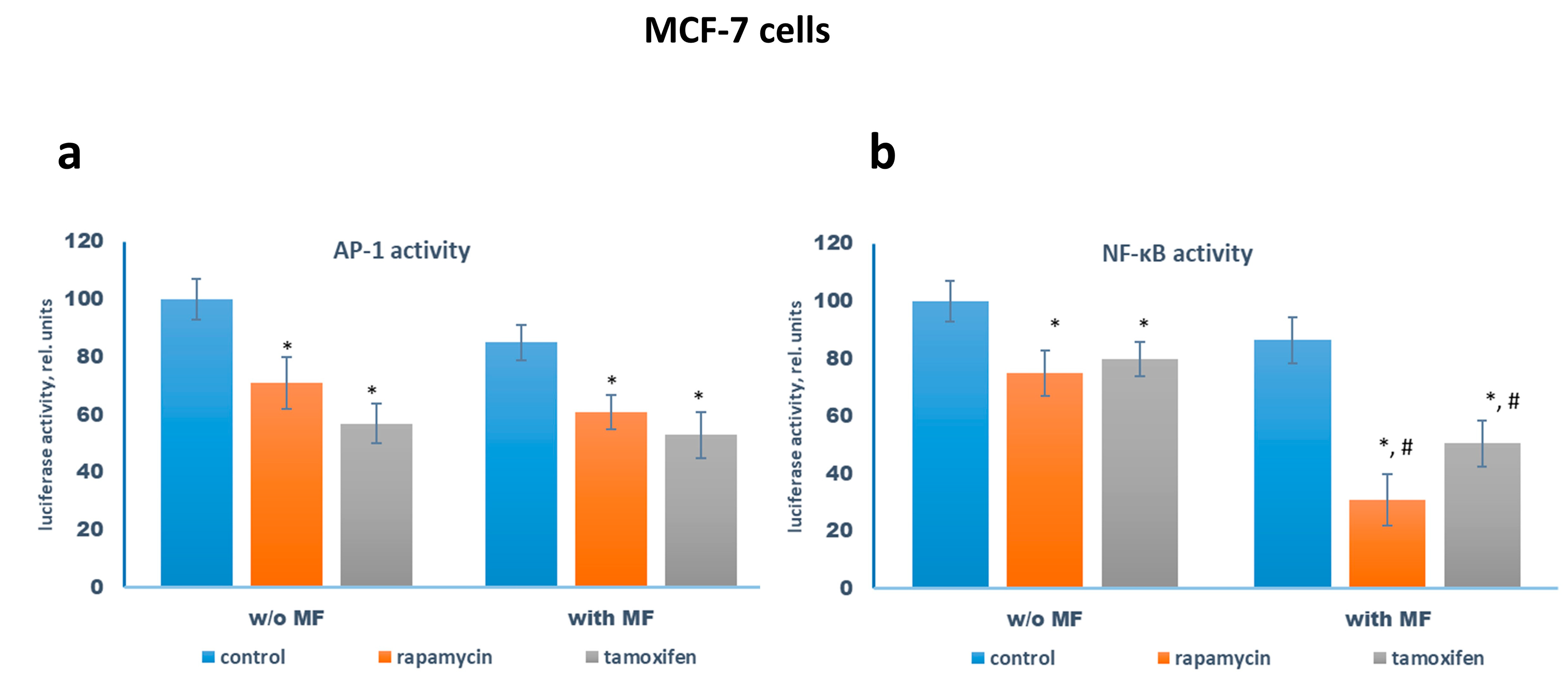
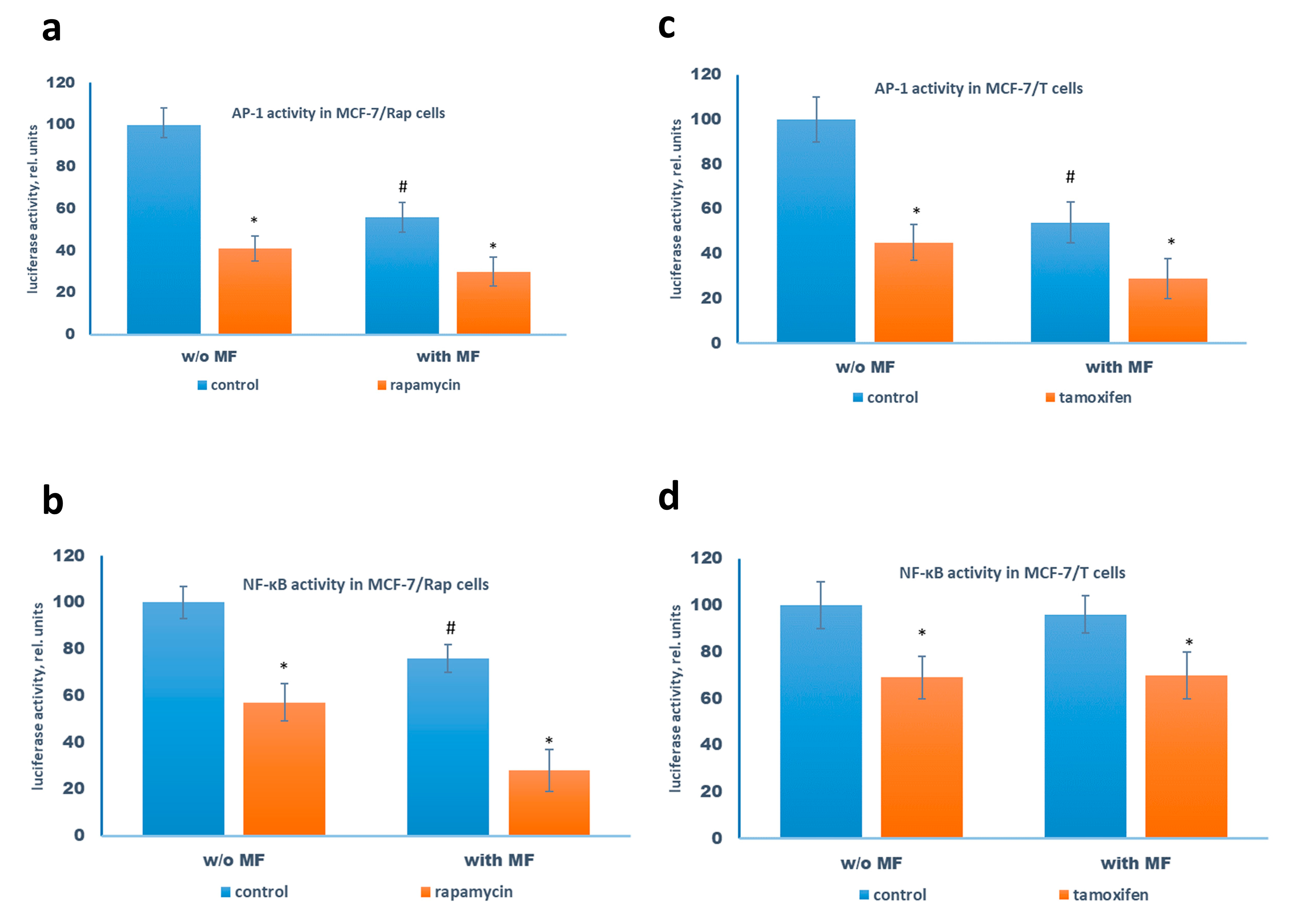
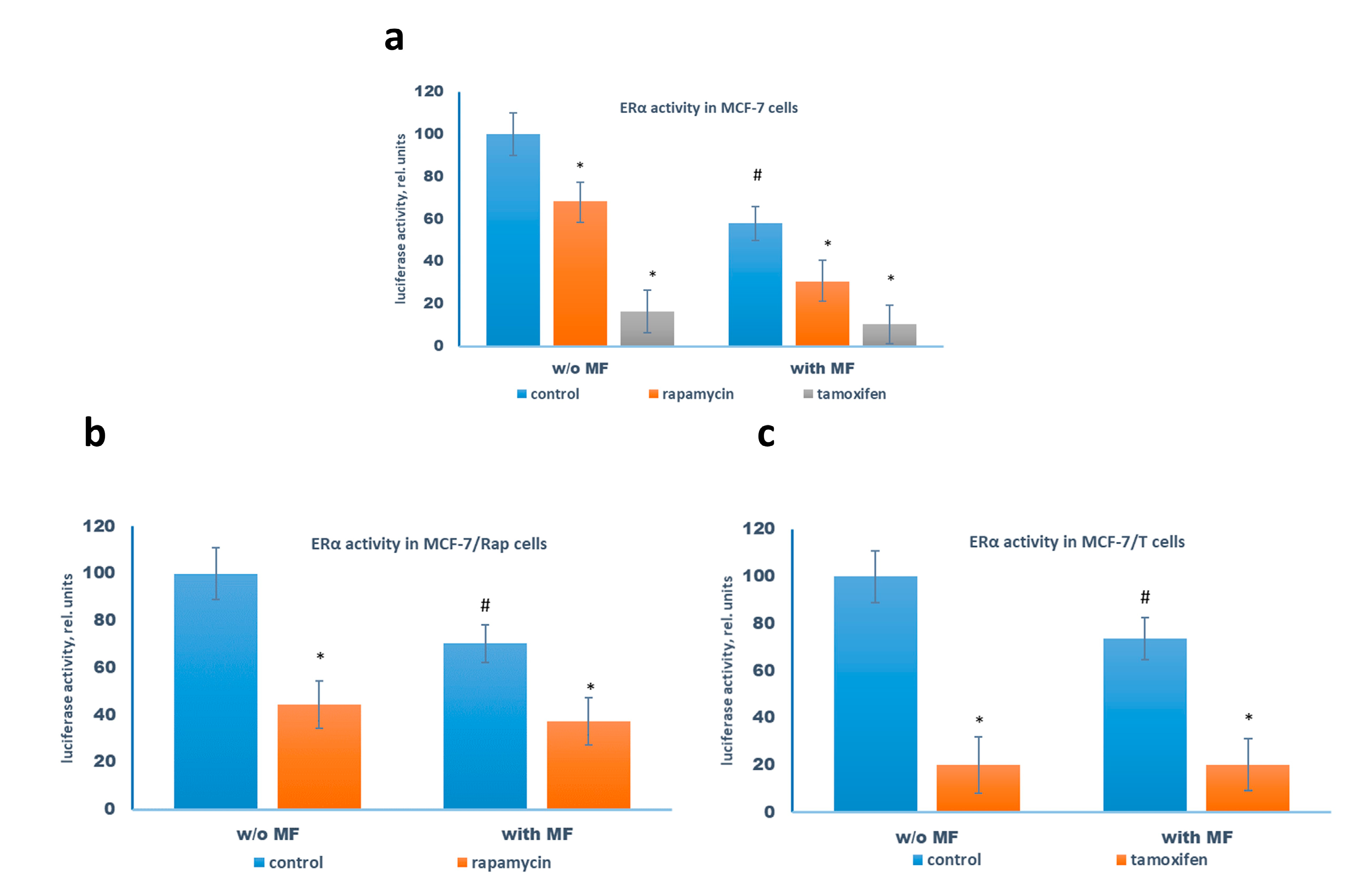
2.3. Metformin and Estrogen Signaling
2.4. Metformin Potentiates the ERα-Independent Tamoxifen Action
3. Discussion
4. Materials and Methods
4.1. Cell Cultures and the Development of Drug-Resistant Sublines
4.2. Transient Transfection and Measurement of Reporter Gene Activity
4.3. Western Blot Analysis of Cell Lysates
4.4. Apoptosis Measurement
4.5. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

References
- Clarke, R.; Liu, M.C.; Bouker, K.B.; Gu, Z.; Lee, R.Y.; Zhu, Y.; Skaar, T.C.; Gomez, B.; O’Brien, K.; Wang, Y.; et al. Antiestrogen resistance in breast cancer and the role of estrogen receptor signaling. Oncogene 2003, 22, 7316–7339. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Lee, C.S.; Tan, P.H. Hormone receptor expression in breast cancer: Postanalytical issues. J. Clin. Pathol. 2013, 66, 478–484. [Google Scholar] [CrossRef]
- Cardoso, F.; Senkus, E.; Costa, A.; Papadopoulos, E.; Aapro, M.; André, F.; Harbeck, N.; Lopez, B.A.; Barrios, C.; Bergh, J.; et al. 4th ESO-ESMO International Consensus Guidelines for Advanced Breast Cancer (ABC 4). Ann. Oncol. 2018, 29, 1634–1657. [Google Scholar] [CrossRef]
- Clarke, R.; Tyson, J.J.; Dixon, J.M. Endocrine resistance in breast cancer—An overview and update. Mol. Cell. Endocrinol. 2015, 418, 220–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, K.; Miyoshi, Y. Mechanism of resistance to endocrine therapy in breast cancer: The important role of PI3K/Akt/mTOR in estrogen receptor-positive, HER2-negative breast cancer. Breast Cancer 2017, 25, 392–401. [Google Scholar] [CrossRef]
- Szostakowska-Rodzos, M.; Trebinska, A.; Grzybowska, E.A.; Fabisiewicz, A. Resistance to endocrine therapy in breast cancer: Molecular mechanisms and future goals. Breast Cancer Res. Treat. 2018, 173, 489–497. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular targeted therapy: Treating cancer with specificity. Eur. J. Pharmacol. 2018, 834, 188–196. [Google Scholar] [CrossRef]
- Konieczkowski, D.J.; Johannessen, C.M.; Garraway, L.A. A Convergence-Based Framework for Cancer Drug Resistance. Cancer Cell 2018, 33, 801–815. [Google Scholar] [CrossRef] [Green Version]
- Bardelli, A.; Corso, S.; Bertotti, A.; Hobor, S.; Valtorta, E.; Siravegna, G.; Sartore-Bianchi, A.; Scala, E.; Cassingena, A.; Zecchin, D.; et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013, 3, 658–673. [Google Scholar] [CrossRef] [Green Version]
- Camidge, D.R.; Pao, W.; Sequist, L.V. Acquired resistance to TKIs in solid tumours: Learning from lung cancer. Nat. Rev. Clin. Oncol. 2014, 11, 473–481. [Google Scholar] [CrossRef]
- Di Nicolantonio, F.; Arena, S.; Tabernero, J.; Grosso, S.; Molinari, F.; Macarulla, T.; Russo, M.; Cancelliere, C.; Zecchin, D.; Mazzucchelli, L.; et al. Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus. J. Clin. Investig. 2010, 120, 2858–2866. [Google Scholar] [CrossRef] [Green Version]
- Citi, V.; Del Re, M.; Martelli, A.; Calderone, V.; Breschi, M.C.; Danesi, R. Phosphorylation of AKT and ERK1/2 and mutations of PIK3CA and PTEN are predictive of breast cancer cell sensitivity to everolimus in vitro. Cancer Chemother. Pharmacol. 2018, 81, 745–754. [Google Scholar] [CrossRef]
- Alimova, I.N.; Liu, B.; Fan, Z.; Edgerton, S.M.; Dillon, T.; Lind, S.E.; Thor, A.D. Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle 2009, 8, 909–915. [Google Scholar] [CrossRef] [Green Version]
- Decensi, A.; Puntoni, M.; Goodwin, P.J.; Cazzaniga, M.; Gennari, A.; Bonanni, B.; Gandini, S. Metformin and cancer risk in diabetic patients: A systematic review and meta-analysis. Cancer Prev. Res. 2010, 3, 1451–1461. [Google Scholar] [CrossRef] [Green Version]
- Schulten, H.J. Pleiotropic Effects of Metformin on Cancer. Int. J. Mol. Sci. 2018, 19, 2850. [Google Scholar] [CrossRef] [Green Version]
- Berstein, L.M. Clinical usage of hypolipidemic and antidiabetic drugs in the prevention and treatment of cancer. Cancer Lett. 2005, 224, 203–212. [Google Scholar] [CrossRef]
- Zakikhani, M.; Dowling, R.J.; Fantus, I.G.; Sonenberg, N.; Pollak, M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006, 66, 10269–10273. [Google Scholar] [CrossRef] [Green Version]
- Scherbakov, A.M.; Sorokin, D.V.; Tatarskiy, V.V.; Prokhorov, N.S.; Semina, S.E.; Berstein, L.M.; Krasil’Nikov, M. The phenomenon of acquired resistance to metformin in breast cancer cells: The interaction of growth pathways and estrogen receptor signaling. IUBMB Life 2016, 68, 281–292. [Google Scholar] [CrossRef]
- Queiroz, E.A.I.F.; Puukila, S.; Eichler, R.; Sampaio, S.C.; Forsyth, H.L.; Lees, S.J.; Barbosa, A.M.; Dekker, R.F.H.; Fortes, Z.B.; Khaper, N. Metformin induces apoptosis and cell cycle arrest mediated by oxidative stress, AMPK and FOXO3a in MCF-7 breast cancer cells. PLoS ONE 2014, 9, e98207. [Google Scholar] [CrossRef]
- Xue, C.; Wang, C.; Liu, Q.; Meng, Q.; Sun, H.; Huo, X.; Ma, X.; Liu, Z.; Ma, X.; Peng, J.; et al. Targeting P-glycoprotein expression and cancer cell energy metabolism: Combination of metformin and 2-deoxyglucose reverses the multidrug resistance of K562/Dox cells to doxorubicin. Tumor Biol. 2016, 37, 8587–8597. [Google Scholar] [CrossRef]
- Ko, G.; Kim, T.; Ko, E.; Park, D.; Lee, Y. Synergistic Enhancement of Paclitaxel-induced Inhibition of Cell Growth by Metformin in Melanoma Cells. Dev. Reprod. 2019, 23, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Ariaans, G.; Jalving, M.; De Vries, E.G.; De Jong, S. Anti-tumor effects of everolimus and metformin are complementary and glucose-dependent in breast cancer cells. BMC Cancer 2017, 17, 232. [Google Scholar] [CrossRef] [Green Version]
- Candido, S.; Abrams, S.L.; Steelman, L.; Lertpiriyapong, K.; Martelli, A.M.; Cocco, L.; Ratti, S.; Follo, M.; Murata, R.M.; Rosalen, P.L.; et al. Metformin influences drug sensitivity in pancreatic cancer cells. Adv. Biol. Regul. 2018, 68, 13–30. [Google Scholar] [CrossRef]
- Uziel, O.; Cohen, O.; Beery, E.; Nordenberg, J.; Lahav, M. The effect of Bortezomib and Rapamycin on Telomerase Activity in Mantle Cell Lymphoma. Transl. Oncol. 2014, 7, 741–751. [Google Scholar] [CrossRef] [Green Version]
- Fraser, C.; Carragher, N.O.; Unciti-Broceta, A. eCF309: A potent, selective and cell-permeable mTOR inhibitor. MedChemComm 2016, 7, 471–477. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, K.E.; Rojo, F.; She, Q.-B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef] [Green Version]
- Berstein, L.M.; Yue, W.; Wang, J.-P.; Santen, R.J. Isolated and combined action of tamoxifen and metformin in wild-type, tamoxifen-resistant, and estrogen-deprived MCF-7 cells. Breast Cancer Res. Treat. 2010, 128, 109–117. [Google Scholar] [CrossRef]
- Kim, J.; Lee, J.; Jang, S.Y.; Kim, C.; Choi, Y.; Kim, A. Anticancer effect of metformin on estrogen receptor-positive and tamoxifen-resistant breast cancer cell lines. Oncol. Rep. 2016, 35, 2553–2560. [Google Scholar] [CrossRef] [Green Version]
- Ramos, P.; Bentires-Alj, M. Mechanism-based cancer therapy: Resistance to therapy, therapy for resistance. Oncogene 2014, 34, 3617–3626. [Google Scholar] [CrossRef]
- Dwarakanath, B.; Jain, V. Targeting glucose metabolism with 2-deoxy-d-glucose for improving cancer therapy. Future Oncol. 2009, 5, 581–585. [Google Scholar] [CrossRef] [Green Version]
- Maschek, G.; Savaraj, N.; Priebe, W.; Braunschweiger, P.; Hamilton, K.; Tidmarsh, G.F.; De Young, L.R.; Lampidis, T.J. 2-deoxy-d-glucose increases the efficacy of adriamycin and paclitaxel in human osteosarcoma and non-small cell lung cancers in vivo. Cancer Res. 2004, 64, 31–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Zhang, J.; Zhou, Z.; Liao, M.-L.; He, W.-Z.; Zhou, X.-Y.; Li, Z.-M.; Xiang, J.-G.; Wang, J.-J.; Chen, H. Synergistic inhibitory activity of zoledronate and paclitaxel on bone metastasis in nude mice. Oncol. Rep. 2008, 20, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Cervantes-Madrid, D.; Romero, Y.; Dueñas-Gonzalez, A. Reviving Lonidamine and 6-Diazo-5-oxo-L-norleucine to Be Used in Combination for Metabolic Cancer Therapy. BioMed Res. Int. 2015, 2015, 690492. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, Y.; Chan, D.K.; Haugrud, A.B.; Miskimins, W.K. Mechanisms by which low glucose enhances the cytotoxicity of metformin to cancer cells both in vitro and in vivo. PLoS ONE 2014, 9, e108444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Zhao, Y.; Ding, Y.; Liu, H.; Liu, Z.; Fodstad, O.; Riker, A.I.; Kamarajugadda, S.; Lu, J.; Owen, L.B.; et al. Warburg effect in chemosensitivity: Targeting lactate dehydrogenase-A re-sensitizes taxol-resistant cancer cells to taxol. Mol. Cancer 2010, 9, 33. [Google Scholar] [CrossRef] [Green Version]
- Shafa, M.H.; Jalal, R.; Kosari, N.; Rahmani, F. Efficacy of metformin in mediating cellular uptake and inducing apoptosis activity of doxorubicin. Regul. Toxicol. Pharmacol. 2018, 99, 200–212. [Google Scholar] [CrossRef]
- Vancura, A.; Bu, P.; Bhagwat, M.; Zeng, J.; Vancurova, I. Metformin as an Anticancer Agent. Trends Pharmacol. Sci. 2018, 39, 867–878. [Google Scholar] [CrossRef]
- Moro, M.; Caiola, E.; Ganzinelli, M.; Zulato, E.; Rulli, E.; Marabese, M.; Centonze, G.; Busico, A.; Pastorino, U.; De Braud, F.G.; et al. Metformin Enhances Cisplatin-Induced Apoptosis and Prevents Resistance to Cisplatin in Co-mutated KRAS/LKB1 NSCLC. J. Thorac. Oncol. 2018, 13, 1692–1704. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Kumar, S. Metformin inhibits human breast cancer cell growth by promoting apoptosis via a ROS-independent pathway involving mitochondrial dysfunction: Pivotal role of superoxide dismutase (SOD). Cell. Oncol. 2018, 41, 637–650. [Google Scholar] [CrossRef]
- Ma, J.; Guo, Y.; Chen, S.; Zhong, C.; Xue, Y.; Zhang, Y.; Lai, X.; Wei, Y.; Yu, S.; Zhang, J.; et al. Metformin enhances tamoxifen-mediated tumor growth inhibition in ER-positive breast carcinoma. BMC Cancer 2014, 14, 172. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Fang, W.; Xia, T.; Chen, Y.; Gao, Y.; Jiao, X.; Huang, S.; Wang, J.; Li, Z.; Xie, K. Metformin potentiates rapamycin and cisplatin in gastric cancer in mice. Oncotarget 2015, 6, 12748–12762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bach, D.-H.; Hong, J.-Y.; Park, H.J.; Lee, S.K. The role of exosomes and miRNAs in drug-resistance of cancer cells. Int. J. Cancer 2017, 141, 220–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assaraf, Y.G.; Brozovic, A.; Gonçalves, A.C.; Jurkovicova, D.; Linē, A.; Machuqueiro, M.; Saponara, S.; Sarmento-Ribeiro, A.B.; Xavier, C.P.R.; Vasconcelos, M.H. The multi-factorial nature of clinical multidrug resistance in cancer. Drug Resist. Updates 2019, 46, 100645. [Google Scholar] [CrossRef] [PubMed]
- Theodossiou, T.A.; Ali, M.; Grigalavicius, M.; Grallert, B.; Dillard, P.; Schink, K.O.; Olsen, C.E.; Wälchli, S.; Inderberg, E.M.; Kubin, A.; et al. Simultaneous defeat of MCF7 and MDA-MB-231 resistances by a hypericin PDT-tamoxifen hybrid therapy. NPJ Breast Cancer 2019, 5, 13. [Google Scholar] [CrossRef] [Green Version]
- Stuart, E.C.; Larsen, L.; Rosengren, R.J. Potential mechanisms for the synergistic cytotoxicity elicited by 4-hydroxytamoxifen and epigallocatechin gallate in MDA-MB-231 cells. Int. J. Oncol. 2007, 30, 1407–1412. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.-Y.; Hung, M.-H.; Wang, D.-S.; Chu, P.-Y.; Su, J.-C.; Teng, T.-H.; Huang, C.-T.; Chao, T.-T.; Wang, C.-Y.; Shiau, C.-W.; et al. Tamoxifen induces apoptosis through cancerous inhibitor of protein phosphatase 2A-dependent phospho-Akt inactivation in estrogen receptor-negative human breast cancer cells. Breast Cancer Res. 2014, 16, 431. [Google Scholar] [CrossRef] [Green Version]
- Velmurugan, B.K.; Wang, P.-C.; Weng, C.-F. 16-Hydroxycleroda-3,13-dien-15,16-olide and N-Methyl-Actinodaphine Potentiate Tamoxifen-Induced Cell Death in Breast Cancer. Molecules 2018, 23, 1966. [Google Scholar] [CrossRef] [Green Version]
- Ma, G.; He, J.; Yü, Y.; Xu, Y.; Yu, X.; Martinez, J.; Lonard, D.M.; Xu, J. Tamoxifen inhibits ER-negative breast cancer cell invasion and metastasis by accelerating Twist1 degradation. Int. J. Biol. Sci. 2015, 11, 618–628. [Google Scholar] [CrossRef]
- Iselt, M.; Holtei, W.; Hilgard, P. The tetrazolium dye assay for rapid in vitro assessment of cytotoxicity. Arzneimittelforschung 1989, 39, 747–749. [Google Scholar]
- Volkova, Y.A.; Antonov, Y.S.; Komkov, A.V.; Scherbakov, A.M.; Shashkov, A.S.; Menchikov, L.G.; Chernoburova, E.I.; Zavarzin, I.V. Access to steroidal pyridazines via modified thiohydrazides. RSC Adv. 2016, 6, 42863–42868. [Google Scholar] [CrossRef]
- Gasparian, A.V.; Yao, Y.J.; Kowalczyk, D.; A Lyakh, L.; Karseladze, A.; Slaga, T.J.; Budunova, I. The role of IKK in constitutive activation of NF-kappaB transcription factor in prostate carcinoma cells. J. Cell Sci. 2002, 115, 141–151. [Google Scholar] [PubMed]
- Reid, G.; Hübner, M.R.; Métivier, R.; Brand, H.; Denger, S.; Manu, D.; Beaudouin, J.; Ellenberg, J.; Gannon, F. Cyclic, proteasome-mediated turnover of unliganded and liganded ERalpha on responsive promoters is an integral feature of estrogen signaling. Mol. Cell 2003, 11, 695–707. [Google Scholar] [CrossRef]
- Scherbakov, A.M.; Komkov, A.V.; Komendantova, A.S.; Yastrebova, M.; Andreeva, O.E.; Shirinian, V.Z.; Hajra, A.; Zavarzin, I.V.; Volkova, Y.A. Steroidal Pyrimidines and Dihydrotriazines as Novel Classes of Anticancer Agents against Hormone-Dependent Breast Cancer Cells. Front. Pharmacol. 2018, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mruk, D.D.; Cheng, C.Y. Enhanced chemiluminescence (ECL) for routine immunoblotting: An inexpensive alternative to commercially available kits. Spermatogenesis 2011, 1, 121–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sorokin, D.; Shchegolev, Y.; Scherbakov, A.; Ryabaya, O.; Gudkova, M.; Berstein, L.; Krasil’nikov, M. Metformin Restores the Drug Sensitivity of MCF-7 Cells Resistant Derivates via the Cooperative Modulation of Growth and Apoptotic-Related Pathways. Pharmaceuticals 2020, 13, 206. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13090206
Sorokin D, Shchegolev Y, Scherbakov A, Ryabaya O, Gudkova M, Berstein L, Krasil’nikov M. Metformin Restores the Drug Sensitivity of MCF-7 Cells Resistant Derivates via the Cooperative Modulation of Growth and Apoptotic-Related Pathways. Pharmaceuticals. 2020; 13(9):206. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13090206
Chicago/Turabian StyleSorokin, Danila, Yuri Shchegolev, Alexander Scherbakov, Oxana Ryabaya, Margarita Gudkova, Lev Berstein, and Mikhail Krasil’nikov. 2020. "Metformin Restores the Drug Sensitivity of MCF-7 Cells Resistant Derivates via the Cooperative Modulation of Growth and Apoptotic-Related Pathways" Pharmaceuticals 13, no. 9: 206. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13090206